
Abstract
Introduction
P
Although several genes have been linked to genetic forms of the disease, transgenic/knockout mouse models have largely failed to reproduce PD pathology or phenotype. Therefore, we have primarily relied on chemical models, such as 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), rotenone, and paraquat, for elucidating mechanism(s) of disease pathogenesis. Importantly, except rotenone where it is seen in some but not all experimental animals, α-syn pathology has been seldom observed in chemical models. The lack of appropriate model systems mimicking key disease pathological events has hampered new drug discovery (3).
Low levels of glutathione (GSH) in SN have been attributed as a cause of selective vulnerability of SNpc DA neurons (44, 51), which is further compounded by the increased presence of iron in melanized neurons (49). Thus, this combination of lowered antioxidant status and increased oxidative load has been suggested to increase thiol oxidation in SNpc DA neurons.
We demonstrate here that perturbation of cellular thiol homeostasis through a single focal delivery of diamide into SN leads to dopaminergic (DA) neurodegeneration, resulting in Parkinsonian phenotype observed as locomotor dysfunction and α-synuclein aggregation in SN neurons. Although oxidative stress has been often implicated in pathogenesis of PD, this is the first demonstration that thiol perturbation alone, including protein thiol oxidation and its sequelae, is sufficient to activate cell death pathway(s), leading to DA neurodegeneration and emergence of PD phenotype. This study provides a paradigm shift in our understanding of pathogenic processes in PD and identifies potential drug target(s) for disease-modifying therapies.
In agreement with this, in the MPTP model, we see a loss of GSH leading to dysfunction of mitochondrial complex I (27). More recently, we have shown that altered redox signaling caused by protein thiol oxidation (PTO) leads to activation of the ASK1 death signaling cascade (46) and attentuation of the Akt-mediated cell survival pathway (16). This cell-specific activation of pathways provide an insight into the loss of selective subset of cells after generalized oxidative stress. If indeed PTO is a leading cause of DA cell death, then exposure to agents that selectively oxidize thiols, such as diamide, should lead to DA loss in SNpc and emergence of PD phenotype.
Diamide, or diazenedicarboxylic acid bis(N,N-dimethylamide), is an azoester compound that was first synthesized by Kosower et al. as a highly cell-permeable agent to oxidize reduced GSH to GSSG without formation of reactive oxygen species (32). It reversibly oxidizes GSH rapidly, within minutes in a stoichiometric manner, utilizing one molar equivalent diamide to form a molar equivalent of thiol disulfide.
Proteins with highly reactive cysteines, such as hemoglobin, thioredoxin (Trx), and tyrosine phosphatase, are also targeted and oxidized by diamide (8, 31, 40). Further, diamide shows specificity, as it reacts very slowly with non-thiol substrates (29). Diamide has been extensively used in cell-free systems, in vitro in cells and in vivo to rapidly oxidize GSH and/or protein thiols (8, 30, 61). Importantly, exposure to diamide in vitro leads to cell death, suggesting that thiol oxidation may have irreversible consequences (8, 36, 45).
In this study, we performed protein thiol homeostasis (PTH) by stereotaxically delivering a single dose of diamide unilaterally, in vivo into SN of mice. We then followed the locomotor behavior of mice and sacrificed them 2 weeks later for histopathological and biochemical assessment. Briefly, we found that this single toxic challenge led to development of a PD phenotype mimicking behavioral, histopathological, and biochemical features, including DA degeneration and α-syn aggregation.
Results
Unilateral diamide injection into mouse SN leads to development of hemiparkinsonism
Motor behavioral assessment was carried out for experimental mice that were divided randomly into two groups, both before and after stereotaxic surgery (Fig. 1A, D and Supplementary Fig. S1C; Supplementary Data are available online at
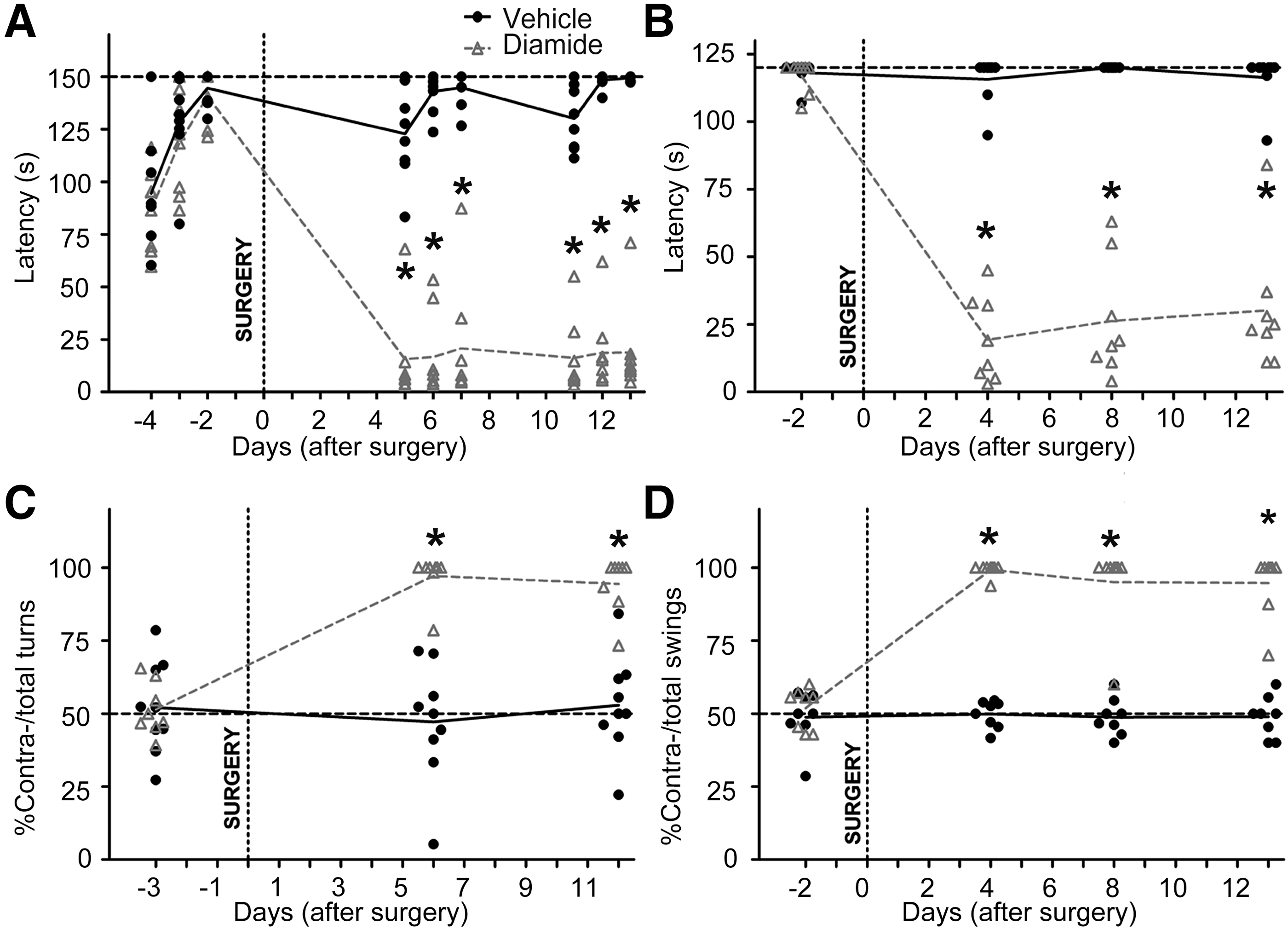
We studied general locomotor deficits in the animals by using the accelerating rotarod (Fig. 1A) and by assessing grip strength (Fig. 1B). The latency to fall in both tests reduced drastically after a diamide injection and was maintained throughout the experimental duration without any improvement with time. Further, the unilateral lesioning paradigm led to asymmetric motor behavior, seen as increased spontaneous contraversive rotations (Fig. 1C). Representative free movement path trajectories are also shown (Supplementary Fig. S1D). This could arise due to the postural change along with robust contralateral hemiparesis in the diamide-injected mice.
Similarly, animals showed a contralateral swing bias (Fig. 1D) in the elevated body swing test (EBST) (6). Interestingly, similar motor deficits were not observed post-surgery when diamide was injected into the motor cortex of mice (Supplementary Fig. S2A, B). Thus, the behavioral battery of tests indicates that a diamide injection into SN leads to development of hemiparkinsonian deficits in mice, which are sustained throughout the experimental period of 2 weeks.
Diamide injection leads to unilateral loss of total GSH in ventral midbrain
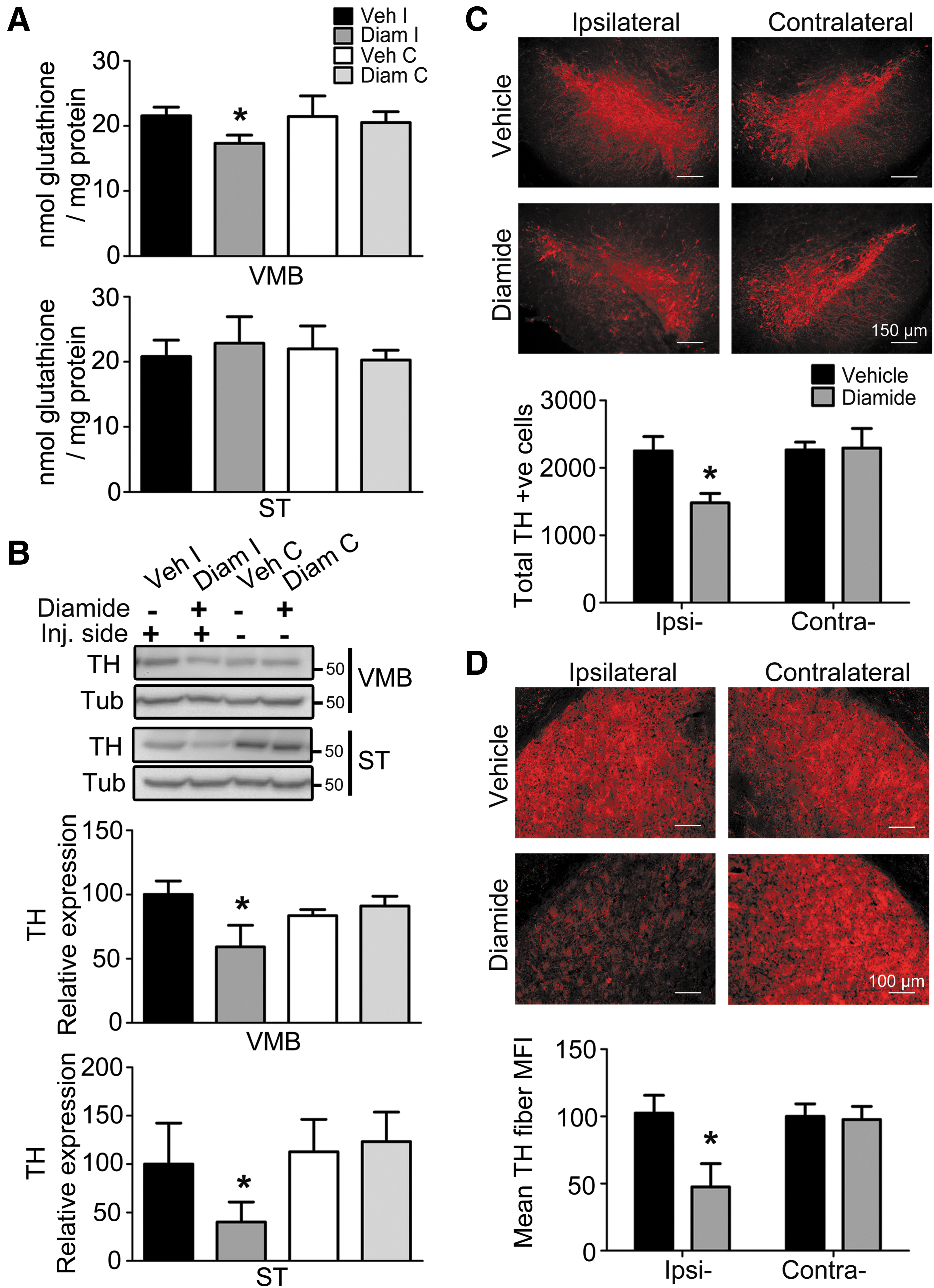
As cellular GSH is one of the major thiol pools that is susceptible to diamide, we examined whether diamide affected total GSH (reduced+oxidized) levels in the cells. We observed a small but significant decrease in total GSH levels, specifically in the diamide-injected ipsilateral ventral midbrain (VMB) but not striatum (ST) (Fig. 2A). Such decreases in GSH levels have been reported in brain tissues from different animal models and PD patients (9, 18, 51, 53). Thus, this model reproduces an important biochemical feature of PD, even though the single dose of diamide was given 2 weeks earlier, indicating its irreversible consequences. Concomitantly, we observed increased lipid peroxidation that was measured as malondialdehyde levels in the diamide-injected VMB but not ST (Supplementary Fig. S3B).
Diamide injection leads to unilateral loss of DA neurons in the SNpc accompanied by striatal fiber loss
To understand whether the hemiparkinsonian behavior arose due to a loss of DA inputs in the basal-ganglia circuitry, we examined the loss of tyrosine hydroxylase (TH; expressed in DA neurons) in SNpc and ST by using immunostaining and protein expression. Stereological immunohistochemical analysis revealed a robust loss of DA neurons only in diamide-injected ipsilateral SNpc (Fig. 2C) along with a loss of TH in striatal fibers (Fig. 2D). The loss of DA neurons was exacerbated in mice that were housed for 4 weeks after the diamide injection (Supplementary Fig. S3A). The immunohistological studies in the 14-day paradigm were further validated biochemically as a loss of TH protein expression in VMB and ST (Fig. 2B).
It is worth mentioning that the extent of depletion of TH protein expression closely matched the loss measured through immunohistochemical analysis. Thus, it appears that behavioral deficits displayed by diamide-injected mice correlate with ipsilateral reduction in DA neurons and their fibers.
Diamide injection leads to neurodegeneration in SNpc
To confirm whether the loss of TH was actually reflected in terms of neuronal death, we performed Fluoro-Jade C (FJC) staining to estimate the extent of degeneration, and we also obtained stereological immunohistochemical data for the pan-neuronal marker, NeuN. Diamide injection clearly increased the number of FJC labeled cells in diamide-injected ipsilateral VMB (Fig. 3A), along with a loss of NeuN-positive cells in SN (Fig. 3B). This was also confirmed biochemically as a loss of Tuj1 expression in VMB (Fig. 3C). Importantly, in control experiments, in which a similar dose of diamide or vehicle was injected into the motor cortex stereotaxically, significant neurodegeneration reflected as a loss of Nissl's staining was not observed beyond the immediate injection tract (Supplementary Fig. S2C).
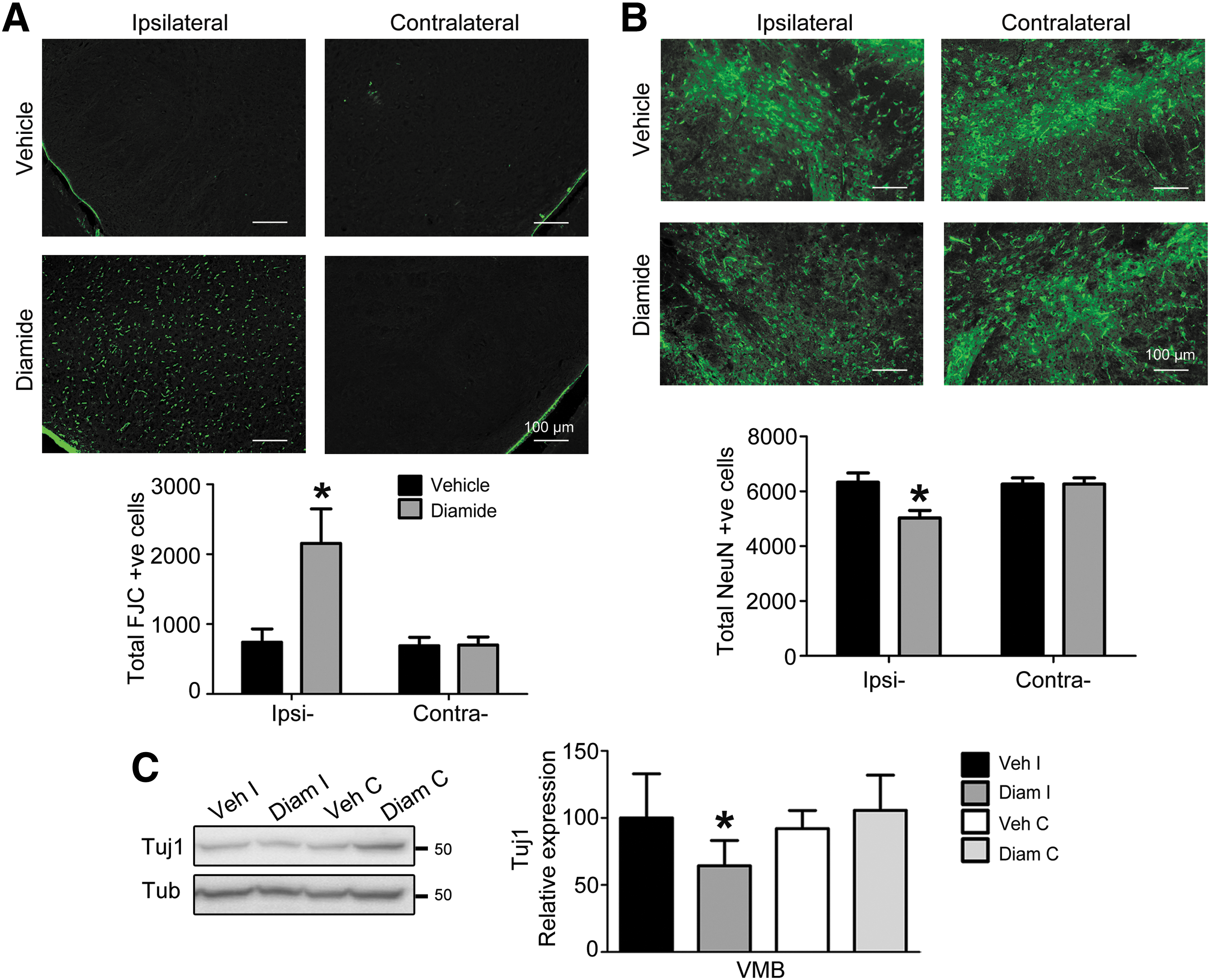
Further, the specificity of degeneration of the neuronal cell type in VMB was investigated biochemically by studying alterations in glutamic acid decarboxylase (GAD) 65/67 levels, a marker for GABAergic (GABA; γ-aminobutyric acid) neurons that are a major neuronal class in SN apart from DA neurons. GAD expression decreased in diamide-injected VMB, but not in ST (Supplementary Fig. S4A, B). Although brain tissues from PD patients and animal models have been primarily assessed for loss of DA neurons, some evidence also exists for limited loss of GABA neurons or expression of GABA markers in SN, especially after exposure to non-DA neurotoxins (10, 25, 33, 38).
Nevertheless, it is important to note that the extent of GAD65/67 loss was lower than that of TH, suggesting enhanced vulnerability of DA neurons over the GABA population to thiol oxidation. Further, neurodegeneration was associated with an extensive increase in glial markers, including astrocytic glial fibrillary acidic protein (GFAP) and microglial ionized calcium-binding adapter molecule (Iba1) in both brain regions (Supplementary Fig. S4C–F), as has been shown earlier in animal models and in PD-affected brain tissue (1, 11, 28, 39, 53).
Diamide injection causes α-syn accumulation in SNpc
Formation of insoluble aggregates in the form of intra-cytoplasmic inclusions (Lewy bodies) is a hallmark of PD. The most commonly used neurotoxin-induced animal models of the disease have not reproduced this key pathological feature. We therefore investigated whether our model replicated this aspect. As seen in Figure 4A, a diamide injection led to selective accumulation of α-syn levels in diamide-challenged ipsilateral SN alone. This accumulation pattern was similar across representative sections taken from five experimental animals, confirming the reproducibility of pathology. On examining the cells at higher magnification, we also observed punctate staining inside the cells, suggesting formation of aggregates (Fig. 4B). This feature was not seen in the motor cortex-injected mice, demonstrating specificity of α-syn aggregation (Supplementary Fig. S2D).

Aggregation of α-syn is accompanied by its increased phosphorylation at the Ser129 residue (52). We also observed increased punctate staining of phospho-α-syn in the diamide-injected SN compared with the non-injected side (Fig. 4C). Further, several diamide-injected SN cells also demonstrated ubiquitin-positive intracellular inclusions compared with the non-injected side (Fig. 4D), suggesting impairment of protein clearance. Thus, a diamide injection into SN reproduces Lewy body-like pathology in the experimental mice, including increased α-syn, phospho-α-syn, and ubiquitin immunoreactivity.
Diamide injection leads to activation of ASK1-p38 MAPK cascade
Activation of the ASK1-p38 mitogen-activated protein kinase (MAPK) cell death signaling cascade is a common feature seen across several PD models. Therefore, we investigated whether diamide activated this pathway. Indeed, ASK1 phosphorylation increased selectively in diamide-injected ipsilateral VMB but not ST (Fig. 5A, B), similar to our earlier observations in the MPTP model (46). This activation of ASK1 was reflected in increased downstream MAPK signaling in the form of p38 MAPK phosphorylation (Fig. 5C, D), but not c-Jun N-terminal kinase (JNK) activation (Fig. 5E, F). This could be due to selective recruitment of p38-mediated stress/cell death pathway in affected neurons, as observed earlier in our laboratory in MPTP-treated mice (26) and in another neurodegenerative disease model elsewhere (22).

Diamide injection activates the ASK1 pathway through a redox-dependent mechanism
ASK1 is activated in a redox-sensitive manner, as its inhibitory association with cytosolic Trx1 is disrupted on Trx1 oxidation (54). It has also been demonstrated that dissociation of Trx1 leads to oxidation of ASK1, which is necessary for its downstream signaling (41, 46). To assess whether these biochemical changes are involved in activation and downstream signaling of the ASK1 pathway, we bilaterally injected mice with diamide or saline on either side, respectively, and harvested brain tissue 14 days post-surgery. As shown in Figure 5A, ASK1 activation was selectively observed in diamide-injected VMB compared with vehicle-injected VMB (Fig. 6A). Such changes were not seen in the striatal groups (ST; Fig. 5B and 6B). Consistently, co-immunoprecipitation studies revealed that Trx1 selectively dissociates from ASK1 in diamide-injected VMB but not ST (Fig. 6C), even though the ratio of total cellular levels of Trx1 to total ASK1 is not altered (Fig. 6D).
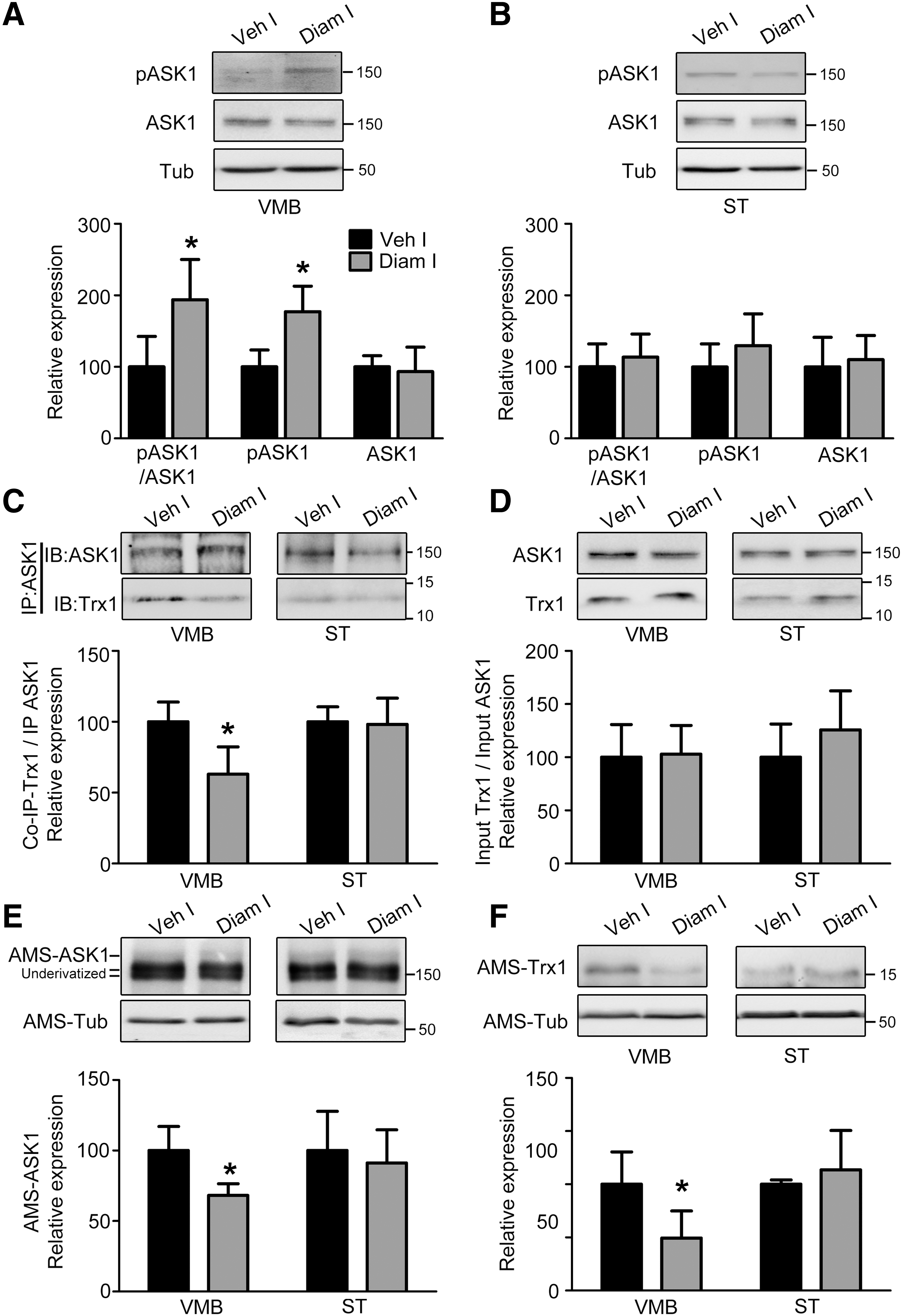
Finally, through redox gel analysis using the free thiol alkylating agent, 4-acetamido-4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid disodium salt (AMS), we examined the redox status of ASK1 and Trx1 in these samples. ASK1 and Trx1 were selectively oxidized in the diamide-injected VMB (Fig. 6E, F), measured as a loss of AMS-ASK1 or AMS-Trx1 (AMS-protein representing reduced protein). These results further corroborated the association study and ASK1 activation profile. Thus, among several potential PTO events induced by diamide, Trx1 and ASK1 oxidation are critical in activating the ASK1-p38 MAPK cascade.
Thus, the in vivo results described demonstrate that a stereotaxic injection of diamide into SN leads to robust and reproducible DA neurodegeneration, α-syn pathology, and locomotor deficits. Further, we also found evidence of increased cell death signaling in this model.
Diamide exposure to Neuro-2a cells induces cell death and robust activation of the ASK1 pathway through a Trx1-dependent mechanism
To examine whether a causal association exists between activation of ASK1-pathway and cell death, we turned to an in vitro model of diamide-induced toxicity in Neuro-2a cells. Diamide at a dose of 300 μM showed robust cell death after a 6 h exposure, which was measured through MTT assay (Supplementary Fig. S5A) and TUNEL assay (Supplementary Fig. S5B). Further, it also led to a loss of cellular GSH levels within 5 min of exposure, which was not sustained till 6 h (Supplementary Fig. S6). Diamide led to rapid ASK1 phosphorylation within 5 min of exposure but returned to baseline after 6 h (Fig. 7A). Although JNK phosphorylation followed a pattern similar to ASK1 activation (Supplementary Fig. S7A), p38 activation was sustained throughout (Fig. 7B), as also confirmed through immunostaining (Supplementary Fig. S5C–H).
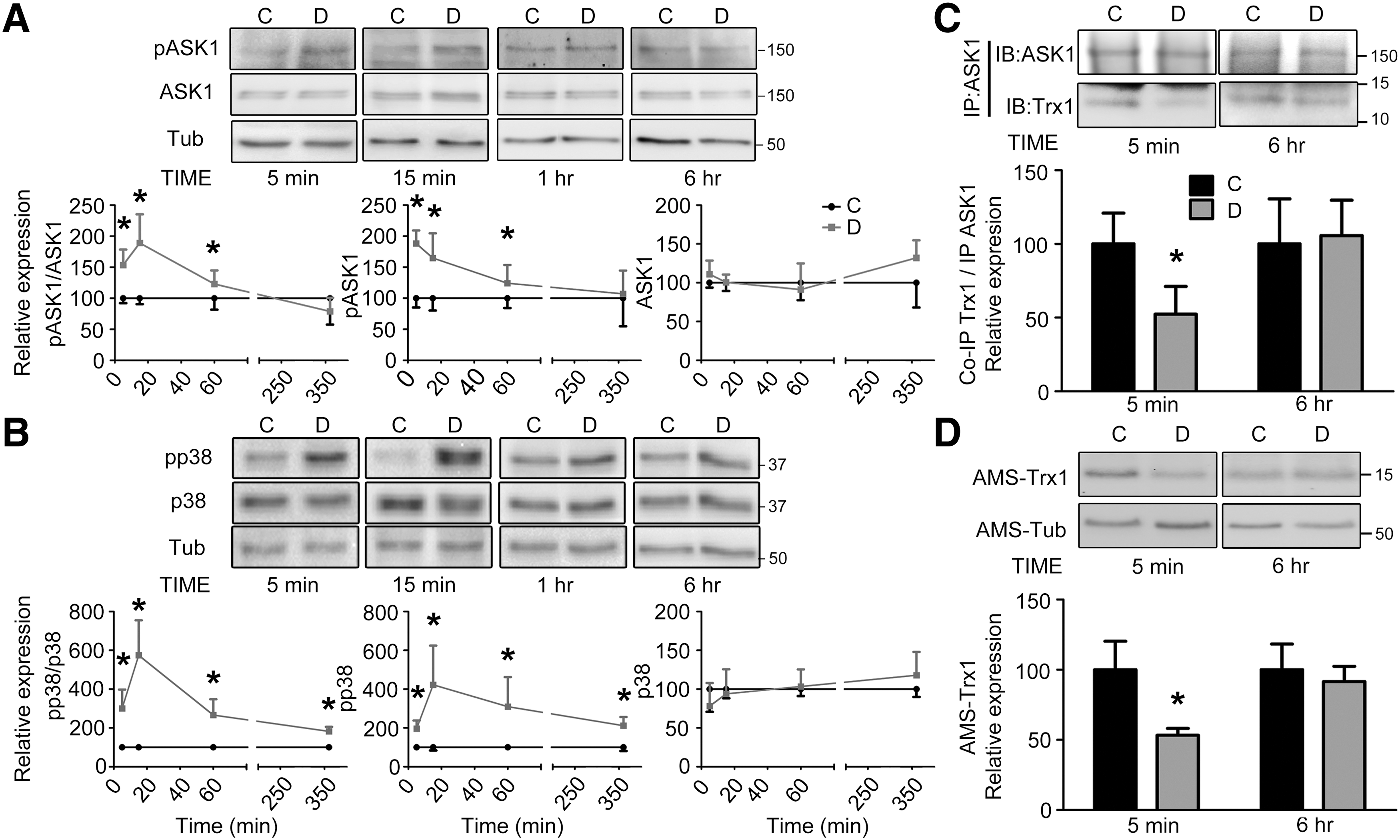
This differential response corroborated with the in vivo MAPK activation profile (Fig. 4). Such differences in temporal profiles of MAPK activation have been reported earlier (17), and they could have arisen due to increased nuclear localization of phospho-p38 compared with phospho-JNK (Supplementary Fig. S5F, H) or through persistent maintenance of intermediary MAP2K phosphorylation (Supplementary Fig. S8).
Further, as observed in vivo, diamide treatment led to decreased Trx1-ASK1 association after 5 min but not 6 h (Fig. 7C). This was despite ratios of total cellular Trx1 levels relative to ASK1 remaining unaltered (Supplementary Fig. S7B). This potentially occurred through increased Trx1 oxidation selectively at the early time-point, similar to our in vivo results (Fig. 7D). We were unable to assess the redox status of ASK1 in vitro, as our method lacked the sensitivity to reliably detect AMS-ASK1 in this system. Thus, diamide triggers the ASK1-p38 MAPK cascade in vitro through mechanisms that are similar to those observed in vivo.
ASK1 is necessary for diamide-induced p38 activation and cell death
To establish whether p38 phosphorylation and cell death are indeed downstream of ASK1 activation, we downregulated ASK1 levels using shRNA (Fig. 8A and Supplementary Fig. S7C). ASK1 downregulation reduced basal as well as diamide-induced p38 phosphorylation after 5 min of diamide exposure (Fig. 8B). Interestingly, there was a small but significant increase in p38 phosphorylation in the ASK1-knocked down cells after diamide exposure for 1 h compared with the respective vehicle-treated group. Nevertheless, the phosphorylation levels were not significantly different from the scrambled vehicle group (Supplementary Fig. S9A, B). This could potentially arise due to residual ASK1 after knockdown getting phosphorylated after diamide exposure over a longer period.

Conversely, overexpression of human wild-type ASK1 (ASK-WT) increased p38 phosphorylation, whereas dominant negative kinase dead mutant ASK1 (ASK-KM; carrying an active site mutation of K709R) suppressed p38 activation (Supplementary Fig. S9C). Further, ASK1 knockdown offered protection from diamide-induced cell death after 6 h of diamide exposure (Fig. 8C). Thus, we conclude that ASK1 is, indeed, an upstream MAP3K that is responsible for p38 phosphorylation and cell death in cells exposed to diamide.
Discussion
The role of PTO in selective death of DA neurons remains unclear in commonly used neurotoxin-induced models of PD. We now demonstrate that a unilateral stereotaxic injection of diamide into SN leads to DA neurodegeneration, α-syn aggregation, and locomotor deficits, indicating that Parkinsonism phenotype can be triggered by a chemical insult that specifically targets protein thiol modification (Figs. 1 –4; summarized in Supplementary Fig. S10). We thereby demonstrate that disruption of PTH triggers the neurodegenerative cascade in SNpc DA neurons. Further, we have demonstrated that activation of the death signaling ASK1-p38 MAPK pathway is a primary event in diamide-induced cell death (Figs. 5 –8).
The single dose of diamide used (250 μg; 1.45 μmol) can potentially oxidize up to ∼75% of adult mouse brain thiols. Adult mouse brain contains ∼40 mg protein with ∼100 nmol thiol equivalents/mg protein (43, 48). Although the initial insult will be local in the SN, diamide is highly lipophilic and could potentially diffuse away from the injected site, creating a scenario akin to “umbra-penumbra” damage in ischemia. Importantly, other experiments in our laboratory with a 10-fold lower dose did not induce motor deficits or changes in marker expression.
Diamide-injected mice showed profound locomotor deficits across different behavioral paradigms examined. Poor performance in rotarod and grip strength tests suggest development of general locomotor problems, whereas the asymmetric motor behavior in open field and EBST indicate hemiparkinsonian features that are consistent with the unilateral injection paradigm. Further, the increased contralateral swings in EBST are in agreement with the 6-OHDA rat model (6).
On the other hand, our mouse model displays spontaneous contralateral rotations unlike the 6-OHDA rodent model (55, 57). Other studies have suggested that this bias is dependent on experimental factors, including choice of neurotoxin, mode and site of lesion, the innate dominant side bias of animal, exposure time, etc. (15, 24, 50, 59), and this may explain the discrepancy observed. These behavioral deficits were associated with substantial DA neuronal loss in ipsilateral SNpc and its fiber innervations in ST (Figs. 1, 2 and Supplementary Fig. S3A).
A single unilateral injection of diamide resulted in a loss of DA neurons (measured as TH-positive neurons) in the ipsilateral SNpc. Death of DA neurons was further corroborated by a loss of NeuN-positive neurons and increased FJC staining (Figs. 2 and 3). These results indicated that the TH loss was due to degeneration of DA neurons and not simply due to altered TH expression. However, it is, indeed, possible that other cells, including GABAergic neurons in the tract, may also be affected, since we observed a loss of GAD65/67 expression, albeit to a lesser extent than TH expression. The loss of DA neurons in SNpc was accompanied by a substantial loss of TH fiber density in the ST. This is in agreement with earlier observations of increased susceptibility of terminals to stress when compared with cell bodies (12).
Several studies from different groups have demonstrated that SNpc DA neurons are particularly vulnerable to oxidative stress that is generated through cell-intrinsic and -extrinsic mechanisms (4, 12, 21). Therefore, SNpc DA neurons appear to be exquisitely vulnerable to redox perturbations. Further, an injection of the same dose of diamide into the motor cortex led to modest degeneration only at or very close to the injection tract, and no behavioral deficits were observed (Supplementary Fig. S2). This indicates that SNpc neurons are more vulnerable to diamide toxicity.
Interestingly, the diamide model showed clear evidence of α-syn aggregation and presence of phosphorylated α-syn within SNpc neurons (Fig. 4), unlike other common neurotoxin-induced PD models using MPTP and 6-OHDA (3). However, such aggregation has been reported in a fraction of the experimental groups after chronic systemic administration of rotenone (2) or paraquat (37). Refinement of the rotenone model through chronic intraperitoneal rotenone administration has shown more consistent α-syn pathology in rats (7). Nevertheless, α-syn aggregation remains a rare observation in toxin-induced models and diamide reproducibly triggers this process. Importantly, increased ubiquitin staining in the diamide-injected SNpc neurons (Fig. 4D) also suggests impairment of the proteasomal degradation machinery and/or formation of insoluble protein aggregates.
Oligomerization and aggregation of α-syn have been shown to be sensitive to oxidative stress, acting through mechanisms including tyrosine nitration, 4-hydroxynonenal adduct formation, and methionine sulfoxidation (19, 20, 58). It remains to be seen whether diamide induces such changes or other mechanisms contribute to its aggregation.
The decrease in total GSH levels (∼20%) seen even 14 days after a single dose of diamide in the ipsilateral VMB indicates that thiol homeostasis remains perturbed for a substantially long period after the initial challenge. This is indeed surprising, since an initial GSH loss (which would occur soon after exposure to diamide) is typically followed by rebound of GSH levels through increased synthesis or uptake (51). Further, astrocytes have higher GSH content compared with neurons (14), and are in a position to synthesize more GSH, thus restoring thiol homeostasis, as seen after transient ischemia (48) and MPTP exposure (51). Indeed, increases in GFAP levels seen after diamide in ipsilateral VMB and ST suggest gliosis (Supplementary Fig. S4C, D), which could also contribute to total GSH content.
Despite existence of such mechanisms, GSH levels continuing to remain significantly lower in VMB indicate that diamide causes irreversible inhibition of the GSH synthesis pathway(s) in neurons and glia. One consequence of this loss of GSH, apart from cell loss, could be its impact on TH expression and activity, both of which are known to directly and indirectly be redox-regulated (13). Indeed, in vitro experiments have demonstrated that TH has redox-sensitive cysteine residues, which on oxidation affect activity (5).
A decrease in GSH levels was also accompanied by increased lipid peroxidation and loss of reduced forms of ASK1 and Trx1 in VMB but not ST. Loss of GSH often leads to formation of protein–protein and protein–GSH mixed disulfides (48). From our data, it appears that there is a coordinated decrease of total GSH coupled with an increase in other oxidative markers, including oxidation of lipids and proteins.
The activation of the death signaling ASK1-p38 MAPK pathway has been seen in both neurotoxin-induced and transgenic PD models (34, 46). Indeed, we saw an increase in phospho-ASK1 levels selectively in VMB but not ST, which was accompanied by an increase in p38 MAPK but not JNK phosphorylation (Fig. 5). Recent work has demonstrated that removal of ASK1 attenuates the Parkinsonism phenotype in different mouse models of PD (23, 34, 35), indicating that this cascade may be a common cell death signaling pathway that is responsible for DA degeneration. The in vitro cell culture experiments confirmed the importance of the ASK1-p38 MAPK death signaling pathway in cell death observed after diamide exposure.
The temporal sustenance of increased p38 phosphorylation beyond that of the ASK1 activation profile and the small increase in p38 phosphorylation even after ASK1 knockdown suggest that a single initial oxidative perturbation can trigger pathogenic pathways if the insult is able to overwhelm the cellular defense system. Although we have demonstrated PTO in the form of Trx1 and ASK1 oxidation to be important for initiating the ASK1 cascade, the role of other mechanisms, such as inflammatory signaling and differential binding of ASK1 regulators, in sustenance of ASK1 activation cannot be ruled out (54). Indeed, an increase in gliosis markers in vivo (Supplementary Fig. S4E, F) indicate ongoing inflammation that may also contribute to cell death.
The well-characterized rodent models of PD include MPTP, 6-OHDA, and paraquat, among others. Although these models replicate the DA cell loss in SNpc, no single model mimics the behavioral (locomotor dysfunction) and pathological hallmarks (α-syn aggregation) of PD. However, in this study, we demonstrate that a single exposure to diamide leads to behavioral dysfunction and α-syn aggregation, in addition to the cardinal feature of DA cell loss in SNpc.
In conclusion, we have developed a novel animal model of PD using a single exposure to diamide leading to sustained locomotor deficits, α-syn aggregation, and degeneration of DA neurons. Moreover, single administration of diamide inducing sustained changes at the biochemical, histological, and behavioral levels emphasizes the importance of maintaining PTH for DA neuronal survival. Finally, due to the relatively simpler molecular mechanism(s) underlying diamide action, we can now attempt to understand why DA neurons are susceptible to thiol oxidation, and also assess the contribution of PTH perturbation and oxidation of thiols of proteins such as thiol disulfide oxidoreductase (TDORs), in regulating pathogenic molecular pathways involved in PD. Further examination of this cross-talk may reveal new targets for designing better disease-modifying strategies.
Materials and Methods
Materials
All phospho-specific antibodies, unless otherwise stated, along with total protein antibodies for JNK, mitogen-activated protein kinase kinase 3 (MKK3), and mitogen-activated protein kinase kinase 4 (MKK4) were procured from Cell Signaling Technology, Inc. α-syn, p38, and ASK1 (for immunoprecipitation) antibodies were purchased from Santa Cruz Biotechnology, Inc. ASK1 antibody for immunoblotting was from Abcam; TH, NeuN, and GAD65/67 antibodies were from Merck Millipore; Tuj1 was from Biolegend; GFAP and ubiquitin were from Dako, Inc.; and Iba1 and phospho-α-syn (Ser129) were from Wako Pure Chemical Industries. Antiserum to Trx1 was a kind gift from Dr. Gary Merrill (Oregon State University, Corvallis, OR).
AMS for redox derivatization, secondary antibodies for fluorescent immunostaining, protein G-coupled magnetic Dynabead suspension, precast 8–16% gradient Tris-glycine gels, and cell culture reagents were purchased from Life Technologies, Inc. Horse-radish peroxidase-labeled secondary antibodies for immunoblotting, mounting medium with DAPI (4′,6-diamidino-2-phenylindole), and reagents for bright field immunostaining were purchased from Vector Laboratories. FJC was procured from Histo-Chem, Inc. Single-stranded cDNA synthesis kit and 2 × SYBR Green master mix were obtained from Applied Biosystems. All other chemicals were procured from Sigma Aldrich or Merck Ltd.
Animal experiments
Animals used for the in vivo experiments were male C57-BL6/J mice (3–4 months age, 25–30 g) obtained from the Central Animal Research Facility of the National Brain Research Centre (NBRC) and the Indian Institute of Science (IISc). All animal experiments were carried out as per the institutional guidelines for the use and care of animals of NBRC and IISc. Animals were randomly divided into either of the two experimental groups. All efforts were taken to minimize animal suffering and number of animals used for experiments. Animals had access to a pellet diet (Lipton India Ltd.) and water ad libitum and were housed in standard housing conditions.
Stereotaxic surgery
Animals were weighed, anesthetized with a ketamine/xylazine mixture, and fixed into the stereotaxic apparatus (Stoelting Co.). Under aseptic conditions, a longitudinal incision was made in the scalp to expose the skull after applying a topical anesthetic. Using the following stereotaxic coordinates: 3.2 mm posterior to bregma; 1.2 mm lateral (either left or right), a hole was drilled into the mouse skull with a 24G needle. With a 30G blunt-tip Hamilton needle and syringe (Hamilton Co.), 250 μg of diamide dissolved in 2 μl of sterile vehicle (0.9% saline) or vehicle alone was injected at a flow rate of 0.2 μl/min at the following coordinates: 3.2 mm posterior to bregma; 1.2 mm lateral to bregma; and 4.5 mm ventral to bregma, based on the mouse brain atlas (for unilateral surgeries targeting SN). The injection site is depicted in a cartoon in Supplementary Figure S1A and B.
The needle was allowed to remain in position for a further 5 min before retraction. The incision was sutured and once the animals recovered from anesthesia, injectable analgesics and topical antiseptic were applied. The animal health was closely monitored over the next 3 days, followed by start of post-surgery behavioral recordings. The animals were euthanized 2 weeks post-surgery for immunohistochemical and biochemical analyses, and 4 weeks post-surgery for experiments described in Supplementary Figure S3A.
For Figure 6 and Supplementary Figure S3B, bilateral injections using the same coordinates were administered, with one side of the SN receiving diamide whereas the other side was administered vehicle. Similarly, for Supplementary Figure S2, bilateral injections of diamide/vehicle were administered to the motor cortices at the following stereotaxic coordinates: 1.7 mm anterior to bregma; 1.5 mm lateral to bregma; and 1.3 mm ventral to bregma.
Behavioral analyses
Behavior was recorded both before and after the surgery (Supplementary Fig. S1C) to examine the development of locomotor deficits after the diamide injection in unilateral SN-injected animals. Grip strength tests and EBSTs were also performed for the bilateral motor cortex-injected group. All behavioral assessment instruments were cleaned with 70% ethanol in between the recording of each animal. The behavioral paradigms are described in detail next: (1) Rotarod analysis: This analysis was carried out to test the ability of the animal to balance itself on a rotating rod at gradually increasing speeds to assess its movement abilities. The recordings were carried out in clusters of three consecutive sessions (days) at a time with each session having three trials, each separated by ∼15 min of rest. Before recording data, the animals were familiarized with the instrument during two sessions having three trials each. During the familiarization, the animals were placed on the static cleaned rod at a height of ∼30 cm of the mildly padded instrument base. The rod started rotating at 5 rpm after placement. The animal was placed back on the rod if it fell off for a total period of 150 s per trial. In the second familiarization session, during the third trial, the rod was rotated at 15 rpm instead to make the animal learn that rotational speed could be altered. For the actual recording sessions, in each trial, the initial rotation started at 5 rpm and increased in steps of 5–25 rpm, finally with each step lasting for 30 s for a total of 150 s. The time to fall off the rod (latency) was recorded for each animal in a given trial. If it did not fall off, the criterion time (150 s) was recorded instead. For representation of the data, the average time from three trials in a session was calculated for every animal. (2) Grip strength test: The animal's gripping ability was measured by assessing the ability of the animal to hold on to an inverted cage lid for a pre-defined period. Each session (day) of recording consisted of three trials of 120 s each with a minimum resting time of 10 min between trials. Briefly, the animal was taken out of its home cage and placed on a cage lid. The lid was briefly shaken to allow the animal to clasp onto the lid bars tightly before inverting the lid and holding it over the animal's cage bedding at a height of ∼30 cm for a criterion time of 120 s. The latency to fall off was recorded in each trial, averaged across three trials for each session, and plotted for analysis. (3) Open-field behavior: This analysis was carried out to observe the natural locomotor behavior of the animal. Individual animals were placed in the center of a 60 × 60 cm open arena situated at a height of ∼50 cm off the ground (a wooden table of indicated dimensions with a white top surface), and their spontaneous movements were recorded with an ANY-maze camera (Stoelting Co.) that was connected to a computer over a period of 15 min for each session on a given day. The video recorded was analyzed with the ANY-maze camera and software (Stoelting Co.) for tracking the path of movement of the mouse. The number of spontaneous contralateral turns (contralateral to the stereotaxically injected side, that is, right turns for an animal injected in the left SN and vice versa) that the animal made in its path divided by the total number of turns (contralateral+ipsilateral) was expressed in percentage to calculate the side preference of turning of the animal. (4) EBST: This analysis was carried out to assess the innate side preference of the animal both before and after the surgery. Each session (day) consisted of one trial lasting for 45 s. Briefly, the animal's movement behavior was recorded while being freely suspended by its tail at a height of ∼30 cm above its home cage against a white background. A blind analysis was carried out to count the number of contralateral or ipsilateral turns (each turn defined as the animal turning its body >15° from the normal on either side after passing the normal). The percentage of contralateral turns to total turns was calculated to identify any preference of the animal.
Sample preparation for biochemical analysis
The animals were killed, brain tissue containing either VMB or ST was dissected out, and the injected (ipsilateral) and non-injected (contralateral) sides of both regions were collected separately. The tissues were homogenized in 0.1 M potassium phosphate buffer (pH 7.4) containing 0.25 M sucrose, 2 mM EDTA, and protease and phosphatase inhibitors. Post-nuclear supernatant (PNS) was prepared by centrifuging the homogenates at 1000 g for 10 min at 4°C. The PNS samples were estimated for protein concentration and used for further experiments as described next: (1) Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting: PNS samples mixed with sample buffer were run on SDS-PAGE under standard conditions. The gels were electrophoretically blotted onto supported nitrocellulose/PVDF membranes. The membranes were blocked with 5% bovine serum albumin in 1 × TBST for 1 h before incubation with primary antibody (prepared in blocking buffer or 1 × TBST) overnight at 4°C. The blots were washed five times with 1 × TBST followed by incubation with horse-radish peroxidase-labeled secondary antibody at room temperature for 1 h. The blots were again washed and developed with Clarity Western ECL substrate (Bio-Rad Laboratories, Inc.) and imaged using the ChemicDoc XRS+ gel documentation system (Bio-Rad Laboratories, Inc.). Protein bands of interest were normalized to corresponding β-tubulin bands for densitometric estimation. Phosphorylation of proteins was measured as phospho-protein to total protein ratio and phospho-protein and total protein normalized to β-tubulin. (2) Co-immunoprecipitation: Co-immunoprecipitation studies were carried out using VMB or ST tissues from bilateral diamide/vehicle SN-injected groups. Approximately 700 μg of PNS protein was suspended in immunoprecipitation buffer containing 50 mM Tris (pH 8.0), 150 mM sodium chloride, 10% glycerol, 0.5% triton X-100, and protease inhibitors to a total volume of 1 ml. Samples were incubated with gentle rocking overnight at 4°C, with ASK1 antibody, after which equilibrated Protein G-coupled magnetic Dynabead suspension (Life Technologies, Inc.) was added and incubated at 4°C for an additional 2 h. The sample mixtures were then centrifuged at 10,000 g for 10 s followed by magnetic separation, and the pellets were washed thrice with immunoprecipitation buffer. Finally, the pellets containing immunoprecipitated complexes were suspended in sample buffer, boiled, resolved using gradient 8–16% Tris-glycine gels (Life Technologies, Inc.), and probed with antibody to ASK1 and Trx1. Co-immunoprecipitated Trx1 band was normalized to immunoprecipitated ASK1 band. Similarly, gels containing input samples were run for estimation of total Trx1 normalized to total ASK1 to assess whether there were any differences in expression of these proteins after diamide exposure. (3) Redox state analysis of proteins: For AMS-derivatization experiments, VMB and ST samples from the bilateral diamide-vehicle SN-injected groups were derivatized with thiol alkylating agent. The reaction mixture contained: AMS (15 mM) in a buffer containing 20 mM Tris (pH 7.4) and 2% SDS for denaturation. Derivatization was performed at room temperature for 4 h with intermittent vigorous agitation, followed by quenching of excess AMS by adding 30 mM reduced GSH in the same buffer for a further 1 h at room temperature. The derivatized and subsequently quenched samples were then mixed with 2 × sample buffer containing dithiothreitol (reducing agent) and boiled for 10 min followed by SDS-PAGE. The AMS-derivatized (reduced) proteins showed electrophoretic mobility retardation when compared with non-derivatized samples. Densitometric estimations of band intensities were done using derivatized β-tubulin (AMS-Tub) for normalization of loading. (4) GSH estimation: We measured total cellular GSH (reduced+oxidized) adapting the enzymatic recycling method of estimation (47, 56). Briefly, equal volumes of 10% sulfosalicylic acid were added to PNS from the brain regions and the protein was precipitated. After centrifugation at 10,000 g for 10 min at 4°C, the supernatant containing acid-soluble low-molecular-weight thiols, including GSH, was used for the assay. A small volume of the supernatant was added to the assay buffer containing 0.8 mM 5-5′-dithio-(2-nitrobenzoic acid), 0.5 U/ml GSH reductase in 0.1 M potassium phosphate buffer (pH 7.4). The reaction was initiated by the addition of 1.2 mM NADPH, and the formation of 2-nitro-5-thiobenzoic acid (TNB) was spectrophotometrically followed at 412 nm at 15 s intervals for a total of 2 min in a plate reader. The average rate of change in TNB absorbance per min was compared against a standard curve obtained from known amounts of pure oxidized GSH. The values were normalized to the protein amount in the original PNS samples and expressed as nmol GSH/mg protein. (5) Malondialdehyde estimation: We measured malondialdehyde levels to examine lipid peroxidation by adapting the fluorimetric thiobarbituric acid reactivity assay (48, 60). Thiobarbituric acid (TBA) assay reagent was prepared by dissolving TBA (0.375% v/v) in a mixture of tricarboxylic acid (15% w/v), 4 mN HCl, and 0.1 mM butylated hydroxyl toluene in ultrapure water. Briefly, a small volume of the PNS was added to the TBA assay reagent and the mixture was vortexed and heated at 60°C for 20 min. The mixture was immediately cooled down on ice and centrifuged at 5000 g for 10 min at room temperature. The fluorescence of thiobarbituric acid-reactive substances in the supernatant was measured against a known standard of 1,1,3,3-tetramethoxypropane at an excitation of 515 nm and an emission of 553 nm. The values were normalized to the protein amount in the original PNS samples and expressed as nmol malondialdehyde/mg protein.
Sectioning and staining of brain tissue
The mice were transcardially perfused with ice-cold 1 × PBS followed by 4% paraformaldehyde (PFA). The fixed brains were cryosectioned in a cryotome (Leica Microsystems) at a section thickness of 25 μm through the ST and midbrain regions. For all quantitative analysis of histological data, blind stereological analysis was carried out by staining every sixth section (sampling section) through a brain region with the respective stain and data were quantified for all sections in an experimentally blind manner. The total measurements were estimated from the systematic sampling method. For α-syn, phospho-α-syn, and ubiqutin staining, representative sections from the midbrain region from five animals from each group were used for the experiment. The different staining protocols are described next: (1) FJC staining: The staining was carried out according to the manufacturer's instructions, with minor modifications. Sampling sections of one control and experimental animal each were processed together on a single slide. Briefly, after treating air-dried midbrain sections with basic alcohol solution, the slides were bleached with 0.06% KMnO4 solution for 7 min followed by washing with water. Subsequently, slides were immersed in 0.0001% FJC in 0.1% glacial acetic acid for 10 min in dark followed by water washes. The sections were air dried, cleared with xylene, and mounted with coverslips using DPX (mixture of distyrene, plasticizer and xylene). Imaging was performed by using the Axio Scope.A1 microscope (Carl Zeiss AG), and blind stereological analysis was carried out to estimate the number of FJC-positive cells in the VMB. (2) Fluorescent immunostaining and analysis: This was performed to assess TH and/or NeuN staining. Sampling sections of one control and experimental animal each were processed together on a single slide. Antigen retrieval was performed on sections that were mounted on slides by using citrate buffer (Vector Laboratories). After blocking/permeabilization with 3% normal horse serum +2% bovine serum albumin +0.1% Triton X-100 in 1 × TBS, the sections were probed with either TH or NeuN primary antibody that was prepared in the same blocking/permeabilization buffer overnight at 4°C. The sections were subsequently washed five times with 1 × TBST, followed by Alexa fluor-conjugated secondary antibody incubation in 1 × TBS. The sections were washed again with 1 × TBST, followed by 1 × TBS. Finally, the sections were mounted with coverslips using Vectashield (Vector Laboratories). Sections processed similarly with replacement of the primary antibodies with naive serum were used as negative controls. The staining was imaged by using the Axioplan (Carl Zeiss; for TH staining in midbrain sections) and Axio Scope.A1 microscope with ApoTome (TH staining in striatal sections; NeuN staining in midbrain sections) and analyzed using blinded stereological measurements. (3) Bright-field immunostaining: This experiment was carried out to assess the pattern of expression of α-syn, phospho-α-syn, and ubiqutin in the VMB. Representative sections from each animal were mounted on to slides and processed together. After antigen retrieval, blocking, and primary antibody incubation as described earlier, the sections were washed with 1 × TBST and incubated with biotinylated-secondary antibody prepared in 1 × TBS. The sections were then incubated with ABC reagent and developed with Nova Red reagent according to the manufacturer's instructions (Vector Laboratories). The sections were air dried, dehydrated with graded alcohol, cleared with xylene, and mounted with coverslips using DPX. Sections processed similarly with substitution of the primary antibody with naive IgG were used as negative controls. The sections were imaged using a Leica DMR microscope (Leica Microsystems).
Cell culture experiments
Plasmids
Constructs overexpressing hemagglutinin-tagged human full-length WT and kinase-dead mutant (KM; K709R) ASK1 constructs cloned into pcDNA 3 were a kind gift from Prof. Hidenori Ichijo from the University of Tokyo (Tokyo, Japan). Plasmids for RNA interference for knocking down mouse ASK1 were designed and cloned in the laboratory by using the mU6pro plasmid background gifted by Prof. David L. Turner from the University of Michigan, Ann Arbor, MI, USA. The sequences of oligonucleotides annealed and used for cloning were as follows: ASK1 shRNA (Top: 5′-TTTGCGGATGCAGTGATCTAATAAGTTCTCTATTAG ATCACTGCATCCGCTTTT-3′; Bottom: 5′-GCCTACGTC ACTAGATTATTCAAGAGATAATCTAGTCACGTAGG CGAAAAAGATC-3′). The scrambled scRNA sequences were (Top: 5′-TTTGTTGGTTACGGGGTATCGATTCAA GAGATCGATACCCCGTAACCAACTTTTT-3′; Bottom: 5′-AACCAATGCCCCATAGCTAAGTTCTCTAGCTATG GGGCATTGGTTGAAAAAGATC-3′).
Transfections and treatments
In vitro experiments were carried out in Neuro-2a cells obtained from the American Type Culture Collection (Manassas, VA, USA). Neuro-2a cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum and penicillin–streptomycin.
Cells were transfected with various plasmids using Lipofectamine LTX according to the manufacturer's instructions. ASK1 downregulation using shRNA was performed for 72 h before the transfected cells were exposed to diamide/vehicle for 5 min or 1 h (for biochemical analysis) and 6 h (for TUNEL analysis). ASK1 overexpression was carried out for 48 h before diamide exposure for 1 h.
Overnight grown cultures or transfected cells were treated with 300 μM diamide or vehicle (0.9% saline) and incubated for varying periods of 5 min, 15 min, 1 h, or 6 h according to the experiment. Cells were immediately harvested with ice-cold 1 × PBS.
Sample preparation
For immunoblotting, cell pellets were washed with 1 × PBS; lysed in 1 × PBS containing 1% Igepal CA-630, 2 mM EDTA, and protease and phosphatase inhibitors using sonication. The lysates were cleared by centrifugation at 10,000 g for 10 min at 4°C. These samples were estimated for protein and run on SDS-PAGE followed by immunoblotting with suitable primary antibodies. Co-immunoprecipitation and redox gel derivatization protocols were performed as described earlier. For GSH estimation, the cells were homogenized in homogenization buffer (as described for brain tissue sample preparation). Total GSH was then estimated from the PNS as described earlier.
Cell death/viability analysis
Cell death or loss of viability of Neuro-2a cells after diamide exposure was assessed using the MTT and TUNEL assays. For MTT, overnight grown cultures were treated with different doses of diamide (0–1200 μM) for 6 h before the replacement of medium with MTT and further incubation for 3 h. The formazan precipitate was dissolved in DMSO, and absorbance was measured at 530 nm in a plate reader. The data were normalized to the untreated group. TUNEL analysis was performed using the In situ cell death detection kit, TMR red (Roche) for non-transfected and ASK1 shRNA/scRNA-transfected cells after 6 h of diamide/vehicle exposure. The protocol followed was as per the manufacturer's instructions. The average number of TUNEL-positive (Red) cells were counted, normalized to the total number of cells (DAPI stained; blue), and expressed as percentage of normalized data.
Immunofluorescent staining
Fluorescent immunostaining was carried out to assess pASK1, pp38, and pJNK expression. Cells treated with 300 μM diamide or vehicle were fixed after 5 min or 6 h of exposure and were fixed with 4% PFA in 1 × PBS before proceeding with immunostaining. Briefly, the cells were blocked and permeabilized in 0.05% Triton X-100 and 3% goat serum +3% bovine serum albumin in 1 × PBS. Suitable antibodies prepared in the same blocking buffer were added and incubated overnight before washing and the addition of Alexa-fluor 488-tagged goat anti-rabbit secondary antibody (Life Technologies). The cells were washed, mounted with DAPI, and subsequently imaged in Axio Scope.A1 with ApoTome optical sectioning.
Quantitative polymerase chain reaction
RNA extracted from ASK1 knockdown cells were extracted and converted to cDNA by using the high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative polymerase chain reactions were performed with this cDNA in the ABI 7500 machine (Applied Biosystems) to assess the extent of ASK1 downregulation and normalized to 18S rRNA levels. Data were analyzed using the comparative threshold cycle (ΔΔCt) method. The primer sequences used were as follows: ASK1 (Forward: 5′-TCCTCAGTCAACTGCCTT TCA-3′; Reverse: 5′-CTCCGGTGAGCTGTTGTAAA-3′) and 18S rRNA (Forward: 5′-GAGAGGGAGCCTGAGAAA CGG-3′; Reverse: 5′-GGTCGGGAGTGGGTAATTTGC-3′).
Statistical analyses
For the SN-injection behavioral analysis, two-way repeated measures (RM) ANOVA was performed followed by Bonferroni's post hoc tests with time and drug group as two factors, whereas one-way RM ANOVA was performed for motor cortex-injected animals. For in vivo experiments, parametric two-way ANOVA followed by Bonferroni's post hoc tests were carried out for histochemical analysis. For histochemical analysis, the injection side and diamide/vehicle group were the two independent variables. For the protein expression analysis, parametric one-way ANOVA followed by Student–Newman–Keuls analysis was performed, as data were not always fully paired.
For all the time-point-based analyses in the in vitro experiments, parametric statistical tests were carried out for the data using Student's t-test to compare differences between saline- and diamide-treated cells. For the multi-group experiments, parametric one-way ANOVA followed by Student–Newman–Keuls analysis was performed. All the statistical tests and visualization of graphs were performed using GraphPad Prism 5.0. All graphs shown represent mean ± standard deviation, except for the behavioral analysis graphs where each animal is individually represented with a trend line connecting means. Values of p < 0.05 were considered statistically significant.
Footnotes
Acknowledgments
The authors are grateful to Prof. Nihar Ranjan Jana and Mr. Shanker Datt Joshi at NBRC, India, and Ms. Aditi Verma and Mr. Sunny Kumar at the Centre for Neuroscience, IISc, for their help. The work was supported by the Department of Science and Technology, India, and NBRC. A.R. received graduate fellowship from the Council of Scientific and Industrial Research, India.
Authors' Contributions
A.R. and M.K. performed the experiments. A.R. analyzed the data and A.R. and V.R. designed the experiments, interpreted data, and wrote the article.
Author Disclosure Statement
The authors declare no conflict of interest.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.