
Abstract
Introduction
P
We first showed that S-nitrosylation of Parkin reduced its ability as a suppressor of dynamin-related protein-1 (Drp1) expression, leading to upregulation of Drp1 in neurotoxin-based Parkinson's disease (PD) models, in vitro and in vivo. Drp1 appears directly pertinent to PD in that its expression is selectively elevated in susceptible neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse models. Our data provide a molecular explanation for the contribution of Drp1 to PD pathogenesis. The discovery of nitric oxide signaling links 1-methyl-4-phenylpyridinium ion (MPP+) neurotoxicity to the malfunction in mitochondrial dynamics affords an opportunity to develop novel therapeutic strategy for sporadic PD.
Mitochondria are dynamic organelles that undergo frequent fission and fusion—processes regulated by machinery involving large GTPases that exert distinct effects; for example, dynamin-related protein 1 (Drp1) for fission, and mitofusin-1/2 (Mfn-1/2) for fusion (4). Mitochondrial dynamics are critical to preserve the health of mitochondrial network and function. Neurons depend largely on mitochondrial function to build membrane excitability and to execute the complicated processes of neurotransmission and plasticity (14). Increasing evidence shows that free radical mediates damage to proteins, lipids, and DNA in PD patients (13), but the relationship between free radical-induced damage and mitochondrial dysfunction is incompletely understood.
Nitric oxide (NO) synthesized by NO synthases (NOSs) serves as a neurotransmitter in the central nervous system, but in excess it mediates neuronal injury, in part, through mitochondrial fission (1). Notably, NO production is essential for S-nitrosylation, which pointed to the possibility that dysregulated S-nitrosylation could contribute to the development of several neurodegenerative disorders. In addition, excess NO can react with superoxide anion (O2 •−) to form highly neurotoxic peroxynitrite (ONOO−), resulting in damage to cellular components (25). Strong evidences have collectively implicated NO stress as an essential contributor to the pathogenesis of PD (13, 40). However, little is known regarding the mechanisms underlying contributions of NO to mitochondrial fragmentation in PD.
Mutations in the Parkin gene are the leading cause of hereditary PD. The identification that Parkin mediates the removal of impaired mitochondria has strengthened the link between PD pathogenesis and mitochondrial dysfunction (23). In this regard, a novel role of Parkin as a regulator of Drp1 was recently reported (32). In addition, nitrosative or oxidative stress can cause S-nitrosylation of Parkin (SNO-Parkin) to impair its E3 ligase activity (6, 38). However, whether these modifications impact mitochondrial dynamics in PD is unclear. Given the critical roles of mitochondrial dynamics in neurodegenerative diseases, we sought to clarify the molecular mechanism that connects Drp1 to PD pathogenesis. We first demonstrated that SNO-Parkin reduces its ability to repress Drp1 expression, leading to upregulation of Drp1 in vitro and in vivo. Drp1 appears directly pertinent to PD in that its expression is selectively increased in susceptible neurons in MPTP-induced PD brains. Furthermore, NO stress, induced by MPP+, triggered Drp1 phosphorylation at serine 616, which results in its recruitment to mitochondria. These events create a death-prone milieu that contributes to the loss of DA neurons, and Drp1-mediated mitochondrial fragmentation can be obviously suppressed by an NOS inhibitor in DA neurons, indicating that NO signaling links MPP+ neurotoxicity to the malfunction in mitochondrial dynamics, in part, underlying the pathogenesis of PD. The discovery of NO signaling pathway affords an opportunity to develop novel therapeutics for PD.
Results
NO is responsible for MPP+-induced Drp1 expression
Previous study showed that MPP+ could induce Drp1 expression in SH-SY5Y cells (35). We sought to confirm and extend these observations regarding the regulation of Drp1 in cellular models of PD. As shown in Figure 1A, the Drp1 protein level was significantly increased in SH-SY5Y cells treated with indicated concentrations (0.5 μM–2 mM) of MPP+ for 24 h, which is consistent with the previous study. No significant differences in overall mitochondrial contents were observed between untreated and MPP+-treated cells, as evidenced by the constant expression levels of COXIV, a mitochondrial marker (Fig. 1A). Real-time quantitative polymerase chain reaction (qPCR) results showed only a slight increase in Drp1 mRNA levels after 12–24 h of exposure to 2 mM MPP+ (Fig. 1B), but the magnitude of increase in Drp1 protein level was greater than that of Drp1 mRNA after MPP+ treatment, suggesting a posttranscriptional contribution to elevated Drp1 levels.
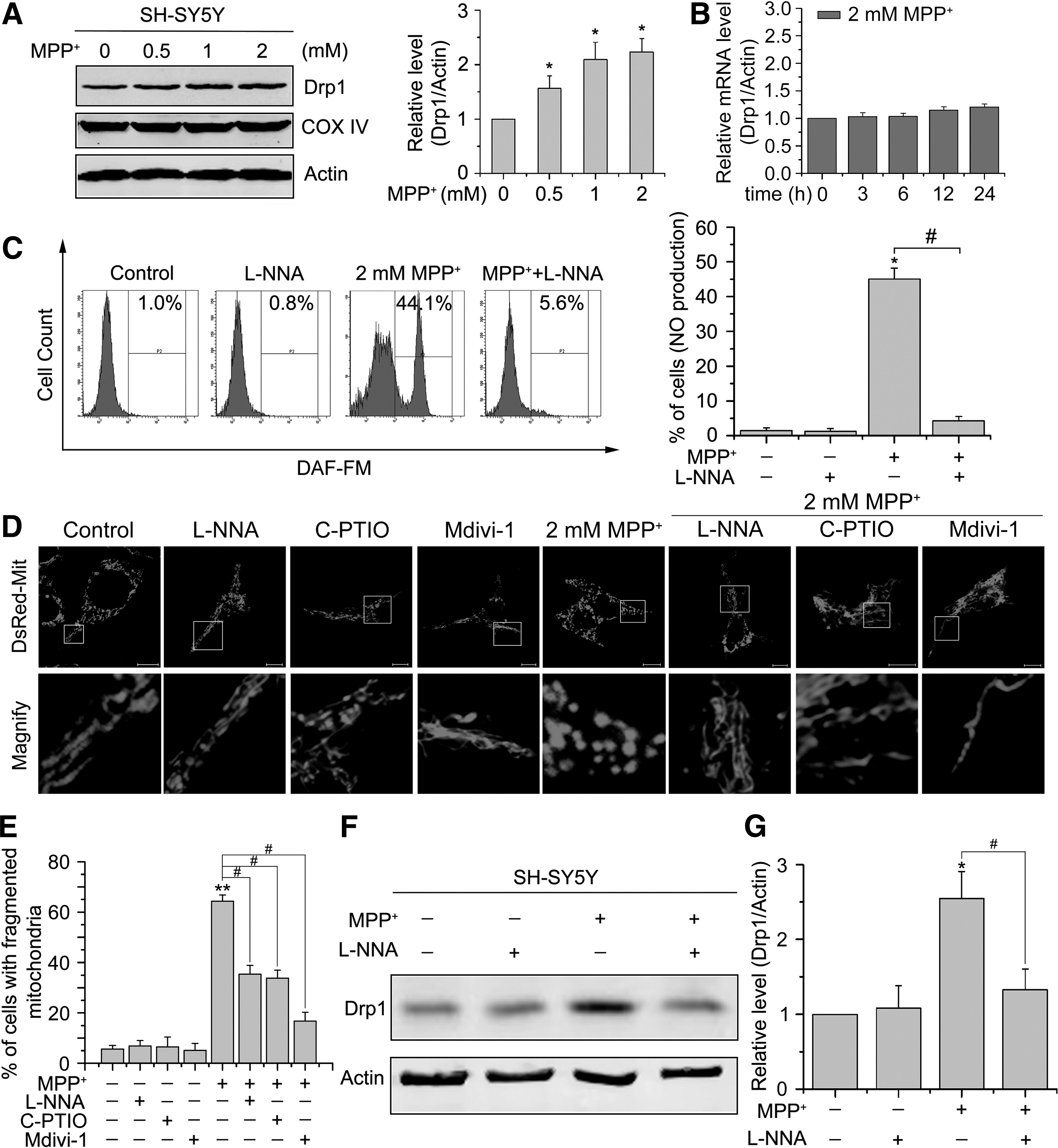
Experimental data have long evidenced the involvement of NO in PD pathogenesis. We therefore examined whether MPP+ treatment induced NO production within cells by using a fluorescence probe, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM) that can detect NO. We found that MPP+ treatment remarkably increased the intensity of DAF-FM in SH-SY5Y cells (Fig. 1C). To further investigate whether increased NO contributes to mitochondrial fragmentation induced by MPP+, SH-SY5Y cells were treated with 1 mM NOS inhibitor N-nitro-l-arginine (L-NNA), which almost completely inhibited MPP+-induced NO production (Fig. 1C). The results showed that 64.3% ± 2.5% of cells displayed fragmented mitochondria after treatment with 2 mM MPP+ for 24 h, which was significantly reduced to 35.4% ± 3.4% in the presence of L-NNA. No obvious fragmentation was found in SH-SY5Y cells treated with L-NNA alone for up to 24 h (Fig. 1D, E). To further address this issue, we also used the NO-specific scavenger carboxy-PTIO (c-PTIO). Likewise, we found that preincubation of SH-SY5Y cells with 40 μM c-PTIO 2 h before 2 mM MPP+ treatment significantly abolished MPP+-induced mitochondrial fragmentation (Fig. 1D, E), further confirming the involvement of NO in MPP+-induced mitochondrial fragmentation.
Since Drp1 is known to be implicated in NO-induced mitochondrial fission in neurons, we therefore determined whether NO signaling is responsible for MPP+-induced Drp1 upregulation. We treated SH-SY5Y cells with 2 mM MPP+ for 24 h in the presence of 1 mM L-NNA or not, and found that MPP+-induced Drp1 expression was strongly inhibited by L-NNA (Fig. 1F, G). These data indicate that NOS-induced synthesis of NO may be the main mechanism for MPP+-induced Drp1 expression.
MPP+-induced mitochondrial fragmentation in primary DA neurons is mediated by NO
As DA neurons in midbrain are specific targets of neurotoxin in PD, we prepared primary cells of midbrain from embryonic day 13 Sprague-Dawley rats and tested whether increased NO contributes to MPP+-induced mitochondrial fragmentation in DA neurons. After 6 days in vitro (DIV6), we exposed the cultured cells to 1 μM MPP+ for 24 h plus 10 μM selective Drp1 inhibitor mitochondrial division inhibitor-1 (Mdivi-1), and cells were fixed and stained. In agreement with the previous study, we focused on mitochondria in the distal segments of axons and axonal branches where mitochondria are separated from each other due to the high density of mitochondria in the soma and proximal segment of axon (35). Double-label immunofluorescence for tyrosine hydroxylase (TH) and COXIV revealed that NOS inhibition significantly attenuated MPP+-induced mitochondrial fragmentation in DA neurons: mitochondrial length decreased from 1.92 ± 0.23 μm in nontreated cells to 0.51 ± 0.04 μm after 24 h of MPP+ treatment, and increased to 1.58 ± 0.21 μm (p < 0.05) in the presence of L-NNA (Fig. 2A, B). Importantly, MPP+-induced reduction of neurite mitochondrial index (total mitochondrial length/neurite length) (35) (0.068 ± 0.0046) was significantly prevented by L-NNA (0.1763 ± 0.0067) (Fig. 2A, C), indicating that NO signaling links MPP+ neurotoxicity to the malfunction in mitochondrial dynamics in DA neurons.

SNO-Parkin decreases its ability to suppress Drp1 expression
Our earlier data indicate that MPP+ induces excessive mitochondrial fission in an NO-mediated manner. We subsequently explore the mechanism through which NO increases Drp1 protein levels. Parkin has been reported to ubiquitinate Drp1 for protease-dependent degradation (32). We initially incubated Parkin-transfected SH-SY5Y cells with a range of concentrations of MPP+ for 24 h and examined the interaction of Drp1 and Parkin using coimmunoprecipitation. We found an obvious decrease in the level of Parkin binding to Drp1 after MPP+ treatment (Fig. 3A), and this decrease was significantly prevented by treatment with L-NNA (Fig. 3B), indicating that endogenous NO attenuated Parkin-mediated repression of Drp1.
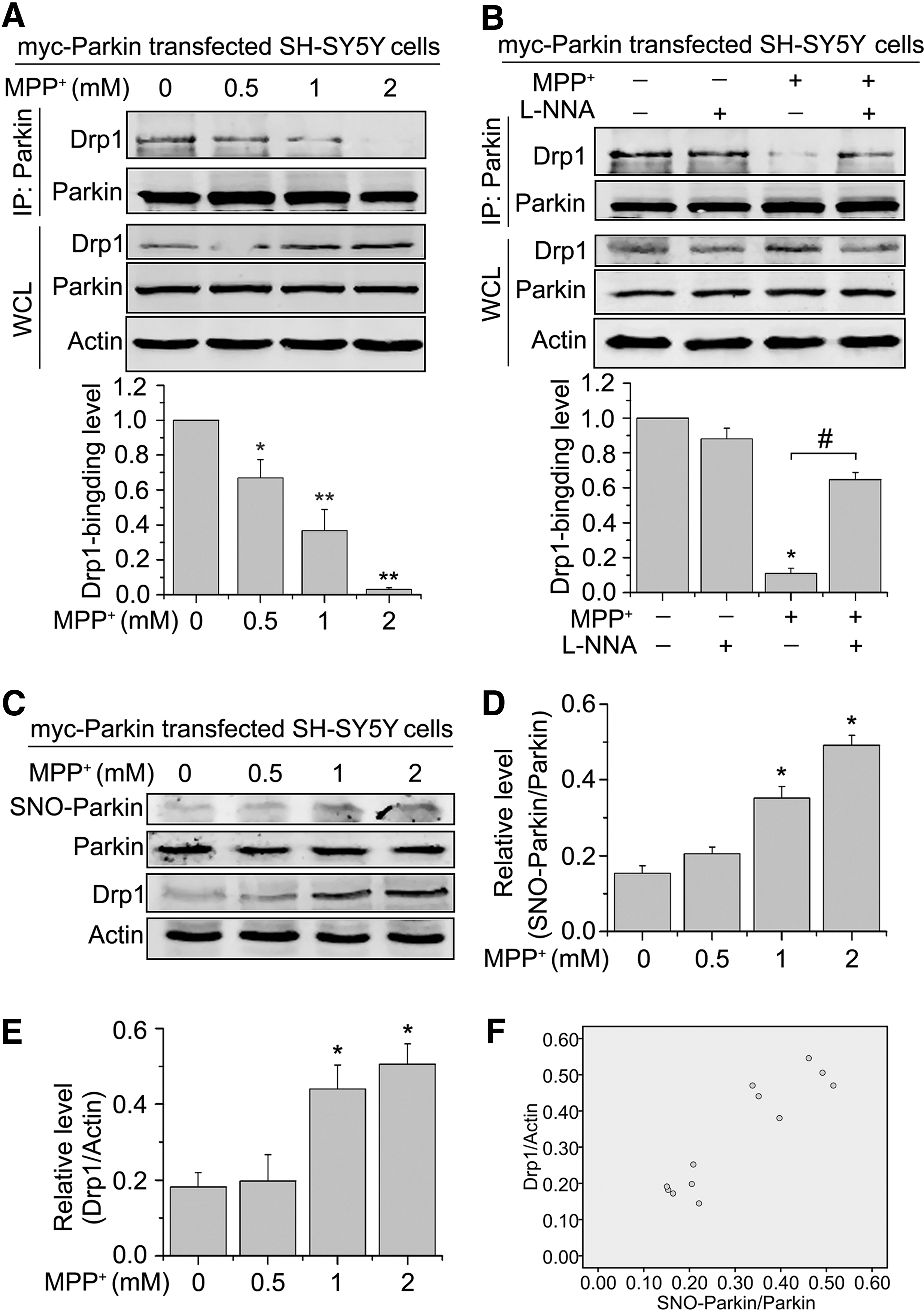
Then, we sought to examine whether Parkin was S-nitrosylated by MPP+ and thereby removed the constraints on Drp1 expression. Parkin-transfected SH-SY5Y cells were treated with a range of concentrations of MPP+ for 24 h and then subjected to the biotin switch assay. Furthermore, we performed Western blotting and quantified the levels of Drp1 in SH-SY5Y cells transfected with Parkin after the 24-h treatment of MPP+. We found that MPP+ treatment led to the generation of SNO-Parkin (Fig. 3C, D). In addition, MPP+ induced a significant increase in the Drp1 protein levels under the same conditions (Fig. 3C, E). Finally, the ratio of SNO-Parkin to total Parkin was highly correlated with the Drp1 level for each treatment using the Pearson correlation coefficient test (r = 0.923) (Fig. 3F). In summary, MPP+ treatment showed an increased SNO-Parkin/Parkin ratio, accompanied by a consistent increase in Drp1 levels. Altogether, these findings suggest that SNO-Parkin reduces its ability to repress Drp1 expression, resulting in elevated Drp1 protein levels.
Drp1 is increased in the ventral midbrain of MPTP-induced PD mouse model
Subsequently, we examined whether the upregulation of Drp1 level observed in cellular PD models also occurs in PD mouse models. Although SNO-Parkin is higher in the midbrain DA neurons of MPTP-treated PD mouse models and PD patients (6, 22, 38, 39), Drp1 levels had not been reported. C57BL/6 male mice received an injection of MPTP (20 mg/kg body weight) once per day for 5 consecutive days (12), and Drp1 protein levels were first detected in postmortem brain tissue from the ventral midbrain, cortex, striatum, and cerebellum. Such a regimen caused an obvious decrease of TH expression after MPTP injection, indicating the loss of DA neurons (Fig. 4A). Moreover, Drp1 was remarkably increased in the ventral midbrain after MPTP administration compared with saline-injected mice (Fig. 4A). In contrast, no significant changes in Drp1 expression were observed in the cortex (Fig. 4B), striatum (Fig. 4C), or cerebellum (Fig. 4D).

To elucidate the source of NO that S-nitrosylates Parkin after MPTP injection, we measured three NOS isoforms in the postmortem brain tissue prepared from saline or MPTP-treated mice. As shown in Supplementary Figure S1A and B (Supplementary Data are available online at
SNO-Drp1 is not increased in vitro and in vivo
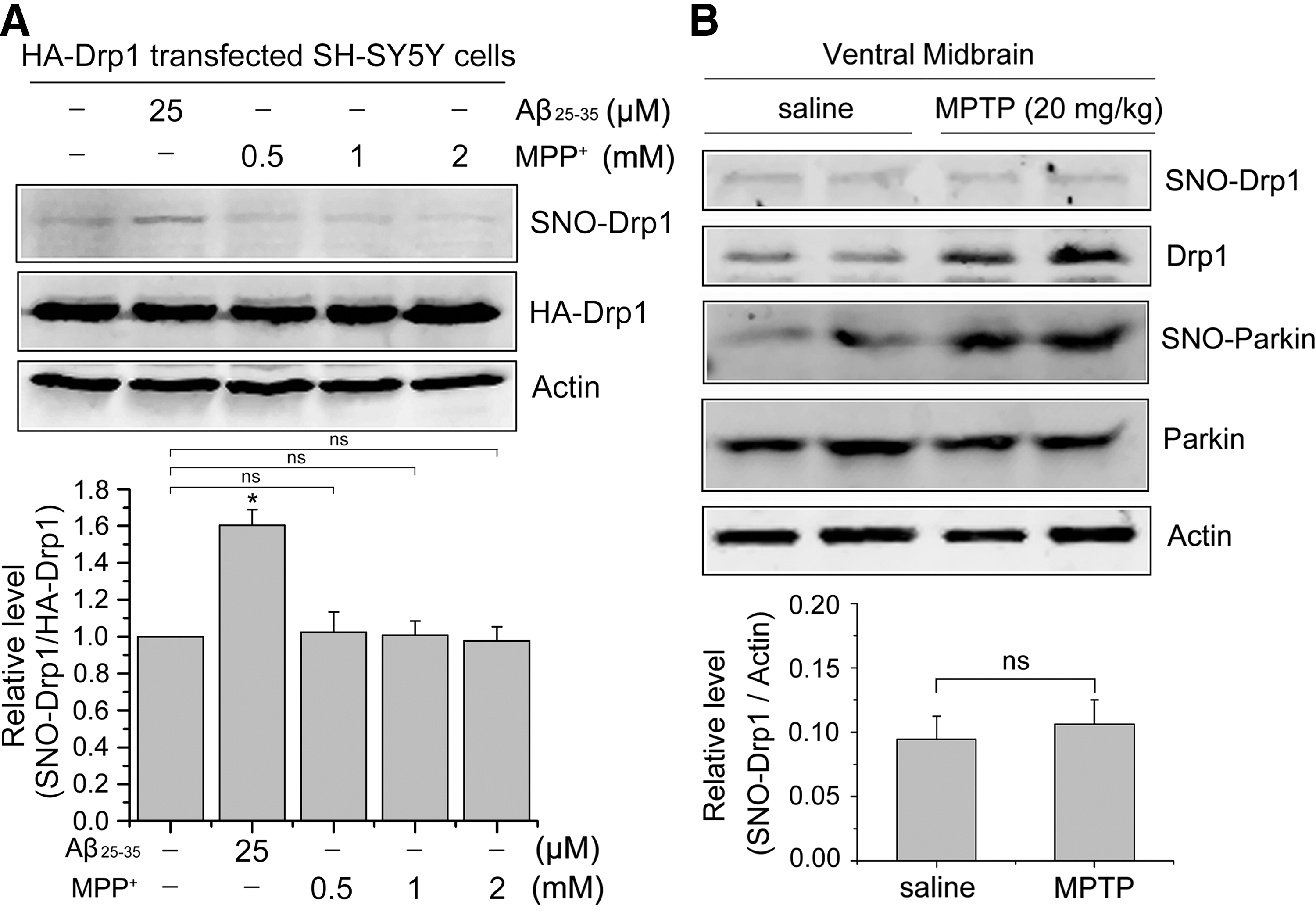
Recent studies reported that S-nitrosylation of Drp1 (SNO-Drp1) activates its GTPase activity, thus accelerating mitochondrial fragmentation and contributing to neuronal injury in neurodegenerative diseases, including Alzheimer's disease and Huntington's disease, in vitro and in vivo (5, 11, 34). To determine whether the level of SNO-Drp1 in PD models is of pathophysiological importance, we also examined SNO-Drp1 in our studies by biotin-switch assay in vitro and in vivo. SH-SY5Y cells transfected with HA-Drp1 were exposed to 25 μM Aβ25-35 or different concentrations of MPP+ for 24 h. The biotin-switch assay of Aβ25-35-treated and MPP+-treated SH-SY5Y cells indicated the presence of increased SNO-Drp1 levels in Aβ25-35-treated cells compared with untreated cells, but not in MPP+-treated cells (Fig. 5A). In MPTP-induced PD mouse model, both SNO-Parkin and Drp1 levels were elevated (Fig. 5B). However, no significant difference in the SNO-Drp1 level was observed in the ventral midbrains obtained from the MPTP-induced PD mouse model relative to controls (Fig. 5B), suggesting that SNO-Drp1 may not be responsible for PD-linked mitochondrial fission.
NO induces Drp1 phosphorylation and translocation to mitochondria
Drp1 normally resides in the cytosol of cells and it is the mitochondrial Drp1 that participates in mitochondrial fission. We initially demonstrated whether NO signaling involved in Drp1 recruitment during MPP+ treatment by measuring the mito-Drp1 levels. SHSY5Y cells were transiently transfected with DsRed-Mit for 24 h and then treated with 2 mM MPP+ for another 24 h. Immunofluorescence for Drp1 showed that MPP+ significantly increased Drp1 colocalization with mitochondria compared with the control cells, and this increase was almost blocked by L-NNA (Supplementary Fig. S2), suggesting the role of NO signaling in Drp1 activation. Given that Drp1 translocation to mitochondria can be induced by Drp1 phosphorylation (3, 10, 29), we then conducted subcellular fractionation experiments and found that MPP+ treatment induced Drp1 phosphorylation at Ser616 and caused its subsequent recruitment to the mitochondria (Fig. 6A, B). Conversely, Drp1 Ser637 was decreased in mitochondrial fraction (Fig 6A, C) in MPP+-treated cells relative to the untreated cells. In addition, we also found increased mito-Drp1 levels after MPP+ treatment, which is consistent with the previous study (35) (Fig 6A, D).
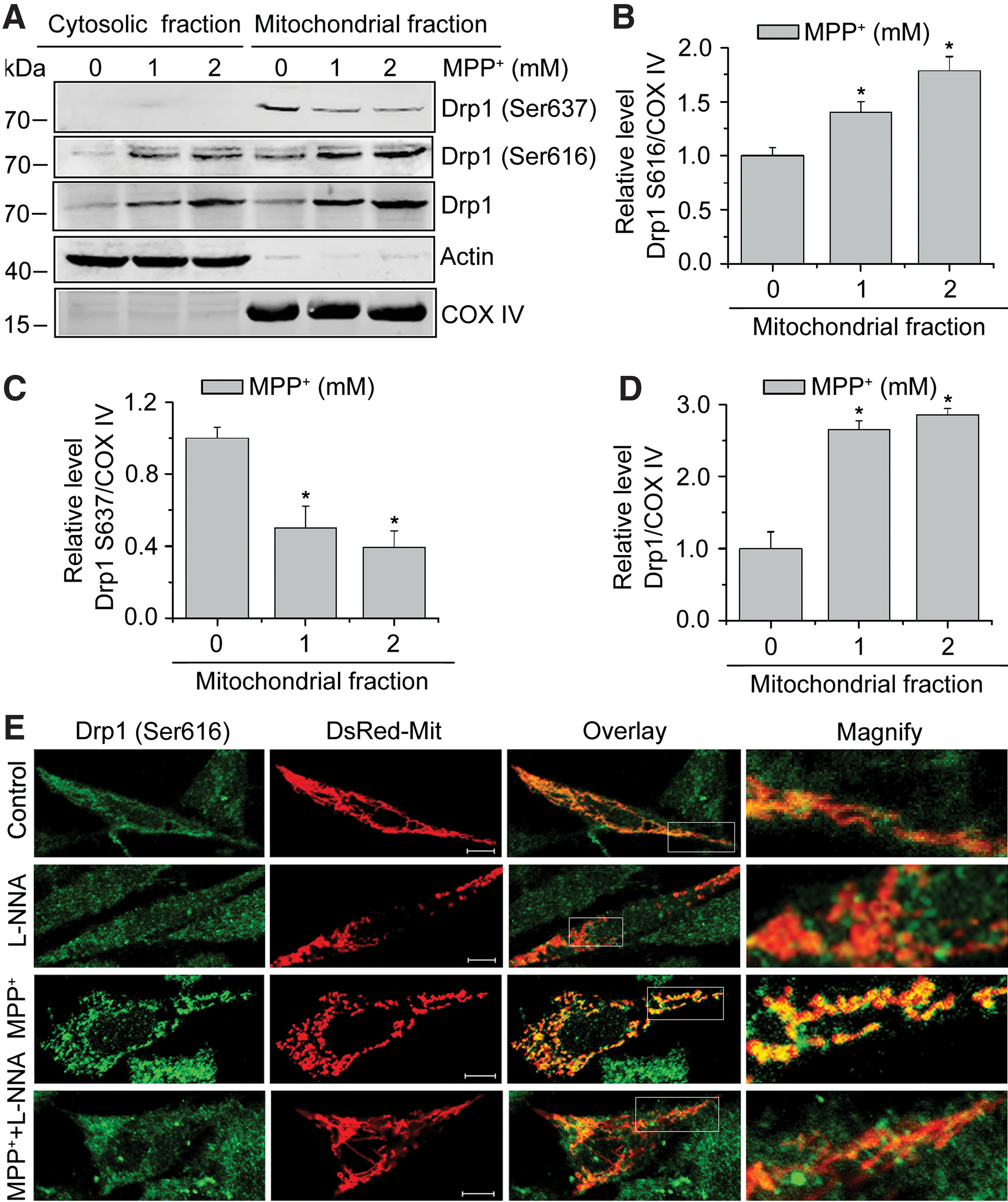
To determine whether NO stress induced by MPP+ stimulates mitochondrial fragmentation by promoting Drp1 Ser616 phosphorylation, SH-SY5Y cells with indicated treatments were examined by confocal microscopy. Immunofluorescence analysis revealed a significant stimulation of Drp1 Ser616 phosphorylation by MPP+ treatment relative to control cells, and this stimulation was significantly attenuated by L-NNA (Fig. 6E). Thus, our results suggest that NO signaling induces the phosphorylation of Drp1 Ser616 and causes its subsequent translocation to mitochondria, offering an alternative mechanism of MPP+-induced mitochondrial fragmentation.
Parkin attenuates Drp1-mediated cell death
Our earlier data show that Drp1 is elevated and activated in PD models, suggesting a critical role of Drp1 in PD. To further explore the role of Drp1 in PD, we examined the apoptotic activity in SH-SY5Y cells expressing HA-Drp1 by flow cytometry (FCM). As shown in Figure 7A, overexpression of Drp1 sensitized cells to death caused by MPP+. Next, we used Drp1 RNAi to knockdown endogenous Drp1 in our cellular models (Fig. 7B). Silencing Drp1 greatly suppressed the accumulation of Bax on the mitochondria, blocked the release of cytochrome c from the mitochondria, and recovered the inner mitochondrial membrane potential (Δψm) in SH-SY5Y cells after treatment with MPP+ (Fig. 7C–E). Collectively, these findings support the notion that Drp1-dependent mitochondrial fragmentation is not simply a consequence of MPP+-induced neuronal cell death.
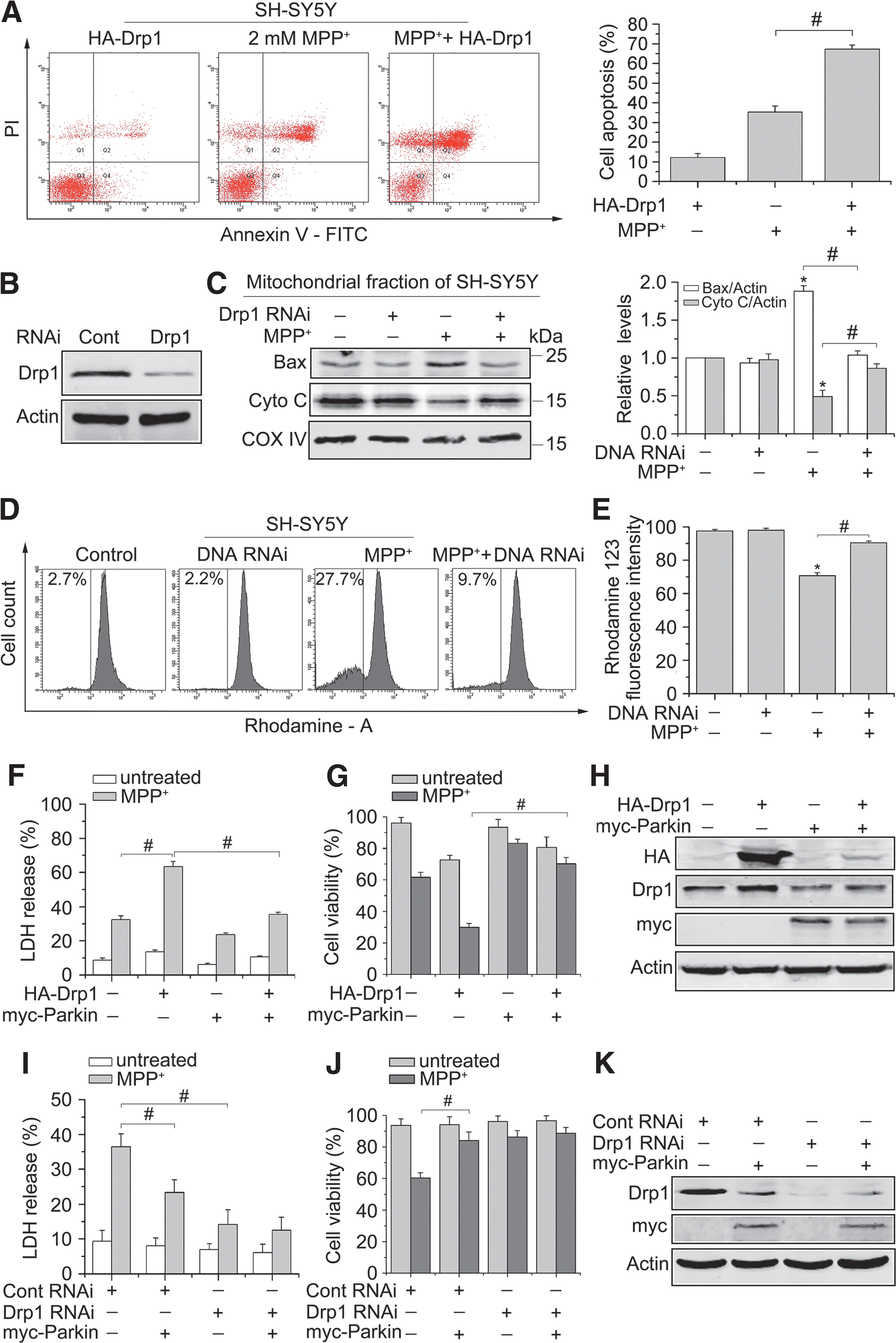
We then asked whether Parkin contributes to Drp1-mediated cell death. Cell death was examined by lactate dehydrogenase (LDH) release, and cell viability was assessed based on the detection of ATP levels in 293T cells transiently transfected with indicated plasmids. Knockdown of Drp1 significantly decreased cell death in response to MPP+ treatment than that of control RNAi-transfected cells, and Parkin overexpression inhibited cell death in control RNAi-transfected cells, suggesting that Parkin was neuroprotective as expected based on prior studies (Fig. 7F–H). Furthermore, we found that Drp1 overexpression sensitized cells to death caused by MPP+, while coexpression of Parkin ameliorated Drp1-induced sensitization of cells to MPP+ (Fig. 7I–K). Taken together, our data imply that Parkin attenuates the death potential of Drp1.
Drp1 inhibition protects against MPP+-induced neuronal cell death in primary mesencephalic cultures
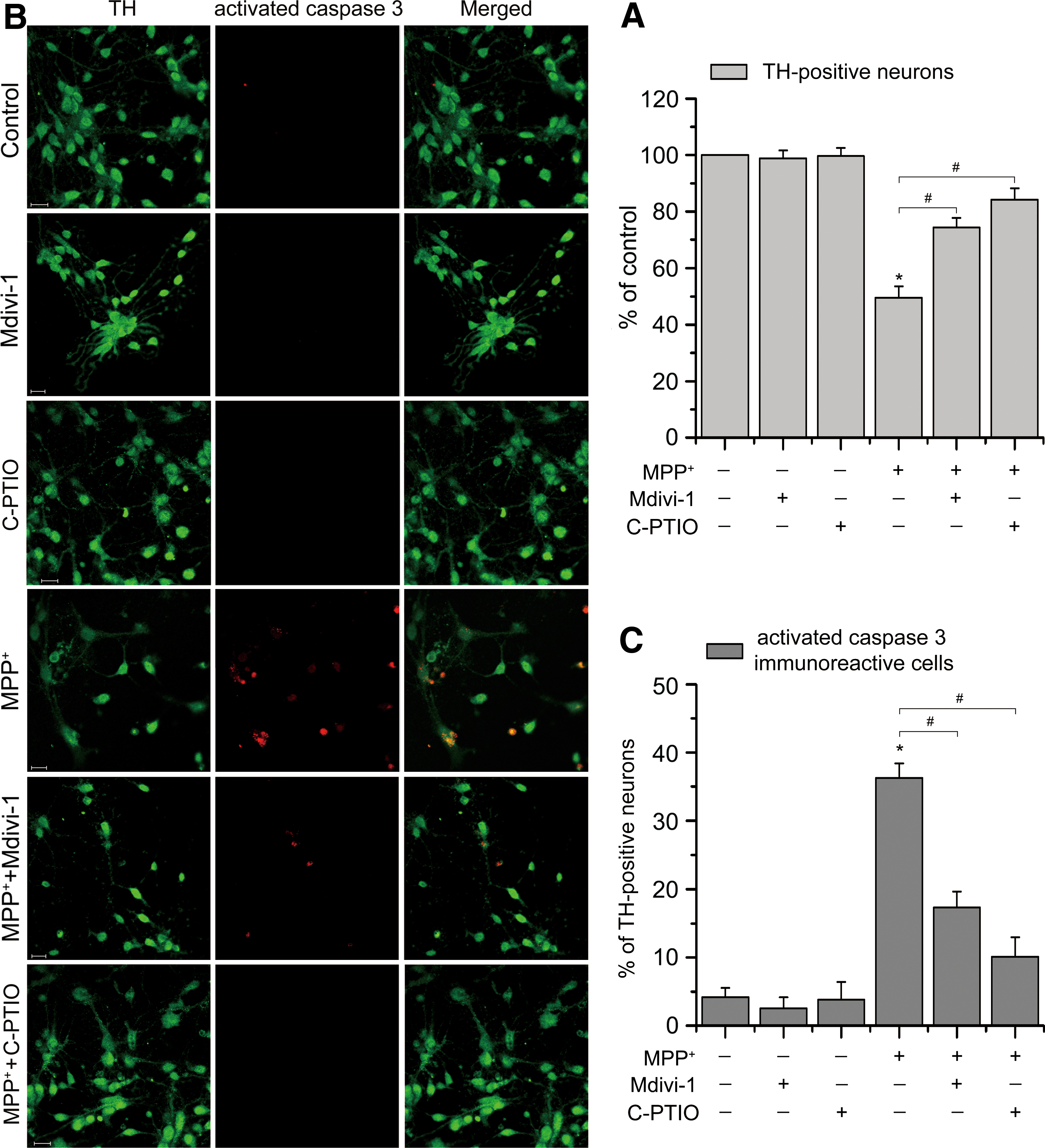
Subsequently, we determined the role of Drp1 in MPP+-induced DA neuronal death. The effect of NO inhibition on MPP+-induced DA neuronal death was also analyzed. As shown in Figure 8A, we found that MPP+ treatment significantly reduced the number of surviving TH-positive neurons to approximately 50% of untreated control, reflecting the loss of DA neurons. Treatment of cultures with Mdivi-1 and C-PTIO caused a dramatic neuroprotection, increased the number of surviving TH-immunoreactive neurons to ∼75% and ∼85% of untreated control, respectively (p < 0.05). Furthermore, double-label immunofluorescence for TH and activated caspase 3 revealed a marked increase in the number of coexpressing DA neurons, from ∼5% in untreated cultures to approximately 36% in MPP+-treated cultures, and this increase was reduced to ∼17% and ∼10% in TH-immunoreactive neurons when coincubated with Mdivi-1 and C-PTIO, respectively (Fig. 8B, C). In conclusion, these findings suggest that inhibition of Drp1-dependent mitochondrial fragmentation protects against MPP+-induced DA neurodegeneration.
Discussion
The molecular chain of events that lead to neurodegeneration in PD remains obscure and any progress in this regard has the possibility to identify potential targets for therapeutic treatments. In this study, we uncover the machinery that connects Drp1 to PD pathogenesis. We showed that SNO-Parkin reduces its ability to repress Drp1 expression, leading to upregulation of Drp1 in PD models, in vitro and in vivo. Concomitantly, both SNO-Parkin and Drp1 protein levels were increased in MPTP-induced PD mouse models, while no significant changes in the level of SNO-Drp1 were found in these mice. In addition, NO induced the phosphorylation of Drp1 Ser616, hence contributing to its activation and subsequent recruitment to mitochondria, offering an alternative mechanism of PD-related mitochondrial fragmentation. Taken together, our findings indicate that NO signaling links MPP+ neurotoxicity to the malfunction in mitochondrial dynamics, in part, underlying the pathogenesis of sporadic PD. These findings have implications for treating PD.
Since a previous study demonstrated that Drp1 expression was enhanced in MPP+-treated cellular models, the Drp1 activity might be tightly linked to the development of PD. During the preparation of this manuscript, an article that was published noted that Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo (27), suggesting a critical role for Drp1 in PD. However, the molecular mechanisms underlying the regulation of Drp1 in PD remain unclear. Over the past few decades, strong evidence has collectively implicated NO stress as a key contributor to the pathogenesis of PD. Our findings provide some clues, suggesting that MPP+ treatment initiates mitochondrial fragmentation in an NO-mediated manner. Moreover, we found that L-NNA, an NOS inhibitor that could almost completely block NO formation induced by MPP+, also prevented MPP+-induced mitochondrial fragmentation in SH-SY5Y cells and DA neurons, confirming a crosstalk between NO production and Drp1-dependent mitochondrial fragmentation induced by MPP+.
Recently, evidence is accumulating that PD-associated genes appear to participate in the regulation of mitochondrial dynamics. For example, dysfunction of human PINK1 and Parkin may result in mitochondrial pathology in PD patients (8, 9). Moreover, the PINK1/Parkin pathway can regulate mitochondrial morphology by mediating the ubiquitination of the profusion factor Mfn or Drp1 (24, 32, 42). These studies indicate that Parkin does play an essential role in mitochondrial dynamics. It is known that Parkin's E3 ubiquitin ligase activity can be impaired by oxidative and nitrosative stress (6, 36, 38), leading to loss of function. We herein showed that MPP+ treatment increased SNO-Parkin in Parkin-overexpressed SH-SY5Y cells, accompanied by consistent increases in Drp1 protein levels. Treatment with MPP+ inhibited the binding ability of Parkin to Drp1, while administration of L-NNA blocked this effect. Therefore, it appears that SNO-Parkin reduces its ability to suppress the expression of Drp1 and thereby removed the constraints on Drp1, leading to elevated Drp1 levels. It will be interesting to investigate whether this type of interaction is also important for governing the Drp1 level in mitochondria. We further observed elevation of SNO-Parkin and Drp1 in the ventral midbrain of MPTP-treated PD mouse model. Our results are consistent with higher levels of SNO-Parkin found in midbrain DA neurons of PD mouse models and PD patients (6, 22, 39) and that we and other groups have observed in cellular PD models (38). Previous studies have shown that MPTP administration specifically increases NO production in the nigrostriatal pathway (17, 25, 26). Consistent with this notion, we also found increased iNOS and nNOS in the ventral midbrain of MPTP-injected mice, perhaps accounting for the region-specific accumulation of SNO-Parkin and Drp1 in PD models. Since MPTP damages DA neurons in the substantia nigra pars compacta, whether Drp1 was selectively increased in this area needs to be further addressed.
Previous studies have shown that knockdown of Parkin results in mitochondrial fragmentation, which is coordinated with enhanced mitochondrial turnover by autophagy (23). It is possible that Parkin has dual roles for mitochondrial dynamics and quality control. On one hand, it is able to keep cellular Drp1 levels under control to prevent mitochondrial fragmentation. On the other hand, once it is recruited onto depolarized mitochondria, Drp1 facilitates Parkin-mediated degradation of damaged mitochondria.
With regard to Drp1 in neurodegenerative diseases, previous studies have reported a role for Drp1 in mitochondrial fragmentation that may contribute to the pathogenesis of neuronal loss. In particular, recent publications proposed a mechanism for Drp1 activation by SNO-Drp1 (5), which activates its GTPase activity and triggers excessive mitochondrial fission. Since the formation of SNO-Drp1 represents an aberrant signaling pathway that predominantly appears under neurodegenerative conditions (2, 5, 11), we also observed a slight but significant SNO-Drp1 level in Aβ25-35-treated HA-Drp1 transfected SH-SY5Y cells. However, no significant changes in the SNO-Drp1 level was found in PD models, in vitro and in vivo, indicating that NO may not mediate mitochondrial fragmentation by directly modulating Drp1 in PD models. So, we hypothesized that there may be other postmodifications of Drp1 to enhance Drp1 activity in PD. We found that Drp1 was dramatically phosphorylated at serine 616, leading to its activation and translocation to mitochondria. We speculated that NO signaling may activate related kinases, which in turn phosphorylate and activate Drp1. However, it remains to be determined the process by which kinases phosphorylate Drp1 in PD.
Together, our data indicate that MPP+-dependent effect on Drp1 operates at two levels: it affects Drp1 and Parkin interaction via augmentation of NO production and Parkin nitrosylation, and it affects Drp1 activation by impinging on Drp1 phosphorylation levels. It would be of great interest to determine how these two pathways interact. Despite this, we cannot exclude possible contributions from other signaling cascades. Together, our data uncover the mechanisms that might account for PD-linked mitochondrial fragmentation. These findings thus indicate that both the product of this pathway (Drp1) and SNO-Parkin implicated in induction appear to be potential targets for therapeutic intervention in PD.
However, Drp1 functions as a two-edged sword, and Drp1 blockade may not only exert beneficial effects but also negatively affect the viability of TH neuron, especially when Drp1 inhibition is pronounced. Identifying appropriate drugs that exert only a moderate effect on Drp1 activity will be essential to avoid excessive neuronal injury associated with complete Drp1 inhibition in neurons.
Materials and Methods
Cell culture and transfection
The human SH-SY5Y neuroblastoma cells and 293T cells were obtained from American Type Culture Collection. These cell lines were authenticated based on viability, recovery, growth, morphology, and isoenzymology by their suppliers. The SH-SY5Y and 293T cells were grown in DMEM (Gibco) supplemented with 10% heat-inactivated fetal calf serum, penicillin (50 U/ml), and streptomycin (50 μg/ml) in 5% CO2 in a humid incubator at 37°C. Transfections were performed with Lipofectamine 2000 reagent (Thermo Fisher–Gibco) according to the manufacturer's instructions. To obtain stable cell lines, cells transfected with indicated plasmids were selected in the presence of 0.8 mg/ml G418 (Sigma-Aldrich) for 2 weeks and the transfectants were enriched.
Primary mesencephalic cultures
The ventral mesencephalon, including substantia nigra, was removed from embryonic day 13 (E13) fetuses of Sprague-Dawley rats as previously described (28). Mixed cultures were plated on poly-L-lysine-coated six-well chamber slides coated with poly-D-lysine (Sigma) in neurobasal medium supplemented with 2% B27 (Thermo Fisher–Gibco), 0.5 mM glutamine, and 2 μM cytosine arabinoside to inhibit glial proliferation. At DIV6, cells were treated with indicated treatments. All cultures were kept at 37°C in a humidified 5% CO2-containing atmosphere.
Antibodies and other reagents
The antibodies used include the following: rabbit polyclonal anti-Drp1 (H-300), mouse monoclonal anti-Parkin (PRK8), and anti-β-actin (Santa Cruz); purified mouse anti-DLP1 (BD Transduction Laboratories); anti-COXIV, rabbit polyclonal anti-Drp1 (Ser616), rabbit polyclonal anti-Drp1 (Ser637), rabbit polyclonal anti-Bax, rabbit monoclonal anti-cytochrome c, rabbit monoclonal anti-cleaved caspase 3, nNOS antibody, iNOS antibody, and rabbit monoclonal anti-eNOS (Cell Signaling Technology); rabbit polyclonal anti-HA (Proteintech Group); mouse monoclonal anti-TH (Millipore); rhodamine 123 obtained from (Molecular Probes) to indicate mitochondrial transmembrane potential. MPP+, MPTP, L-NNA, C-PTIO, Mdivi-1, Aβ25-35 were purchased from Sigma-Aldrich.
Microscopy and quantification of mitochondrial morphology
DsRed and FITC fluorescence were monitored by LSM510 confocal microscopy (Carl Zeiss) equipped with a Plan-Neofluar 40 × /1.3 numerical aperture oil differential interference contrast objective. Excitation wavelength and detection filter settings for each of the fluorescent indicators were as follows: DsRed (He-Ne laser, Ex. 543 nm, Em. LP 615 nm); FITC (Art laser, Ex. 488 nm, Em. BP 500–550 nm). For quantification of mitochondrial morphology, SH-SY5Y cells were transfected with DsRed-Mit for mitochondrial visualization, and mitochondrial shape changes were performed by evaluating which cells displayed fragmented mitochondria after addition of the indicated dose of MPP+ for 24 h as previously described (16, 41). For primary neurons, mitochondrial morphology was quantified as described before (35).
NO detection by DAF-FM staining
DAF-FM diacetate (Beyotime, China) is an NO indicator for quantifying micromolar concentrations of NO. For detection of NO, cells were mixed with 5 μM DAF-FM for 30 min after treatment with indicated MPP+ for 24 h in the presence of 1 mM L-NNA or not. After incubation, the DAF-FM solution was removed, and cells were maintained in a medium to enable NO production. DAF-FM intensity was quantified in a FACSCanto II cytofluorimeter (19).
Real-time qPCR analysis
Total RNA was extracted from cells using the RNAiso Plus (Takara, Japan). ReverTra Ace® qPCR RT Master Mix with gDNA Remover (Toyobo) was used to synthesize cDNA. The following primers were used: Drp1: 5′-GATGCCATAGTTGAAGTGGTGAC-3′ and 5′-CCACAAGCATCAGCAAAGTCTGG-3′; β-actin: 5′-CCAGAGGCGTACAGGGAT AG-3′ and 5′-CCAACCGCGAGAAGATGA-3′. The cDNA was loaded into capillary tubes with SYBR Green qPCR Master Mix and then incubated in the LightCycler for an initial denaturation at 95°C for 10 s, followed by 40 cycles, each cycle consisting of 95°C for 5 s, 60°C for 10 s, and 72°C for 10 s. The expression of β-actin was measured to normalize targeted gene expression.
Biotin-switch assay
Analysis of SNO-Parkin and SNO-Drp1 by the biotin-switch assay was performed as described previously (30, 38). Briefly, cell lysates were prepared in HEN buffer (100 mM HEPES, 1 mM EDTA, and 0.1 mM neocuproine) with 1% Triton X-100 and 0.1% sodium dodecyl sulfate. Free thiols were blocked by incubation with 20 mM methyl methanethiolsulfonate (Sigma) for 30 min at 50°C. Cell extracts were then precipitated with acetone and resuspended in HEN buffer with 1% sodium dodecyl sulfate (SDS). SNO was selectively reduced by ascorbate to reform the thiol group and subsequently biotinylated with N-[6-(biotinamido)hexyl-3′-(2′-pyridyldithio)-propionamide] (biotin-HPDP; Soltec Ventures). The biotinylated proteins were pulled down with streptavidin–agarose beads (Thermo Fisher–Invitrogen) and detected by Western blotting. Total protein as a loading control was quantified by standard Western blotting analysis. Relative levels of S-nitrosylated proteins were determined by a comparison of S-nitrosylated proteins to the total amount of protein for each sample.
Animals and MPTP injection
The present study was performed in accordance with the guidelines of the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council). It was approved by the Institutional Animal Care and Use Committee of our university (South China Normal University, China). C57BL/6 mice were purchased from the Guangdong Medical Laboratory Animal Center, China. MPTP-induced model of PD was performed as described previously with slight modifications (12). Briefly, 8-week-old male C57BL/6 mice, weight 220–250 g, received intraperitoneal injections of MPTP in saline (20 mg/kg body weight) or saline alone once per day for 5 consecutive days. Animals were sacrificed by decapitation 7 days after the last injection, and samples were processed for analysis.
Immunofluorescence
Cells on 22-mm culture glasses were fixed with 3.7% paraformaldehyde for 10 min at room temperature, permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) for 15 min, and then blocked with 1% bovine serum albumin for 45 min, followed by overnight incubation with indicated primary antibodies at 4°C, three washes with PBS, and 2 h at room temperature with FITC-conjugated secondary antibodies (1:50; Proteintech Group). Images were acquired with a laser scanning confocal microscope (Zeiss LSM 510 META).
Subcellular fractionation
Subcellular fractions from cells were prepared as previously described (33).
Western blotting and coimmunoprecipitation
Western blotting was performed as previously described (18). Briefly, equivalent proteins were loaded on SDS-PAGE, transferred to the polyvinylidene difluoride membrane (Millipore), and blotted with indicated primary antibodies, followed by Alexa Fluor-conjugated secondary antibodies (Abcam). Detection was performed using the Odyssey infrared imaging system (LI-COR).
For coimmunoprecipitation, add an appropriate amount of the specific antibody to the cell lysates and gently rock at 4°C for 1–3 h. The immunocomplexes were captured by the addition of protein A-Sepharose (50% slurry) (Roche Applied Sciences) mixed at a 1:1 ratio overnight at 4°C. The beads were washed three times and collected by centrifugation at 12,000 rpm for 5 s. After the final wash, the beads were resuspended with SDS sample buffer, boiled for 5 min, and analyzed by Western blotting.
Transfection with siRNA for Drp1
Small interfering siRNA for Drp1 or negative control was obtained from GenePharma. Cells at 60–70% confluency were transfected with siRNA of Drp1 or control siRNA using Lipofectamine 2000 according to the manufacturer's protocol. For siRNA experiments, the cells were treated with specific siRNA for 48 h first, and then, the treated cells were used to perform various experiments as desired.
Cell apoptosis analysis
Quantification of apoptosis by FCM was performed as described previously (21, 37). Briefly, we used annexin V–FITC (0.1 μg/ml) and PI (0.5 μg/ml) for cell apoptosis analysis. Cell death was measured in a FACSCanto II cytofluorimeter (BD).
FACS analyses of ΔΨm
Quantification of mitochondrial membrane potential (ΔΨm) by FCM was performed as described previously (18).
Measurement of cell death and cell viability
Cell death and viability were analyzed by the Cytotoxicity Detection Kit (LDH; Roche) and ATP Determination Kit (Invitrogen), respectively.
Statistical analysis
Data are expressed as mean ± standard error of the means. Statistical analysis was applied using Student's t-test. Differences were considered statistically significant at p < 0.05. Representative data are shown for all other cases.
Footnotes
Acknowledgments
We thank Dr Y. Gotoh (University of Yokyo, Yayoi, Tokyo, Japan) for kindly providing the pDsRed-Mit plasmid; Dr Alexander M. van der Bliek (David Geffen School of Medicine at UCLA, USA) for kindly providing pHA-Drp1 plasmid; Prof. Zhou (Nankai University, China) for kindly providing myc-Parkin plasmid. This work was supported by the National Basic Research Program of China (2011CB910402; 2010CB732602), the Program for Changjiang Scholars and Innovative Research Team in University (IRT0829), the National Natural Science Foundation of China (31470072; 61405061), and the Natural Science Foundation of Guangdong Province, China (2014A030313419).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.