
Abstract
Significance:
Soluble guanylyl/guanylate cyclase (sGC) is the primary receptor for nitric oxide (NO) and is central to the physiology of blood pressure regulation, wound healing, memory formation, and other key physiological activities. sGC is increasingly implicated in disease and is targeted by novel therapeutic compounds. The protein displays a rich evolutionary history and a fascinating signal transduction mechanism, with NO binding to an N-terminal heme-containing domain, which activates the C-terminal cyclase domains.
Recent Advances:
Crystal structures of individual sGC domains or their bacterial homologues coupled with small-angle x-ray scattering, electron microscopy, chemical cross-linking, and Förster resonance energy transfer measurements are yielding insight into the overall structure for sGC, which is elongated and likely quite dynamic. Transient kinetic measurements reveal a role for individual domains in lowering NO affinity for heme. New sGC stimulatory drugs are now in the clinic and appear to function through binding near or directly to the sGC heme domain, relieving inhibitory contacts with other domains. New sGC-activating drugs show promise for recovering oxidized sGC in diseases with high inflammation by replacing lost heme.
Critical Issues:
Despite the many recent advances, sGC regulation, NO activation, and mechanisms of drug binding remain unclear. Here, we describe the molecular evolution of sGC, new molecular models, and the linked equilibria between sGC NO binding, drug binding, and catalytic activity.
Future Directions:
Recent results and ongoing studies lay the foundation for a complete understanding of structure and mechanism, and they open the door for new drug discovery targeting sGC. Antioxid. Redox Signal. 26, 107–121.
Introduction
N
Impaired NO signaling can lead to hypertension and atherosclerosis, and it can contribute to heart attack and stroke (8, 17). Targeting of NO signaling has long been a treatment goal for cardiovascular disease, beginning more than 150 years ago with administration of amyl nitrite (10) and nitroglycerin (71) to relieve symptoms of angina pectoris, although the mode by which these compounds act (release of NO) was not discovered until many years later. More recently, sGC has become a primary target for drug discovery (28).
sGC, the focus of this review, is generally composed of two homologous subunits, α and β, with each containing four recognizable domains that share sequence and structural similarity to bacterial proteins. There is a single heme moiety in the heterodimer that is coordinated to a histidine in the N-terminal domain of the β subunit (His 105 in the human protein). During signaling, NO binding to heme leads to the formation of a pentacoordinated Fe-NO complex with proximal histidine bond breakage (26, 103, 116) and stimulation of cyclase activity. Despite much study, how NO binding leads to cyclase activation remains poorly understood, although tantalizing new details are emerging. Likewise, new small-molecule stimulators and activators of sGC have been discovered, opening new doors for drug discovery in the treatment of cardiovascular diseases. However, where they bind and how they function remains unclear.
Here, we describe recent studies on sGC with a particular focus on discoveries since 2010 that reveal structure and provide insight into the signal transduction mechanism. Excellent reviews on sGC function (24, 36), physiology (12, 77), and drug targeting (28, 30) have recently appeared, and they provide summaries of earlier studies and topics beyond the scope of the present review. We refer the reader to these articles for additional information.
Genes and Domain Structure
Genes
sGC displays a fascinating evolutionary history. Insects, worms, algae, and invertebrates employ sGC, but bacteria, fungi, and higher plants apparently do not (29, 91). All sGC proteins are dimeric since the active site forms at the interface of two catalytic domains, one from each subunit. Each sGC subunit is composed of four recognizable domains—a heme domain, a Per-ARNT-Sim (PAS) domain, a coiled-coil signaling helix, and a cyclase domain—that are fused together into a single polypeptide chain of 600–700 amino acids (Fig. 1). Each sGC domain has a bacterial homologue, but the intact sGC gene is more recent. Certain insect and all vertebrate sGC proteins contain one β chain and one α chain in which the ability to bind heme has been lost. Two potential catalytic sites have been reduced to one functional site that utilizes residues from both subunits. Although the other pseudosymmetric site is non-catalytic, it may still bind ligands and serve a regulatory function.
sGC may have first arisen in green algae. For example, the unicellular flagellate Chlamydomonas reinhardtii, a well-studied model organism, contains at least three genes coding for possible sGC proteins, CYG12, CYG15, and CYG56. These genes vary considerably in length—coding for proteins of 991, 619, and 1721 amino acids—but each contains all of the expected sGC functional domains. All three have higher similarity with the β chain of vertebrate sGC than with the α chain, and all have the residues needed for forming a functional homodimeric cyclase. That these proteins can, in fact, form functional homodimers has been demonstrated through bacterial expression of the cyclase domains of both CYG12 (119) and CYG56 (20). Thus, sGC appears to have begun as a homodimeric protein.
In Caenorhabditis elegans (worm), there were a series of gene duplications yielding seven β-subunit-like genes, all of which appear to retain heme domains. These, too, vary in length, particularly at the C-terminus. Although not well characterized, the C. elegans sGC proteins appear to be oxygen sensors rather than NO sensors and to function in worm neurons to detect and avoid environments that are too rich or too poor in oxygen (79, 128). Most, but not all, of the residues that are considered to be necessary for homodimeric activity are intact in these genes, suggesting that the corresponding proteins may also function as homodimers. However, genetic knockout studies indicate that they may function in heterodimeric pairs.
Gene duplication to yield the sGC α subunit and α/β heterodimer was well established once insects crawled into existence, and many examples of such proteins are known. We have characterized an α/β sGC from Manduca sexta, the hawkmoth, where the protein is required for odor detection and neural activity (18, 40, 72). Manduca sGC behaves much like vertebrate α1/β1 sGC and is considered an α1/β1 homologue (32, 33, 41, 42, 80, 81). Atypical sGC proteins are also abundant in insects. These proteins are similar to the typical β subunit but may have lost heme binding and appear to function as homodimers (67).
In vertebrates, both the α and β subunits have been duplicated, yielding subunits α1, α2, β1, and β2 (24). The α1/β1 heterodimer is equivalent to the insect α/β dimer and is widely expressed in mammalian tissues. The α1/β1 heterodimer is the most thoroughly studied of the sGC proteins and is the primary focus of this review. An α2/β1 heterodimer is prevalent in nerve cells, but a role for the β2 subunit has yet to be reported, although it retains all of the residues needed for cyclase activity and may function as a homodimer. Alternative splice variants for the human protein have also been detected (60, 96). These are described in more detail elsewhere in this Forum.
Heme-binding domain
A ferrous b-type heme resides in the N-terminal domain of the β subunit and is coordinated through a histidine residue, His 105 in α1/β1 heterodimers (Fig. 2). This domain was first identified as a “Heme NO Binding” domain (45), or an HNOB domain, and this nomenclature persists throughout most sequence databases. Bacterial homologues of the HNOB domain were later discovered to bind oxygen as well as NO, leading to the suggested change in name to H-NOX for heme-nitric oxide oxygen binding (78). H-NOX is the most common name in the literature and is used here. Sensor of nitric oxide (SONO) has been suggested for the domain (73) and is also still in use.

The α1 subunit has an extra ∼65-residue N-terminal extension that is predicted to be intrinsically disordered. No function has been assigned to these residues, although one report indicates that Ser 64 of the human protein may be a target for phosphorylation by protein kinase G (PKG), which is stimulated by cGMP, leading to feedback inhibition (127). Removal of this domain from Manduca sGC leads to greater stability in recombinantly expressed proteins but has little other consequence (42).
Following this N-terminal string of residues of the α subunit is an “H-NOX” domain that has lost the ability to bind heme and is thus perhaps best referred to as a pseudo-H-NOX domain. This is the least conserved portion of α1/β1 sGC but, nonetheless, appears to have a regulatory role in signal transduction and also appears to retain an H-NOX fold (42). Removal of this domain in recombinant Manduca sGC leads to enhanced CO, and presumably NO, binding to heme (80). Interestingly, one of the splice variants for human α1/β1 sGC is missing this domain (96), which may affect its cellular location (50).
PAS domain
Following the H-NOX domain is a short linker and a PAS domain (also called HNOBA in sGC for HNOB-associated domain). The linker length is conserved in the α1/β1 heterodimeric sGC proteins, but it is quite variable in many β-only sGC proteins. The role of the PAS domain in sGC function is unclear but may enhance dimer formation (55), dampen NO affinity to heme (33), and/or participate in heat shock protein 90 (hsp90)-assisted heme insertion into the β subunit (90).
PAS domains are common in nature and are found both as stand-alone proteins and as components in multi-domain signaling proteins (65). Many PAS domains bind ligands or cofactors such as heme or photoactivatable chromophores and are involved in sensing and signal transduction. Some are connected to coiled-coil signaling helices much like the arrangement in sGC. The first crystal structure for an sGC-related PAS domain was for a domain from a Nostoc punctiforme signal transduction histidine kinase (STHK), which revealed a typical PAS domain fold and a potential dimer interface, but no ligand-binding pocket (55). Notably, this domain is attached to a putative coiled-coil signaling helix (55), much like that predicted for sGC, suggesting that an evolutionary link may exist between sGC and more ancient STHK proteins. The crystal structure of the Manduca α1 PAS domain displayed a similar fold and a small pocket in the location where ligands often bind to PAS domains (Fig. 3); however, no ligand-binding activity has been discovered for sGC PAS domains.

Coiled-coil domain
Following the PAS domain is a predicted short linker and a coiled-coil domain comprising two helices, one from each subunit. PAS-coiled-coil arrangements are common in signaling proteins, particularly in bacterial STHK proteins such as those from N. punctiforme, leading to the suggestion that such domains play a functional role in signal transduction (2). Coiled-coil helices have a characteristic amphipathicity with a solvent-exposed hydrophilic face and a hydrophobic face buried in the coiled-coil interface. Generally, there is a repeating heptad motif (two turns of an α helix) in which bulky hydrophobic residues such as leucine and isoleucine appear at the “a” and “d” positions of the heptad of each helix, allowing for excellent “knobs-in-holes” packing at the coiled-coil interface (2). The external surfaces are often charged, and electrostatic interactions may be important for proper coiled-coil formation and alignment.
In α1/β1 sGC, the predicted coiled-coil region is ∼50 amino acids long and capped on each end by one or more prolines. Coiled-coil helices can have either parallel or antiparallel chains and the correct orientation can be difficult to predict based on sequence alignment alone, due to the repetitive nature of the heptad repeat. A crystal structure of the rat β1 sGC coiled-coil strand revealed a homodimeric coiled coil in an antiparallel arrangement (54). The authors argued that this was likely an artifact based on electrostatic considerations and that the α1/β1 heterodimeric coiled coil was likely parallel (54). This conclusion was later confirmed by chemical cross-linking analyses that identified cross-links that were only consistent with parallel helices (33). A model for the α1/β1 coiled coil is shown in Figure 4.

Cyclase domains
Signaling through formation of cyclic nucleotides is found in all kingdoms of life and includes not only cAMP and cGMP formation but also formation of the cyclic dinucleotides c-di-GMP and c-di-AMP (37). Cyclic AMP formation occurs in all life forms, whereas cGMP formation is common in animals but is more sporadic in lower eukaryotes, plants, and bacteria.
The sGC cyclase domains are members of the class III cyclase family, the largest cyclase family, and each contains a cyclase homology domain (CHD) (100). The active site in class III cyclases forms at the interface of two CHDs, which can either reside in a single polypeptide or come together through homodimerization or heterodimerization, the arrangement found in α1/β1 sGC. This dimeric arrangement leads to two potential active sites; however, in most cyclases containing two different CHDs, only one of the possible active sites remains catalytically active (Fig. 5). Despite loss of catalytic residues, the other “pseudosymmetric” site may retain a functional binding pocket that binds regulatory ligands. For example, forskolin, a natural product that stimulates cAMP production, binds to the pseudosymmetric site of adenylyl cyclase (107, 125).

Several crystal structures of adenylyl cyclase domains with bound ligands are known and have led to a model for cyclase domain catalysis in which the ribose 3′ hydroxyl performs an in-line nucleophilic attack on the 5′ α phosphate, leading to pyrophosphate displacement and release (39, 100). Two metal ions are required for the reaction, which can be either Mg2+ or Mn2+, with Mg2+ being of greater importance in vivo.
Two crystal structures of heterodimeric α1/β1 sGC cyclase domains have been reported (1, 94), but, unfortunately, neither structure includes ligands. The overall fold in both structures is similar to that of adenylyl cyclase, with the exception of a small change in the dimer interface that leaves the active site misaligned for efficient catalysis. Catalytic activity for the truncated cyclase domains is quite low, consistent with misalignment of the individual domains, leading to the suggestion that small changes in the H-NOX, coiled-coil, or PAS domains might have large effects on catalytic rates (94). Interestingly, the N-terminal “dorsal” face of the cyclase, which is away from the catalytic pocket, is highly conserved in α1/β1 sGC and may provide a functionally important interface with other domains (94). The pseudosymmetric site is collapsed in the cyclase crystal structures and is not capable of binding a ligand the size of forskolin, even after realignment to accommodate GTP binding (1). This site has been suggested as a binding site for ATP (106), which acts as an inhibitor of sGC and appears to have a distinct allosteric binding site (14, 23, 88, 106). For ATP to bind to the pseudosymmetric site, however, additional rearrangement of the domain interface would be needed.
Signal Transduction
Although considerable information is available on sGC domain structure, how these domains are arranged to yield a sensing and signaling unit is much less clear. Binding of NO to the heme domain leads to a signaling cascade that ultimately increases the catalytic rate by as much as 200 fold. NO binding both lowers K m for GTP and increases k cat (16, 21, 47, 82, 95). A hallmark of NO binding is the release of the proximal histidine from heme coordination, yielding a five-coordinate nitrosyl species, but whether histidine release is simply correlated with activation or is the leading edge of the signaling cascade remains unknown. CO binding, which leads to a six-coordinate species with proximal histidine attached, can also fully activate sGC, but only in the presence of sGC-stimulating compounds, suggesting that proximal histidine breakage is not required for full sGC activity. Additionally, it remains unclear as to how many NO molecules are required for full stimulation, with some data indicating that two or more molecules are required (106). Other complications include the potential roles for nucleotide-based allostery and post-translational modifications.
In this section, we review recent data that are beginning to unravel the domain arrangement in sGC and the signal transduction mechanism. Taken together, these data lead to two models for signal transduction. The first is a model in which the β1 H-NOX domain inhibits the cyclase domain through direct contact. The second is a model in which signaling is transduced from the β1 H-NOX domain through the coiled coil, leading to cyclase domain rearrangement and catalytic activation.
Domain arrangement
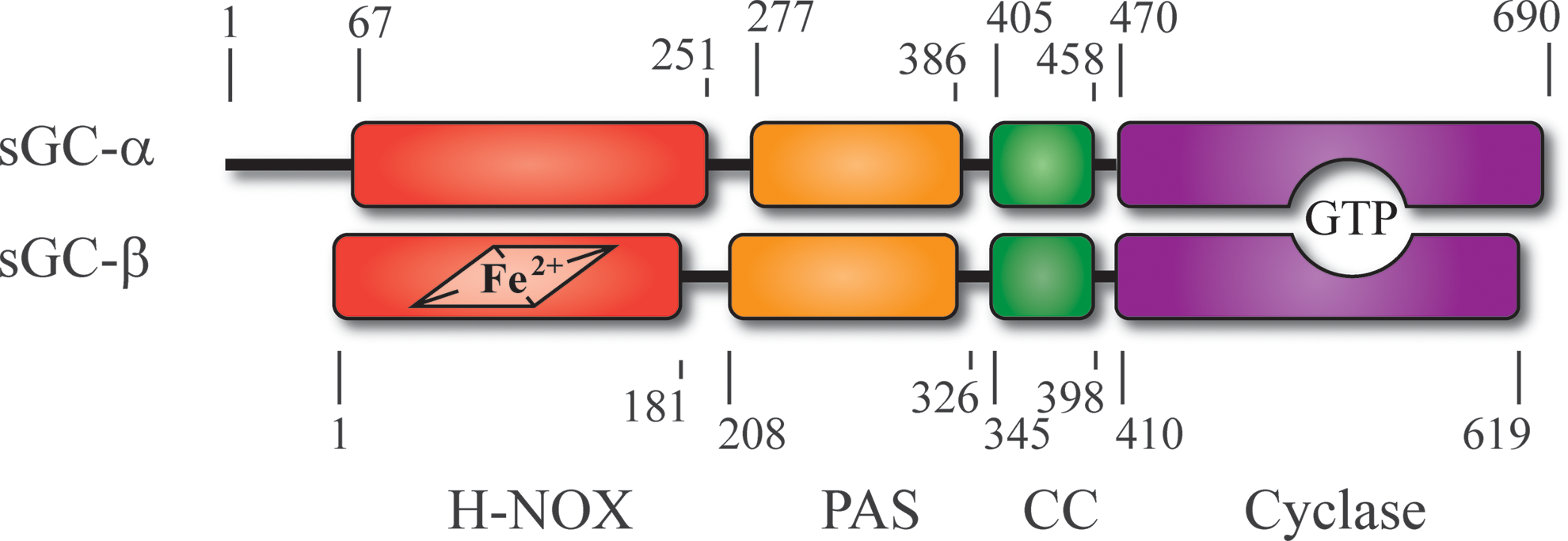
Our group uncovered domain–domain contacts among the H-NOX, PAS, and coiled-coil domains by using chemical cross-linking of a truncated form of M. sexta sGC lacking the cyclase domains, referred to as Ms sGC-NT (33). In this study, amine–amine and amine–carboxylate chemical cross-links were introduced into the recombinant protein, and they were identified by high-resolution tandem mass spectrometry. Both inter-subunit and intra-subunit cross links were identified in this way. The first major conclusion from these data was that the coiled-coil domain was indeed formed from parallel helices, as predicted based on electrostatic considerations (54). The second major conclusion was that the α1 H-NOX and PAS domains were in contact with the β1 H-NOX domain, and that all of these domains appeared to assemble onto the coiled-coil domain (Fig. 6).
We obtained a molecular envelope for Ms sGC-NT by using small-angle X-ray scattering (SAXS) and analytical ultracentrifugation (AUC), revealing an elongated molecule with approximate dimensions of 115 × 90 × 75 Å. The scattering profiles in these experiments indicated that Ms sGC-NT was well folded but exhibited some flexibility. Little change was observed in this envelope on addition of NO, CO, or stimulator compounds, suggesting that the conformational changes that occur on ligand binding are small (on the order of a few angstroms) for the truncated protein. We constructed an energy-minimized molecular model by using homology models for each domain and distance restraints from the cross-linking and SAXS data. The resulting model had the coiled coil parallel to the long axis of the molecular envelope with the other domains assembled, on average, onto one coiled-coil face (Fig. 6). The other coiled-coil face was open, providing a potential site for the cyclase domains.
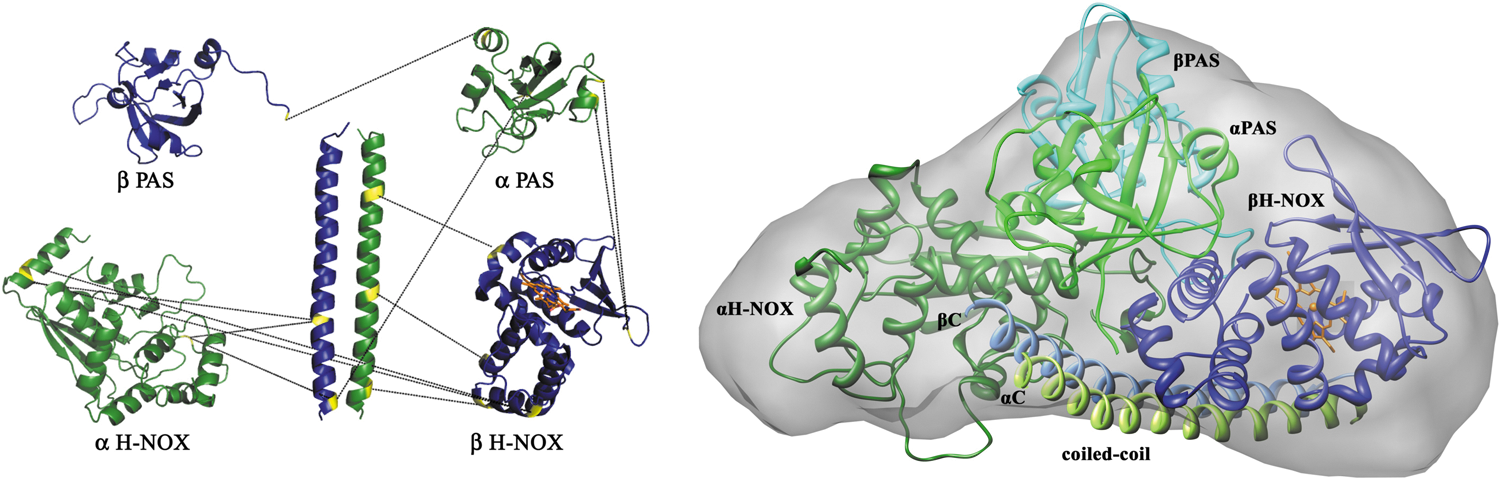
Campbell et al. generated a molecular model for the full-length protein from rat by using negative-stain single-particle electron microscopy (13). Their images indicate that sGC may exhibit a wide range of conformations (Fig. 7). The α1/β1 H-NOX and PAS domains occupy an elongated envelope similar to that derived from SAXS analysis. However, these domains appear to occupy multiple orientations with respect to the coiled-coil domain. In some images, they lie alongside of the coiled coil, similar to the SAXS model arrangement, but in other images they appear perpendicular to the coiled-coil domain (Fig. 7). The cyclase domains appear at the opposite end of the coiled coil and perpendicular to the helix axis. A similar distribution of conformations was observed in both the presence and the absence of NO binding or on binding of a non-hydrolyzable GTP analog.
Full models were constructed by using the molecular envelopes provided by electron microscopy (EM), homology models for each domain, and the constraints provided by the previously published cross-linking data. In their modeling, the authors assumed that not all cross-links need to occur at the same time; rather, they are derived from all conformations. In some of the resulting models, the β1 H-NOX is in direct contact with the cyclase domain, where it might directly inhibit cyclase activity, but in others there is no contact. Thus, the models rule out neither proposed activation mechanism–release of direct H-NOX inhibition of cyclase or indirect domain rearrangement.
In a third approach, Behrends and colleagues used Förster resonance energy transfer (FRET) to examine domain proximities. In one experiment, yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP) were fused onto the N- and C-termini of α1/β1 human sGC in various combinations, and FRET efficiency was measured. The data indicated efficient energy transfer between a construct with YFP fused to the α1 C-terminus and CFP fused to the β1 N-terminus (α1-YFP/CFP-β1), leading the authors to conclude that the β1 H-NOX and α1 cyclase are near one another (38). In a second approach, FRET was measured between endogenous tryptophan residues and a fluorescent GTP analog (11). In these studies, NO binding induced enhanced FRET between the fluorescent GTP analog in the active site and a tryptophan residue in the β1 H-NOX domain, or between the fluorescent GTP and a tryptophan in the linker connecting the α1 coiled-coil domain and the α1 cyclase domain. The authors conclude that the β1 H-NOX and the coiled coil are near the cyclase domains and that NO-induced movements force the catalytic domains into the active conformation. These data are more consistent with a compact sGC conformation than with a more extended conformation, and with relatively small NO-induced conformational changes taking place.
Underbakke et al. used hydrogen–deuterium exchange mass spectrometry to assess domain interfaces and to identify residues with altered solvent exposure on NO binding (112, 113). Major changes in hydrogen–deuterium exchange rates were found for the β1 H-NOX domain and the cyclase domain on NO binding to full-length rat sGC, as expected. These changes included the H-NOX F-helix, which contains proximal His 105, and active site catalytic residues α1 Asp 485 and α1 Asp 529 (rat numbering). Interestingly, substantial changes were also observed for the coiled-coil helix domain and for the highly conserved N-terminal dorsal face of the catalytic domains, suggesting that signal transduction may proceed through these regions of the protein. However, the authors conclude that these data do not rule out either of the primary models for NO stimulation: relieving of a direct inhibitory interface between H-NOX and cyclase domains, or rearrangement of the cyclase domains to the active conformation through changes in the coiled-coil domain.
Taken together, these data indicate that sGC forms a compact signaling complex containing H-NOX and PAS domains, a long coiled-coil domain with parallel helices, and a compact cyclase domain. The functional arrangement of these three regions remains unclear, as do the magnitudes of NO-induced conformation changes. Most techniques employed so far suggest that NO-induced changes in conformation are small in scale. However, images using electron microscopy clearly show that large changes in sGC conformation can occur. Caveats exist with all of the experiments addressing domain arrangement and dynamics. Studies with truncated Ms sGC-NT are lacking the cyclase domains, which may influence dynamical behavior. The SAXS and AUC analyses are low-resolution techniques and reveal an average conformation. Multiple coiled-coil orientations might not be readily detected by these approaches, since the coiled coil contributes only ∼11% of the total Ms sGC-NT mass. The electron microscopy is of sGC coated with uranyl formate and dried, which could be harsh. The full-length recombinant protein samples used likely contain a mixture of heme-loaded and heme-free (apo) proteins, based on the ratio of absorbances at 430 nm (heme Soret) and 280 nm (measuring protein) generally reported, which could also affect conformational mobility. The A 430/A 280 should be ∼1 for the full-length protein (27, 62) but is often much less than 1 for the recombinant proteins.
Of the models most commonly presented, we favor one in which signal transduction occurs through NO-induced changes in the coiled-coil helices. Such a model would allow signal transduction in a compact or an extended domain arrangement, and in a static or highly flexible molecule, as implied by the SAXS and EM data. In support of this model, small changes in coiled-coil length yield large changes in ligand affinity (described later) whereas ligand binding results in little change to SAXS and AUC molecular envelopes for Ms sGC-NT, or conformation distribution for full-length sGC as seen by EM.
Coiled-coil helices are increasingly discovered to play an active role in signal transduction and for this reason are sometimes referred to as signaling helices. Despite their widespread occurrence, the underlying mechanisms governing signal transduction through signaling helices are largely unknown (2). Signaling through coiled-coil helices has precedent in bacterial two-component systems, from which the sGC PAS-coiled-coil arrangement likely evolved, and in related bacterial proteins. One example is the sensor histidine kinase YF1, which appears to signal via a change in coiled-coil helix cross-over angle (25). A second example is for the transcriptional regulator DhaR, which appears to signal through a rotation of coiled-coil helices (98). In both examples, activation occurs through small changes in domain orientation rather than through large structural rearrangements. These data make clear that signal transduction through the coiled coil is a viable possibility for sGC. Determination of whether it is the correct mechanism awaits the results of high-resolution structural studies.
Linked equilibria
It has long been known that NO binding to the heme in sGC enhances GTP affinity for the cyclase domain, leading to a lower K m value (114). NO also enhances binding of other nucleotides, including product cGMP, which leads to increased product inhibition (57). The reverse should also be true, but the demonstration of enhanced NO binding in the presence of nucleotide has been more challenging to directly demonstrate due to the complicated nature of NO binding. One challenge is the very high affinity of NO for heme after proximal histidine release (87, 110). A second is the possibility that multiple NO binding sites reside in the protein (14, 110). Another complication is an apparent second binding site for nucleotides, which induce additional allosteric regulation (14, 16, 88, 106, 121). Nucleotide binding clearly changes NO-binding behavior, but precisely how remains unclear. Published conclusions are influenced by the time scale of measurement, which spans picoseconds to seconds, and the concentration of NO used, which is often in the micromolar range, yielding a robust signal, whereas binding affinity is in the nanomolar to picomolar range. These factors paired with the inherent reactivity of NO make key NO binding and release measurements difficult to acquire, and data interpretation challenging and controversial (14, 87, 89, 110).
Despite these challenges, recent studies make it clear that individual sGC regulatory domains modulate heme affinity for distal pocket ligands such as NO and CO. The inherent affinity of the β1 H-NOX domain for CO, and presumably NO, is high in the absence of other domains, displaying a value as low as K d = 200 nM for Ms sGC (80). Binding affinity is reduced as additional domains are included: CO-binding affinity decreases ∼10 fold for an Ms sGC construct containing the α1 PAS and coiled-coil domains, and ∼250 fold for an Ms sGC-NT construct possessing an intact α1 H-NOX domain in addition to the α1 PAS and coiled-coil domains (80). A similar trend is observed with bovine sGC, although CO binding to the bovine protein is, in general, weaker than to the Manduca protein (80). Much of the increased affinity derives from an enhanced on rate for bimolecular binding to the isolated heme domain and an increased rebinding (geminate binding phase) on dissociation (122).
Taken together, these data suggest that the other domains in sGC either induce a strained heme geometry that binds gaseous ligands less well or induce an open heme pocket that facilitates ligand escape. Addition of allosteric stimulators such as nucleotides or stimulating compounds such as YC-1 (described in a later section) relieve this strain or close the heme pocket, allowing for tighter binding of NO and CO.
Coiled-coil domain influence on NO binding
Kinetic measurements also provide insight into the mechanism behind sGC signal transduction, particularly with respect to the coiled-coil domain. In stopped-flow kinetic measurements with full-length sGC, the transient six-coordinate NO complex can be detected and the rate of its decay to a five-coordinate species can be measured, leading to a rate constant of ∼10 s−1, depending on temperature, NO concentration, and other experimental variables (110, 123, 126). For the β1 H-NOX alone, decay of the six-coordinate intermediate is accelerated and too fast to be observed in rapid-mixing experiments (126). Interestingly, truncated versions of Ms sGC containing the full coiled-coil domain also display slow proximal His 105 release, again with a rate constant of ∼10 s−1 (42). However, a 10-residue truncation of the coiled coil speeds up proximal His 105 cleavage to >100 s−1 such that it can no longer be observed by stopped-flow spectrophotometry (33). These data are consistent with signal transduction occurring through coiled-coil rearrangement, such as occurs in the bacterial two-component systems discussed earlier.
Heme Domain Properties
Trapping gas with ferrous heme
Ferrous [Fe(II)] heme can bind to numerous gaseous ligands, including O2, CO, and NO. Oxidation to ferric [Fe(III)] heme leads to loss of O2 and CO binding, and to reduced NO-binding affinity. Globins use ferrous b-type heme for O2 transport and storage, whereas the nitrophorins use ferric b-type heme for NO transport and storage during feeding by certain bloodsucking insects (66, 83, 115, 117, 118). A hallmark of sGC is the use of ferrous b-type heme to tightly bind NO but discriminate against O2, which could lead to an unwanted reaction with NO in the heme pocket. Oxygen binding to sGC has been described, but only at a temperature of 77 K, and only yielding partial saturation in the absence of stimulating compounds (56).
Two factors are likely responsible for the low oxygen affinity of sGC heme. First, oxygen binding is inherently weak to ferrous heme. For example, oxygen binding to an imidazole-capped model small-molecule heme displays K d = 5 μM (15), a value about 103 fold larger than for CO binding, and 106 fold larger than for NO binding (86). In myoglobin, O2 binding is improved about 100 fold through hydrogen bonding to the distal-pocket histidine [reviewed in Ref. (109)], but no such group exists in sGC. Tt H-NOX, however, does bind O2 and binding is stabilized through a distal-pocket tyrosine (78). Introduction of a distal-pocket tyrosine into sGC led to mixed results, with binding apparently occurring for the isolated H-NOX domain but not in the full-length protein (9, 22, 58). However, oxygen binding could be detected in full-length sGC on addition of a second distal-pocket mutation in a position suggested by homology modeling of the Drosophila sGC Gyc-88E, an atypical sGC that does bind oxygen (22). Thus, the absence of an appropriate hydrogen-bond donating group likely contributes to the lack of oxygen binding in the wild-type protein.
The second factor limiting oxygen binding is an overall decreased affinity for all gaseous ligands through changes in the heme pocket or heme geometry. Tsai et al. have proposed a “sliding scale rule” for ferroheme proteins such that the affinity ratios of O2:CO:NO are always about 1:103:106 for imidazole-coordinated heme (109, 111). Using the sliding scale model, Tsai et al. predict binding of O2 to full-length human sGC to have K d = ∼1 M. Since the concentration of O2 in air-saturated solutions at room temperature and atmospheric pressure is ∼250 μM, significant binding should not occur and, indeed, binding is not seen even in fully oxygenated solutions (102).
As previously noted, one likely role of the other sGC domains is to modulate the H-NOX domain affinity for NO and other gaseous ligands, including O2, leading to overall lower affinity for all ligands (80, 122). For example, the inherent CO affinity for the β chain H-NOX domain is much higher when isolated than when placed in the full-length protein (80). Importantly, although O2, CO, and NO have lower affinity to six-coordinate full-length sGC than to other ferroheme proteins, NO binding leads to proximal histidine release and an ∼104-fold increase in NO affinity, resulting in picomolar K d values. This high-affinity state for NO appears to be disproportionately enhanced through a change in conformation that stabilizes proximal histidine displacement.
The mechanism behind the overall tuning of gaseous ligand affinity could be through changes in heme geometry, changes in protein conformation that alter the ability for ligands to escape, or a combination of both. Inducing a conformation with heme iron pulled below the heme plane (toward the proximal pocket) would reduce ligand affinity, whereas placing the heme iron into or above the heme plane (toward the distal pocket) would increase ligand affinity. This mechanism is used to alter oxygen affinity in hemoglobin on switching between R and T states. The bacterial H-NOX proteins have decidedly non-planar heme geometries, which lends support to this model, and heme flattening is suggested to be key for signal transduction in these proteins (70). Alternatively, blocking escape from the heme pocket, leading to geminate recombination rather than escape from the protein, could also enhance affinity. This mechanism is used by the nitrophorins, which transport NO and enhance binding at low pH by blocking the distal pocket with two conformationally mobile loops (6, 117). Both binding of compound YC-1 to sGC (41) and removal of the α1 chain (122) lead to enhanced geminate recombination, but whether this is due to a change in heme geometry, a blocking of ligand escape, or both is yet to be determined.
Reduction potential
sGC is designed to function as a ferrous heme protein, which allows it to sense very low NO concentrations, in the picomolar range (4, 120). At these concentrations, NO side reactions are minimized and sGC becomes the primary NO receptor. Ferric heme can also bind NO, but binding affinity drops to the nanomolar to micromolar range (108). To preserve picomolar NO affinity, the sGC heme is highly stabilized in the ferrous state, considerably more so than the same heme in hemoglobin or myoglobin. The midpoint potential measured for truncated Manduca sGC was +234 mV (32), whereas that for the full-length bovine protein was measured as +187 mV (56). The midpoint potential for the bacterial Tt H-NOX was measured as +167 mV, indicating that the stabilized ferrous state for this domain is highly conserved.
Heme reduction potentials are set by the binding pocket in heme proteins and can vary by nearly a volt overall. How this is accomplished remains incompletely understood, but two factors have been investigated. First, the electrostatic environment is important, with ferric heme, having a formal charge of +1, often stabilized through nearby negative charges. For example, the Rhodnius nitrophorins, which are ferric heme proteins, display a midpoint potential of about −300 mV and have a heme pocket surrounded by numerous Glu and Asp residues. Mutation of these residues shifts the midpoint potential to be more positive (ferrous) (7). H-NOX domains generally have quite hydrophobic heme pockets, which are consistent with stabilizing the ferrous state.
A second factor considered to influence the midpoint potential is heme planarity. Heme distortion is often conserved in heme proteins and greater distortion, particularly heme “ruffling,” has been correlated with more negative (ferric) midpoint potentials (97). The nitrophorins, for example, display among the most distorted protein-bound hemes known (85) and are stabilized in the ferric state (3, 99). However, the bacterial H-NOX proteins also display highly distorted heme geometries but are highly stabilized in the ferrous state, just the opposite of the nitrophorins. Mutations designed to test the role of heme distortion in setting the reduction potential are intriguing but have not resolved the issue. Mutations designed to flatten the nitrophorin heme led to more positive (ferrous) midpoint potentials for the nitrosyl complexes; however, results for the aqua complexes were mixed (99). Mutations that flatten the Tt H-NOX heme led to more negative (ferric) midpoint potentials (74, 75), leading the authors to suggest that heme distortion stabilizes the ferrous state, the opposite of that proposed for the nitrophorins. Binding pocket hydrophobicity and heme distortion in sGC are likely to be critical for function; however, understanding how these factors influence the ferrous state remains to be uncovered.
sGC as a Drug Target
Unwittingly, sGC has long been a target for drugs that deliver NO for the treatment of cardiovascular disease, beginning with organic nitrates such as glyceryl trinitrate (nitroglycerin) in the 1860s to treat angina pectoris [reviewed in Ref. (19)]. Organic nitrates, which are metabolized to release NO, remain a staple treatment for relieving the acute symptoms of congestive heart failure, coronary artery disease, and arterial hypertension; however, nitrate tolerance rapidly reduces the effectiveness of these compounds, possibly in part through a mechanism leading to sGC heme oxidation (46).
More recently, compounds that directly enhance sGC activity have been discovered. The first of these was the compound YC-1 (Fig. 8), a benzylindazole derivative that can inhibit platelet activation through stimulating sGC (49). YC-1 and related compounds stimulate sGC activity two- to fourfold in the absence of NO, but they act synergistically with CO or NO to achieve several hundred-fold activation (31, 104). The compounds require an intact heme for activity and are commonly referred to as sGC stimulators [reviewed in Refs. (28, 30)]. YC-1 binding can also overcome inhibitory phosphorylation of sGC (82). YC-1 optimization led to numerous related stimulator compounds, including BAY 41-2272, which has seen extensive use for investigating stimulator mechanism, and compound BAY 63-2521 (riociguat), a YC-1 derivative that has recently completed phase III clinical trials (5, 34, 35, 64) and is now FDA approved for treatment of pulmonary hypertension (marketed as Adempas). However, despite much work, where YC-1 and related stimulator compounds bind to sGC and how they increase catalytic activity remains unknown, although it now appears likely that binding is directly to the β1 H-NOX domain, as discussed next.

A second class of compounds, referred to as sGC activators, target heme-depleted sGC, which can occur in response to inflammation and oxidative stress (30). Prominent among these are compounds BAY 58-2667 (cinaciguat, Fig. 8) (93) and HMR1766 (ataciguat) (92). Activators are proposed to occupy the heme cavity in sGC after heme oxidation and loss, thereby both stimulating activity and reducing proteasomal degradation (63). A crystal structure of cinaciguat bound to the heme-depleted bacterial Nostoc H-NOX domain (61) provides strong evidence in support of this model (Fig. 9). In the structure, cinaciguat occupies the heme pocket and folds into a heme-like conformation with its two carboxylate substituents arranged much like that of the two heme propionates in the native structure. Helix αF, which contains the proximal histidine, occupies a conformation that is considered to occur in NO-activated sGC, which is consistent with cinaciguat acting as an sGC activator.

Although the mechanism underlying activator function seems clear, how stimulator compounds bind to and stimulate sGC remains a mystery. Photoaffinity labeling of rat sGC with a BAY 41-2272 analog revealed cross-links to Cys 238 and Cys 243 in the α1 H-NOX domain (101), suggesting that this domain contains the compound binding pocket, an attractive possibility since homology modeling suggested that a remnant of the heme-binding pocket might be retained in the domain (42). A second attractive possibility is for binding to occur in the pseudosymmetric site of the cyclase domains, much as forskolin binds in adenylyl cyclase (51), and this suggestion remains popular (57, 68, 121) despite strong data indicating that binding occurs elsewhere.
The most compelling data indicate that stimulator compounds bind to the N-terminal portion of the β1 chain, most likely to the H-NOX domain itself. Binding of the compounds to full-length sGC (i) enhances CO binding (48); (ii) leads to greater geminate recombination in photolysis experiments (124); (iii) leads to a small shift in the heme Soret absorption band for the CO complex (48, 124); and (iv) leads to changes in the resonance Raman vibrational spectra (43, 53, 59, 76). Importantly, all four binding indicators occur with full-length sGC as well as with truncated sGC lacking the cyclase domains or the entire α1 subunit. For example: • Binding of stimulator PF-25 (84) to Ms sGC β1 1-380, which contains the β1 H-NOX, PAS, and coiled-coil domains, but lacks both cyclase domains and all of the α1 chain, was directly measured by using surface plasmon resonance (SPR) (80). The measurements revealed PF-25 K
d values of 7 μM (+NO) and 92 μM (−NO). • CO-binding affinity for an Ms sGC-NT construct containing α1 272-450 (PAS-CC) and β1 1-380 (H-NOX-PAS-CC) is enhanced 11 fold by YC-1 and 31 fold by BAY 41-2272 (80), indicating that functional binding does not require the α1 H-NOX domain or either of the cyclase domains. • Binding of either YC-1 or BAY 41-2272 to CO-ligated Ms sGC-NT constructs induces an ∼2 nm shift in the Soret band (41, 80), allowing for compound affinity to the CO complexes to be measured via compound titration. K
d values for YC-1 ranged between 600 and 800 nM using this approach, depending on construct, and between 80 and 90 nM for BAY 41-2272. • Binding of YC-1 or BAY 41-2272 to the CO complex of Clostridium botulinum H-NOX (Cb-SONO) (124) also exhibited a shift in Soret band maximum, suggesting that binding may be conserved in all H-NOX domains. • Binding of YC-1 or BAY 41-2272 to Ms sGC-NT (41), bovine β1 H-NOX (residues 1–200) (124), and Cb-SONO (H-NOX) (124) leads to faster rebinding of CO and increased geminate recombination after flash photolysis. • Resonance Raman spectra for YC-1 ligated protein (CO-complex) are similar for the full-length bovine protein and a construct containing only residues β1 1-385 (H-NOX-PAS-CC) (43).
In contrast, one recent report examined binding of YC-1 and related stimulator compounds to the isolated cyclase domains by SPR, and found binding to occur (68). These measurements were for a single compound concentration (100 μM) and displayed rapid association and dissociation kinetics, which did not allow for affinity constants to be determined. In a separate study, YC-1 was unable to stimulate catalytic activity in the isolated cyclase domains (23), suggesting that the binding to the cyclase domains seen by SPR was non-specific rather than functional, at least for the isolated domains. Despite the possibility that some level of binding may occur in the cyclase domains, overall, the data strongly indicate that functional binding for stimulator compounds is in the H-NOX domain.
Linked equilibria for drug binding
A concern with some of the stimulator binding studies is the high concentration of YC-1 used (as high as 200 μM), which could result in non-specific binding and generate non-specific spectral changes. However, only nanomolar quantities of YC-1 or BAY 41-2272 are needed to induce a shift in the Ms sGC-NT-CO Soret absorption band, providing confidence that a specific binding event occurs. We developed a linked-equilibria analysis to extract dissociation constants for both CO and compound binding to Ms sGC-NT constructs, as well as the allosteric enhancement factor exerted on one ligand through binding of the other (80). As expected, CO binding led to tighter binding of YC-1 or BAY 41-2272, just as YC-1 or BAY 41-2272 binding led to tighter CO binding. K d values ranged from 9 to 21 μM for YC-1, and from 2 to 17 μM for BAY 41-2272, depending on the construct used. Cooperativity factors, which measure the degree to which binding of one ligand enhances binding of the other, were ∼17 for the weaker binding YC-1, and ∼135 for the tighter binding BAY 41-2272. Using similar logic and catalytic rate enhancements, Roy et al. extracted dissociation constants for BAY 41-2272 binding to full-length sGC in the presence or absence of NO, which yielded K d values of 11 μM and 20 nM, respectively (87). Thus, the success of a particular stimulator compound correlates with its ability to induce an sGC conformation with a high affinity for NO, CO, and GTP.
Conclusions
Soluble guanylyl cyclase is a heterodimeric protein comprising α and β chains in animals. Both subunits have four domains, each with bacterial homologues. A crystal structure of the full protein has yet to be obtained but domain arrangement is becoming clear from SAXS, chemical cross-linking, FRET and EM studies. Binding of NO to a ferrous heme in the β subunit increases both active site affinity for substrate GTP and overall catalytic rate. EM studies indicate that sGC is a flexible molecule, but signal transduction may require only small rearrangements that are transmitted from the heme domain through the coiled coil to the active site, which resides between two cyclase domains and may require only small adjustments for activation or inhibition. A promising family of stimulatory compounds bind near or directly to the heme domain, leading to enhanced affinity for NO and CO, and enhanced catalysis. Open questions include the overall structure of sGC; the signal transduction mechanism; the number of NO molecules needed for full activation; regulation by ATP and other nucleotides; and the role of post-translational modification in sGC function. sGC is a highly sought-after therapeutic target for overcoming cardiovascular disease. Although much has been learned, unlocking the full potential of sGC as a drug target awaits the answers to these questions.
Footnotes
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01 GM117357 and P30 CA023074 to W.R.M., and T32 GM008804 to J.W.), the American Heart Association and the AHA Phoenix Heart Ball (14GRNT20080006 to W.R.M.), and a contract from Ironwood Pharmaceuticals (100003104 to W.R.M.). The authors are grateful to Melody Campbell, Bridget Carragher, and Michael Marletta for ![]() .
.