
Abstract
Aims:
Cathepsin S is highly expressed in various cancer cells, and it has protumoral effects, including promotion of migration, invasion, and neovascularization. In this study, we show that inhibition of cathepsin S could sensitize cancer cells to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis.
Results:
An inhibitor of cathepsin S (Z-FL-COCHO; ZFL) markedly induced apoptosis in human renal cancer cells treated with TRAIL. In contrast, combined treatment with ZFL and TRAIL had no effect on normal cells. ZFL downregulated Bcl-2 expression at the transcriptional level in a p53-dependent manner, and overexpression of Bcl-2 also markedly blocked apoptosis induced by combined treatment with ZFL and TRAIL. In addition, ZFL induced downregulation of c-FLIP, and overexpression of c-FLIP blocked the apoptosis induced by ZFL plus TRAIL. Moreover, ZFL increased the expression of Cbl, an E3 ligase of c-FLIP, in a p53-dependent manner, and knockdown of Cbl markedly prevented c-FLIP downregulation and the apoptosis induced by ZFL plus TRAIL. Interestingly, ZFL induced p53 expression via production of mitochondrial reactive oxygen species (ROS). We also demonstrated that downregulation of cathepsin S by small interfering RNA sensitized TRAIL-mediated apoptosis in Caki cells.
Innovation:
These results reveal the importance of cathepsin S on resistance against TRAIL, and inhibition of cathepsin S activity plays a crucial role in TRAIL-mediated cell death of cancer cells.
Conclusion:
Our results indicated that inhibition of cathepsin S stimulates TRAIL-induced apoptosis through downregulation of Bcl-2 and Cbl-mediated c-FLIP by ROS-mediated p53 expression. Antioxid. Redox Signal. 27, 215–233.
Introduction
C
Downregulation of cathepsin S expression induces apoptosis in human hepatocellular carcinoma cells (37), and treatment with an inhibitor of cathepsin S (6r and Z-FL-COCHO [ZFL]) induces cell death via xanthine oxidase-dependent reactive oxygen species (ROS) production in nasopharyngeal carcinoma (15). Furthermore, the specific inhibitor of cathepsin S (ZFL) induces autophagy, and an autophagy inhibitor reduces the apoptosis induced by ZFL in glioblastoma cells (42). In addition, inhibition of cathepsin S by 6r or ZFL induces autophagy and subsequent apoptosis through activation of the epidermal growth factor receptor-mediated extracellular signal-regulated kinase signaling pathways in glioblastoma cells (4). Furthermore, interferon-gamma-mediated upregulation of cathepsin S by radiation is involved in radio resistance in human breast carcinoma cells (29). Although it has been reported that cathepsin S is critically involved in cancer cell survival and resistance, its role in renal carcinoma cells remains unknown.
Cathepsin S has been shown to be highly expressed in malignant cells and high expression levels of cathepsin S have been associated with tumor progression and poor outcome. However, the roles of cathepsin S are not fully elucidated in cancer cells. Our results suggested that cathepsin S modulates reactive oxygen species production via regulation of mitochondrial functions and induces tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through a p53-dependent downregulation of Bcl-2 expression and Cbl-mediated c-FLIP expression in human cancer cells. These results indicated that inhibition of cathepsin S induced sensitivity against TRAIL.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) specifically induces apoptosis in cancer cells but not in normal cells. The anticancer activity of TRAIL is mediated through extrinsic pathways for apoptosis. TRAIL binds to death receptors (DRs) and then forms the death-inducing signaling complex with caspase-8. Activated caspase-8 activates caspase-3 directly or indirectly, thus inducing apoptosis. Cancer cells highly express DRs, whereas normal cells highly express decoy receptors (1). Therefore, TRAIL is considered to be a safe and efficient anticancer drug.
However, there are several limitations to the use of TRAIL or the TRAIL receptor antibody. First, the half-life of human soluble TRAIL is notably short, measuring ∼30 min (18). Second, agonistic monoclonal antibodies induce apoptosis via clustering of FcγRs. However, high concentrations of endogenous IgG prevent the clustering of agonistic antibodies at physiological concentrations in cancer patients (10). Third, many cancer cells have inherent TRAIL resistance or have acquired TRAIL resistance by repeat TRAIL exposure. Multiple mechanisms for the causes of TRAIL resistance in cancer cells have been proposed, including downregulation of the DRs and/or upregulation of antiapoptotic proteins (c-FLIP, antiapoptotic Bcl-2 family proteins [Bcl-2, Bcl-xL, and Mcl-1] and inhibitor of apoptosis proteins [cIAP1, cIAP2, and XIAP]) (17, 19, 23, 36, 43). Therefore, many studies have investigated methods to overcome TRAIL resistance and increase TRAIL sensitivity in cancer cells.
In our study, we examined whether inhibition of cathepsin S sensitizes TRAIL-mediated apoptosis, and we investigated the molecular mechanisms involved in the sensitization of human renal carcinoma Caki cells to TRAIL-mediated apoptosis by cathepsin S inhibition.
Results
Inhibition of cathepsin S sensitizes TRAIL-mediated apoptosis
Cathepsin S is highly expressed in multiple malignant cells (6, 7, 40), and anticancer effects of cathepsin S inhibition on glioblastoma and hepatocellular carcinoma cells (4, 37, 42) were reported. Therefore, we investigated whether inhibition of cathepsin S sensitizes TRAIL-mediated apoptosis. Treatment with cathepsin S inhibitor (Z-FL-COCHO [ZFL]) alone up to 2 μM or treatment with 50 ng/ml TRAIL did not induce apoptosis, but combined treatment with ZFL and TRAIL markedly induced apoptosis and poly (ADP-ribose) polymerase (PARP) cleavage, which is a substrate of caspase-3 (Fig. 1A). We next examined whether combined treatment with ZFL and TRAIL has synergistic effects. Isobologram analysis after the combined treatment with various concentrations of ZFL and TRAIL indicated that ZFL and TRAIL synergistically induced apoptosis (Fig. 1B). We observed typical apoptotic morphologies, including blebbing, apoptotic bodies, and chromatin condensation in Caki cells treated with ZFL and TRAIL (Fig. 1C). In addition, we detected DNA fragmentation induced by ZFL plus TRAIL (Fig. 1D). In addition, combined treatment with ZFL and TRAIL induced cytochrome c release from the mitochondria to the cytosol (Fig. 1E). We next investigated whether combined treatment with ZFL and TRAIL induces caspase-dependent apoptosis. As shown in Figure 1F, neither ZFL alone nor TRAIL alone induced caspase-3 activation, but combined treatment with ZFL and TRAIL markedly induced caspase-3 activation. Furthermore, the pan-caspase inhibitor, z-VAD-fmk, completely blocked apoptosis, PARP cleavage, and caspase-3 cleavage (Fig. 1G). Therefore, our data suggested that combined treatment with ZFL and TRAIL induces caspase-dependent apoptosis in human renal carcinoma Caki cells.

ZFL-mediated Bcl-2 downregulation is associated with the enhancement of TRAIL-mediated apoptosis
To investigate the molecular mechanism of ZFL-mediated TRAIL sensitization, we first examined the expression of apoptosis-related proteins. Among the tested antiapoptotic proteins, the protein levels of cIAP2 and Bcl-xL were not altered by ZFL treatment (Fig. 2A). In contrast, the Bcl-2 protein levels were reduced in a dose-dependent manner in ZFL-treated cells (Fig. 2A). Bcl-2 expression was downregulated after 9 h of ZFL treatment (Fig. 2B). To confirm the functional importance of the Bcl-2 reduction in the sensitizing effect of ZFL on TRAIL-mediated apoptosis, we utilized Caki cells overexpressing Bcl-2 (Caki/Bcl-2). The induction of apoptosis and PARP cleavage resulting from the combined treatment with ZFL and TRAIL were effectively blocked in Bcl-2-overexpressing Caki cells (Fig. 2C, D).

We next examined whether ZFL modulates Bcl-2 expression at the transcriptional level. As shown in Figure 2E, ZFL markedly reduced Bcl-2 mRNA expression within 3 h. Furthermore, ZFL treatment inhibited the promoter activity of Bcl-2 in cells transfected with the Bcl-2 promoter construct (Fig. 2F). Because p53 has been shown to negatively regulate Bcl-2 mRNA levels (2, 21, 39), we next investigated the possible involvement of p53 in Bcl-2 downregulation. Interestingly, we found that ZFL treatment increased not only p53 mRNA levels but also p53 protein levels (Fig. 2G). In addition, small interfering RNA (siRNA)-mediated p53 knockdown prevented the ZFL-induced downregulation of Bcl-2 (Fig. 2H). Furthermore, pretreatment with pifithrin-α, a p53 inhibitor, reversed ZFL-mediated downregulation of Bcl-2 expression (Fig. 2I). The importance of p53 in ZFL-mediated regulation of Bcl-2 was also confirmed using HCT116 isogenic cell lines differing in p53 status. Although ZFL treatment reduced Bcl-2 protein levels in wild-type (WT) HCT116 cells, in parallel with p53 upregulation, it did not affect Bcl-2 expression in p53−/− HCT116 cells (Fig. 2J). Furthermore, siRNA-mediated p53 knockdown prevented ZFL plus TRAIL-induced apoptosis and PARP cleavage in Caki cells (Fig. 2K), and combined treatment with ZFL and TRAIL did not induce apoptosis and PARP cleavage in p53−/− HCT116 cells (Fig. 2L). Taken together, these results indicated that the upregulation of p53 induced by ZFL is involved in Bcl-2 downregulation, thereby contributing to the stimulation of TRAIL-mediated apoptosis.
Downregulation of c-FLIP is important for ZFL plus TRAIL-mediated apoptosis
TRAIL induces apoptosis through the extrinsic apoptotic pathway. Therefore, we investigated whether ZFL modulates the expression of the DRs and c-FLIP. ZFL did not change the expression levels of DR4 and DR5. c-FLIP levels were markedly reduced by ZFL treatment in a dose-dependent manner (Fig. 3A). ZFL induced the downregulation of c-FLIP expression within 6 h, and it caused a further gradual decrease of c-FLIP expression for up to 24 h (Fig. 3B). To investigate the significance of c-FLIP downregulation, we treated c-FLIP-overexpressing cells with ZFL and TRAIL. When c-FLIP was overexpressed, the induction of apoptosis and PARP cleavage caused by ZFL and TRAIL were markedly blocked (Fig. 3C, D). Therefore, these data indicated that downregulation of c-FLIP may contribute to ZFL-mediated TRAIL sensitization.

We next investigated whether ZFL modulates c-FLIP expression at the transcriptional level. c-FLIP mRNA expression was not changed by ZFL treatment (Fig. 3E). Therefore, we investigated whether ZFL modulates c-FLIP expression at the post-translational level. Caki cells were treated with or without ZFL in the presence of 20 μg/ml cycloheximide (CHX), an inhibitor of de novo protein synthesis, for various time periods. ZFL significantly reduced c-FLIP stability compared with CHX alone (Fig. 3F). Because degradation of c-FLIP is known to be modulated mainly by the ubiquitin–proteasome pathways (9), we investigated whether proteasome inhibitors (MG132 and lactacystin) reverse ZFL-mediated c-FLIP downregulation. Both proteasome inhibitors reversed c-FLIP downregulation induced by ZFL (Fig. 3G). Furthermore, ZFL progressively increased proteasome activity after 12 h of treatment (Fig. 3H). Therefore, these data suggested that ZFL reduces c-FLIP expression via proteasome activity at the post-translational level.
ZFL induces Cbl expression in a p53-dependent manner
When c-FLIP is degraded via the ubiquitin–proteasome pathways, ubiquitination of c-FLIP by E3 ligase is important (44). We found that the protein expression of Cbl, one of the E3 ligases that targets c-FLIP, was markedly increased in a dose-dependent manner in ZFL-treated cells (Fig. 4A). In addition, upregulation of Cbl protein and mRNA was detected within 3 h of ZFL treatment (Fig. 4B). To investigate the significance of Cbl upregulation in cell death induced by ZFL plus TRAIL, Caki cells were infected with the lentivirus containing the nontargeting small hairpin RNA (shRNA) or Cbl-targeting shRNA. Downregulation of Cbl markedly reduced apoptosis and PARP cleavage induced by combined treatment with ZFL and TRAIL (Fig. 4C, D). Furthermore, shRNA-mediated knockdown of Cbl reversed the downregulation of c-FLIP induced by ZFL (Fig. 4D). Therefore, these data indicated that ZFL induces the downregulation of c-FLIP via induction of Cbl expression.

Because the Cbl promoter has a putative p53-binding site and ZFL induces p53 expression, we examined whether p53 regulates Cbl expression. Using the same lysates as shown in Figure 2J, ZFL induced Cbl expression in p53 WT HCT116 cells but not in p53−/− HCT116 cells (Fig. 4E). Furthermore, when we used the same lysates shown in Figure 2H, siRNA-mediated knockdown of p53 in Caki cells also blocked the upregulation of Cbl expression (Fig. 4F). Our data indicated that p53 modulates ZFL-induced Cbl expression.
ZFL induces p53 expression via mitochondrial ROS production
ROS are important signaling molecules for TRAIL sensitization (37, 38), and an inhibitor of cathepsin S has been shown to increase intracellular ROS levels (40, 42). Therefore, we examined whether ZFL induces ROS production in human renal carcinoma Caki cells. As shown in Figure 5A, ZFL increased intracellular ROS production. ROS scavengers (N-acetyl-

ROS production is mainly modulated by NADPH oxidase and the mitochondrial electron transport chain (35). To investigate the source of ROS production induced by ZFL, we employed NADPH oxidase inhibitors (diphenyleneiodonium [DPI] and apocynin) and a mitochondrial complex I inhibitor (rotenone) (16, 28). Interestingly, the NADPH oxidase inhibitors had no effect on ZFL-mediated ROS production (Supplementary Fig. S1A; Supplementary Data are available online at
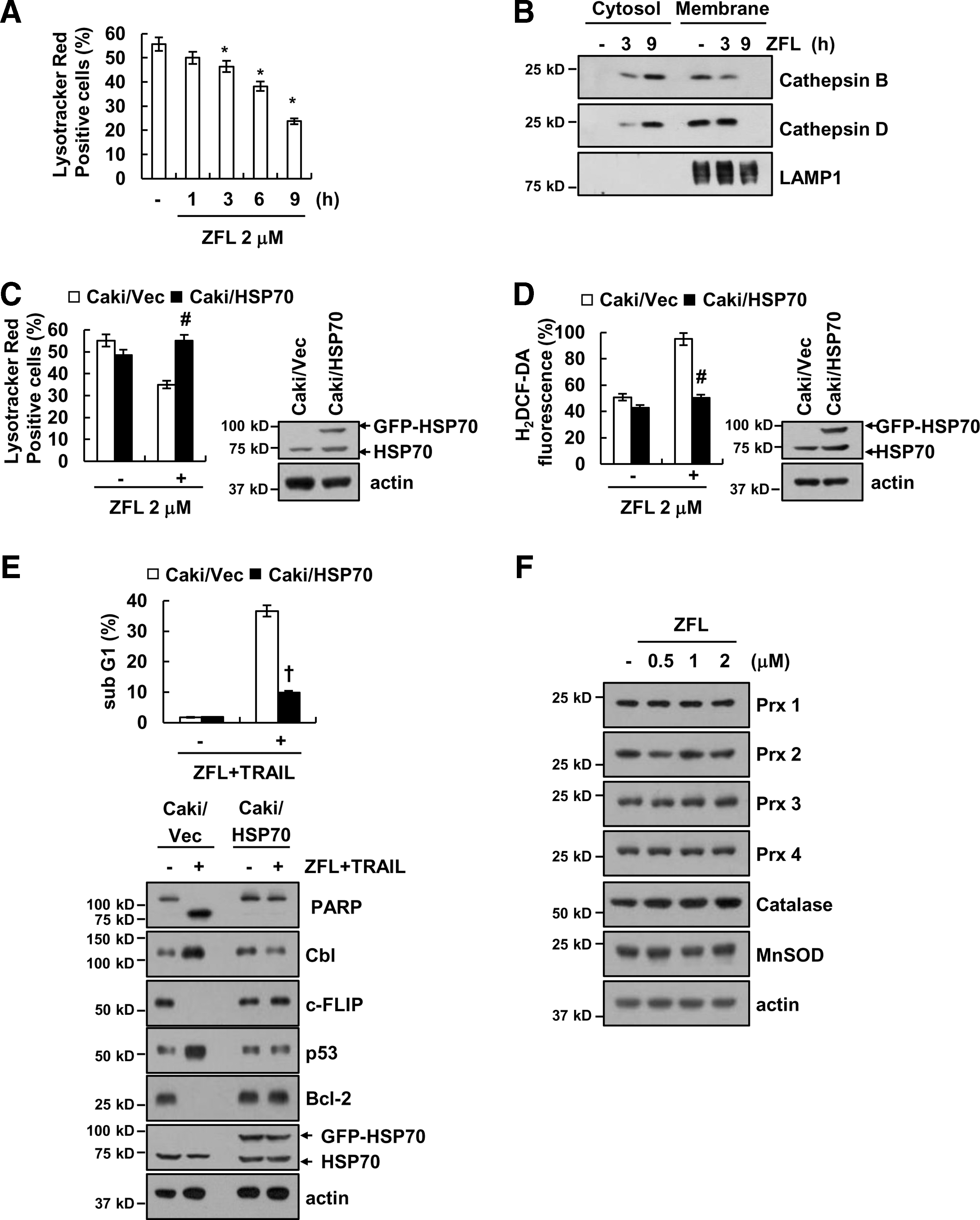
Next, we investigated how ZFL affects mitochondria and ROS production. First, we examined whether ZFL induces lysosomal membrane permeability (LMP), which is involved in mitochondria dysfunction (12, 14). ZFL markedly induced loss of lysosomal membrane integrity and released cathepsin B and D into cytosol (Fig. 6A, B). Furthermore, we investigated whether ZFL-induced LMP is related with ROS production. Nylandsted et al. reported that HSP70 could inhibit LMP (24). Overexpression of HSP70 inhibited induction of LMP, and then markedly blocked ROS production in ZFL-treated cells (Fig. 6C, D). Ectopic expression of HSP70 also inhibited apoptosis and downregulation of c-FLIP and Bcl-2 expression and upregulation of Cbl and p53 expression in ZFL plus TRAIL-treated cells (Fig. 6E). In addition, we also examined whether ZFL increases ROS production through modulation of cellular redox status by downregulation of antioxidant enzymes expression. As shown in Figure 6F, ZFL did not affect catalase, MnSOD, and peroxiredoxin (Prx) 1–4 expression. However, we could not rule out the possibility of involvement of other antioxidant enzymes.
Knockdown of cathepsin S sensitizes Caki cells to TRAIL-mediated apoptosis
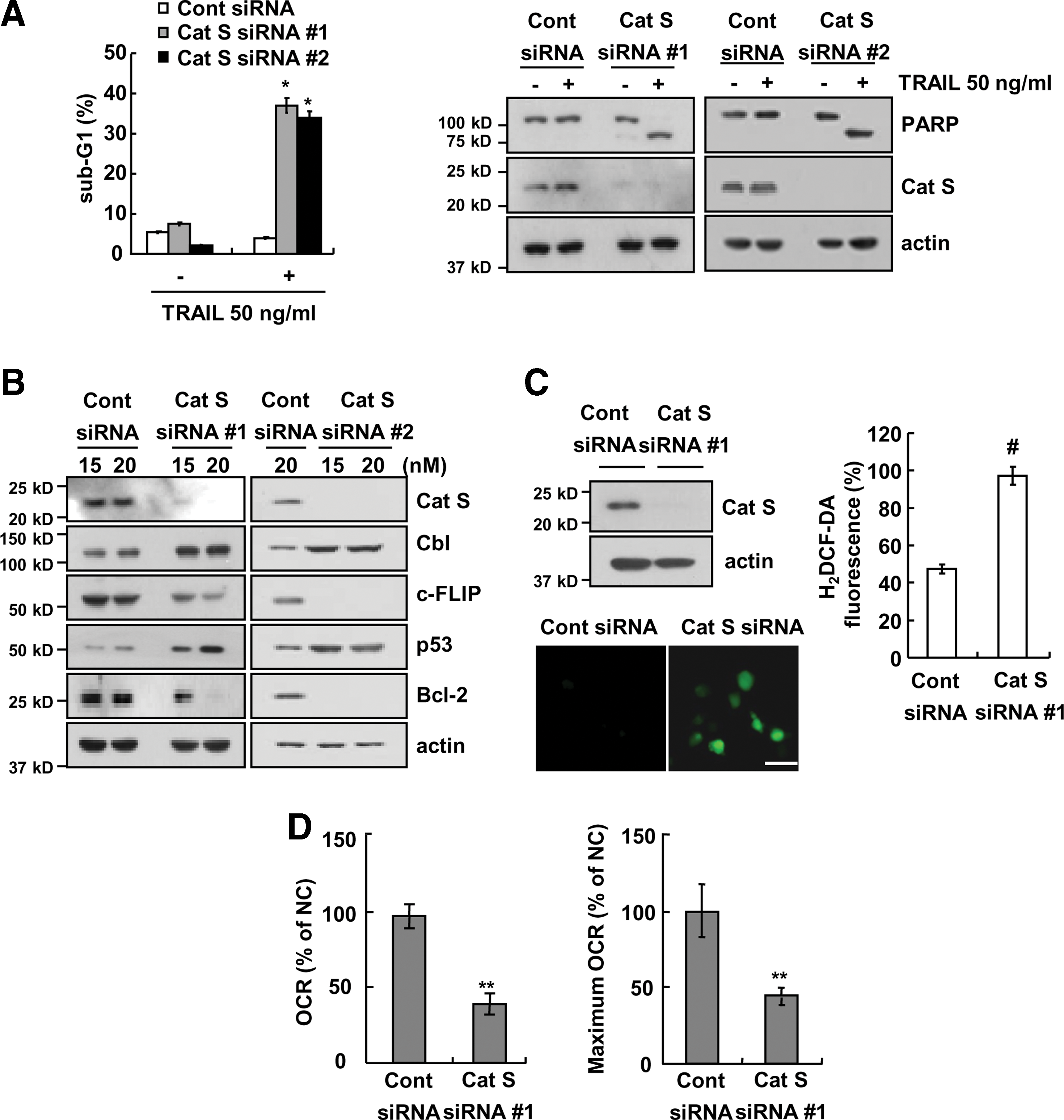
We investigated whether the effect of ZFL on TRAIL sensitization is dependent on the inhibition of cathepsin S. As shown in Figure 7A, downregulation of cathepsin S by siRNA sensitized cells to TRAIL-mediated apoptosis and induced PARP cleavage in TRAIL-treated cells. We next investigated whether the downregulation of cathepsin S modulates c-FLIP, Bcl-2, p53, and Cbl similar to the effect of ZFL treatment. We found that downregulation of cathepsin S inhibited c-FLIP and Bcl-2 expression but induced Cbl and p53 expression (Fig. 7B). Furthermore, downregulation of cathepsin S also induced intracellular ROS production (Fig. 7C). The oxygen consumption rate (OCR) and maximum OCR induced by cathepsin S siRNA were lower than the rates induced by control siRNA (Fig. 7D). Collectively, these results indicated that both pharmacological and genetic inhibition of cathepsin S can sensitize Caki cells to TRAIL-mediated apoptosis. In this process, the ROS-mediated upregulation of p53 plays a critical role in Bcl-2 downregulation at the transcriptional level and Cbl-mediated downregulation of c-FLIP at the post-translational level.
The effect of combined treatment with ZFL and TRAIL on other cancer cells and normal cells
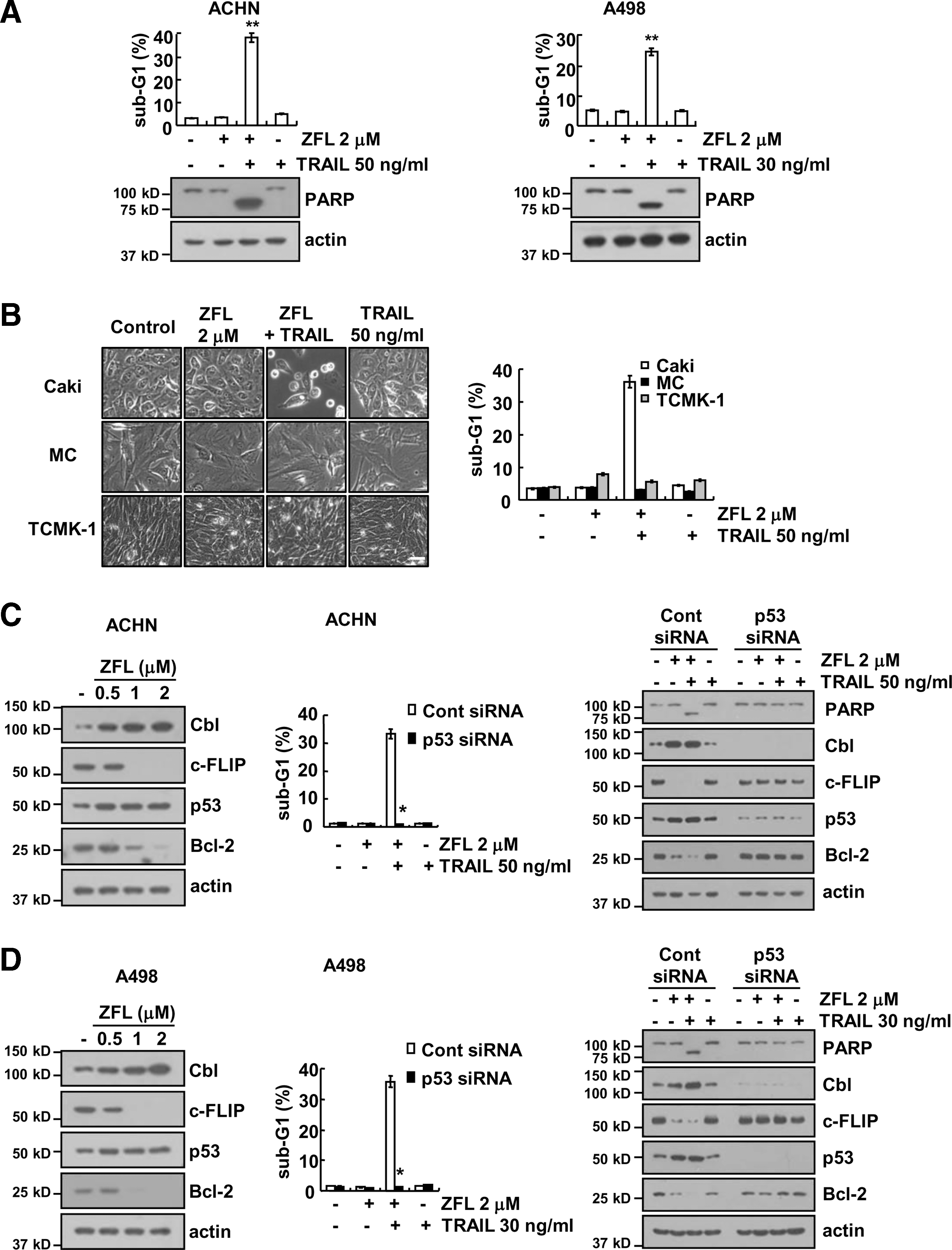
We next examined whether the sensitizing effect of ZFL on TRAIL-mediated apoptosis is restricted to Caki cells. We found that combined treatment with ZFL and TRAIL markedly induced apoptosis in other renal carcinoma cells (ACHN and A498) (Fig. 8A). The PARP cleavage was also effectively induced by combined treatment with ZFL and TRAIL in these cancer cells (Fig. 8A). These results suggested that ZFL may overcome TRAIL resistance in various renal cancer cells. In contrast, combined treatment with ZFL and TRAIL did not induce morphological changes and apoptosis in normal human mesangial cells and normal mouse kidney cells (TCMK-1) (Fig. 8B). Furthermore, ZFL induced upregulation of Cbl and p53 expression and downregulation of c-FLIP and Bcl-2 expression, and downregulation of p53 blocked ZFL-mediated TRAIL sensitization and prevented the ZFL-induced downregulation of Bcl-2 and c-FLIP and upregulation of Cbl expression in ACHN and A498 cells (Fig. 8C, D). Therefore, our data indicated that ZFL may selectively sensitize cancer cells to TRAIL-mediated apoptosis while sparing normal cells.
Combined treatment with ZFL and TRAIL inhibits tumor growth in vivo
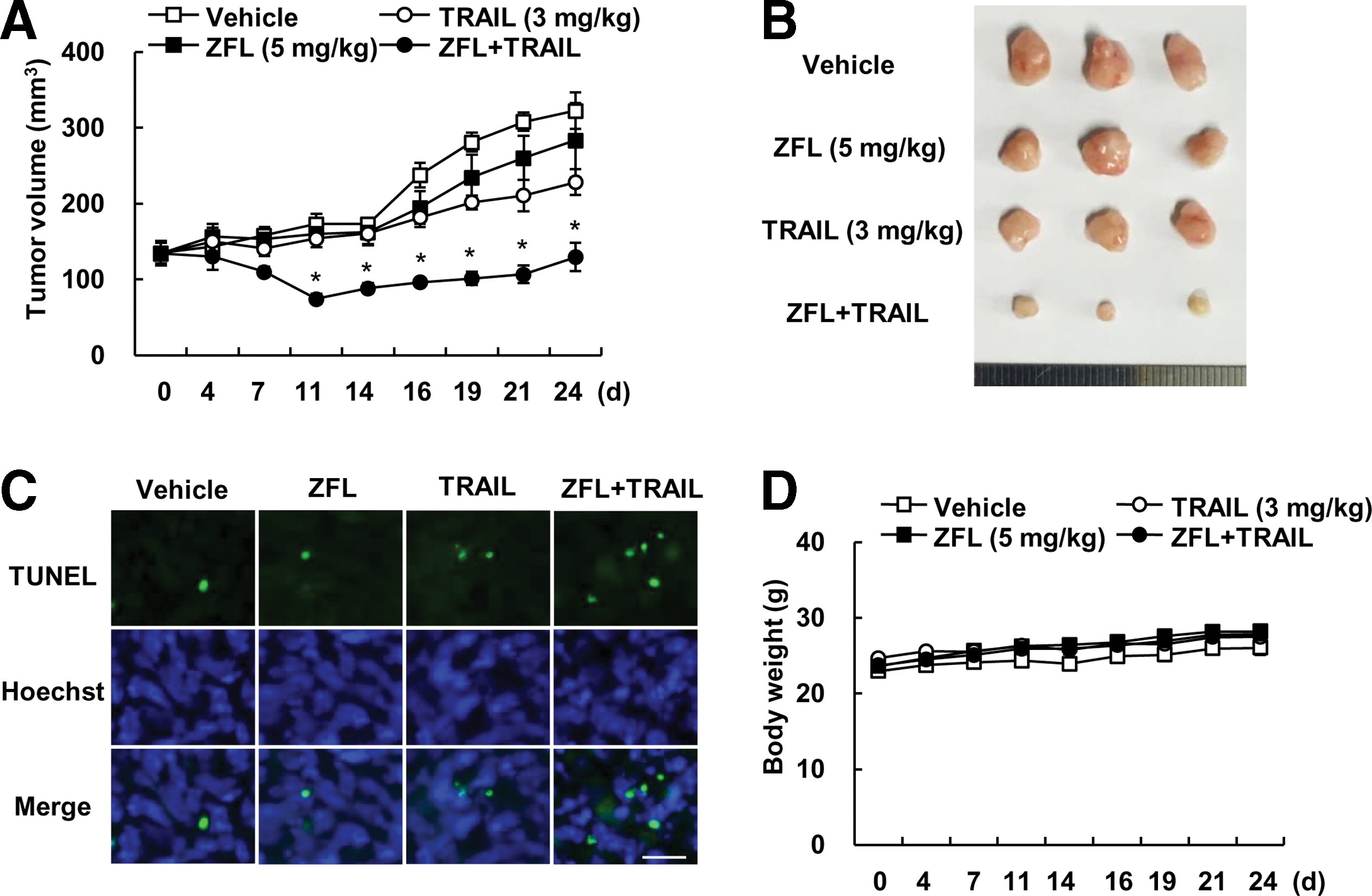
Next, we examined the anticancer effect of combined treatment with ZFL and TRAIL in vivo xenograft model. Mice bearing tumors were treated with ZFL alone, TRAIL alone, and combined with ZFL and TRAIL. Combined treatment with ZFL and TRAIL markedly inhibited tumor growth, compare with that of vehicle, ZFL alone, and TRAIL alone (Fig. 9A, B). Terminal deoxynucleotide transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) analysis also indicated that ZFL plus TRAIL induced cell death (Fig. 9C). Combined treatment with ZFL and TRAIL did not affect the body weight (Fig. 9D). These data suggest that combined treatment with ZFL and TRAIL inhibits tumor growth and induces apoptosis in vivo.
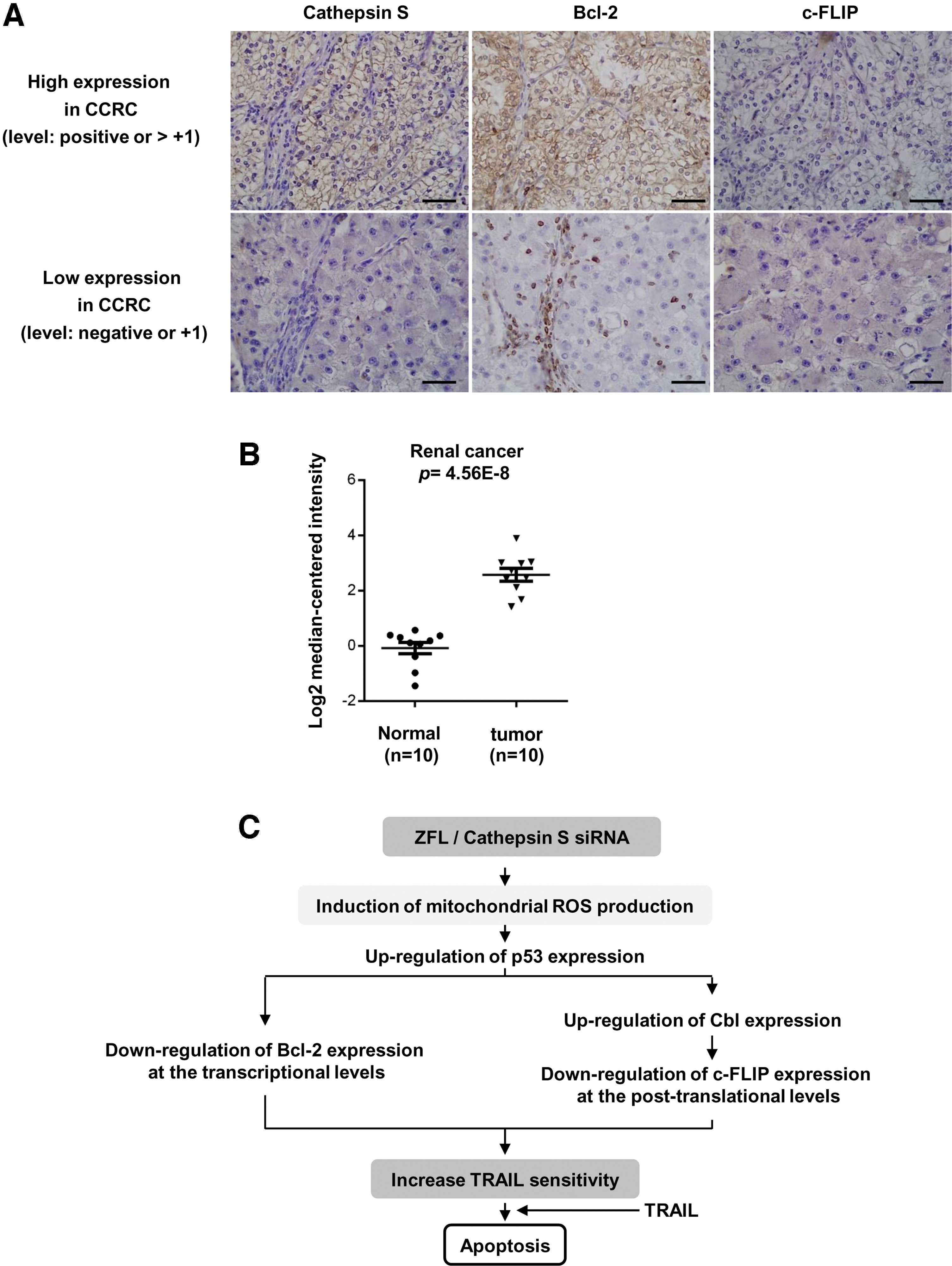
The expression levels of cathepsin S are correlated with those of Bcl-2 in clear cell renal cell carcinoma patients
Finally, we examined the expression levels of cathepsin S, Bcl-2, and c-FLIP in the tissues of clear cell renal cell carcinoma (CCRCC) patients by immunohistochemistry (Table 1). Representative results are shown in Figure 10A. In tumor tissues with an elevated expression of cathepsin S, Bcl-2 proteins were highly stained, but the c-FLIP were moderately stained. In contrast, in tumor tissues with a low expression of cathepsin S, Bcl-2 expression was detected in infiltrated immune cells but not in tumor cells. In addition, c-FLIP expression was not detected in these tissues. We further observed that mRNA levels of cathepsin S were significantly increased in renal cancer tissues than in normal tissues based on an analysis of the Oncomine database (
n indicates number. The data represent the number of patients (percentage). The bold values indicate a statistically significant difference (p < 0.05).
Discussion
In this study, we demonstrated that ZFL enhances TRAIL-mediated apoptosis in cancer cells but not in normal cells. These findings suggested that inhibition of cathepsin S may be an attractive strategy for TRAIL sensitization. We showed that inhibition of cathepsin S transcriptionally downregulated Bcl-2 and post-translationally downregulated c-FLIP via Cbl to sensitize cells to TRAIL-mediated apoptosis. In both events, the ROS-induced upregulation of p53 played a critical role (Fig. 10C).
Although cathepsin S is mainly localized in lysosomes, it is also located in the cytosol, cell surface, and extracellular spaces. Although most cathepsins are only active in an acidic environment, cathepsin S is active in a relatively broad pH range (approximately pH 7) (30). Therefore, cathepsin S might have multiple functions in cancer cells compared with other cathepsins. In our studies, inhibition of cathepsin S induced TRAIL sensitization, and ROS production played a critical role in these functions. Huang et al. showed that inhibition of cathepsin S induces ROS production via xanthine oxidase but not by NADPH and the mitochondria electron transport chain in nasopharyngeal carcinoma (15). However, we found that inhibition of cathepsin S increased intracellular ROS levels via the mitochondria electron transport chain (Fig. 5D), and inhibition of cathepsin S induced mitochondria dysfunctions in human renal carcinoma Caki cells (Fig. 7D).
The apical point of the TRAIL sensitization mechanism induced by ZFL or knockdown of cathepsin S is mitochondrial dysfunction followed by mitochondrial ROS production. Deficiency of lysosomal enzymes (cathepsin L, cathepsin E, and cathepsin S) induces mitochondria dysfunction (25, 26, 34). There are two possible causes of mitochondria dysfunction. First, the mitochondria dysfunction results from failure in the removal of damaged mitochondria. Tsukuba et al. reported that cathepsin E deficiency causes autophagy impairment and is involved in the accumulation of damaged mitochondria followed by production of ROS (34). Pan et al. also reported that cathepsin S deficiency increases abnormal accumulation of autophagosomes that contain damaged mitochondria in macrophages (25). Because mitochondria turnover is dependent on the autophagy process, inhibition of cathepsin function may inhibit the autophagy process, resulting in the accumulation of damaged mitochondria. Consistent with these results, we also detected ZFL-mediated mitochondria dysfunctions, but ZFL did not increase LC3 II conversion, p62 downregulation, and autophagy flux in Caki cells (Supplementary Fig. S2A, B). Therefore, it remains to be elucidated whether novel mechanisms independent of autophagy inhibition are involved in ROS production induced by cathepsin S inhibition. Second, impairment of mitochondrial respiratory chain components might be associated with mitochondria dysfunction. Although the mechanism of mitochondria function modulated by cathepsin is not fully understood, Petermann et al. reported that deficiency of cathepsin L in myocardium reduces the expression of respiratory chain-related proteins, including NADH-ubiquinone oxidoreductase 24 kDa subunit, ubiquinol-cytochrome c reductase core protein (UCRC) 1, cytochrome c oxidase subunit Va, and ATP synthase beta chain (26). We also investigate whether ZFL induced downregulation of expression of respiratory chain-related proteins, such as NDUFA9, Flovoprotein, UQCRC2, cytochrome c oxidase subunit 2 (COX II), and ATP synthase α-subunit (ATP5A). However, ZFL had no effect on expression of respiratory chain-related proteins (Supplementary Fig. S2C). Finally, LMP is also associated with mitochondria dysfunction (12, 14). As shown in Figure 6A and B, ZFL markedly induced loss of lysosomal membrane integrity and released cathepsin B and D into cytosol. Overexpression of HSP70 blocked ZFL-mediated induction of LMP, and then markedly inhibited apoptosis and downregulation of c-FLIP and Bcl-2 expression and upregulation of p53 expression through downregulation of ROS production in ZFL plus TRAIL-treated cells (Fig. 6C–E). Therefore, induction of LMP by ZFL could be one of the candidates to explain the mitochondrial dysfunction.
In addition, we found that ZFL induced downregulation of c-FLIP expression at the post-translational level via reduction of protein stability (Fig. 3F) and that proteasomal activity plays a critical role in the downregulation of c-FLIP expression in ZFL-treated cells (Fig. 3G, H). We investigated the effect of ZFL on protein expression of two critical proteasome subunits, PSMA5 and PSMD4/S5a (11), and we found that ZFL had no effect on either of these proteins (data not shown). Therefore, further investigation is required to understand how ZFL induces proteasome activity. Recently, Cbl and Itch have been shown to act as E3 ligases for c-FLIP (3, 44). As shown in Figures 4A and 7B, ZFL treatment and cathepsin S knockdown markedly increased Cbl expression, and downregulation of Cbl blocked ZFL-mediated TRAIL sensitization (Fig. 4C, D). Our results demonstrated that cathepsin S modulates c-FLIP expression via Cbl expression.
p53 is an important transcription factor in cancer cells. Multiple stresses, including DNA damage, nutrient deprivation, oxidative stress, and endoplasmic reticulum stress, cause p53 to be stable and active, and thereby resulting in modulation of target gene expression. In our study, inhibition of cathepsin S using ZFL and its siRNA increased p53 expression at the transcriptional level via mitochondrial ROS production. p53 has dual functions in the TRAIL sensitization induced by cathepsin S inhibition. (i) We showed that p53 inhibits Bcl-2 mRNA expression in ZFL- or cathepsin S siRNA-treated Caki cells. First, knockdown of p53 blocked the ZFL-mediated downregulation of Bcl-2 expression (Fig. 2H). Second, a p53 inhibitor (pifithrin-α) reversed the inhibition of Bcl-2 expression in ZFL-treated cells (Fig. 2I). Finally, ZFL reduced Bcl-2 expression in p53 WT HCT116 cells but not in p53−/− HCT116 cells (Fig. 2J). Thus, we demonstrated that ZFL modulates Bcl-2 expression via the direct or indirect upregulation of p53. (ii) p53 also inhibits c-FLIP expression via induction of Cbl expression in ZFL- or cathepsin S siRNA-treated Caki cells. Because the promoter of Cbl has a putative p53-binding site, we investigated whether p53 modulates Cbl expression. Interestingly, ZFL increased Cbl expression in p53 WT HCT116 cells but not in p53−/− HCT116 cells (Fig. 4E). In addition, downregulation of p53 also did not alter Cbl expression (Fig. 4F). ZFL also induced downregulation of Bcl-2 and c-FLIP expression and upregulation of p53 and Cbl expression in ACHN and A498 cells (Fig. 8C, D). Furthermore, downregulation of p53 blocked ZFL-mediated TRAIL sensitization and prevented the ZFL-induced downregulation of Bcl-2 and c-FLIP and upregulation of Cbl expression in ACHN and A498 cells (Fig. 8C, D). However, ZFL did not upregulate other p53-target proteins and mRNA (p21, PUMA, Noxa, Bax, and DR5) (Supplementary Fig. S3). Our data demonstrated for first time that p53 regulates Cbl expression. However, we cannot exclude the possibility that p53 may modulate Cbl expression indirectly.
Collectively, these results suggest that the inhibition of cathepsin S induces mitochondrial dysfunction and increases intracellular ROS production. The ROS-mediated upregulation of p53 induced the downregulation of Bcl-2 at the transcriptional level and the Cbl-mediated downregulation of c-FLIP expression. Therefore, the combination of cathepsin S inhibition and TRAIL may be a novel and effective strategy for cancer therapy.
Materials and Methods
Cell culture and materials
Human renal carcinoma (Caki [ATCC HTB-46], ACHN [ATCC CRL-1611], and A498 [ATCC HTB-44]), human colon carcinoma (HCT116 [ATCC CCL-247]), and the mouse kidney (TCMK-1) were obtained from the American Type Culture Collection. HCT116 p53-deficient cells were kindly provided by Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD). NHMCs were purchased from Lonza (CC-2559). All cell lines tested negative for mycoplasma contamination. The lines were authenticated by standard morphological examination using microscopy. The culture medium used throughout these experiments was Dulbecco's modified Eagle's medium or Roswell Park Memorial Institute (RPMI) containing 10% fetal bovine serum, 20 mM HEPES buffer, and 100 μg/ml gentamycin. The polymerase chain reaction (PCR) primers were purchased from Macrogen. The recombinant human TRAIL and z-VAD-fmk were purchased from R&D System, and NAC, Z-FL-COCHO, and Trolox were obtained from Calbiochem. GST-TRAIL cDNA plasmid was a gift from Dr. Kim YS (Ajou University, Korea). pEGFP hsp70 was a gift from Lois Greene (Addgene plasmid No. 15215) (41). Anti-cytochrome c (556433) and anti-Bax (554104) antibodies were purchased from BD Biosciences. Anti-Bcl-2 (sc-783), anti-p53 (sc-126), anti-cathepsin S (sc-6505), anti-Cbl (sc-170), anti-DR4 (sc-7863), anti-Bcl-xL (sc-634), anti-PUMA (sc-19187), anti-p21 (sc-397), anti-cIAP2 (sc-7944), anti-Lamp1 (sc-5570), anti-cathepsin B (sc-13985), anti-cathepsin D (sc-6486), anti-SQSTM1(p62) (sc-28359), and anti-HSP70 (sc-24) antibodies were purchased from Santa Cruz Biotechnology. Anti-DR5 (8074S), anti-PARP (9542S), anticleaved caspase-3 (9661S) antibodies were obtained from Cell Signaling Technology. Anti-pro-caspase-3 (ADI-AAP-113) and anti-c-FLIP (ALX-804-961-0100) antibodies were obtained from ALEXIS Corporation. Anti-NDUFA9 (A21344) antibody was purchased from Molecular Probes. Anti-Noxa (OP180) and anti-MnSOD (06-984) antibodies were purchased from Millipore Corporation. Anti-Prx1 (LF-PA0086), anti-Prx2 (LF-PA0091), anti-Prx3 (LF-PA0030), and anti-Prx4 (LF-PA0009) antibodies were purchased from AbFrontier. Anti-catalase (ab16731) antibody was obtained from Abcam. Anti-LC3 (PD014) antibody was purchased from MBL. Anti-actin antibody and other chemicals were obtained from Sigma Chemical Co..
Flow cytometry analysis
For flow cytometry, the cells were resuspended in 100 μl of phosphate-buffered saline (PBS), and 200 μl of 95% ethanol was added while the cells were being vortexed. Then, the cells were incubated at 4°C for 1 h, washed with PBS, resuspended in 250 μl of 1.12% sodium citrate buffer (pH 8.4) with 12.5 μg of RNase, and incubated for an additional 30 min at 37°C. The cellular DNA was then stained by adding 250 μl of a propidium iodide solution (50 μg/ml) to the cells for 30 min at room temperature. The stained cells were analyzed by fluorescent-activated cell sorting on a FACScan flow cytometer to determine the relative DNA content, which was based on the red fluorescence intensity.
Western blot analysis
Cells were washed with cold PBS and lysed on ice in modified radioimmunoprecipitation assay buffer (50 mM Tris–HCl, pH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM Na3VO4, and 1 mM NaF) containing protease inhibitors (100 μM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 2 mM EDTA). Lysates were centrifuged at 13,000 g for 15 min at 4°C, and the supernatant fractions were collected. Proteins were separated by SDS-PAGE and transferred to an Immobilon-P membrane. Specific proteins were detected using enhanced chemiluminescence. Uncropped image for the figures are provided in Supplementary Figures S4–14.
Determination of synergy
The possible synergistic effect of ZFL and TRAIL was evaluated using the isobologram method. In brief, the cells were treated with different concentrations of ZFL and TRAIL alone or in combination. After 24 h, XTT assay was employed to measure the cell viability using WelCount Cell Viability Assay Kit (WelGENE). Relative survival was assessed and the concentration effect curves were used to determine the IC50 (the half-maximal inhibitory concentration) values for each drug alone and in combination with a fixed concentration of the second agent (33).
4′,6′-Diamidino-2-phenylindole staining for nuclei condensation and fragmentation
To examine cellular nuclei, the cells were fixed with 1% paraformaldehyde on glass slides for 30 min at room temperature. After the fixation, the cells were washed with PBS and a 300 nM 4′,6′-diamidino-2-phenylindole solution (Roche) was added to the fixed cells for 5 min. After the nuclei were stained, the cells were examined by fluorescence microscopy.
DNA fragmentation assay
After treatment with ZFL, TRAIL, or ZFL plus TRAIL, Caki cells were lysed in a buffer containing 10 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, and 0.5% Triton X-100 for 30 min on ice. Lysates were vortexed and cleared by centrifugation at 10,000 g for 20 min. Fragmented DNA in the supernatant was extracted with an equal volume of neutral phenol:chloroform:isoamyl alcohol mixture (25:24:1) and analyzed electrophoretically on 2% agarose gels containing 0.1 μg/ml of ethidium bromide.
Analysis of cytochrome c release
Cells were harvested, washed once with ice-cold PBS, and gently lysed for 2 min in 80 μl ice-cold lysis buffer (250 mM sucrose, 1 mM EDTA, 20 mM Tris–HCl [pH 7.2], 1 mM DTT, 10 mM KCl, 1.5 mM MgCl2, 5 μg/ml pepstatin A, 10 μg/ml leupeptin, and 2 μg/ml aprotinin). Lysates were centrifuged at 12,000 g at 4°C for 10 min to obtain the supernatants (cytosolic extracts free of mitochondria) and the pellets (fraction that contains mitochondria). The resulting cytosolic fractions were used for Western blot analysis with an anticytochrome c antibody.
Asp-Glu-Val-Asp-ase (DEVDase) activity assay
To evaluate DEVDase activity, cell lysates were prepared after their respective treatments with TRAIL in the presence or absence of ZFL. Assays were performed in 96-well microtiter plates by incubating 20 μg of cell lysates in 100 μl of reaction buffer (1% NP-40, 20 mM Tris–HCl, pH 7.5, 137 mM NaCl, and 10% glycerol), containing a caspase substrate (Asp-Glu-Val-Asp-chromophore-p-nitroanilide [DVAD-pNA]) at 5 μM. Lysates were incubated at 37°C for 2 h. Thereafter, the absorbance at 405 nm was measured with a spectrophotometer.
Reverse transcription PCR
Total RNA was isolated using the TriZol reagent (Life Technologies), and the cDNA was prepared using M-MLV reverse transcriptase (Gibco-BRL) according to the manufacturers' instructions. The following primers were used for the amplification of human c-FLIP, Bcl-2, Cbl, p53, p21, PUMA, Noxa, Bax, and DR5, and actin: c-FLIP (sense) 5′-CGG ACT ATA GAG TGC TGA TGG-3′ and (antisense) 5′-GAT TAT CAG GCA GAT TCC TAG-3′; Bcl-2 (sense) 5′-GGT GAA CTG GGG GAG GAT TGT-3′ and (antisense) 5′-CTT CAG AGA CAG CCA GGA GAA-3′; Cbl (sense) 5′-GCT GCT ACG TCC CTA ATC CC-3′ and (antisense) 5′-ATC CAC CAC CCT CTA GGC AT-3′; p53 (sense) 5′-GAA GAC CCA GGT CCA GAT GA-3′ and (antisense) 5′-CTC CGT CAT GTG CTG TGA CT-3′; p21 (sense) 5′-CCC AGT GGA CAG CGA GC-3′ and (antisense) 5′-ACT GCA GGC TTC CTG TGG GC-3′; PUMA (sense) 5′-GTC CTC AGC CCT CGC TCT-3′ and (antisense) 5′-CAC CTA ATT GGG CTC CAT CT-3′; Noxa (sense) 5′-GTG CCC TTG GAA ACG GAA GA-3′ and (antisense) 5′-CCA GCC GCC CAG TCT AAT CA-3′; Bax (sense) 5′-ACC AAG AAG CTG AGC GAG TGT C-3′ and (antisense) 5′-TGT CCA GCC CAT GAT GGT TC-3′ and DR5 (sense) 5′-AAG ACC CTT GTG CTC GTT GT-3′ and (antisense) 5′-GA CAC ATT CGA TGT CAC TCC A-3′; and actin (sense) 5′-GGC ATC GTC ACC AAC TGG GAC-3′ and (anti-sense) 5′-CGA TTT CCC GCT CGG CCG TGG-3′. The PCR amplification was carried out using the following cycling conditions: 94°C for 3 min followed by 17 (actin) and 25 cycles (c-FLIP, Bcl-2, Cbl, and p53) or 28 cycles (p21, Noxa, Bax, DR5, and PUMA) of 94°C for 45 s, 58°C for 45 s, 72°C for 1 min, and a final extension at 72°C for 10 min. The amplified products were separated by electrophoresis on a 1.5% agarose gel and detected under UV light.
DNA transfection and luciferase assay
Transient transfection was performed in six-well plates. One day before the transfection, Caki cells were plated at ∼60–80% confluence. The Bcl-2/-3254 promoter plasmid was transfected into the cells using Lipofectamine™ 2000 (Invitrogen). To assess the promoter-driven expression of the luciferase gene, the cells were collected and disrupted by sonication in lysis buffer (25 mM Tris-phosphate, pH 7.8, 2 mM EDTA, 1% Triton X-100, and 10% glycerol), and aliquots of the supernatant were used to analyze the luciferase activity according to the manufacturer's instructions (Promega).
Lentiviral particle production and viral transduction
For the knockdown experiments using Cbl-targeting shRNA, HEK293TN cells were transfected with the plasmid containing the nontargeting shRNA (SHC002 V; Sigma-Aldrich) or the plasmid containing the Cbl-targeting shRNA (TRCN0000039723; Sigma-Aldrich), together with the pMD2.G (the envelope plasmid) and pPsAX2.0 (the packaging plasmid) plasmids, using TransIT-2020 transfection reagents (Mirus Bio LLC) according to the manufacturer's instructions. After 48 h of lentiviral particle production, Caki cells were infected with the filtered lentiviral medium (derived from HEK293TN cultures) supplemented with 5 μg/ml polybrene.
Small interfering RNA
The p53 siRNA used in this study was purchased from Santa Cruz Biotechnology. The cathepsin S siRNA No. 1 and No. 2 were purchased from Santa Cruz Biotechnology and Dharmacon, Inc. (ON-TARGETplus), respectively. The NDUFA9 siRNAs (ON-TARGETplus) were obtained from Thermo Scientific Dharmacon, Inc. The green fluorescent protein (control) siRNA was purchased from Bioneer.
Proteasome activity assay
Chymotryptic proteasome activities were measured with Suc-LLVY-AMC (chymotryptic substrate; Biomol International). Lysate from ZFL-treated cells was prepared. A mixture containing 1 μg cell lysate protein in 100 mM Tris–HCl (pH 8.0), 10 mM MgCl2, and 2 mM ATP was incubated at 37°C for 30 min with 50 μM Suc-LLVY-AMC. Enzyme activity was measured with a fluorometric plate reader at an excitation wavelength of 380 nm and an emission wavelength of 440 nm.
Measurement of ROS
Intracellular accumulation of ROS was determined using the fluorescent probes 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and Mitosox Red. Caki cells were treated with ZFL, and then cells were stained with the H2DCFDA fluorescent dye or Mitosox Red for an additional 10 min. Then, cells were trypsinized and resuspended in PBS, and fluorescence was measured at specific time intervals with a flow cytometer (Becton–Dickinson) or fluorescence microscope (Zeis).
Measurement of lysosomal membrane permeabilization
Lysotracker red (Molecular Probes, Inc.) uptake by lysosome is proportional to its membrane potential. Caki cells were treated with ZFL for the indicated time periods, then cells were incubated with 2.5 μM lysotracker red for 30 min at 37°C. After treatment, cells were washed several times in PBS. The cells were then trypsinized and resuspended in PBS, and fluorescence was measured at specific time intervals with a flow cytometer.
Fractionation of cytosol and membrane extracts
Cells were washed with ice-cold PBS, then resuspended in cytosol extraction buffer (250 mM sucrose, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and 20 mM HEPES) containing a 250 μg/ml digitonin and left on ice for 10 min and then lysate was centrifuged at 13,000 g for 90 s. The supernatant (cytosol) was transferred to a new tube and pellets (membrane fraction) were suspended with lysis buffer. Lysates were centrifuged at 13,000 g at 4°C for 15 min to obtain the supernatant fractions as the membrane extract.
Endogenous cellular OCR
Endogenous cellular OCR was measured using a Mitocell equipped with a Clark oxygen electrode (782 Oxygen Meter; Strathkelvin Instrument) or a XF24 Extracellular Flux Analyzer (Seahorse Bioscience) according to the protocol provided. For measurement of endogenous cellular respiration with Mitocell, cells were washed with TD buffer (0.137 M NaCl, 5 mM KCl, 0.7 mM Na2HPO4, and 25 mM Tris–HCl; pH 7.4) and collected by trypsinization. After resuspending the cells in medium without phenol red, the cells were transferred to the chamber of the Mitocell equipped with a Clark oxygen electrode (782 Oxygen Meter; Strathkelvin Instrument) for measurement of endogenous cellular OCR. OCRs were measured to obtain maximum respiration rate, and its specificity for mitochondrial respiration was confirmed by adding 5 mM KCN.
Animal
Male BALB/c-nude mice, aged 5 weeks, were purchased from the Central Lab Animal, Inc. All the mice were allowed 1 week to acclimatize to the surroundings before the experiments, and were kept at 25°C ± 2°C, with a relative humidity of 55% ± 5% and a 12 h light–dark cycle. The study protocol was approved by the IRB Keimyung University Ethics Committee.
In vivo xenograft model
Each mouse was subcutaneously injected on each flank with Caki cells (2 × 106). After tumors had grown after ∼2 weeks, 28 mice were randomly divided into four treatment groups: (i) vehicle alone, (ii) ZFL alone, (iii) GST-TRAIL alone, and (iv) ZFL plus GST-TRAIL. ZFL and GST-TRAIL were administered at 5 and 3 mg/kg, respectively. ZFL was prepared in 20% DMSO and 80% PBS (pH7.4), and GST-TRAIL was prepared with PBS (pH 7.4). The mice received an intraperitoneal (i.p.) injection of ZFL and GST-TRAIL. Treatment was administered three times a week for 24 days. The tumor size was measured three times a week using a Vernier's caliper (Mytutoyo Co.) to measure two perpendicular diameters, and the tumor size was calculated using the equation (length × width2)/2. The animals were sacrificed by cervical dislocation, and the tumors were collected for histological analysis. The tumors were fixed in 30% formalin, embedded in optical cutting temperature compound (Miles, Inc.), and cut into 20 μm sections using a cryostat (SLEE International, Inc.).
TUNEL assay
Apoptosis in tumor cells was detected by TUNEL assay. It was performed using the ApopTag Fluorescein In situ Apoptosis Detection Kit (Millipore) as the manufacturer's protocol.
Patient and immunohistochemical detection
We studied 140 patients who underwent radical nephrectomy for CCRCC at Keimyung University Dongsan Medical Center between 1997 and 2007. From each tissue microarray block, 4-μm thick sections were deparaffinized, rehydrated, and quenched with hydrogen peroxide. The primary antibodies used were specific for cathepsin S, c-FLIP, and Bcl-2 (1:1600; Zymed). The sections were observed with diaminobenzidine and counterstained with hematoxylin. The expression levels were semiquantitatively assessed and classified into negative and positive groups. The negative group was defined as cases in which the number of cells stained was less than 5%, whereas the positive group included cases in which the number of cells stained exceeded 5%. Associations of cathepsin S expression with c-FLIP and Bcl-2 expression were evaluated using the Chi-square test. A two-sided p value <0.05 was considered statistically significant. PASW ver. 22.0 (SPSS, Inc.) was used for statistical analysis. The study protocol was approved by the IRB Keimyung University Ethics Committee.
Analysis of cathepsin S expression in renal cancers using public data sets
Cathepsin S expression was analyzed in various tumor types using the Oncomine database (
Statistical analysis
The data were analyzed using a one-way ANOVA and post hoc comparisons (Student–Newman–Keuls) using the Statistical Package for Social Sciences 22.0 software (SPSS, Inc.). We determined the sample size on the basis of the smallest effect we wish to measure.
Footnotes
Acknowledgment
This work was supported by an NRF grant funded by the Korea Government (MSIP) (2014R1A5A2010008 and NRF-2016R1A2B2013393).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.