Abstract
Significance:
Alzheimer's disease (AD) is a multifactorial neurodegenerative disorder and represents one of the most disabling conditions. AD shares many features in common with systemic insulin resistance diseases, suggesting that it can be considered as a metabolic disease, characterized by reduced insulin-stimulated growth and survival signaling, increased oxidative stress (OS), proinflammatory cytokine activation, mitochondrial dysfunction, impaired energy metabolism, and altered protein homeostasis.
Recent Advances:
Reduced glucose utilization and energy metabolism in AD have been associated with the buildup of amyloid-β peptide and hyperphosphorylated tau, increased OS, and the accumulation of unfolded/misfolded proteins. Mammalian target of rapamycin (mTOR), which is aberrantly activated in AD since early stages, plays a key role during AD neurodegeneration by, on one side, inhibiting insulin signaling as a negative feedback mechanism and, on the other side, regulating protein homeostasis (synthesis/clearance).
Critical Issues:
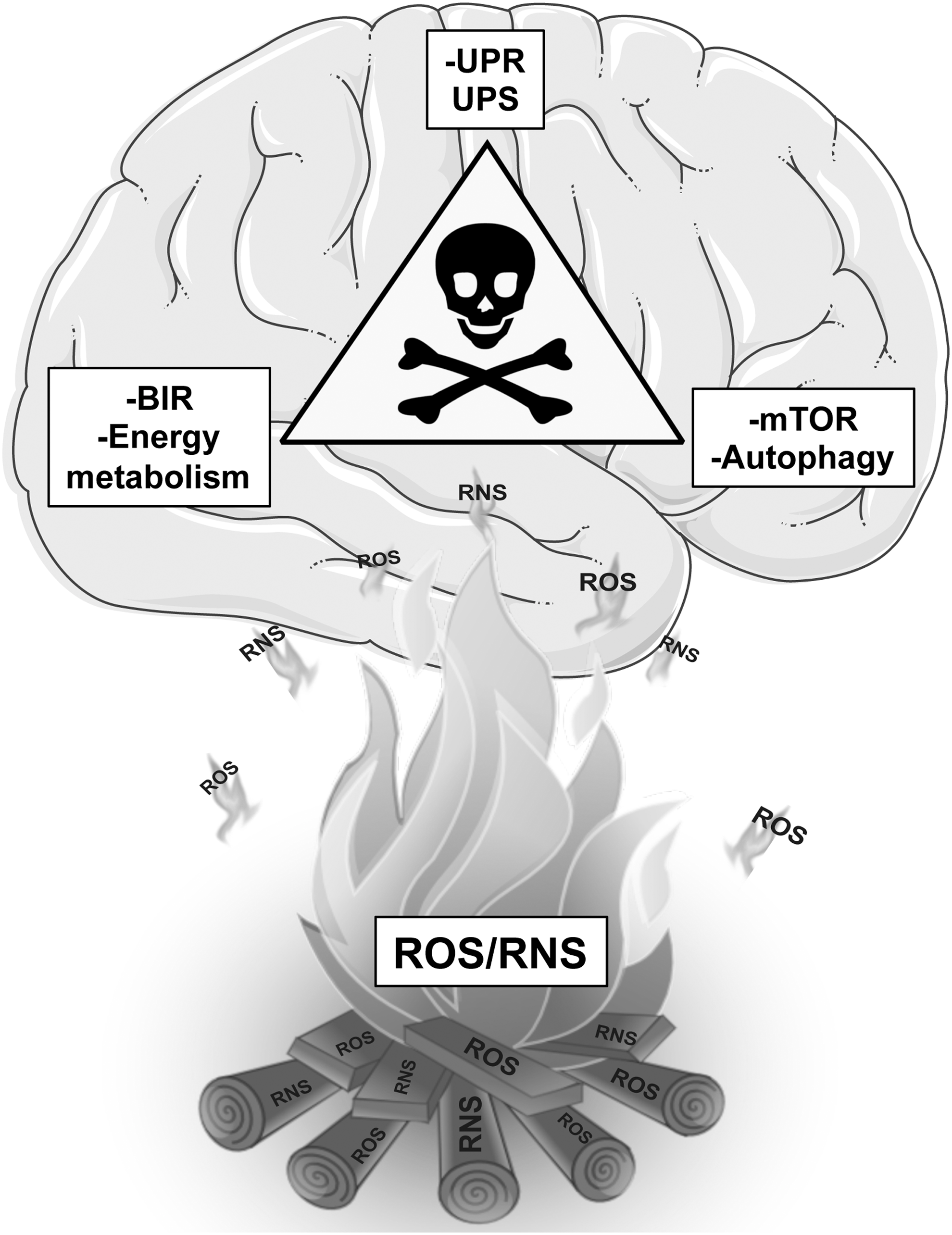
It is likely that the concomitant and mutual alterations of energy metabolism–mTOR signaling–protein homeostasis might represent a self-sustaining triangle of harmful events that trigger the degeneration and death of neurons and the development and progression of AD. Intriguingly, the altered cross-talk between the components of such a triangle of death, beyond altering the redox homeostasis of the neuron, is further exacerbated by increased levels of OS that target and impair key components of the pathways involved. Redox proteomic studies in human samples and animal models of AD-like dementia led to identification of oxidatively modified components of the pathways composing the triangle of death, therefore revealing the crucial role of OS in fueling this aberrant vicious cycle.
Future Directions:
The identification of compounds able to restore the function of the pathways targeted by oxidative damage might represent a valuable therapeutic approach to slow or delay AD. Antioxid. Redox Signal. 26, 364–387.
Introduction
Alzheimer's disease
A
During the degenerative process, the brain of AD patients undergoes morphological and functional changes, including loss of synapses, disturbed neurotransmission, and decreased metabolism, all contributing to neuronal death (248). AD is often preceded by three stages of progression characterized by a measured increase of AD hallmarks initiating from preclinical AD to amnestic mild cognitive impairment (MCI) and early AD (EAD) (172, 202).
The amyloid cascade hypothesis proposes that increased production and decreased clearance of aberrant Aβ peptide secondary to β and γ secretase-induced cleavage of amyloid precursor protein (APP), including Aβ oligomers and formation of C-terminal fragments of APP, are the primary originating causes for synaptic flaws and subsequent cognitive complications of AD (27, 31). Indeed, increased Aβ formation, on one side, disrupts cell-to-cell communication and activates immune cells, which trigger inflammation, and on the other side, leads to increased lipid peroxidation and subsequent oxidative stress (OS) at membrane levels, thus causing overall neuronal death (102). The importance of APP, and consequently Aβ, in AD pathogenesis has clearly appeared from patients with familial AD (FAD) and Down syndrome (DS) that present genetic alteration of the genes involved in APP cleavage (41).
FAD patients are linked directly to highly penetrant, autosomal, dominant genetic mutations in the APP and presenilin 1 and 2 (PS1, PS2) genes, while DS patients develop AD pathology prematurely due to the extra copy of chromosome 21, which contains the gene for APP. In this regard, several authors have shown that by the age of 40, the majority of individuals with DS have evidence of brain changes characteristic of AD, with deposition of senile plaques and NFTs (191, 193). However, despite that AD has been extensively studied in the last decades, the etiology of sporadic AD is not yet well understood and several other hypotheses have been proposed to support changes occurring in AD such as the cholinergic hypothesis, the mitochondrial cascade hypothesis, and the tau hypothesis.
Recent human and preclinical studies have provided convincing evidence that AD is also a metabolic disease (13, 29). AD shares many features in common with systemic insulin resistance diseases, including reduced insulin-stimulated growth and survival signaling, increased OS, proinflammatory cytokine activation, mitochondrial dysfunction, and impaired energy metabolism (71). The brain, although accounting for only 2% of the total body weight, consumes about 20% of glucose (231); therefore, glucose metabolism is essential for healthy brain function, and a reduction of glucose utilization may lead to increased OS, altered neuron homeostasis, increased neuronal death, and eventually brain dysfunction and memory loss (3, 62, 70, 128). However, in addition to being crucial in controlling glucose levels, insulin in the brain is deeply involved in development and maintenance of cognitive performances (such as learning and memory) by regulating neurogenesis, synaptic plasticity, lipid metabolism, transmitter receptor trafficking, cerebral blood flow, protein homeostasis, inflammatory responses, and OS (13, 29).
The triangle of death
AD brains exhibit defective insulin signaling, altered levels and/or aberrant activation of components of the insulin signaling pathway, and decreased sensitivity to insulin, all summarized in a condition known as brain insulin resistance (BIR) (12, 68, 70). Deficits in cerebral glucose utilization and reduced energy metabolism were shown in AD (128, 233, 260a). The impairment in brain insulin/insulin growth factor (IGF) signaling in AD is directly associated with increased accumulation of Aβ, phosphorylated tau, reactive oxygen species/reactive nitrogen species (ROS/RNS), and proinflammatory and proapoptotic molecules (12, 69 –71).
Insulin signaling is mutually related, through the phosphoinositide-3-kinase (PI3K)/AkT axis, with the mammalian target of rapamycin (mTOR) pathway recognized as being involved in cellular senescence, organismal aging, and age-dependent diseases. mTOR is a serine–threonine kinase that functions as an intracellular energy sensor and plays a particularly important role in regulating energy balance (65, 72). Several studies report that the aberrant and sustained activation of PI3K/Akt/mTOR signaling is an early feature of AD and results in alteration of multiple pathways, including glucose metabolism and energy production, and of the regulation of protein synthesis/clearance (65, 182, 184, 192, 254). Insulin signaling is both an upstream and a downstream signal of mTOR. Indeed, under sustained insulin activation, mTOR is able to inhibit the insulin receptor substrate-1 (IRS-1), thus uncoupling the PI3K/Akt axis from the insulin signal (51, 217). When this feedback mechanism falls outside the control of the neuron, BIR occurs, a phenomenon characteristic of AD (43, 184).
In addition, mTOR signaling is tightly linked to neurodegeneration through its role in controlling protein homeostasis that appears to be particularly important in the brain (99, 113, 135, 179). Protein homeostasis is maintained by a network of cellular mechanisms that control folding, expression levels, cellular localization, and interactions of proteins from their synthesis through their degradation (76). The unfolded protein response (UPR) and molecular chaperones ensure the proper folding of proteins throughout the life of neurons (125, 221, 258). When refolding strategies fail, misfolded/unfolded proteins are transferred to cellular degradation pathways (15, 16) comprising the ubiquitin–proteasome system (UPS) and autophagy (135, 147, 166, 252). AD is characterized by abnormal accumulation of aggregated proteins in postmitotic neurons, and autophagy, one of the best-characterized downstream pathways regulated by mTOR, has been largely implicated in this phenomenon (99, 179, 188). Several studies reported that reduced autophagy in AD brain and animal models, which leads to the accumulation of Aβ and/or p-tau aggregates, is likely caused by the hyperactivation of mTOR (42, 76, 179, 195, 212). Therefore, the worsened activity of autophagy, accompanied with impairment of the UPS, allows the overproduction, aggregation, and lower clearance of Aβ and p-tau; increased levels of OS; and oxidative damage and dysfunction of mitochondria (76, 166, 192). These aberrations, in turn, contribute to impairment of the abovementioned metabolic pathways controlled by insulin and mTOR.
Within this scenario, the concomitant and mutual alterations of energy metabolism–mTOR signaling–proteostasis network represent a self-sustaining chain of detrimental events that lead to the degeneration of neurons and to development and progression of AD. Intriguingly, this triangle of death, beyond altering the redox balance of the neuron, is strongly fueled by the increased levels of OS. Indeed, evidence supports that major elements of glucose metabolism, insulin signaling, mTOR pathway, autophagy, UPR, and UPS are targeted and oxidatively modified by increased OS during AD and AD-like dementia, thus contributing to boost the mutual aberrant cross-talk, which finally leads to neuronal death (30, 33).
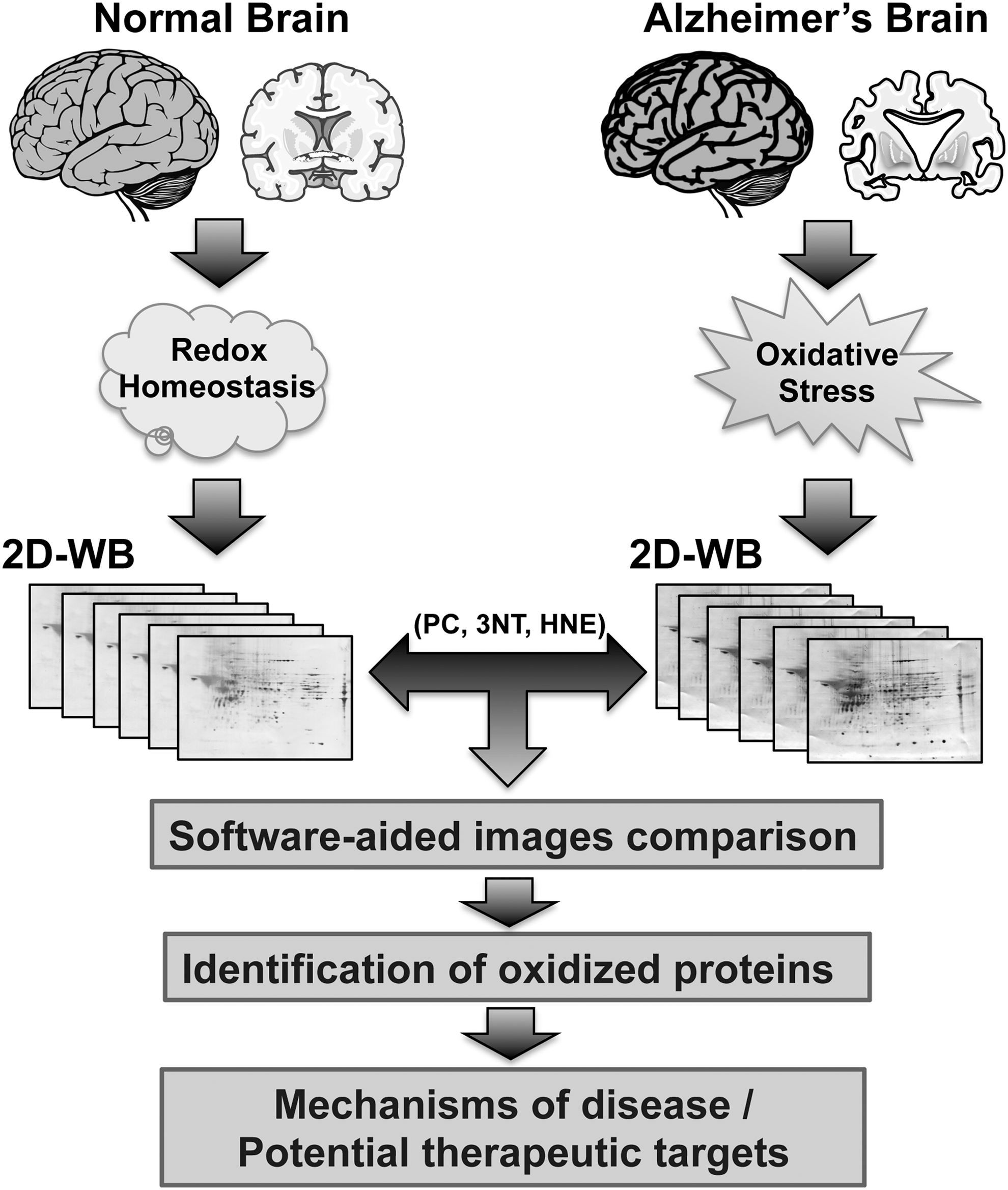
Since 2002, the year of the first use of redox proteomics, methods pioneered in our laboratory represent a promising approach to unravel and highlight the brain targets of oxidative damage, thereby increasing understanding of the molecular mechanisms involved in the AD neurodegenerative process and potential strategies of intervention (36). This review article summarizes redox proteomic results, collected in human samples and animal models of AD-like dementia, with respect to oxidatively modified proteins that are components of the pathways composing the triangle of death and that participate in maintaining such aberrant vicious cycle (Table 1).
3-NT, 3-nitrotyrosine; AD, Alzheimer's disease; DS, Down syndrome; EAD, early Alzheimer's disease; HNE, 4-hydroxynonenal; MCI, mild cognitive impairment; PC, protein carbonyl; PCAD, preclinical Alzheimer's disease; TCA, tricarboxylic acid; UPR, unfolded protein response; UPS, ubiquitin–proteasome system.
Redox Proteomics in AD-Like Dementia
OS and oxidative markers in neurodegenerative diseases
OS indicates a condition in which ROS overwhelm the cellular antioxidant defense activity by increasing production and/or by a decrease in the detoxification of ROS. The principal source of free radicals is the mitochondrial oxidative phosphorylation (Ox Phos) pathway due to the electron leakage that causes the formation of free radical superoxide (O2 •−) from the electron transport chain (93, 230, 232). The high lipid content of nervous tissue, together with its elevated aerobic metabolic activity, makes the brain exceptionally vulnerable to OS that targets and damages all biological molecules (carbohydrates, nucleic acids, lipids, and proteins) (40). Oxidation of proteins often leads to modifications in the secondary and tertiary structures, including dissociation of subunits, unfolding, exposure of hydrophobic residues, aggregation, and backbone fragmentation (16). Proteins can be oxidized by direct ROS attack, by secondary oxidation products such as reactive aldehydes (malondialdehyde [MDA] and 4-hydroxynonenal [HNE]), formed as final by-products of lipid peroxidation that bind to proteins via Michael addition, or by glycoxidation reactions (26, 160, 208). Protein carbonyl (PC) levels are one of the most abundant indices of protein oxidation that may result in loss of function of the affected protein (149, 246). Furthermore, the reaction of O2 •− with the free radical nitric oxide (•NO) produces peroxynitrite, a powerful pro-oxidant molecule. Proteins can undergo nitration in the presence of peroxynitrite and CO2 by modification of tyrosine residues to produce 3-nitrotyrosine (3-NT) (39). Protein nitration, as well as carbonylation, inactivates proteins or enzymes by altering their protein structure. In addition, by inhibiting phosphorylation of Tyr residues due to the steric effect of 3-NT bound to enzymes, protein nitration may drastically affect several regulatory pathways with pathological consequences (33).
The oxidative damage to proteins is of particular interest in age-related neurodegenerative diseases because in the majority of cases, it is a nonreversible phenomenon that alters functionality of neurons (36). Some proteins show susceptibility to one of these types of oxidative modifications due to the intracellular localization (i.e., membrane proteins), the protein sequence (i.e., amino acids susceptible to oxidation), and the tridimensional structure, among others. For example, among all amino acid side chains, cysteine and methionine are the most susceptible to oxidation because they contain reactive sulfur atoms (224).
Furthermore, another key event that affects susceptibility to different oxidative modifications is the release of different oxidant species in the close vicinity of the peptide/protein. At the level of the membrane, lipid peroxidation with production of toxic aldehyde (HNE-bound proteins) is likely to occur and may target membrane proteins. In addition, increased levels of nitric oxide as well as variation in pH and concentration of carbon dioxide are particularly harmful for cysteine, methionine, and tyrosine residues (i.e., methionine sulfoxide and protein nitration). Taken together, it is quite complicated to predict the susceptibility of a protein to undergo carbonylation, HNE modification, or nitration without considering its environment (tissue, intracellular localization, and also exogenous factors).
Generally, oxidation of proteins could affect different processes, including protein expression and gene regulation, protein turnover, cell signaling, apoptosis, and necrosis, eventually leading to the loss of cell homeostasis and function (26, 30, 73). Beyond altering protein structure and function, one of the major consequences of protein oxidation is the formation of large protein aggregates, which are often toxic to cells if allowed to accumulate. Deposits of aggregated, unfolded/misfolded, and oxidized proteins amass normally as function of time both outside and inside the cell and are recognized to be one of the leading events in a broad range of neurodegenerative diseases. The protein quality control system (PQC) offers a critical protective role in the cell throughout the degradation of damaged proteins. The decreased functionality of such protective machinery secondary to oxidative modification may leave the cell incapable of efficiently removing damaged/dysfunctional proteins, resulting in their accumulation and in the alteration of neuronal protein homoeostasis (85, 137, 256). Although increased protein oxidation could yield, in some cases, to protein gain of function, which translates into increased protein binding capacity and/or increased enzyme activity, when the gain of function of a protein is not accompanied by compensatory mechanisms in the cell, it may result in the alteration of cell, physiological status, and in cell toxicity.
The redox proteomic approach
Proteomic platforms allow defining an accurate and detailed profile of specific protein alterations occurring in a system [extensively revised in Butterfield et al. (33, 36)]. To collect insights into the status of the proteome challenged by diseases or therapies, the use of this technique represents a valuable tool. The use of redox proteomics is currently performed on in vitro or in vivo samples to identify specific oxidative modification to proteins (28). Among different techniques available, the gel-based redox proteomic approach takes advantage of immunochemical detection methods for the recognition of distinctive OS markers. Indeed, the two-dimensional blots obtained after isoelectrofocusing-SDS-PAGE are probed with primary antibodies against a specific oxidative modification (PC, HNE-, or 3-NT-bound) and incubated with a secondary antibody for detection/quantification and analysis (30, 36, 37). Spot-matching software are used to compare spot density changes in samples by pixel detection analysis, allowing for multiple comparisons of gels and blots at once. Spots located within the gel map with altered density are then excised from the gel, digested with trypsin, and identified using two different approaches that include peptide mass fingerprinting using matrix-assisted laser desorption–ionization time-of-flight (MALDI-TOF) mass spectrometry and sequence tags using nanoelectrospray ionization tandem mass spectrometry (nano-ESI-MS/MS) (33). Due to the availability of human and other species genomic databases, raw MS data, no matter the MS platform, can be searched against protein databases using search tools such as MASCOT and SEQUEST (89) (Fig. 1). In addition, a further aspect worth considering in performing proteomics is the status/quality of the brain sample at disposal. Indeed, several factors such as brain pH, postmortem interval (PMI), and isolation/storage methods could affect post-translational modifications of brain proteins, thus resulting in biased redox proteomic data. Brain tissue pH is considered to reflect postmortem tissue integrity, and extended PMIs as well as improper storage lead to altered pH and increased oxidation of brain proteins (55).
Results obtained by redox proteomic studies on postmortem brain tissue allowed identifying proteins with differential oxidation ratio between two or more sets of samples, but they did not allow obtaining the quantitative measurement of the amount of oxidative modification targeting a specific protein so far. Recent methodological advancement in the detection of protein oxidation draws the possibility to quantify the amount of oxidative modification targeting a specific protein, therefore allowing to establish, in the near future, a threshold of oxidation that triggers protein structure/function alteration.
As demonstrated by several authors, the increase of OS is one of the main contributors to the development of age-related neurodegenerative disorders (26, 111, 181). The brain of AD patients experiences more oxidative damage than a normal brain, demonstrating an increased propensity to OS and a relatively low level of naturally occurring antioxidants (159, 201). Among others, Aβ peptides, altered mitochondrial function, and the presence of trace metal ions, such as iron and copper, have been identified as possible sources of OS (93, 138, 159, 199). In agreement with the Aβ-induced OS hypothesis, OS is the result of Aβ insertion into the membrane bilayer, thus causing the release of ROS (31, 41) and initiating lipid peroxidation and protein oxidation in AD pathology. Regions of the brain rich in Aβ peptide demonstrated increased levels of protein oxidation, while regions poor of Aβ presence, such as cerebellum, do not (118). Elevated levels of carbonylation, HNE and 3-NT bound to proteins, free HNE, and MDA have been described not only in the brain but also in cerebrospinal fluid (CSF), blood, and urine of AD patients when compared with healthy controls (74, 77, 81, 200). The use of redox proteomics performed in AD, EAD stages, and AD-like dementia brain samples and models revealed a number of oxidatively modified brain proteins potentially related to the degenerative process (Fig. 1). Most of the identified proteins are involved in energy production, synaptic plasticity, and protein homeostasis consistent with the notion that alteration of the key protein regulating these pathways is involved in AD progression and pathogenesis (30, 33, 36). The results of proteins, part of such pathways, are discussed in the following paragraphs.
Energy Metabolism and Insulin Signaling
Dysfunction of glucose utilization represents a key aspect in the development of neurodegenerative disorders (83, 86, 165). Uncontrolled progressive weight loss and abnormal glucose tolerance are common metabolic dysfunctions observed in AD, HD, and PD, which appear to negatively impact overall prognosis through different mechanisms not yet elucidated. It is noteworthy that the disease loci of AD, HD, and PD often involve the hypothalamus, a key regulatory brain region for global energy homeostasis (45).
Although for long time it has been thought that brain energy metabolism mainly relies on oxidative metabolism, further studies demonstrated that the activity-dependent increase in blood flow and glucose utilization was only marginally matched by parallel increases in oxygen consumption (14, 94, 95). These observations opened the way to further studies demonstrating that the brain metabolic needs are met, at least in part, by nonoxidative glucose metabolism, such as glycolysis (92). In fact, while neurons rely on oxidative metabolism to meet their high energy needs, astrocytes do not because their profile is highly glycolytic (14).
In this picture, defects in energy metabolism observed in AD rely on a dysregulation of complex network coupling defects of the glycolytic pathway, the oxidative metabolism, and insulin signaling, the latter also known to regulate glucose uptake in the brain (11, 13, 18, 29, 68). These pathways are strictly related to each other, and trying to shed light on causative links possibly explaining which event precedes the other is not easy. Despite this heterogeneity, one of the aspects, which strongly emerges in several studies and that seems to be a common link, is the neurotoxic effect mediated by increased oxidative/nitrosative stress (NS) levels (10, 26, 199). As noted above, neurons and astrocytes, although complementary, differ in terms of metabolic pathways and needs—the former more oxidative and the latter more glycolytic—and consequently, the impairment of the enzymes deputed to glucose metabolism and energy production has a great impact on their vitality (14). Furthermore, neurons are vulnerable to excitotoxic stress and therefore any increased stress or reduced repair mechanism can accumulate over time, contributing to the development of neurodegenerative disorders as it leaves neurons more exposed to toxic influences (14, 68). In that picture, redox proteomic analyses of AD, MCI, or EAD human brain samples revealed the oxidative modification of several key proteins that belong to the metabolic pathways cited above (33).
From a molecular point of view, glycolysis starts once glucose uptake from the extracellular space takes place thanks to the activity of several glucose transporters (GLUTs). The association of cerebral glucose transport dysfunction with AD pathology has been investigated. Structurally, GLUTs show a high degree of homology and share common sequence features, although they exhibit tissue-specific localization and substrate specificities (i.e., fructose, galactose, mannose, glucosamine, xylose, dehydroascorbic acid, urate, and myoinositol) (19). Among GLUTs, the brain expresses five GLUTs (GLUT1, GLUT2, GLUT3, GLUT4, and GLUT8) differently distributed in brain areas (19). AD patients show decreased GLUT1 and GLUT3 expression, especially in the cerebral cortex (227). The dentate gyrus of hippocampus also exhibits decreased GLUT3 expression (115). Significant decline in expression levels, but without statistical changes in mRNA levels of GLUT1 in the human AD brain, suggests a post-transcriptional regulation mechanism (169). Interestingly, increased OS levels were shown to modulate the subcellular redistribution of GLUT1, decreasing its content at the plasma membrane, thus suggesting a possible molecular mechanism (91).
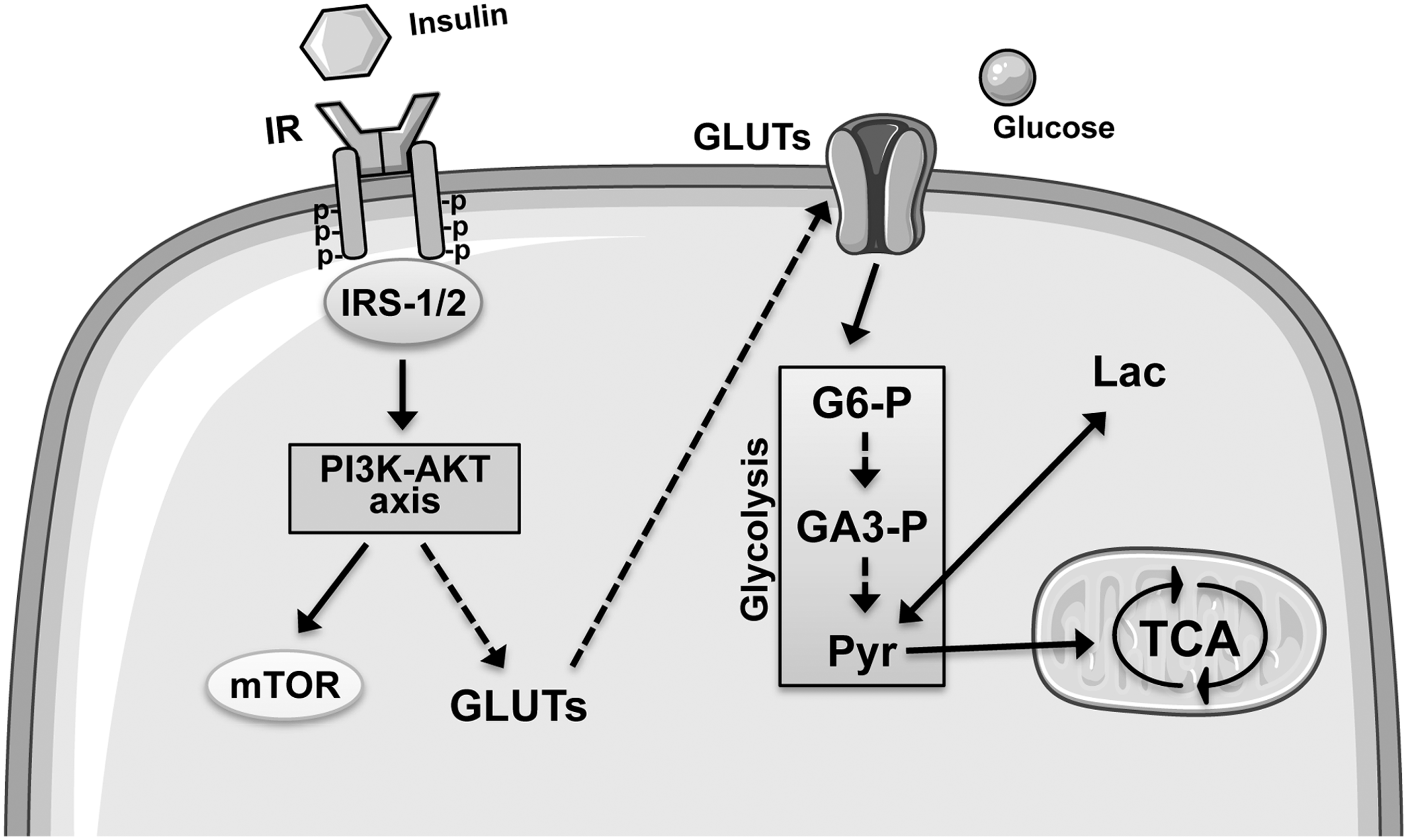
Once in the cytoplasm, glucose is phosphorylated by the hexokinase (HK) to produce glucose-6-phosphate (G6-P) (165). Then, G6-P can follow three different metabolic pathways: (i) glycolysis, (ii) the pentose phosphate pathway (PPP), and (iii) glycogenesis (in the astrocytes) (165). Glycolysis is used to produce ATP and NADH under anaerobic conditions by transforming glucose to pyruvate and lactate (165). Indeed, once produced, pyruvate can be converted into lactate by the enzyme, lactate dehydrogenase (LDH), or enter in the mitochondria where, under aerobic conditions, it is metabolized through the tricarboxylic acid (TCA) cycle and Ox Phos, leading to the production of ATP and CO2 (165) (Fig. 2). In parallel with glycolysis, G6-P could enter in the PPP to generate reducing equivalent in the form of NADPH as well as ribose 5-phosphate (R5-P). R5-P can alternatively undergo a series of isomerizations, transaldolations, and transketolations that result in the production of fructose-6-phosphate (F6-P) and glyceraldehyde-3-phosphate (GA3-P) (both intermediates in glycolysis) (165). Finally, in astrocytes, G6-P can also be used to store glucosyl units as glycogen (165).
By following the glycolytic pathway, the first protein found to be oxidized is fructose bisphosphate aldolase (FBA). This enzyme catalyzes a reversible reaction that splits fructose 1,6-bisphosphate into the triose phosphates, dihydroxyacetone phosphate and GA3-P. The first data about FBA activity in AD date back to early 1980s when a dysfunction of this enzyme was observed in autopsy temporal lobes (21, 132, 164). Interestingly, in 2005, FBA was reported to be an autoimmune target, suggesting that the aldolase autoantibodies detected might enter brain cells, bind the enzyme, and hinder its function, leading to reduced glucose utilization (170). Quite recently, proteomic studies reported decreased FBA protein levels in the hippocampus of AD subjects (238). Similarly, increased carbonylation and increased nitration were found in the inferior parietal lobule (IPL) of EAD and MCI subjects, respectively (209, 242), highlighting the OS-induced impairment of this protein.
The second protein in the glycolytic pathway is glyceraldehyde-3-phosphate dehydrogenase (GAPDH). This enzyme catalyzes the sixth step of glycolysis, during which a molecule of GA3-P is converted to 1,3-bisphosphoglycerate with production of NADH (130). Although GAPDH also possesses other functions, including DNA/RNA binding, gene transcription, and protein interaction (34), its role in the glycolytic process is unquestionable. Reduced activity of GAPDH in AD was reported in early studies (141, 163). Interestingly, further studies reported that increased OS-induced disulfide bonding occurred in the structure of GAPDH with subsequent protein aggregate formation, which may have relevance to the pathophysiology of AD (64). Impaired GAPDH activity was then additionally proved by showing that GAPDH is subject to many different types of oxidative modifications in AD brain, modification that drastically affects its structure and function, including S-glutathionylation (177), S-nitrosylation, and nitration (167, 168, 243), and direct or indirect reaction with ROS (23). These OS/NS-induced GAPDH modifications would preclude GAPDH membrane binding, which (under normal conditions) prevents modification of active-site Cys residues, essential to glycolytic activity (98). Furthermore, inhibition of GAPDH would cause the cell to shift its reliance on glycolysis to the pentose phosphate shunt, which produces NADPH in lieu of NADH. This switch could permanently uncouple the production of ATP and pyruvate from glycolysis (58, 222), thereby contributing to the growing anaerobic environment found in AD brain (98). Intriguingly, oxidatively modified GAPDH accelerates Aβ amyloidogenesis, subsequently leading to mitochondrial dysfunction and neuronal cell death, key features of AD pathogenesis (131).
α-Enolase (ENO1), one of the three enolase isoforms, is a glycolytic enzyme that catalyzes the conversion of 2-phosphoglycerate to phosphoenolpyruvate. ENO1 has been identified as differentially expressed and a target of different kinds of oxidative/nitrosative post-translational modifications, rendering it one of the top 15 most frequently identified differentially expressed/oxidized proteins in AD (35). These observations further suggest that this enzyme is strongly impaired along the progression of AD pathology. Several redox proteomic studies from the Butterfield laboratory reported both increased oxidation and nitration of ENO1 in different brain areas of AD (32, 49, 50, 208, 209, 239, 243) and MCI subjects (38, 49, 244). Similar results have been also obtained in mitochondria isolated from peripheral lymphocytes from patients with AD and MCI (237, 240), suggesting the possibility that oxidatively modified ENO1 could be part of a panel of proteins useful as biomarker for MCI and AD. ENO1 also was found to be oxidatively modified in the brain from animal models of AD pathology, including mice and dogs (187, 216, 225). Interestingly, impairment of ENO1 would affect neuronal Ca2+ homeostasis by reducing ATP production and the consequent ATP content required, for example, for the functioning of the plasma membrane Ca2+-ATPase. This event would be then associated with an increased release of Ca2+ from endoplasmic reticulum (ER), provoking the emptying of ER store (157). Disruptions in the release and storage of intracellular Ca2+, which are consequences of moderate changes in energy metabolism, may contribute to the cell death process, including death of neuronal cells.
Phosphoenolpyruvate resulting from ENO1 activity is then converted to pyruvate with phosphorylation of ADP to ATP through the activity of pyruvate kinase (PK), which is therefore an enzyme responsible for energy production in the glycolytic pathway under anaerobic conditions (165). PK is significantly nitrated in the hippocampus of MCI subjects (38). Subsequently, in vitro experiments demonstrated increased PK oxidation following Aβ exposure in neuronal cells (241), and these data have been further confirmed both in aged rat brain (194) and human IPL MCI samples also reporting reduced enzyme activity (207).
The last step of the anaerobic glycolytic pathway is the reduction of pyruvate to lactate mediated by LDH. LDH dysfunction and subsequent reduced glucose metabolism are highly observed in PET scans of MCI and AD brain. Redox proteomic studies demonstrated increased oxidation (207) and increased nitration (50) of LDH in MCI and AD brain, respectively, together with reduced LDH activity (207).
Oxidative impairment of ENO1, PK, and LDH represents an especially strong neurotoxic stress because by reducing their activities, a block of energy production occurs in the brain. Indeed, whether (on one side) ENO1 and PK activities are required for the production of ATP along the glycolytic pathway and (on the other side) pyruvate production is essential because it can be transported to the mitochondria where it enters the TCA cycle and is further broken down to produce considerably more ATP through Ox Phos, the impairment of LDH activity impacts lactate production (especially in astrocytes), which according to recent findings can be used as an energy substrate for neurons for oxidative-derived ATP production (14). Reduced glucose utilization due to OS/NS-induced impairment of glycolytic enzymes would be further associated with increased glucose levels, especially in the early phases of AD (260). According to recently discovered mechanisms, increased glucose levels would lead to an increased extracellular glutamate release, thus resulting in N-methyl-D-aspartate receptor activation and increased neuronal [Ca2+], neuronal nitric oxide synthase stimulation, and consequent NS (1). NS would be then responsible for S-nitrosylation and inactivation of the insulin-degrading enzyme known to reduce Aβ when functional. Similarly, increased glucose levels can evoke rapid changes in neuronal excitability through inhibition of KATP channels, which result in increased Aβ production (156). Increased Aβ levels, in turn, might mediate similar effects, amplifying this vicious cycle (1) and promoting mitochondrial damage (41).
In the mitochondria, malate dehydrogenase (MDH) catalyzes the reversible oxidation of malate to oxaloacetate by NAD+ through the TCA cycle. MDH links glycolysis to the electron transport chain by transferring NADH to NADH dehydrogenase (complex I) through the malate–aspartate shuttle, resulting in the production of ATP (36). Interestingly, MDH was found both significantly oxidized and nitrated in the hippocampus and IPL of MCI (244), EAD (208), and AD (145) subjects. In contrast to other oxidatively modified proteins, these modifications are associated with an increased MDH activity, consistent with elevated MDH activity in late-stage AD (145). Since the activity of MDH rises also during normal aging processes (25, 186), findings in MCI and EAD can further strengthen the hypothesis that oxidative modification of brain MDH could promote conformational changes resulting in elevated activity and mitochondrial dysfunction in AD.
The evidence discussed above supports the concept that modification of glycolytic and TCA cycle enzymes may disrupt neuronal energy metabolism and ion homoeostasis, thereby impairing ion-motive ATPases, signal transduction, membrane lipid asymmetry (8), and glucose and glutamate transporters (146), thus pointing to altered energy metabolism as a common and key theme in AD neurodegeneration.
The impairment of energy metabolism enzymes in AD is further corroborated by recent findings obtained by a proteomic approach that demonstrated the presence of CSF autoantibodies targeting key players of energy metabolic pathways, including glycolysis and the TCA cycle, found oxidatively modified in AD brain studies, including GAPDH and PK (78). Indeed, it is conceivable that proteins undergoing oxidative modification become dysfunctional, prone to aggregation, and toxic to neurons, which induce clearance systems and immune responses, thus producing auto-IgGs targeting oxidized brain proteins. These data suggest a potential casual sequence between oxidative damage at the brain level, autoantibody presence in CSF, and reduced energy metabolism of AD patients (78).
Reduced glucose uptake and utilization in AD brain (18, 29, 68) closely parallels with BIR (3, 29). Of importance, while in peripheral tissues, insulin mainly regulates glucose uptake (235), in the brain, this hormone also plays neurotrophic functions (117). Accordingly, impaired insulin signaling and glucose uptake in AD patients were observed in brain areas related to cognitive performances such as the hippocampus, cortex, and choroid plexus (84).
On a molecular level, insulin and/or IGF-1 greatly affect the activities of the central nervous system (CNS) by regulating key processes, including among these energy homeostasis, neuronal survival, longevity, and learning and memory (97, 198). Insulin and IGF-1 bind to the tyrosine kinase receptors, IR and insulin growth factor-1 receptor (IGF-1R), which are highly distributed in two of the main brain areas affected by AD pathology, that is, the hippocampus and cerebral cortex (97, 198).
Following their activation, IR and IGF-1R recruit IRS-1 (97, 198), which leads to the activation of two main signaling pathways: (i) the PI3K pathway, which, among other functions, is involved in the maintenance of synaptic plasticity and memory consolidation (127), Aβ-induced memory loss (56), synthesis of NO, which in turn plays a role in learning and memory processes (47); and (ii) the mitogen-activated protein kinase (MAPK) cascade, which is responsible both for the induction of several genes required for neuronal and synapse growth, maintenance, and repair processes and for serving as a modulator of hippocampal synaptic plasticity that underlies learning and memory (3) (Fig. 2).
Several reports highlight a close connection between insulin resistance and defects in energy metabolism driven by OS (29, 62, 70, 262). AD patients show reduced brain IR sensitivity (214, 250), hypophosphorylation of the IR, and downstream second messengers such as IRS-1 (233, 250) and attenuated insulin and IGF-1R expression (233). Indeed, increased OS levels in AD promote multiple effects, including the inhibition of cellular energy production, as described above, and the reduction of both insulin secretion and sensitivity (100, 171). In turn, defective insulin signaling-associated impairment in glucose uptake and utilization leads to a vicious cycle, in which reduced energy production is associated with increased ROS and RNS that contribute to oxidative/nitrosative damage in mitochondria (70, 176).
Interestingly, studies on humans showed that intranasal insulin administration slows AD-associated cognitive decline, thus strengthening the role of BIR in AD onset and progression. A single dose of intranasal insulin acutely improved memory in memory-impaired older adults with AD or MCI and also improved memory and cognitive function with multiple treatments of patients with AD or MCI (210). Furthermore, intranasal insulin administration to MCI and AD subjects facilitated recall on two measures of verbal memory in memory-impaired adults and also differentially modulated plasma β-amyloid for memory-impaired subjects and normal controls by promoting an increased Aβ40/42 ratio in the former (63). In addition, based on PET findings, the authors also provided direct evidence for a higher 18F fluorodeoxyglucose uptake in the parietotemporal, frontal, precuneus, and cuneus regions of the CNS following intranasal insulin compared with placebo administration (63).
Whether BIR precedes the augmentation of OS/NS levels, or vice versa, is still under evaluation in AD. Recent data collected in both wild-type and 3xTg-AD mice support a role for OS-induced hypothesis of insulin resistance in AD (12). Indeed, based on these results, the increase of OS/NS levels precedes the molecular events (reduced IR levels/activation and increased IRS inactivation) responsible for BIR during both normal aging and AD (12). In addition, the progressive worsening of insulin resistance along the progression of AD correlates with increased OS levels, DNA damage, and protein oxidation demonstrated by HNE, PC, and 3-nytrotyrosine 3-NT accumulation (70).
To summarize, the Aβ/OS-induced (247) impairment of (i) energy metabolism and (ii) insulin signaling would increase neuronal vulnerability to synapse damage by ultimately leading to memory impairment in AD (20, 69, 154, 250). To discriminate which, between impairment of the glycolytic pathways and the dysfunction of oxidative metabolism, plays a major role in terms of defective energy metabolism is hard to say based on the data we collected so far. In this study, we can speculate by saying that because astrocytes outnumber neurons in the brain (175), this difference in terms of quantity also would affect the final outcomes, possibly suggesting a major role for the glycolysis. Notwithstanding, neurons display limited defense mechanisms against OS compared with astrocytes, rendering them more susceptible to OS/NS-induced damage (14), which finally would account for a major role for the oxidative metabolism dysfunction. Despite that, the observed increase in mitochondrial OS/NS levels can be blunted by astrocytes, which being more resistant than neurons could provide the antioxidants defense to avoid further damage (14). Therefore, once again, we are in front of a vicious cycle, which requires further analyses.
It is interesting to discuss that in response to reduced glucose metabolism, mammalian cells promote the synthesis and utilization of ketone bodies. Ketone bodies are well-recognized circulating energy sources for tissues in times of fasting or prolonged exercise (2). Metabolism of ketone bodies supplies a fraction of needed ATP that may partly compensate for the deficiency in glucose metabolism in AD patients. However, this is possible because glucose metabolism is not compromised in AD to a degree inhibiting ketone body metabolism (121).
Furthermore, recent data demonstrate that the ketone body beta-hydroxybutyrate (bOHB) has a broad range of signaling and regulatory effects, including inhibition of many protein deacetylases, and a role as a ligand for at least two cell surface receptors. In addition, metabolism of bOHB in target tissues can alter the intracellular balance of acetyl-CoA, succinyl-CoA, and NAD, all of which have their own emerging regulatory functions.
Taken together, these data suggest that a ketogenic diet may help to improve mental function in AD.
mTOR and Related Protein Homeostasis
The signaling pathways controlled by mTOR represent an appealing objective of study with respect to both aging and energy balance since these pathways have the potential to affect a large number of processes that could be critical in age-related degenerative phenomena (184). mTOR is not only expressed at high levels in the brain, mainly in neurons, but also in glial cells. During embryonic development, mTOR signaling functions as a powerful neuronal survival and division signal in response to growth factors and hormones, including IGF-1 and insulin (123). During brain development, mTOR promotes extension of neurites (dendrites and axons) and activation of mTOR results in increased dendrite branching, higher numbers of immature filopodia-like protrusions in dendrites, and decreased density of mature dendritic spines (249, 255). In adult brain, the mTOR pathway evolves to control synaptic plasticity and processes underlying memory and learning, having significant functional impact on neuronal polarity, neurotransmission, protein homeostasis, metabolic pathways, and stress responses (7, 72, 123, 182).
In the control of synaptic plasticity, mTOR is crucial because it coordinates the regulation of protein synthesis/degradation, through the on/off switch of translation and autophagy, to avoid both the loss of protein expression balance and accumulation of toxic protein aggregates that might result in brain degeneration (113, 166). The regulation of protein synthesis/degradation is both nutrient and energy dependent and occurs through well-characterized downstream targets of mTOR, consisting of cap-binding initiation factor eIF4E 1 (4EBP1), p70 ribosomal S6 kinase (p70S6K), and unc-51-like kinase 1 (ULK1) complex (190) (Fig. 3). Interestingly, data from postmortem human AD brains indicate the hyperphosphorylation of mTOR along with those of p70S6K and eIF4E in the hippocampus and in other brain areas (104, 150, 189, 245, 254). In addition, mTOR hyperactivation correlates with Braak stages and/or cognitive severity of AD patients (190).

Consistent with these results, numerous studies also demonstrated the impairment of an mTOR upstream signaling pathway, that is, the PI3K/Akt axis, in AD brain (5, 150, 161, 189, 190, 192, 197, 249, 259). Under normal conditions, PI3K binds to IRS-1 and converts phosphatidylinositol-4,5-phosphate (PIP2) to phosphatidylinositol-3,4,5-phosphate (PIP3) (182). Increased PIP3 levels recruit Akt to the membrane, where the latter is activated by phosphorylation and inactivates TSC2, a negative regulator of mTOR (96). A primary negative feedback exists between mTOR and insulin signaling. This mechanism acts through mTOR-mediated serine phosphorylation of IRS-1 to induce IRS-1 inactivation and degradation under sustained mTOR activation by uncoupling the PI3K/Akt axis from the insulin and IGF-1Rs (192) (Fig. 3). In agreement with this mechanism, persistent activation of neuronal mTOR signaling in MCI and AD brain was reported to contribute to IRS-1 inhibition, halting the normal activation of PI3K/Akt by insulin (12, 44, 109, 183, 254).
As mentioned above, the primary impact of mTOR on neuronal function is mediated by regulation of protein synthesis and protein degradation pathways. Studies on AD brain and mouse models demonstrated that alteration of mTOR, and its downstream signals, is a key event in the neurodegenerative process being directly involved in the accumulation of Aβ and p-tau peptides, which in turn, after aggregation, contribute to the hyperactivation of mTOR pathways (46, 152, 188, 190, 263). So far, multiple hypotheses have been formulated to explain the chain of events regarding mTOR signaling and AD hallmarks. Interestingly, redox proteomic analysis of AD postmortem brain samples demonstrates that the oxidative modification of key components of the protein synthesis and autophagy machinery, as result of Aβ-induced increase of OS, might contribute to the aberrant functionality of the mTOR pathway and consequently to the deposition of senile plaques and NFTs (30, 33, 192).
Protein synthesis
mTOR controls a number of components involved in the initiation and elongation stages of translation, in particular (via repressor proteins) the 4EBP1 and p70S6K (150, 189). Phosphorylation of 4EBP1 by mTOR results in its dissociation from eIF4E, promoting assembly of the eIF4F complex and enabling eIF4E to promote cap-dependent translation (213). Similarly, the stimulation of S6K1 activity by mTOR leads to increases in mRNA biogenesis, cap-dependent translation and elongation, and the translation of ribosomal proteins through the regulation of the activity of many proteins through phosphorylation, such as S6K1 aly/REF-like target, programmed cell death 4 (PDCD4), eukaryotic elongation factor 2 kinase (eEF2K), eIF4B, and ribosomal protein S6 (155).
In addition, mTOR negatively regulates eEF2 kinase, thus activating eEF2 that mediates the translocation step of elongation in which the ribosome moves relative to the mRNA by one codon and the peptidyl-tRNA shifts from the A site to the P site (60). Redox proteomic studies demonstrated that the elongation factor 1A (EF1A) and the eukaryotic initiation factor 2α (eIF2α) are HNE modified in the IPL of MCI brain (207). EF1A (also known as EF-Tu) and eIF2α are intimately involved in the protein synthesis machinery and have been demonstrated to be downstream signals of mTOR (251). Human EF1A is a crucial translation factor that mediates peptide elongation by promoting GTP-dependent binding of aminoacyl tRNA to the ribosome. Levels of EF1A were reportedly sensitive to inhibitors of PI3K, mTOR, and extracellular signal-regulated kinase (ERK) (6). eIF2a works in concert with mTOR as part of the two major mechanisms for the regulation of protein production rates. eIF2a is phosphorylated under stress conditions to prevent recycling of the eIF2 complex and to inhibit formation of the 43S translation initiation complex (87). This mechanism allows cells to promptly reduce protein synthesis during stress conditions even if growth signaling and/or nutrient availability are not limiting. Phosphorylation of eIF2a downregulates mTOR activity (205), while rapamycin (mTOR inhibitor) was able to prevent eIF2α phosphorylation in hepatic cells (257). The oxidative modifications of both EF1A and eIF2α could significantly compromise protein synthesis and overall neuronal protein homeostasis, thereby contributing to the development of the neurodegenerative process.
Autophagy
With respect to protein homeostasis, increasing evidence suggests that the mTOR/autophagy axis plays a critical role in the removal of toxic/aggregate proteins and impaired organelles that could damage cells during stress. Altered autophagy is reported in various human pathologies, including neurodegenerative diseases, cancers, and lysosomal storage disorders (99, 119, 188). Autophagy is an evolutionarily conserved pathway in which several autophagy-related (Atg) proteins coordinate the activation of three different steps, initiation, elongation, and maturation, which are followed by fusion with lysosomes to form the autolysosome (101). Initiation of autophagy is triggered by activation of the ULK1 kinase complex, which in turn causes the activation of another complex that comprises (among other proteins) the class III PI3 kinase, Vps34, and the protein, Beclin-1 (206). The other protein complex involved in this stage of autophagosome formation is the ULK1/Atg1-Atg13-FIP200/Atg17-Atg101 complex that plays an important role in Atg protein recruitment and autophagosome synthesis (120). The following reaction involves the conjugation of microtubule-associated protein 1 light chain (LC3) to the lipid phosphatidylethanolamine (PtdEn). LC3 is cleaved at its C-terminus by Atg4 to form the cytosolic LC3-I, which is conjugated with PtdEn through the action of Atg7 (E1-like) and Atg3 (E2-like) to generate LC3-II (76). LC3-II is a specific indicator of autophagosome formation widely used as marker to analyze autophagy function (143). Autophagosomes are then transferred along microtubules in a dynein-dependent manner to lysosomes, where membrane fusion forms the autolysosome.
mTOR is the main negative regulator of autophagy. Previous data showed that under starvation conditions or rapamycin treatment, mTOR-mediated phosphorylation of Atg13 and ULK1 is inhibited, thus leading to activation of ULK1 and ULK1-mediated phosphorylations of Atg13, FIP200, and ULK1 itself that normally trigger autophagy initiation (261). Reduced autophagic flux is thought to be implicated in AD neurodegeneration (184, 188, 192), supporting a role in AD for the hyperactivation of the PI3K/Akt/mTOR axis (43, 129, 133, 182, 195, 253, 254). Recent studies in the brain of subjects with AD or AD-like dementia showed reduced markers of autophagosome formation (e.g., LC3-II/I) together with mTOR hyperactivation (75, 179, 188, 195, 254). However, other studies reported the accumulation of autophagic vacuoles in AD (153, 180), suggesting that due to the defective autophagosomal maturation occurring in AD, neurons might attempt to upregulate autophagic flux. In addition, an AD stage-dependent effect has been proposed, with impaired autophagy occurring early in disease and dysregulated overcompensation in advanced AD (188).
Several studies demonstrated that the hyperactivity of mTOR signaling in AD enhances Aβ accumulation (46, 188) by inhibiting induction of the autophagy clearance system (44, 152, 158, 195, 263). In parallel, it was shown that accumulation of Aβ is one of the principal causes of aberrant mTOR signaling. Indeed, preventing Aβ accumulation in the 3xTg-AD mouse model was sufficient to restore the mTOR signaling. In line with these findings, injection of Aβ in the brain of wild-type mice increases the mTOR pathway (44, 158).
Moreover, the final step of the autophagic flux seems also to be affected in AD neurodegeneration. Indeed, degradation of the autophagosome cargo by the lysosomal hydrolases is intimately dependent on the activity of two proteins: the vacuolar [H+] ATPase (V0-ATPase) and cathepsin D (CTSD), which have been found to be oxidized by redox proteomics in DS brain (75, 195, 254). The V0-ATPase pump is required for acidification of the newly formed autolysosomes and for activation of lysosomal hydrolases and degradation of substrates (166, 261). CTSD participates in the degradation of proteins and processing of precursor proteins (15, 105).
Therefore, the aberrant function of both V0-ATPase pump and CTSD strengthens the hypothesis that dysfunction of autophagy significantly contributes to AD-like neurodegeneration. In addition, a recent report showed that V0-ATPase is necessary for amino acids to activate mTOR, thus suggesting that V0-ATPase is an active component of the mTOR pathway (264). Taken together, these results suggest that the alteration of autophagy may disturb both Aβ degradation and APP processing, allowing the buildup of toxic aggregates (179, 188).
Other interesting in vitro redox proteomic studies reported the carbonylation of GFAP in synaptosomes treated with Aβ (1 –42) as a model of AD (22, 134). GFAP is recognized as a member of the chaperone-mediated autophagy (CMA) pathway, a highly specific degradation pathway induced by deletion of the lysosome-associated membrane protein type 2A (LAMP-2A). CMA in involved in the translocation of unfolded proteins directly across the limiting membrane of the lysosome (261), and GFAP directly interacts at the lysosomal membrane either with LAMP-2A or with the EF1A (9). Since CMA is activated as part of the cellular response to OS required for targeting oxidized proteins to lysosomes, oxidation of GFAP might contribute to disrupt the complex network involved in autophagy processes (140).
Overall, the above discussion highlights the close connection between the altered proteostasis network controlled by mTOR and the development of AD-like dementia. In particular, redox proteomic data suggest that increased OS, through oxidative modification of autophagy components, might represent the link between accumulation of Aβ and alteration of mTOR signaling pathways, and vice versa.
Unfolded Protein Response
A key component of PQC in the ER is the UPR (221). UPR is initiated upon disturbance of ER homeostasis and involves the activation of three transmembrane proteins in the ER membrane that serve as sensor proteins: the double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK), the activating transcription factor 6 (ATF6), and the inositol-requiring kinase 1 (IRE1) (256). IRE1, PERK, and ATF6 pathways coordinate an intricate network of stress signals to the cytoplasm and the nucleus that result in overall inhibition of protein translation and control the expression of specific transcription factors and other rapid effects on protein synthesis (54, 126, 258) (Fig. 4). In this way, the UPR is finely connected to the proteolytic machinery of the cell, including the proteasome (219) and autophagy (17, 185). Although many mechanistic details and additional regulatory pathways are still unknown, the core signaling of the mammalian UPR has been revealed (221).
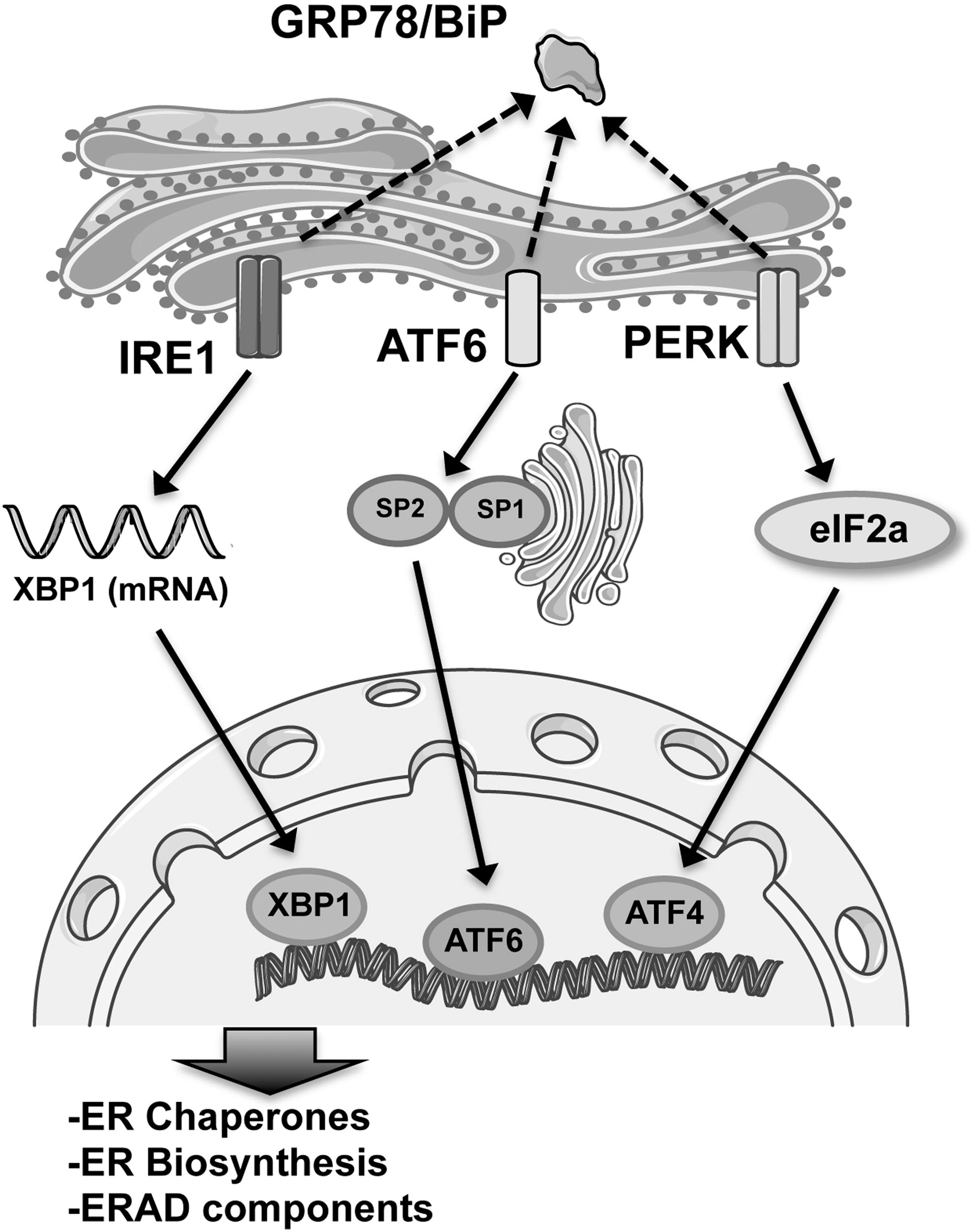
All the three arms of the UPR are activated when the chaperone immunoglobulin-binding protein (BiP/Grp78), normally bound on the luminal side of each protein, dissociates to aid in the folding of accumulated unfolded proteins in the ER lumen. When unfolded proteins accumulate, BiP/Grp78 binds to them to keep correct protein folding and is thereby released from PERK, IRE1, and ATF6, which are consequently activated (252) (Fig. 4). Sustained ER stress can lead to the activation of the apoptotic cascade and ultimately to cell death. Thus, despite the protective role of the UPR to restore cellular homeostasis, prolonged ER stress is considered to contribute to the development of pathological conditions (218).
Activation of PERK occurs through specific phosphorylation, which in turn phosphorylates the eIF2α. Conversely, p-eIF2α is the less active form and leads to a reduction in global protein levels, thus decreasing the load of new proteins inside the ER (114). In addition, activation of PERK promotes the nuclear import of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), which activates the transcription of antioxidant enzyme genes to promote cellular survival (258).
IRE1 presents two genes with homolog sequences: IRE1a and IRE1b. IRE1a is expressed ubiquitously, whereas the expression of IRE1b is limited to gut epithelial cells (110). Once activated, IRE1a produces a kinase and endoribonuclease and catalyzes the correct splicing of the transcription factor X box-binding protein 1 (XBP1), which in turn induces a subset of UPR target genes involved in ER protein synthesis and folding, endoplasmic reticulum-associated protein degradation (ERAD), autophagy, and redox metabolism. IRE1 also activates the preapoptotic c-Jun N-terminal kinase (JNK), the apoptosis signal-regulating kinase (ASK1), and the caspase 4. The IRE1-JNK pathway is also required for activation of autophagy after ER stress (162).
Upon accumulation of unfolded proteins in the ER, ATF6 is released from BiP/Grp78 and is trafficked to the Golgi apparatus where it is cleaved by site 1 and site 2 proteases at the transmembrane site. The cytoplasmic part of ATF6, an active transcription factor known as ATF6 p50 (or nATF6), migrates to the nucleus to activate UPR gene expression (116).
It is well established that accumulation of misfolded/damaged proteins is a common feature of neurodegenerative diseases, and aberrant activation of the UPR has been extensively studied in both in vitro and in vivo models (221). Specific markers for UPR activation are increased in AD brain tissue compared with nondemented control brain (124). BiP/Grp78 expression levels are increased in the hippocampus and temporal cortex from AD brain, and several studies have shown the increased presence of phosphorylated PERK, IRE1, and eIF2α in AD neurons (112, 125, 236). These markers were detected either in healthy neurons or in neurons with hyperphosphorylated tau protein, but are almost absent from NFT-containing neurons. Overall, the levels of BiP/Grp78 and the occurrence of p-PERK in AD neurons correlate well with the presence of abnormally phosphorylated tau and the Braak staging (a method used both in research and for clinical diagnosis to classify AD pathology with respect to the location of the tangle-bearing neurons and the severity of changes) for NFTs (24, 124). These observations indicate that the UPR is involved in the early stages of AD pathology. Activation of the UPR is observed in neurons in AD brain that show diffuse immunohistochemical staining for p-tau, suggesting that UPR activation parallels early tau pathology before deposition of NFTs (178). Recent in vitro studies also show that Aβ can induce ER stress and activation of the UPR (61).
However, data obtained in the cortex of aged Tg2576 mice—a Tg mouse model of AD that undergoes extensive Aβ burden and cognitive deterioration—did not show induction of ER stress markers, suggesting that plaques alone are not sufficient for activation of UPR (148).
Redox proteomic studies reported increased HNE modification of BiP/Grp78 in DS patients with AD pathology (79). It is likely that irreversible oxidation of BiP/Grp78 affects its three-dimensional structure, causing the inability to bind to the misfolded proteins, and contributes to dysfunction of the UPR system (79). Accordingly, oxidation of eIF2α found in MCI IPL supports these notions (207). Indeed, eIF2α (as described above) is involved in the PERK arm of UPR and its impairment of reduced activity could lead to further UPR defects and accumulation of unfolded/misfolded proteins.
In addition, in response to environmental stress, induction of chaperones is a key factor in cell survival and in repairing cellular damage. ATP-dependent molecular chaperones can be grouped in four large and ubiquitous families: HSP100 proteins, HSP90 proteins, HSP70 proteins, and HSP60 proteins. Moreover, there are also ATP-independent chaperones, including small heat shock proteins (sHSPs). Accumulation of chaperones is the response of the affected neurons to eliminate Aβ and Tau (76). By immunohistochemistry and Western blot analysis, it was shown that the expression levels of a number of HSPs, particularly HSP27 and HSP70, were elevated in affected regions from AD brain, possibly indicating the response to activated glia and stressed neurons (59, 112). It is interesting to note that protein levels of HSP27, HSP32, HSP60, HSP70, and HSP90 were also found to be increased already in the hippocampus and IPL of MCI patients (80). However, redox proteomic studies showed that HSP70, although being induced in response to Aβ-mediated neuronal toxicity, is carbonylated in AD brain, possibly with reduced neuroprotection (239). Several other HSPs have been identified to be oxidatively modified in AD, including HSP90 and HSP60 (4, 23). Impairment of these proteins could contribute to proteasomal overload and dysfunction, as it is observed in AD brain (139). A further study demonstrated that in Aβ-treated synaptosomes, HSPs are oxidatively modified (22), thus accounting for the vulnerability of HSPs to Aβ-induced OS.
Another first-line defense against Aβ toxicity is the protein disulfide isomerase (PDI) family of proteins that catalyze the oxidation–reduction and isomerization of intra- and intermolecular disulfide bonds to ensure the correct folding of secretory proteins before their further modification and transport in the ER. The neuroprotective role of PDIA3 against Aβ aggregation in AD has been documented (90). Moreover, the Lipton group showed that active site thiols of PDI were S-nitrosylated, inhibiting its isomerase and chaperone activities, in the brains of virtually all cases of sporadic AD (174). Formation of S-nitrosylated PDI led to accumulation of misfolded and polyubiquitinated proteins, resulted in prolonged UPR activation, and thus participated in persistent ER stress.
Ubiquitin–Proteasome System
The proteasomal system is located both in the cytosol and the nucleus, and it is specialized for the removal of more than 70–80% of intracellular proteins. The UPS not only digests misfolded, oxidized, or damaged proteins but also removes proteins involved in a plethora of intracellular processes, such as signal transduction, cell cycle regulation, and cell death, and also gene transcription (137). Indeed, the UPS exerts specialized activities, depending on its localization and/or on time-dependent regulation.
The majority of damaged/unfolded proteins are destined for degradation to the proteasomal system after being conjugated with ubiquitin, a small protein with 76 amino acids, which is covalently bound through the formation of an isopeptide bond between the ɛ-amino group of a lysine residue of the substrate and the C-terminal carboxylate (151). This conjugation is achieved through three different types of enzymes: E1 (ubiquitin-activating enzyme) hydrolyzes ATP and forms a thioester bond between itself and ubiquitin; E2 (ubiquitin-conjugating enzyme) receives ubiquitin from E1 and forms a similar thioester intermediate with ubiquitin; and E3 (ubiquitin ligase) binds both E2 and the substrate and transfers the ubiquitin to the substrate (106). In some specific conditions and cell types, a fourth conjugation factor, known as the ubiquitin chain elongation factor E4, is necessary together with the E1, E2, and E3 enzymes for efficient polyubiquitination (144) (Fig. 5).

Polyubiquitin chains are then recognized by the proteasome, a multicatalytic complex indicated as the 26S proteasome. The 26S particle comprises ∼60 subunits and therefore is ∼50–100 times larger than the typical proteases that function in the extracellular environment (e.g., in digestion, blood clotting) and differs in critical ways. The 26S complex comprises a central barrel-shaped 20S proteasome with a 19S regulatory particle at either or both of its ends (147).
The 20S proteasome has a cylindrical structure comprising two outer alpha-rings and two inner beta-rings. These stacked rings include two noncatalytic outer rings called α-rings and two catalytic inner rings called β-rings. These three active subunits (beta1, beta2, and beta5) have caspase-like activity that cleaves substrates behind acidic residues, trypsin-like activity that cleaves substrates after basic residues, and chymotrypsin-like activity that cleaves substrates behind hydrophobic residues, respectively (136). The 19S proteasome component contains at least 18 subunits, with a base comprising 6 ATPases that possess a chaperone-like activity and a lid comprising 8 subunits that recognize the polyubiquitin signals. The 19S proteasome binds and unfolds ubiquitinated proteins and opens the entry gate of the 20S proteasome to allow proteins in the central cavity (136) (Fig. 5).
Oxidation of a protein induces several reversible or irreversible modifications in the three-dimensional structure of a native protein, including amino acid modification, fragmentation, or aggregation, and significantly increases its susceptibility to proteolysis (16, 40). Oxidation of proteins causes the exposure of hydrophobic moieties to the surface via partial unfolding that is recognized by the proteasome (53, 66, 67). While the 26S proteasome degrades polyubiquitinated proteins, the 20S proteasome by itself seems to be sufficient to degrade nonubiquitinated oxidatively modified proteins in an ATP-independent manner; however, the exact mechanism is still unclear (106, 136, 137).
Experimental evidence indicates that disturbance of UPS plays a major role in AD. The activity of proteasome is significantly decreased in the brain of AD (139). It has been shown that proteasome inhibition leads to the accumulation of Aβ and tau, both proteins that are themselves degraded by the proteasome. For example, inhibition of proteasome with lactacystin and MG132 leads to increased production of Aβ42 in NT2 cells, likely by inhibiting APP-carboxy-terminal fragment (CTF) degradation by proteasome (229). However, the same group also showed decreased secreted Aβ40 level in response to MG132 treatment (229). The different effects of proteasome inhibition on the production of Aβ40 and Aβ42 are the basis for the hypothesis that Aβ40 and Aβ42 are generated from different enzymes or γ-secretases (122, 142).
Nonetheless, proteasome is involved in APP and APP-CTF metabolism and affects APP amyloidogenic and nonamyloidogenic processing. In addition, evidence exists for both ubiquitin-dependent and -independent mechanisms of proteasome-mediated APP degradation. APP ubiquitination can serve not only as a proteasomal degradation signal but also as a signal for subcellular trafficking of APP (88). Therefore, age-related dysfunction of UPS could contribute to dysregulated amyloidogenesis in aging.
Besides Aβ accumulation, the perturbation of UPS correlates with other AD hallmarks such as tau hyperphosphorylation and defective autophagy. Recently, Myeku et al. (173) observed in the brain of a mouse model expressing the P301 L tau mutation a progressive increase of ubiquitylated proteins and decreased proteasome activity, which were correlated with increasing tau aggregation in the early disease stage. In these mice, 26S proteasomes were physically associated with tau and were less active in hydrolyzing ubiquitinated proteins. Treatment with a drug able to restore proteasome activity was shown to lower the levels of aggregated tau and improve cognitive function in mice with tauopathy.
Increased OS/NS levels were suggested as one of the major factors contributing to the impairment of UPS in AD (37). Indeed, the levels of oxidized proteins in AD correlate with loss of activity of the 20S proteasome (139, 196). Studies from the Davies and Grune groups showed that if from one side moderately oxidized proteins were preferentially recognized and degraded by the proteasome, severely oxidized proteins cannot be easily degraded and, instead, inhibit the proteasome (107, 211). Accordingly, experimental data indicate that proteins broadly oxidized are more resistant to degradation by 20S proteasomes (215, 228). As a whole, the overload of mutated/oxidized substrates together with oxidative damage to UPS may lead to accumulation of abnormal proteins and to selective degeneration of neurons.
The efficiency of the proteasome system is also regulated by ubiquitinylation and deubiquitinylation reactions. As the presence of ubiquitin chains prevents substrates from entering the proteasome due to spatial restrictions, deubiquitinating enzymes (DUBs) play an essential role in determining the rate of protein clearance in cells. The recycling of ubiquitin is critical for the brain, which has a fixed amount of ubiquitin. Among deubiquitinating enzymes, ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) is one of the most abundant proteins in the brain and regulates the timing and the pattern of ubiquitinylation of brain proteins. It is also one of the main enzymes that play a role in maintaining free ubiquitin levels in neurons. Several studies from the Butterfield group (48, 239) and others (57, 82, 103, 108, 226) demonstrated that UCH-L1 was oxidatively modified in AD brain and its activity was reduced. Recent studies demonstrated that UCH-L1 is oxidatively modified also in DS brain, prior and after development of AD neuropathology (75, 79). A report from Saito et al. showed that the E3 ubiquitin ligase—HRD1—was precipitated by OS, but not by Aβ and tau protein (220). Furthermore, the finding that the heat shock cognate 71 (HSC71) undergoes oxidative modifications highlights the link between folding and degradation machineries that once impaired by oxidative damage, is an attempt for neuronal integrity (49).
Cecarini et al. also demonstrated that despite any changes in the levels of proteasome complex subunits—isolated from control, MCI, and AD brains—a significant reduction in proteasome-mediated degradation of oxidized proteins was measured in MCI and AD subjects (52). The decreased proteolytic activities were associated with the increase of oxidative modifications such as carbonylation, HNE conjugation, and neuroprostane conjugation. Indeed, by adding a reducing agent to proteasome complexes, it was possible to restore its proteolytic activity in both MCI and AD samples, thus confirming the role for OS as a causative factor (52).
Conclusions
AD and AD-like dementia are multifactorial diseases mainly characterized by increased accumulation of Aβ peptide, hyperphosphorylation of tau, increased OS, reduced glucose utilization, impaired mitochondrial functionality, and altered protein homeostasis. Despite extensive knowledge concerning the molecular mechanisms potentially involved in progression of the disease, the initiating events are still largely unknown and several hypotheses have been suggested.
Defects of energy metabolism in AD are one of the earliest events occurring during the degenerative process. Reduction in glucose metabolism has been extensively investigated, and PET scanning represents a robust tool for AD diagnosis. Redox proteomic data highlighted the role of the oxidation of key enzymes of glycolysis and the TCA cycle in the alteration of glucose utilization in the brain that eventually leads to the dysfunction of mitochondria and further leakage of ROS. In parallel, the appearance of BIR contributes to alter not only brain metabolism but also neuronal growth, synaptic maintenance, and neuroprotection, thus resulting in increased Aβ accumulation, tau hyperphosphorylation, increased OS, and inflammation. mTOR is tightly linked to insulin signaling since (on the one hand) it controls the modulation of the PI3K/AkT axis, while (on the other hand) it regulates the hyperactivation of insulin signaling.
Experimental results in AD support the observation of hyperactivation of mTOR in the brain, which contributes to the establishment of BIR. Increased mTOR activity in AD results in impaired protein homeostasis evinced by the decline of protein synthesis and autophagy function. The alterations of protein synthesis and autophagy, other than by mTOR, are further amplified by the oxidation of constituents of protein translation processes and autolysosome formation, thereby increasing cell damage and accumulation of toxic aggregates. The UPR and UPS in AD are crucial systems working in concert with autophagy for the monitoring of protein quality and promoting refolding or degradation of misfolded/oxidized proteins. However, despite that these pathways represent important lines of defense against accumulation of oxidative injury in the neuron in AD, components of UPR and UPS are themselves targets of ROS, resulting in impaired detoxifying function and promoting a vicious cycle that fosters accumulation of OS and damage to biological components.
Overall, by analyzing the results obtained in AD and DS brain by redox proteomics, we propose a key role in AD pathogenesis for a self-sustaining system of aberrant interactions, involving deficient energy metabolism, mTOR activation, and inefficient protein degradation pathways (Table 1). Such alterations are mutually related to each other, representing a triangle of death for neurons leading to neurodegeneration. Interestingly, a prominent role in the self-sustenance of this triangle is played by the increased OS that is both a detrimental effect, produced by mitochondrial defects and altered proteostasis, and a background signal that fuels and contributes to the exacerbation of neuronal damage, targeting metabolic enzymes and components of the protein degradation machinery (Fig. 6). Within this scenario, novel therapeutic approaches for AD could take advantage of the redox proteomic identification of potentially valuable target for AD treatment. Therefore, one or more compounds able to target the components of the triangle of death might lead to the recovery of their mutual interactions and subsequent restoration of the functionality of pathways controlling neuronal metabolism and protein homeostasis and thus may eventually slow or delay cognitive decline.