
Abstract
Significance:
Oxidative stress is one mechanism whereby tobacco smoking affects human health, as reflected by increased levels of several biomarkers of oxidative stress/damage isolated from tissues and biological fluids of active and passive smokers. Many investigations of cigarette smoke (CS)-induced oxidative stress/damage have been carried out in mammalian animal and cellular models of exposure to CS. Animal models allow the investigation of many parameters that are similar to those measured in human smokers. In vitro cell models may provide new information on molecular and functional differences between cells of smokers and nonsmokers.
Recent Advances:
Over the past decade or so, a growing number of researches highlighted that CS induces protein carbonylation in different tissues and body fluids of smokers as well as in in vivo and in vitro models of exposure to CS.
Critical Issues:
We review recent findings on protein carbonylation in smokers and models thereof, focusing on redox proteomic studies. We also discuss the relevance and limitations of these models of exposure to CS and critically assess the congruence between the smoker's condition and laboratory models.
Future Directions:
The identification of protein targets is crucial for understanding the mechanism(s) by which carbonylated proteins accumulate and potentially affect cellular functions. Recent progress in redox proteomics allows the enrichment, identification, and characterization of specific oxidative protein modifications, including carbonylation. Therefore, redox proteomics can be a powerful tool to gain new insights into the onset and/or progression of CS-related diseases and to develop strategies to prevent and/or treat them. Antioxid. Redox Signal. 26, 406–426.
Introduction
C
Recent results of a large prospective study of smoking and its relationship with mortality suggest that CS causes 25% of deaths among humans 35–69 years old in the United States and that smokers die, on average, more than a decade before nonsmokers (72). Mortality among current smokers is two to three times as high as that among persons who never smoked. Most of this excess mortality is due to 21 common diseases that have been formally established as caused by CS (32, 36). Moreover, almost 20% of excess mortality of current smokers is due to additional diseases, among which are breast and prostate cancer, intestinal ischemia, renal failure, and hypertensive heart disease (32).
Despite many epidemiological studies that have established a correlation between CS and smoke-related diseases, the exact cellular and molecular mechanisms of injury remain not completely clear. This is particularly so because over 7000 chemical species constitute CS, many of which can act as oxidants, proinflammatory molecules, carcinogens (or a combination of these), or tumor promoters (37, 119). In particular, free radicals, aldehydes, ketones, and other reactive species can induce oxidative damage in almost all biological macromolecules, compromising their structure and/or function and, consequently, highly contributing to CS-related diseases. Long-term exposure to CS reactive species can result in oxidative burden in the respiratory system as well as in the entire organism, both in humans and in animal models (42, 124, 157). Oxidative burden can be due either to direct oxidative damage/modification of macromolecules or to indirect pathways, such as inflammation and antioxidant depletion.
Reactive Species of CS and CS-Induced Oxidative Stress
CS is rich in reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive carbonyl species (RCS). Water-soluble reactive species may circulate through the body fluids and generate sustained oxidative stress in different organ systems (20, 145, 157). The oxidant burden in smokers' lungs is further enhanced by the reactive species released from alveolar macrophages and neutrophils, which migrate in greater amount in smokers' lungs compared with nonsmokers (114) (Fig. 1). The massive production of reactive species during inflammation plays an important defense role, but when inflammation becomes chronic, it induces persistent activation of inflammatory cells that persistently induce oxidative damage. CS also exposes plasma, red blood cells (RBCs), and leukocytes in the pulmonary microvasculature to excess of oxidants, either directly by diffusion into the blood or indirectly from the reactive species produced by activated inflammatory cells in the lung and peripheral leukocytes (114).

Whole CS consists of a tar phase and a gas phase, both rich in ROS and RNS (119). In particular, gas-phase smoke contains a constant level of nitrogen-, carbon-, and oxygen-centered radicals: that is, radical species are continuously produced and destroyed following gas-phase radical reactions (33). Among RNS, nitric oxide (NO•), which is neither particularly reactive nor toxic, combines within a few seconds with molecular oxygen in air to form the brown gas nitrogen dioxide (NO2 •), a far more reactive free radical and nitrating species. NO2 • reacts rapidly with other CS chemicals to form highly reactive nitroso-carbon-centered radicals, which react instantaneously with molecular oxygen to form peroxyl radicals (112). The latter react with CS gas-phase NO• to form alkoxyl radicals and NO2 •, thereby creating a continuous cycle (112). NO• may be converted to several more reactive RNS, such as NO2 •, N2O3, N2O4, and ONOO− (peroxynitrite). In cells exposed to the gas phase of CS, RNS damage DNA and nitrate protein tyrosine residues. NO• in CS reacts with superoxide anion radical derived from the reducing dihydroxybenzenes in the particulate phase of CS (e.g., hydroquinone and catechol) to form ONOO− (99). ONOO− readily forms in vivo, reacting rapidly with intracellular carbon dioxide to form metastable nitrating, nitrosating, and oxidizing intermediates (132), as well as with molecules localized in the cellular vicinity of its formation. As ONOO− is short-lived in living tissues (∼10−2 s) and therefore difficult to measure directly, nitration of Tyr residues to 3-nitrotyrosine is widely used as a biomarker of ONOO− generation in vivo.
Furthermore, harmful chemicals of CS are highly reactive RCS, including α,β-unsaturated aldehydes, such as 4-hydroxy-trans-2-nonenal (HNE) and acrolein, and dialdehydes, such as malondialdehyde (MDA) and glyoxal (60, 85). Carbonyl emissions of mainstream CS differ significantly among different brands of cigarettes (106). Acrolein and HNE can also be generated during inflammation following lipid peroxidation (55). α,β-Unsaturated aldehydes occur at low-micromolar concentrations in saliva and airway secretions of healthy humans and are augmented up to 10-fold in heavy smokers (3, 4, 106). The unsaturated carbonyl group of α,β-unsaturated aldehydes allows them to bind covalently to nucleophilic amino acids, often resulting in protein carbonylation (37, 39 –41).
The effects of CS-induced oxidative stress can be demonstrated by analyzing individual biomarkers of oxidative stress/damage isolated from tissues and biological fluids such as bronchoalveolar lavage (BAL) fluid and blood of active and passive (or involuntary) smokers—the latter are individuals who do not smoke, but are exposed to secondhand (or environmental) tobacco smoke—in comparison with nonsmokers (130). Secondhand smoke is a mixture of sidestream smoke (the smoke released from the smoldering cigarette or other smoking device) and mainstream smoke (the smoke exhaled by a smoker).
Protein Carbonylation
Protein carbonylation involves the insertion of carbonyl groups into amino acid residues (133). It is (usually) an irreversible covalent modification and is considered a major hallmark of severe protein oxidative damage and of oxidative stress-related diseases and conditions (44, 46, 49, 135). Carbonylated proteins (PCOs) are typically dysfunctional or nonfunctional and may be the cause of subsequent cell, tissue, and even whole organ dysfunction (15, 44). It is likely that protein dysfunction following carbonylation may depend on the quantitative level and/or location of the protein-bound carbonyl group(s). However, at present, specific rules, if any, linking protein carbonylation and resulting dysfunction have not yet been codified, probably because a limited number of studies specifically investigated, at the same time, the level of carbonylation, the specific amino acid residue(s) carbonylated, the type of carbonylation (i.e., primary, or direct, carbonylation or secondary, or indirect, carbonylation), and the eventual resulting protein dysfunction. Although most PCOs are dysfunctional or nonfunctional, which affects their metabolic activity, in some cases, protein carbonylation may activate signaling pathways, such as the NRF2/KEAP1 system that controls phase II antioxidant enzyme expression and the thioredoxin/ASK1 system that controls the c-Jun N-terminal kinase (JNK) (43).
Protein carbonyls include aldehydes and ketones inserted within the protein structure at different sites and formed via different mechanisms (Fig. 1), which have been excellently discussed in other reviews (12, 37, 49, 98, 133). Usually, PCOs cannot be reversed by enzymatic repair activities of cells and are generally removed by proteasome degradation activities (73, 74) (Fig. 2). The 20S proteasome is the primary pathway for the removal of oxidized proteins (74). CS can also induce degradation of highly oxidized proteins, including PCOs, via the ubiquitin–proteasome system, as shown, for instance, in different cultured cells and in animal models in the case of Akt protein (77) and histone deacetylase 2 (1). However, cells have a limited capacity to eliminate PCOs, mainly because the proteasome itself may experience oxidation of its subunits, resulting in its inactivation (44, 74).

A thiol-dependent decarbonylation mechanism has been hypothesized to be associated with receptor-dependent protein carbonylation in cultured pulmonary vascular smooth muscle cells and in murine heart homogenates (151 –153). Although no enzyme capable of reversing protein carbonylation has been identified at present, recent evidence suggests the existence of some heat-labile biological component, which may catalyze such thiol-dependent protein decarbonylation (152). Anyway, thiol-dependent decarbonylation may not necessarily require enzymatic activity; indeed, especially protein Michael addition products may decompose via retro-Michael addition, followed by addition of α,β-unsaturated carbonyls to small-molecular-weight thiols, such as glutathione (GSH).
Since protein carbonylation correlates well with oxidative damage, PCOs are currently regarded as the most general and widely used marker of severe protein oxidation and an indirect way of evaluating oxidative stress both in vitro and in vivo. A number of assays, including redox proteomic techniques, are available for the quantification and identification of PCOs, which have been exhaustively described in several review articles (7, 12, 24, 30, 38, 48, 49, 53, 59, 69, 121, 139, 148, 149). Many of the assays for PCO detection are based on derivatization of the carbonyl group with 2,4-dinitrophenylhydrazine (DNPH), which produce a stable 2,4-dinitrophenyl (DNP) hydrazone that can be detected by various methods, including quantitative and semiquantitative immunoassays based on anti-DNP antibodies.
PCOs in Human Smokers
CS has been reported to increase protein carbonylation in different tissues and body fluids of smokers, including saliva, BAL fluid, parenchymal lung tissue, plasma, RBC membrane, and skeletal muscles. Furthermore, increased levels of HNE-modified proteins occur in airway and alveolar epithelial cells, endothelial cells, and neutrophils of smokers with COPD compared with individuals without COPD (115).
CS-induced protein carbonylation in saliva
The primary body fluids exposed to inhaled CS reactive species are the saliva and the respiratory tract lining fluid. Aldehydes from CS are easily dissolved in the saliva during smoking (125). Measurements of salivary PCOs between smokers and nonsmokers by the one-dimensional polyacrylamide gel electrophoresis (1D-PAGE)/Western blot immunoassay with anti-DNP antibodies suggested that exposure to CS caused a significant increase in PCO levels, the major salivary proteins, amylase, acidic proline-rich proteins, and lysozyme, being the ones most carbonylated (118). Acrolein was the main RCS involved in the CS-induced salivary PCO formation and in the diminution of salivary lactate dehydrogenase activity following carbonylation (8). Specifically, exposure to CS induced a 34% reduction in lactate dehydrogenase activity. Treatment with 10 μmol acrolein (an unsaturated aldehyde) reduced lactate dehydrogenase activity by 61%, while treatment with 200 μmol acetaldehyde (a saturated aldehyde) caused no substantial effect. The OxyBlot assay showed that carbonyl content of lactate dehydrogenase increased fourfold following treatment with CS and sevenfold following treatment with acrolein, whereas no significant increase in carbonylation was observed when the enzyme was treated with acetaldehyde (8).
CS-induced protein carbonylation in the respiratory system
PCO levels are increased in BAL fluid of older smokers without lung diseases (101, 136). BAL fluid is obtained during fiberoptic bronchoscopy by washing the epithelial lining of the lung. It is thought to reflect most faithfully the protein composition of the airways, especially the presence of peptides/proteins specifically secreted by different cell types of the lungs and the airways, proteins involved in pulmonary host defense (immune response, inflammation, antioxidant proteins), and those participating in tissue repair and proliferation. Specifically, BAL fluid contains proteins diffused from serum across the air–blood barrier and proteins produced by pulmonary T cells, alveolar macrophages, bronchial epithelial cells, alveolar type I and type II cells, and Clara cells. Since albumin constitutes about 50% of total proteins in BAL fluid (103), which is representative of components in alveolar space, albumin is likely the main reservoir of free sulfhydryl groups in BAL fluid, as it is in plasma. Thus, the Cys34 thiol moiety of albumin may be the most important component of the antioxidant system in BAL fluid. Sodium dodecyl sulfate (SDS)-PAGE and Western blot analysis with anti-DNP antibodies proved that albumin is the main PCO in the BAL fluid of older long-term smokers, even without lung diseases (101, 136).
COPD is characterized by chronic bronchitis and lung parenchymal destruction (emphysema). Parenchymal tissue from current smokers with COPD contained lower levels of total albumin, but had proportionally greater levels of carbonylated albumin (Alb-CO), compared with individuals with normal lung function (67). Decreased albumin levels in current smokers with COPD could contribute to increased oxidant burden and, consequently, to oxidative tissue injury. Lung tissue from current smokers also contained lower levels of highly Alb-CO compared with lung tissue from ex-smokers and nonsmokers. The mean concentration of Alb-CO in lung tissue was identical in ex-smokers and nonsmokers (67). This suggests that smoking cessation could prevent, or at least limit, albumin carbonylation, facilitating the reinstatement of the pulmonary oxidant/antioxidant balance.
Total protein carbonylation levels were significantly greater in the bronchi of patients with COPD, with and without lung cancer, ∼70% of whom were active smokers, compared with non-COPD control individuals, 75% of whom were never smokers (18). Patients with both COPD and lung cancer exhibited greater levels of bronchial MDA-protein adducts, a type of indirect protein carbonylation (Fig. 1) (12, 29), than patients with only lung cancer and control individuals, both with and without COPD. In addition, bronchial MDA-protein adduct levels were significantly higher in patients with lung cancer than in healthy controls. Blood levels of protein carbonylation were significantly greater in all patients affected by lung cancer and/or COPD compared with healthy individuals, particularly in those with both pathologies (18).
In COPD, protein carbonylation is also a hallmark of limb and respiratory skeletal muscles. Patients with severe COPD showed greater protein carbonylation in the vastus lateralis (quadriceps) and in the diaphragm than controls (19, 89, 120). In the diaphragm, PCO levels correlated with the severity of the disease, whereas HNE-protein adducts, higher in the patients than in the controls, correlated with the respiratory muscle strength (16). Identification of diaphragm PCOs by redox proteomics revealed carbonylation of proteins involved in ATP production and contraction; in particular, carbonylation levels in myosin heavy chain were fivefold greater in severe COPD patients than in controls (89). Furthermore, muscle and blood protein carbonylation levels were correlated. In general, carbonylation of specific muscle proteins could partially contribute to skeletal muscle dysfunction in patients with severe COPD (15). Cachectic patients too, affected by either severe COPD or lung cancer (most of whom were active smokers), exhibited higher total protein carbonylation in both the vastus lateralis and blood (indicative of systemic oxidative stress) than healthy controls (113).
CS-induced protein carbonylation in the circulatory system
Amount of plasma PCOs is higher in smokers than in matched nonsmokers. Elevated amounts of carbonylated fibrinogen in smokers compared with nonsmokers were measured by Western blot assay, whereas no difference was seen between heavy (range 21–143 pack-years) and light (range 0.3–4.3 pack-years) smokers (108). (Note: A pack-year is a quantification of cigarette smoking over a long period of time. It is defined as 20 cigarettes smoked daily for 1 year. For instance, one pack-year is equal to smoking one pack per day for 1 year, or two packs per day for half a year; 1.5 packs of cigarettes smoked daily for 20 years are equal to 30 pack-years). The increase in plasma content of PCOs was also confirmed in smokers of hand-rolled cigarettes and smokers of manufactured filter cigarettes compared with healthy nonsmokers (78).
A large population-based study proved that, in smoking women, plasma levels of PCOs, measured by means of a noncompetitive ELISA method, were significantly increased among current smokers and former smokers compared with nonsmoking women and may be associated with breast cancer risk (122). Although plasma PCO levels did not significantly correlate with cigarette smoking status (i.e., current vs. former smokers) or duration in smokers (mean age [±SD], 67 [± 5] years) recruited in the ELCAP program, smokers had higher carbonyl levels than nonsmokers (160).
We showed drastic decrease in albumin Cys34 thiol and marked carbonylation when purified albumin was exposed to whole-phase CS extract (CSE) (37). Mass spectrometry analysis detected acrolein and crotonaldehyde (α,β-unsaturated aldehydes) Michael adducts at Cys34, Lys525, Lys351, and His39 and a Schiff base with acrolein at Lys541 and Lys545 (37). Even though higher amounts of variously oxidized proteins were measured in blood and/or plasma of smokers, no study found Alb-CO in plasma (88, 108). Considering that Alb-CO occurs in various fluids and tissues of smokers other than blood or plasma (67, 101, 136) and that RBCs contain GSH at the concentration of ∼3 mM, whereas plasma GSH concentration is in the low micromolar range (63), we investigated whether RBCs protected both purified albumin and total human plasma proteins from CS-induced oxidation (41). GSH acted as an antioxidant only at supraphysiological concentrations, implying that, under physiological conditions, plasma GSH fails to prevent CS-induced albumin carbonylation and oxidation of its Cys34 sulfhydryl group. Differently, human RBCs protected both albumin and total human plasma proteins from CS-induced carbonylation and thiol oxidation (41). The high GSH concentration in human RBCs could at least partly explain why Alb-CO was found within extravascular fluids (e.g., in the BAL fluid) and parenchymal lung tissue in smokers (67, 101, 136), but not within plasma (88, 108).
CS-induced protein carbonylation in the skeletal musculature
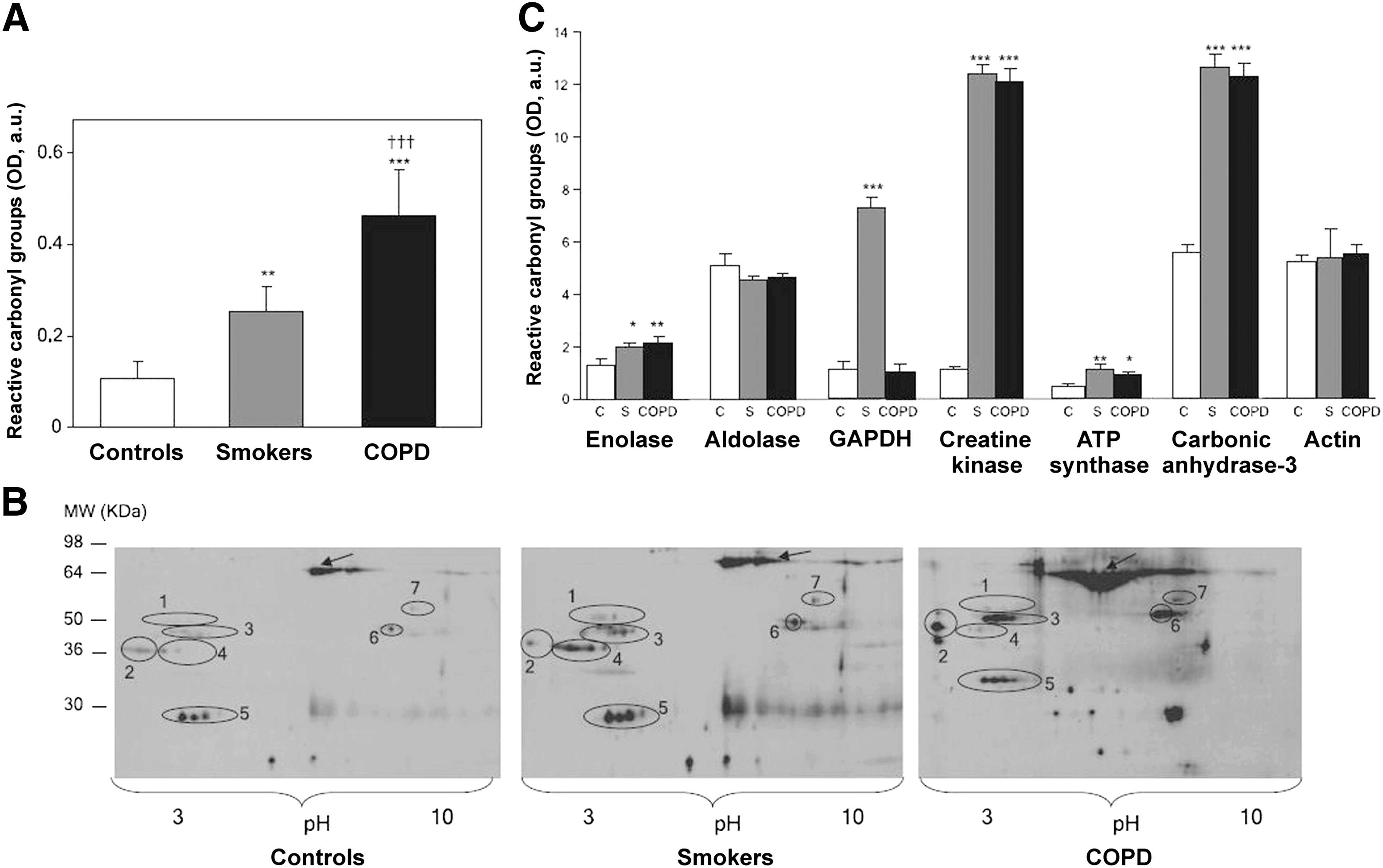
Barreiro et al. (15, 20) elucidated that chronic cigarette smoking causes direct oxidative modifications of skeletal muscle proteins, without triggering muscle inflammation. Total protein carbonylation level was significantly increased in the quadriceps (vastus lateralis) of healthy smokers (>20 pack-years) and even more in patients with COPD than in control individuals. Detection of PCO by means of redox proteomics and identification of specific PCOs by mass spectrometry showed that glycolytic enzymes β-enolase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), creatine kinase, carbonic anhydrase-3, and ATP-synthase were more carbonylated in the vastus lateralis of smokers and patients with COPD than in the quadriceps of control subjects (Fig. 3) (15, 20). Furthermore, the quadriceps of smokers showed higher MDA-protein adduct levels compared with controls. Differently, only the vastus lateralis of patients with severe COPD showed a reduction of about 30% in creatine kinase activity compared with healthy control subjects (15, 20). The oxidation of specific muscle proteins may contribute to skeletal muscle dysfunction in smokers, as suggested by a significant mild decrease in quadriceps muscle force in the healthy smokers compared with nonsmokers (20) and by greater peripheral muscle fatigue shown by healthy smokers compared with nonsmokers (156).
PCOs in Mammalian Animal Models of Exposure to CS
Animal models of exposure to CS allow the investigation of many parameters (biomarkers of oxidative stress, mechanical lung functions, and morphologic assessments) that are similar to those measured in human smokers. Most studies in mammalian animal models exposed to CS have been performed in mice and guinea pigs inhaled via a smoking chamber. However, the studies differed by the cigarette brand, the amount of CS inhaled, and the duration of exposure to CS, ranging from days to months.
Thanks to their relatively short life span, mice are favorite animals of investigators doing research on CS-induced oxidative stress-related diseases. Common to all these studies is the development of lung inflammation, whose characteristics, however, vary by the chosen method of exposure to CS and mouse strains; indeed, resistant and sensitive strains to exposure to CS have been identified (145). At least some of the differences between mouse strains following exposure to CS can be explained by difference(s) in the inflammatory response. Two CS-sensitive mouse strains have been used for investigating effects of exposure to CS, namely AKR/J (supersusceptible strain) and C57BL/6 (mildly susceptible strain). The great sensitivity of AKR/J mice to CS is supposed to be due to a strong upregulation of Th1 cytokines, indicative of a type 1 T helper (Th1) cell-adaptive inflammatory response and suggestive of an autoimmune component in emphysema development, as well as to increased macrophages, polymorphonuclear leukocytes, and T cells in the lungs (66). The similarity between this response and the one seen in humans highlights the importance of the AKR/J strain as an animal model for emphysema. Alveolar macrophages also increase in the C57BL/6 strain (57). Alveolar macrophages from C57BL/6J mice release a higher amount of proinflammatory cytokines, matrix metalloproteinases, and ROS when exposed to CS than ones from resistant strain mice (147).
CS-induced protein carbonylation in the mouse model
In mice exposed to CS, protein carbonylation is enhanced in the BAL fluid (127, 137), in the lung (76, 80, 92, 159), in the hippocampus (138), and the skeletal musculature (see below). In the BAL fluid of C57BL/6 mice exposed to CS for 60 min/day for up to 10 consecutive days, total PCO, Alb-CO, and the count of neutrophils and macrophages were significantly increased (137). Whole lung extracts from C57BL/6 mice exposed to CS showed decreased levels and, concurrently, increased carbonylation of sirtuin1 compared with air-exposed mice (31). Immunoprecipitation of sirtuin1 from mouse lung extracts showed a reduction in free cysteine residues (as detected by labeling with maleimide-PEO2-biotin, followed by immunoblot with streptavidin conjugated to horseradish peroxidase) on sirtuin1 following exposure to CS compared with control mice. In mouse lungs exposed to CS, total protein carbonylation and lipid peroxidation products, HNE and MDA, were increased in sirtuin1 heterozygous knockout mice compared with wild-type mice. These responses were lessened by overexpression or pharmacological activation of sirtuin1 (159). These findings suggest that sirtuin1, a type III histone/protein deacetylase mainly involved in inflammation, stress resistance, and cellular senescence/aging (71), protects lungs of CS-exposed mice from protein carbonylation.
CS and aging contribute a lot to the development of pulmonary emphysema. In senescence marker protein-30 (SMP30) knockout (SMP30Y/−) mice and the related wild-type (SMP30Y/+) mice exposed to CS for 8 weeks, levels of PCO and MDA in the lung, total GSH amount in the BAL fluid, and extent of apoptosis of lung cells were measured, in addition to anatomic and histopathologic analysis of the lungs (127). SMP30 is a multifunctional protein highly conserved among humans, rats, and mice, protecting cells from age-associated deterioration (54). Exposure to CS generated marked airspace enlargement and parenchymal destruction in the SMP30Y/− mice, but not in the SMP30Y/+ mice. PCO, MDA, total GSH, and apoptosis were significantly increased in the CS-exposed SMP30Y/− mice, suggesting that SMP30 protects murine lungs from aging- and CS-associated oxidative stress (127). Protein disulfide isomerase (PDI)—a redox-sensitive key protein in the formation of disulfide bonds within the endoplasmic reticulum (ER), which transfers oxidative equivalents from Ero1p to newly synthesized proteins (58), and which is upregulated in lungs of smokers (75)—was found to be extensively carbonylated in lung extracts of C57BL/6 mice exposed to CS from one cigarette, predominantly in complexes with client proteins (76). Usually, most of PDI is incorporated into a multiprotein complex that is formed during protein folding by the transient and reversible binding of client proteins to ER chaperones and isomerases. The ER quality control machinery senses when a client protein achieves its native conformation and releases it from the chaperone-PDI complex for export from the ER. Client proteins that fail to fold properly remain in these multiprotein complexes called high-molecular-weight protein complexes (HMWCs). Kenche et al. (76) examined whether CS enhances HMWC formation by analyzing complexes formed by PDI on nonreducing gels. Mice exposed or not to the smoke of one cigarette were subject to euthanasia at various times postexposure and lung lysates were analyzed. Results demonstrate that CS induced an increase in PDI-containing HMWCs, which was most evident 12 h postexposure. Such findings suggest that PDI-associated proteins were unable to achieve active conformation, thus remaining tied to the chaperone because carbonylated PDI was unable to promote disulfide bond formation. Therefore, acute exposure to CS in vivo modifies PDI and affects oxidative protein folding within the ER, which in turn disturbs ER homeostasis and induces ER stress response (76). Defective protein folding because of chaperone malfunction is one characteristic of aging cells (123). Therefore, PDI carbonylation may be one of the mechanisms by which CS brings about premature cellular senescence by inducing ineffective chaperones.
A redox proteomic approach was employed to identify PCOs within respiratory and limb muscles of supersusceptible AKR/J mice exposed to chronic CS (17). In susceptible mouse strains, chronic exposure to CS leads to the development of lung emphysema and inflammation in a similar way to what happens in patients with COPD (66), who commonly show skeletal muscle abnormalities, which can lead to reduced exercise capacity, poor quality of life, and increased mortality (138). In this study, AKR/J mice were exposed to CS for 6 months and then PCO, HNE-, and MDA-protein adducts were detected in the diaphragm and gastrocnemius with specific antibodies after 2D-PAGE (17). PCO levels were significantly higher in the muscles of the mice exposed to CS than in those of the control mice. The amount of HNE-protein adducts was significantly increased only in the gastrocnemius of the CS-exposed mice compared with the controls, whereas no significant increase in the amount of MDA-protein adducts was detected. The identified carbonylated muscular proteins are metabolic (pyruvate kinase, succinyl CoA transferase-1, α-enolase, creatine kinase, fructose 1,6-biphosphate aldolase A, GAPDH, carbonic anhydrase-3, triosephosphate isomerase, aldolase reductase, ATP-synthase) or structural (actin) proteins, besides the ubiquitous albumin. These findings are similar to those gleaned from previous human studies, where almost the same proteins resulted to be oxidatively modified in the quadriceps (vastus lateralis) and the diaphragm of patients with severe COPD (15), as well as in the quadriceps of smokers (20).
CS-induced protein carbonylation in the guinea pig model
Guinea pigs exposed to CS had plasma PCO concentrations more than 30 times greater than unexposed animals (105). The lungs of guinea pigs exposed to CS for 7–21 days, five cigarettes (two puffs/cigarette)/animal/day, show infiltration of inflammatory cells, morphometric change, and alveolar enlargement compared with air-exposed animals (13, 14). Exposure to CS also increased PCOs in whole lung lysates (14) (Fig. 4). Programmed cell death substantially contributes to CS-induced lung damage (5, 51, 141). Apoptotic cells exhibit increased oxidative damage, which further contributes to cell death (142). In guinea pigs, PCO formation starts even after exposure to CS from only one cigarette and increases considerably after exposure to CS from two and three cigarettes. However, after exposure to CS from three cigarettes, guinea pig lungs did not show any apoptosis, suggesting that PCO formation precedes apoptosis (13, 14).

Respiratory (diaphragm) and limb (gastrocnemius) muscles of guinea pigs chronically exposed to CS (seven cigarettes/day, 5 days/week) for up to 6 months showed increased total PCO levels compared with control animals as early as 3 months of exposure (20). Specifically, CS-exposed guinea pigs showed higher carbonylation levels of creatine kinase in the diaphragm and of enolase, aldolase, GAPDH, creatine kinase, actin, and tropomyosin in the gastrocnemius than the corresponding muscles of control animals. Moreover, chronic exposure to CS also caused a significant increase in MDA-protein adducts in both muscles. Creatine kinase activity was markedly lessened in both diaphragm and gastrocnemius muscles of guinea pigs exposed to CS for 4 and 6 months compared with the enzyme activity of their respective controls (20).
Oxidative modifications of muscle proteins may impair their function, eventually rendering them more susceptible to proteolysis, which in turn would result in muscle loss and dysfunction in smokers and patients with COPD. In guinea pigs, CS-induced oxidative damage in the skeletal muscles occurred before the typical bronchiolar and parenchymal alterations induced by CS in the lungs (20), suggesting that exposure to CS induces systemic effects on several tissues. Guinea pigs exposed to CS showed enhanced protein carbonylation also in the heart (50, 97, 105), leading to myocardial injury, as revealed by histological analysis showing thrombus and fibrous tissue formation in the left ventricle and by release of cardiac troponin-T and -I in the serum (50). It is noteworthy that oxidative protein damage occurred at the early stage of myocardial injury before inflammation and apoptosis (50).
PCOs in Cultured Mammalian Cell Models of Exposure to CS
CS is a complex mixture of noxious chemicals that can variously modify many cellular pathways and activate numerous (often many concurrent) cellular responses. Obviously, no in vitro study can reproduce such a complex phenomenon as in vivo exposure to CS, which induces inflammatory processes, airway changes, and lung parenchymal destruction (emphysema). In vitro cellular models allow researchers to screen cellular responses to CS and to carry out mechanistic studies in a relatively simple system. The purpose of an in vitro study is therefore to simplify the experiment and to limit variables.
Fibroblasts or epithelial cells are the most frequently used cells in studies of exposure to CS. Alveolar macrophages are also used. These cells can be either human or animal primary cell cultures or commercial cell lines, such as human HaCaT keratinocyte cell line (from adult human skin), human type II alveolar epithelial cells (A549, from an adenocarcinoma), human bronchial epithelial cell line (BEAS-2B, from normal human bronchial epithelium, SV-40 transformed), human urinary bladder carcinoma cell line (ECV-304, a misidentified cell line for long presumed to be an endothelial cell line), or a continuous cell line of murine alveolar macrophages (MH-S).
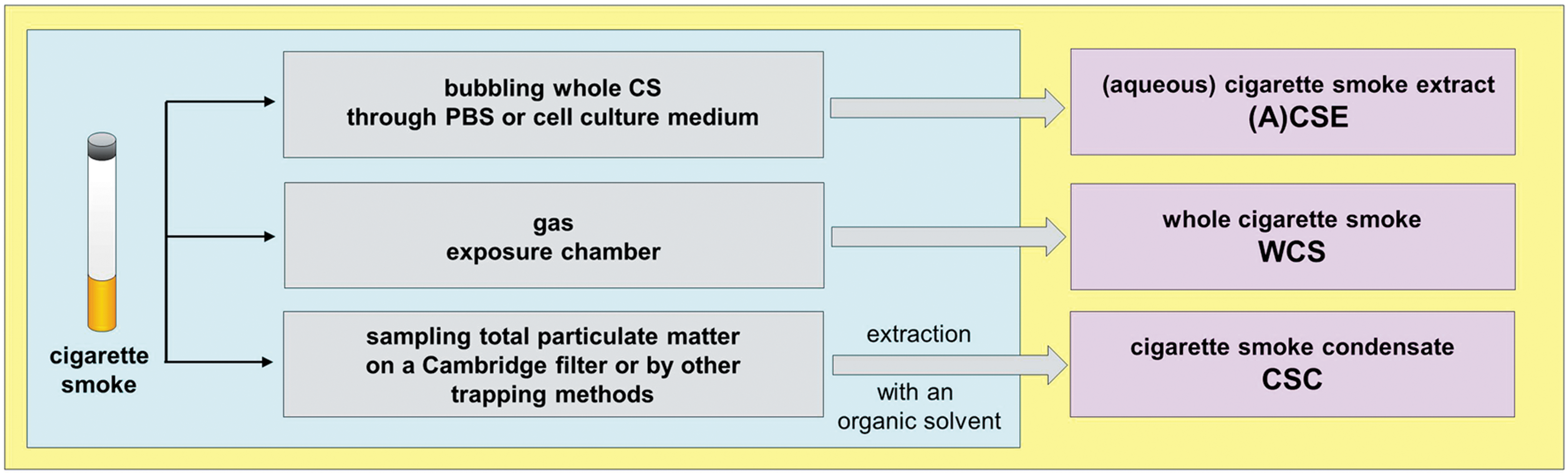
Commonly, in vitro cell models are exposed to whole phase (mainstream) CS (WCS or smoking chamber model), to a specific fraction of CS (i.e., CSE or CS condensate [CSC]), or any of CS individual components (Fig. 5).
CS-induced protein carbonylation in human oral cavity cells
All the cells within the oral cavity are exposed first to CS in active smokers. Epithelial cells of the oral mucosa act as the first line of defense and are essential for maintaining host, first of all periodontal, homeostasis. Numerous studies demonstrated detrimental effects of CS on the oral cavity tissues, ranging from simple tooth staining to inflammatory conditions, to periodontal disease, and to oral cancer (109, 116). CS components, which are readily dissolved in saliva, can potentially penetrate into oral cavity tissues and increase protein carbonylation.
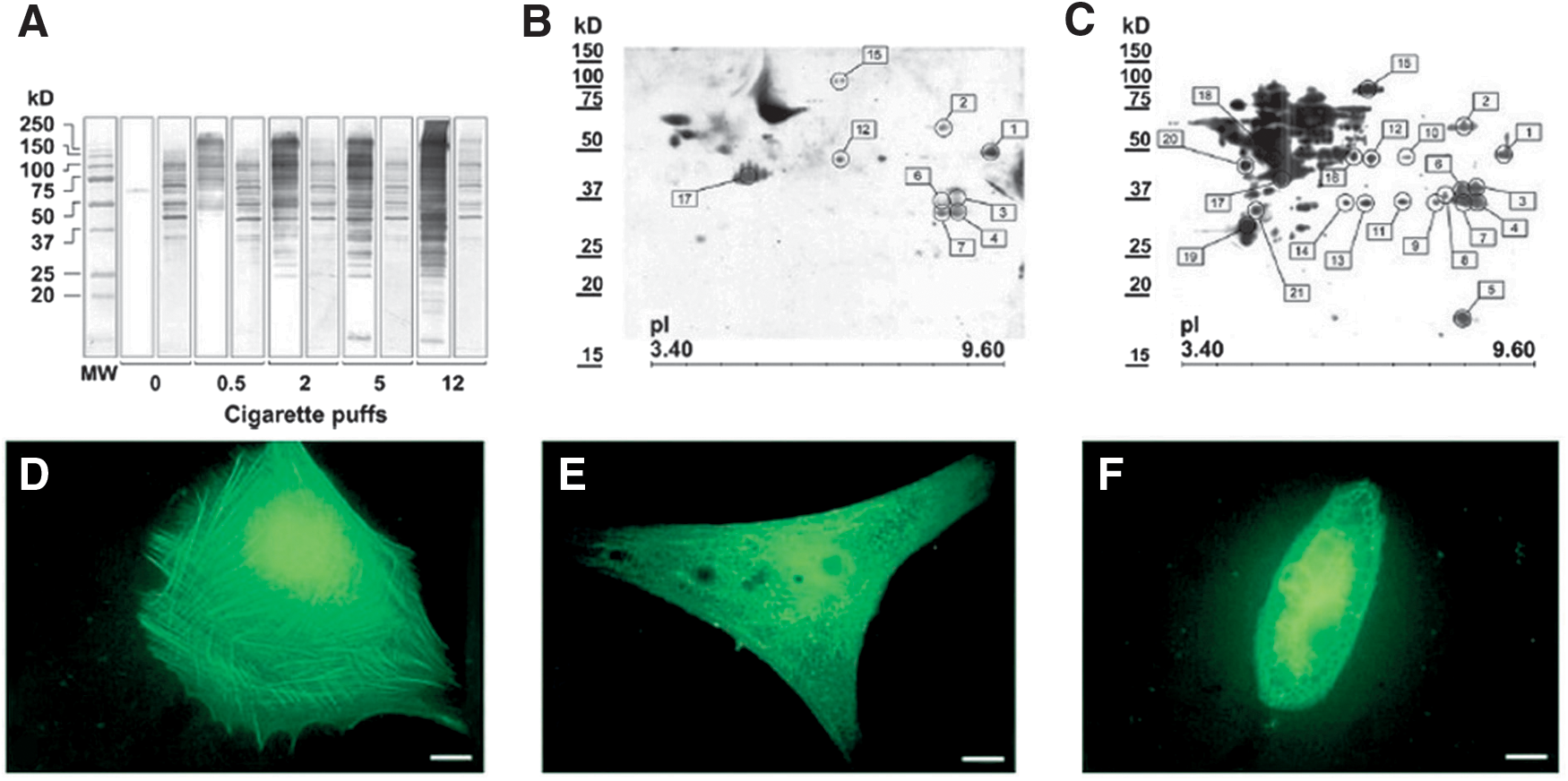
The effects of WCS on oral epithelial cells or fibroblasts have been explored only recently. A study examined protein carbonylation in the human HaCaT keratinocyte cell line exposed to WCS, saturated (acetaldehyde), or α,β-unsaturated (acrolein) aldehydes (9). HaCaT are in vitro spontaneously transformed keratinocytes from normal human skin, which are essentially immortal (>140 passages, i.e., highly proliferating cells), maintain full differentiation capacity, and are nontumorigenic (25). Therefore, they are broadly used as a model for epithelial tissue studies (61). Dose-dependent PCO formation was observed in HaCaT cells exposed to WCS or acrolein: intracellular PCOs increased 5.2 times after cell exposure to WCS and 2.7 times after exposure to 1 μmol of acrolein. Only a minor elevation of PCOs was observed upon exposure to acetaldehyde, indicating that α,β-unsaturated aldehydes from CS traverse cell membranes and induce intracellular PCO formation (9).
A further study confirmed that intracellular protein carbonylation induced by WCS and acrolein in HaCaT keratinocytes is dose and time dependent and increases after an exposure time as short as 10 min, thus suggesting that even a brief exposure to WCS and its aldehydic constituents can be potentially harmful (10).
Exposure to WCS and acrolein (but not to acetaldehyde) also led to increased ROS production in HaCaT keratinocytes (9), which is consistent with results from other studies on CS performed in the adenocarcinomic human type II alveolar epithelial cell line, A549 (79, 146), primary human gingival fibroblasts (HGFs) (40), and the human ECV-304 cell line (65). The increase in ROS measured in HaCaT cells exposed to WCS was expected given its high levels of ROS and other reactive species. Differently, a similar increase in intracellular ROS generation upon cell exposure to acrolein was unexpected, suggesting that acrolein may induce production of cellular ROS via a secondary mechanism. The same study also demonstrated lower GSH levels and no glutathione disulfide (GSSG) elevation in HaCaT cells exposed to acrolein, but not in those exposed to acetaldehyde. It was previously proposed that, following cell exposure to CS, GSH-α,β-unsaturated aldehyde adducts are formed thanks to the detoxifying function of GSH, thus preventing GSH oxidation to GSSG (40, 86). Indeed, acrolein double bond can react with the thiol group of GSH, diminishing its cellular levels and hence its antioxidant function; consequently, intracellular ROS are elevated in HaCaT keratinocytes exposed to acrolein. As acetaldehyde is devoid of double bonds, it does not react with GSH, which can carry out its antioxidant function (9). Findings in HaCaT keratinocytes thus reveal that α,β-unsaturated aldehydes are at least partly responsible for CS-induced intracellular protein carbonylation and ROS elevation, probably because of their covalent conjugation with GSH (9).
Another study showed increased levels of PCO, HNE-, and acrolein-protein adducts in HaCaT cells exposed to WCS (134) (Fig. 6).

In the gingival connective tissue, fibroblasts are the main cell type and are essential in remodeling and maintaining structures, extracellular matrix homeostasis, tissue repair, and wound healing. We investigated how the exposure to WCS can affect cultured primary HGFs at the proteome level (40), focusing on PCOs induced by increasing number of WCS puffs. HGFs showed a basal level of protein carbonylation, which increased in a puff-dependent way (Fig. 7A). By redox proteomic analysis, we identified 21 PCOs (Fig. 7C), nine of which were slightly or moderately carbonylated also in HGFs not exposed to WCS (Fig. 7B). Among the PCOs, we identified cytoskeletal proteins such as actin, cofilin-1, and elongation factor 1-α, enzymes involved in energy production such as α-enolase, GAPDH, fructose 1,6-bisphosphate aldolase, and pyruvate kinase isozyme M1/M2, and several annexin family proteins (annexin A1, A2, and A5) involved in many physiological roles, among which were membrane scaffolding, exocytosis, endocytosis, signal transduction, and rearrangement of the actin cytoskeleton (40). Carbonylation of actin and some actin-binding/remodeling proteins could impair the dynamic and structural role of the actin-based cytoskeleton, thus explaining the CS-induced morphological alterations observed in HGFs (Fig. 7D–F). Furthermore, carbonylation of enzymes of energy metabolism can damage HGFs, leading to an impaired glycolytic pathway and decreased ATP production.
CS-induced protein carbonylation in human airway epithelial cells
In airway epithelial cells, GSH plays a critical role for the defense from endogenous and exogenous oxidants, including CS, and inflammatory injury (23), as also highlighted in a human bronchial epithelial cell line (16HBE) exposed to CSE (21). Accordingly, many inflammatory pulmonary diseases are characterized by changes in lung GSH metabolism. GSH is an effective antioxidant and an important detoxifying agent of many reactive electrophilic constituents of CS because of its reactivity with a great number of different molecules. In most mammalian cells, the cytosolic concentration of GSH is 1–10 mM, accounting for >98% of total GSH (47). GSH can also bind sensitive protein sulfhydryl groups (a process known as S-glutathionylation, which gives rise to S-glutathionylated proteins) under conditions of mild oxidative stress to protect them against irreversible oxidation and to store itself intracellularly. Under physiological conditions, reversal of protein S-glutathionylation can be catalyzed by the enzyme glutaredoxin (Grx), which can also catalyze, when thermodynamically favorable, S-glutathionylation of specific proteins (47).
Using the human alveolar type II cell-derived A549 adenocarcinoma cell line as a model of human type II alveolar epithelial cells (83), given that freshly isolated type II alveolar epithelial cells from human lung tissue are often obtained with limited purity or yield, a study showed that CSE attenuated Grx1 expression and increased Grx1 S-glutathionylation, thereby decreasing its activity (81). Using mass spectrometry, authors demonstrated indirect carbonylation of recombinant Grx1 by CSE and acrolein, which correlated with decreased enzyme activity. Carbonylation of Grx1 was also shown in human lung epithelial A459 cells exposed to CSE (81). Some of the target proteins of S-glutathionylation regulate apoptosis (45), which in turn might contribute to the development of COPD and pulmonary emphysema (51). Overall, mounting evidence points to Grx1 as a potential candidate for therapeutic interventions aimed at enhancing survival of cells exposed to CS. Restoring Grx1 content in the CS-exposed lungs might enhance cell survival and potentially help to prevent the development of CS-induced emphysema.
A study conducted in the human type II alveolar epithelial cell line, A549, and in the human bronchial epithelial cell line, 16HBE, exposed to CSE demonstrated the involvement of the ubiquitin–proteasome system in CS-induced cell damage (146). Exposure of A549 cells to CSE induced time- and dose-dependent cell necrosis, increased levels of intracellular ROS, PCO, and polyubiquitinated proteins, and inactivation of the proteasome activity without alteration of proteasome expression. Exposure to high CSE concentrations inhibited all the chymotrypsin-like, caspase-like, and trypsin-like proteasomal activities in A549 cells, whereas only the trypsin-like activity was inhibited at low and nontoxic concentrations of CSE. CSE-induced proteasome inhibition was also observed in 16HBE cells, which showed higher sensitivity to CSE than A549 cells. Inhibition of only the trypsin-like proteasomal activity was revealed at nontoxic concentrations of CSE, whereas inhibition of the chymotrypsin-like activity was only observed for the highest and toxic CSE concentration (which was nontoxic in A549 cells) (146).
These findings are partially in line with results from different types of cultured cells proving that CS induces misfolding and oxidation (including carbonylation) of specific cellular proteins, impairing their function and making them prone for ubiquitin-mediated proteasomal degradation (91). Augmented levels of insoluble ubiquitinated proteins were detected in lungs of COPD patients with smoking history and in an acutely CS-exposed mouse model of COPD (96). In contrast, proteasome expression and activity are both downregulated in the lungs of patients with COPD and inversely correlate with lung function; decreased proteasome expression also correlates with diminished expression of the antioxidant transcription factor, Nrf2 (87). As a whole, results revealed a direct correlation between smoking and emphysema status of patients with COPD with decreased proteasome activity (87).
CS-induced protein carbonylation in other human epithelial cells
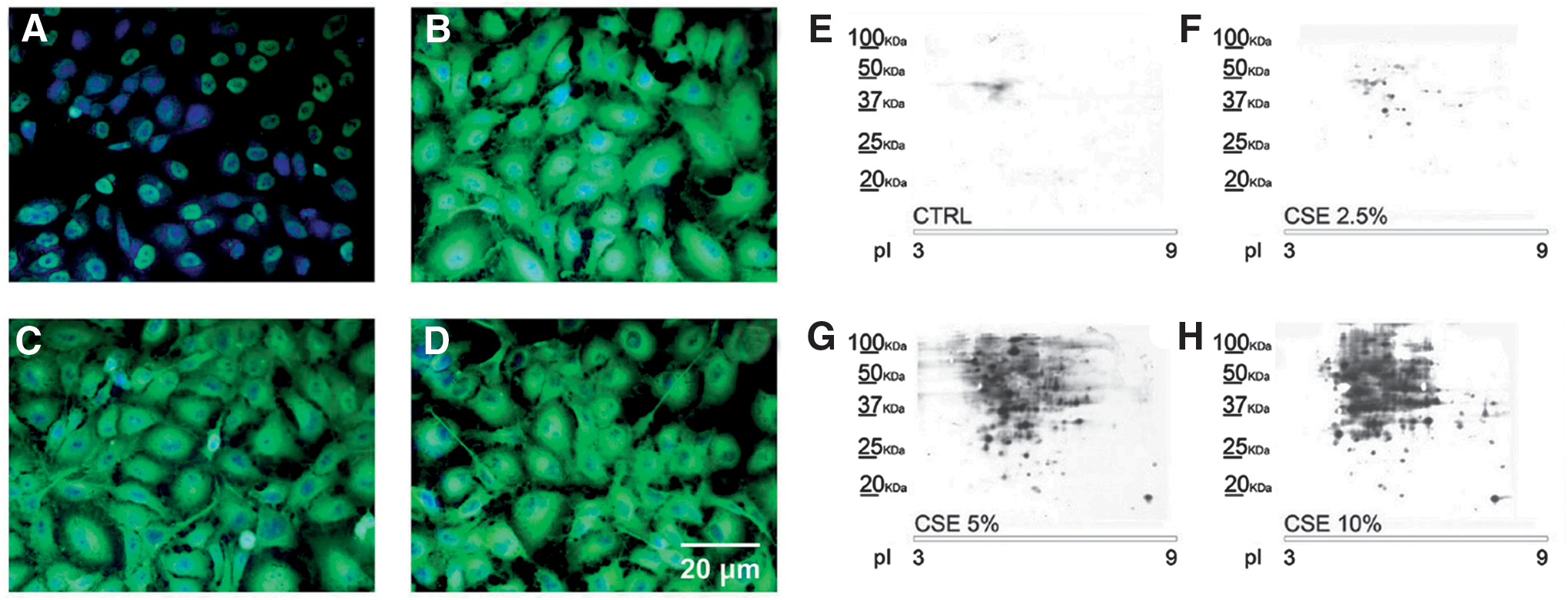
We carried out immunofluorescence and redox proteomic analysis with anti-DNP antibodies to show cellular distribution of protein carbonylation (Fig. 8A–D) and to identify PCOs (Fig. 8E–H), respectively, in the human urinary bladder carcinoma cell line, ECV-304, exposed to CSE (65). The main PCOs were cytoskeletal proteins, glycolytic enzymes, xenobiotic metabolizing and antioxidant enzymes, and ER proteins. The ECV-304 cell line was for long used as a model for human endothelium because it was originally believed to be a spontaneously transformed human normal endothelial cell line. Actually, it is derived from the human bladder carcinoma T24 cell line (2, 28), although ECV-304 cells display some human endothelial characteristics, such as the expression of ICAM-1 (28, 82).
Carbonylation of enzymes involved in carbohydrate metabolism was also found in smokers and CS-exposed rodents (15, 20) and primary HGFs (40). Carbonylation of cytoskeletal/muscle-specific proteins was also found in muscles of healthy smokers, patients with severe COPD, rodents (15, 20), and primary HGFs (40) exposed to CS. All these findings from different cell types suggest that CS can induce oxidative damage of glycolytic enzymes and cytoskeletal/muscle-specific proteins and that this is not a cell type-specific effect.
CS-induced protein carbonylation in murine inflammatory cells
Alveolar macrophages, neutrophil, and eosinophil granulocytes are increased in the lungs of smokers (70). Phagocytosis of alveolar macrophages is impaired in chronic lung diseases such as COPD, where exposure to CS provokes significant oxidative damage (22). Intracellular protein carbonylation in MH-S murine alveolar macrophages exposed to CSE was examined by SDS-PAGE/Western blot immunoassay with anti-DNP antibodies (26, 52). MH-S cells are a noncancer immortalized cell line derived from an adherent cell-enriched population of mouse alveolar macrophages (obtained by BAL) after transformation with simian virus 40, which retain many morphologic and functional characteristics of primary alveolar macrophages (90). Thus, MH-S cells are a useful model for the in vitro study of alveolar macrophages. Exposure of MH-S cells to CSE induced dose- and time-dependent carbonylation of many cytosolic, nuclear, and membrane proteins within 10 min that inversely correlated with the lipopolysaccharide (LPS)-induced expression of TNF-α. Marked protein carbonylation also affected MH-S cell pseudopodia, which failed to phagocytose bacterial cells, although their formation and extension were not impaired, at least morphologically, by exposure to CSE. CSE induced high protein carbonylation levels and inhibited the LPS-induced expression of TNF-α in primary murine alveolar macrophages as well. These results prove that CSE-induced protein carbonylation occurs rapidly and extensively in alveolar macrophages, where localized carbonylation of pseudopodia was associated with impaired bacterial phagocytosis (26).
Limitation and Congruence of In Vivo and In Vitro Studies of Exposure to CS
Limitations of studies in smokers
PCOs have been detected in different tissues and cells in human smokers (Table 1). A limitation in several studies on human smokers concerns the relatively small number of subjects studied. Larger population studies will be needed to delineate the action of CS on PCO formation in smokers and the role of PCOs in the susceptibility to CS-induced human diseases. Findings from human studies were due, in large part, to the possibility of researchers to access human biological material allowing for comparisons of tissue samples between smokers and nonsmokers. However, given that (for obvious ethical reasons) it is not always possible to obtain human specimens from a population of healthy smokers and healthy nonsmokers, the design of experimental mammalian animal and in vitro cell models of exposure to CS appears to be justified.
1D-PAGE, one-dimensional polyacrylamide gel electrophoresis; BAL, bronchoalveolar lavage; COPD, chronic obstructive pulmonary disease; DNP, 2,4-dinitrophenyl; PCO, carbonylated proteins; RBC, red blood cell.
Limitations of studies in mammalian animal models
Various cigarette brands, ranging from standard research-grade cigarettes to commercial ones, with and without filter, and different amounts of CS have been used in animal and cultured cell models of exposure to CS.
Although humans, mice, and guinea pigs share many basic physiological/biochemical processes, animal models of exposure to CS pose various problems. One of the problems is the different manner of exposure to CS experienced by humans and mammalian animal models. First, mice and guinea pigs are obligate nose breathers; therefore, their nares and upper respiratory tract show a very different pattern of particle filtration than that experienced by mouth breathers, such as human smokers. Second, even though mice and guinea pigs have a much smaller respiratory surface than humans, in various studies, animals were exposed to a higher number of cigarettes than humans. Particularly mice, although widely used, have several limitations, including strain differences in the tendency to develop pulmonary emphysema and in the blood-mediated metabolism of thiol oxidants (62, 158). Furthermore, CS-exposed mice develop only mild structural and mechanical lung alterations (57).
The guinea pig shows several advantages that are helpful in the study of the CS effects. Guinea pigs exhibit a small airway remodeling, manifested as airway wall thickening and, mainly, increases in thick collagen fibers, as well as goblet cell metaplasia, which are absent in mouse models of exposure to CS (155). Guinea pigs also show correlation between emphysema and lung volume alterations, which are well established in human smokers (155). Therefore, both in the guinea pig model of chronic exposure to CS and in smokers, CS-induced alterations of pulmonary function are a result of both emphysema and airway remodeling (155). Guinea pigs also develop airway inflammation, associated with an increase in the number of alveolar macrophages and activation of the ERK and JNK MAP kinases and of cathepsin K, a powerful elastase and collagenase, as happens in smokers with emphysema (64). However, the guinea pig model has some limitations too. For example, Barreiro et al. showed that exposure to seven cigarettes (24 h, 5 days/week) for up to 6 months did not induce pulmonary emphysema in guinea pigs (20). These results are congruent with a previous investigation (6), but are in contrast with other previous studies in which guinea pigs chronically exposed to CS showed anatomical and functional changes leading to small airway remodeling and emphysema associated with lung inflammation (13, 14, 34, 95). Differences in the doses of CS and its main active chemicals might explain the incongruities among studies in relation to the development of emphysema in guinea pigs chronically exposed to CS (20, 154).
Limitations of studies in mammalian cell models
Primary cells may be damaged during isolation and may be isolated with limited purity and yield. By contrast, immortalized cell lines often show alterations in their signaling pathways or may have acquired mutations with change in the genotype and phenotype.
In vitro cell models lack circulation; even when appropriate constituents are present in culture medium, their concentrations may be (even very) different from those occurring in vivo; in vivo cells are not normally bathed in antibiotics, whereas unless optimal sterile conditions can be kept constant (e.g., using laminar flow hoods), it is necessary to add antibiotics and sometimes antimycotic agents as well into the culture media; the endocrine environment is very different; and the types and rates of nutrient and O2 supply, as well as of CO2 and metabolite removal, are different. Additionally, in vitro studies mainly use CSE, whereas human smokers and animal models are exposed to CS inhalation.
The fact that CSE differs from CS is a limitation. However, in vivo cells (except keratinocytes) are not exposed to CS as a whole, but rather to constituents of CS that dissolve into biological fluids.
Evaluation of definite concentrations of specific components of CS, such as acrolein and HNE, is also questionable. Even though the biological response of individual components can be determined, it is very likely that the response to the complex, dynamic, and reactive mixture of CS is not the sum of multiple independent responses.
Congruence between smokers and animal and cell models of exposure to CS
An animal or cell culture model of exposure to CS is congruent with the smoker's condition only when we fully understand both the animal/cell culture model and the CS-induced human diseases and when the smoker and the model thereof are substantially congruent in all important biological respects. Animal/cell models can help in the identification of possible molecular pathways, but their applicability to the human condition is often uncertain.
First, no individual nonhuman mammalian animal model is totally comparable with humans regarding disease progression and metabolism. In vitro studies can be useful for investigating structure–activity relationships and mechanisms through which CS constituents may impact on cellular or molecular processes. However, a major concern is that cell culture studies may either overestimate (e.g., because of cell treatment with too high doses of CS or CSE) or underestimate (e.g., because in vitro systems may lack cellular factors that enhance the effects of CS in vivo) the in vivo effects of smoking. The major purpose of animal models and in vitro cell models is therefore to evaluate hypotheses relating to molecular pathways of CS-induced human disease, but only studies in smokers will serve as decisive tests of any of these hypotheses. Yet, despite all these limitations, studies using mammalian animal and cell models provided (and, likely, will continue to provide) valuable clues.
There is substantial congruence between some studies in human smokers, animal models, and in vitro cell models of exposure to CS regarding (i) carbonylation of glycolytic enzymes and cytoskeletal/muscle-specific proteins and (ii) increased levels of polyubiquitinated proteins and impaired proteasome activity. Carbonylation of glycolytic enzymes (Fig. 9), creatine kinase, carbonic anhydrase-3, ATP synthase, and actin was more marked in the quadriceps of healthy smokers and patients with severe COPD and in respiratory and limb muscles of guinea pigs exposed to CS (15, 20). Healthy smokers also showed reduced functionality of their quadriceps muscle compared with control subjects (20). However, chronic exposure to CS did not induce muscle inflammation in either healthy smokers or guinea pigs (15, 20). Glycolytic enzymes, carbonic anhydrase-3, skeletal α-actin, and proteins involved in energy production and distribution were highly carbonylated in respiratory and limb muscles of AKR/J mice chronically exposed to CS, which concurrently also developed lung emphysema (17). In both AKR/J mouse and guinea pig models, CS effects on muscle proteins occurred simultaneously in both respiratory (diaphragm) and limb (gastrocnemius) muscles, suggesting that chronic exposure to CS could exert direct deleterious systemic effects on the skeletal musculature (17, 20). Carbonylation of glycolytic enzymes and actin was also observed in primary HGFs (40) and ECV-304 cells exposed to CSE (65). All these studies suggest that CS-induced carbonylation of actin and enzymes involved in carbohydrate metabolism is common in both smokers and animal models exposed to CS and that this is not a cell-type-specific effect.
Exposure to CS increases levels of PCOs and polyubiquitinated proteins (Fig. 2) and impairs proteasome activity in the lungs of C57BL/6 mice, in human type II alveolar epithelial cells A549, and human bronchial epithelial cells 16HBE (146). Coherently, increased amounts of insoluble polyubiquitinated proteins were detected in the lungs of acutely CS-exposed C57BL/6 mice and in the lungs of individuals with smoking-related COPD, which suggested their involvement in the development of pulmonary emphysema (96). Moreover, reduced expression and activity of proteasome in the lungs inversely correlated with lung function in patients with COPD (87). Several studies have demonstrated ubiquitin-mediated proteasomal degradation of oxidized/carbonylated and misfolded proteins in different cell types exposed to CS (91). Overall, all these studies in different cell types and mammalian animal species, including humans, suggest that CS induces extensive protein carbonylation, resulting in misfolding and subsequent impairment of protein homeostasis (or proteostasis), which in turn could have a crucial role in the pathogenesis of emphysema and COPD (96).
Conclusion
Each type of study has limitations and strengths. Understanding the pros and cons of a given animal or cellular model offers insight into the validity of researchers' conclusions and is essential for the progress of our knowledge of the pathogenesis of CS-related diseases.
Despite clear evidence of CS-induced protein carbonylation in human smokers and in models thereof, more studies are needed in human smokers as well as in animal and cellular models thereof to better understand the role of oxidative stress and protein oxidation, including carbonylation, in the etiology of CS-related diseases.
Clearly, quitting smoking (or, obviously, absolutely not smoking) is the best prevention and the most effective method to reduce deleterious effects of firsthand CS. Stopping smoking before age 40 reduces the risk of dying from CS-related causes by about 90% and cessation by age 45–54 reduces chance of dying prematurely by about 67% (72). Generally, smokers who quit smoking, regardless of their age, improve substantially their life expectancy compared with those who continue to smoke (72). However, quitting smoking may not be sufficient to repair all the damages caused by long-term exposure to CS (84). Given that oxidative damage, as shown by PCO levels, also continues after smoking cessation (101), the relationship between CS, PCOs, how long you quit smoking, and inflammation needs further study both in smokers and in mammalian animal/cellular models of exposure to CS.
While the correlation between protein oxidation, including protein carbonylation, and human disease is widely recognized, ascertaining whether oxidation is a consequence or a primary cause of a certain oxidative stress-related disease remains a challenge. Is the increase in PCOs causal or consequential to disease onset? Protein carbonylation is mostly associated with adverse/unfavorable consequences for the protein activities or functions. Thus, protein carbonylation is the most widely used biomarker of cell damage caused by oxidative stress and is linked to various oxidative stress-associated diseases. The identification of protein targets is crucial for the understanding of the mechanism by which PCOs accumulate and potentially affect cellular functions. Recent progress in redox proteomics allows the enrichment, identification, and characterization of specific oxidative protein modifications, including carbonylation. Therefore, redox proteomics can be a powerful tool to gain new insights into the onset and/or progression of oxidative stress-related diseases, including those related to CS, and to develop therapeutic approaches to prevent and/or treat such illnesses.
Footnotes
Acknowledgment
Some authors (I.D.D., A.M., and G.C.) of this article acknowledge the financial support they received from Fondazione Ariel (