
Abstract
Aims:
New drugs are needed to treat flatworm infections that cause severe human diseases such as schistosomiasis. The unique flatworm enzyme thioredoxin glutathione reductase (TGR), structurally different from the human enzyme, is a key drug target. Structural studies of the flatworm Echinococcus granulosus TGR, free and complexed with AuI-MPO, a novel gold inhibitor, together with inhibition assays were performed.
Results:
AuI-MPO is a potent TGR inhibitor that achieves 75% inhibition at a 1:1 TGR:Au ratio and efficiently kills E. granulosus in vitro. The structures revealed salient insights: (i) unique monomer–monomer interactions, (ii) distinct binding sites for thioredoxin and the glutaredoxin (Grx) domain, (iii) a single glutathione disulfide reduction site in the Grx domain, (iv) rotation of the Grx domain toward the Sec-containing redox active site, and (v) a single gold atom bound to Cys519 and Cys573 in the AuI-TGR complex. Structural modeling suggests that these residues are involved in the stabilization of the Sec-containing C-terminus. Consistently, Cys→Ser mutations in these residues decreased TGR activities. Mass spectroscopy confirmed these cysteines are the primary binding site.
Innovation:
The identification of a primary site for gold binding and the structural model provide a basis for gold compound optimization through scaffold adjustments.
Conclusions:
The structural study revealed that TGR functions are achieved not only through a mobile Sec-containing redox center but also by rotation of the Grx domain and distinct binding sites for Grx domain and thioredoxin. The conserved Cys519 and Cys573 residues targeted by gold assist catalysis through stabilization of the Sec-containing redox center. Antioxid. Redox Signal. 27, 1491–1504.
Introduction
T
The crystal structures of Echinococcus granulosus thioredoxin glutathione reductase (egTGR), free and in complex with gold, provide insights to understand TGR reaction mechanism and its inhibition, and to improve flatworm TGR-based drug design. A single oxidized glutathione (GSSG) binding site is present in egTGR, and different sites of the thioredoxin reductase domain interact with the Grx domain and thioredoxin. Cys519 and Cys573 residues constitute a primary TGR site for binding gold and play a key role in positioning the flexible Sec-containing redox center close to N-terminal active site disulfide. Cys519 and Cys573 sites are the ideal target for designing promising gold prodrug compounds through scaffold adjustments.
The thioredoxin and glutathione systems are vital to several thiol-dependent redox pathways, including deoxyribonucleotide biosynthesis, protein folding, and antioxidant defense, and key to redox regulation, signaling, and homeostasis (25). In contrast to their mammalian hosts and free-living flatworms, parasitic flatworms possess linked thioredoxin–glutathione systems instead of canonical thioredoxin and glutathione systems (52). This unique array of thiol-based redox pathways provides a pharmacological target. The selenoenzyme thioredoxin glutathione reductase (TGR, EC 1.8.1.B1), a natural fusion of thioredoxin reductase (TR) and glutaredoxin (Grx) domains (44), is the single core enzyme for thioredoxin- and glutathione-dependent pathways.
This bottleneck enzyme for flatworm parasites functionally replaces both canonical TR (EC 1.8.1.9) and GR (EC 1.8.1.7) (1, 2, 36, 39), and constitutes a validated drug target. RNAi studies and assays with specific inhibitors have shown that TGR is essential for the trematodes Schistosoma mansoni (31), Schistosoma japonicum (42) and the cestode Echinococcus granulosus (11). Furthermore, different TGR inhibitors have been shown to partially cure flatworm experimental infection in mice (31, 38, 41, 42).
Gold(I) compounds, such as auranofin (Fig. 1), are potent inhibitors of pyridine nucleotide thiol–disulfide oxide reductases, such as eukaryotic TR, GR, and TGR, and have attracted considerable attention as antitumor and antiparasitic agents (23, 35). Due to the unique thiol-based redox array present in parasitic flatworms, the use of gold-based therapy against flatworm infections has been successfully explored. Auranofin kills cestode and trematode flatworms (11, 31, 33, 42) and is able to cure schistosome infection in a mouse model (31, 42).
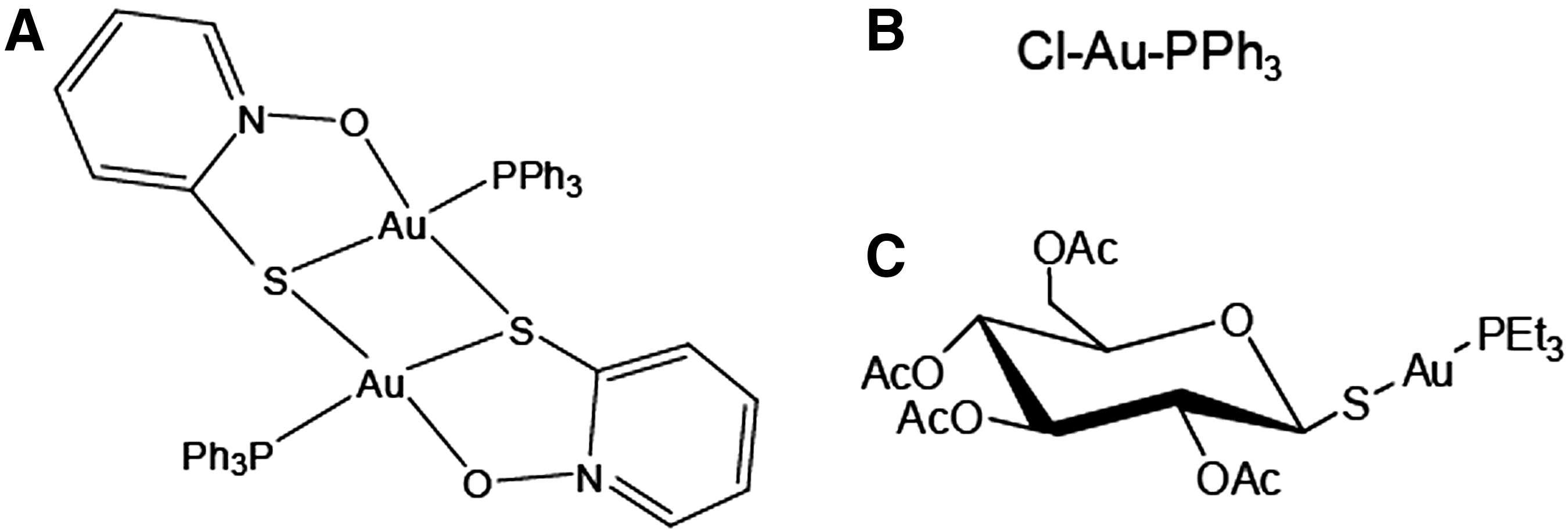
The inhibition of S. mansoni TGR (smTGR) by auranofin has been studied in detail. Using X-ray crystallography it has been shown that the actual enzyme inhibitor is AuI, released from auranofin in a reaction that is thought to be catalyzed by TGR (5). This study found that gold can bind at three different TGR sites, including the Cys154-Cys159 redox active site and an additional pair of Cys residues (Cys520-Cys574). Other AuI phosphine derivatives have been found to be irreversible inhibitors of human pyridine nucleotide thiol–disulfide reductases, and for human GR it was found that gold was the actual inhibitor with a linear S-AuI-S coordination at the enzyme CX4C redox active site (48). However, little is known about structures of medicinally relevant gold compounds.
A gold(I) triphenylphosphine [AuICl(PPh3)] complex with pyridine-2-thiol N-oxide (MPO) as coligand, AuI 2(MPO)2(PPh3)2 (AuI-MPO, Fig. 1), has previously been described as a potent antiproliferative agent against trypanosomatids (49).
We report the inhibition of the flatworm parasite E. granulosus TGR (egTGR) by AuI-MPO, AuICl(PPh3), and auranofin (Fig. 1). AuI-MPO efficiently inhibits egTGR and kills E. granulosus protoscoleces. We solved the structure of free egTGR and AuI-egTGR complex, providing structural and enzymatic insights into egTGR catalysis and inhibition by AuI.
Results
Overall structure of egTGR
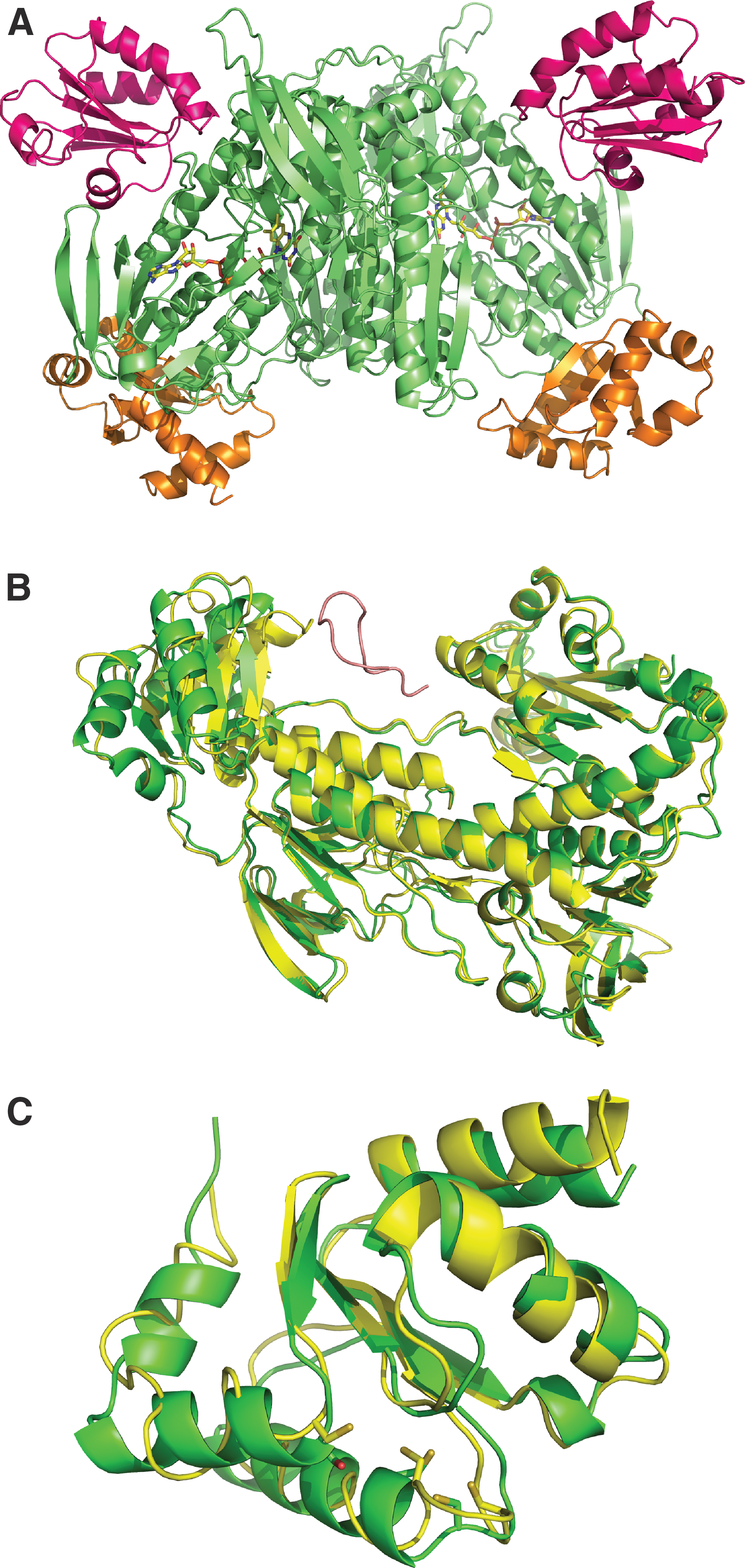
The crystal structure of the full-length form of egTGR, in which the C-terminal penultimate Sec was replaced with Cys, was solved to 2.7 Å resolution. The crystals obtained belong to the space group of P43212. The overall structure of egTGR is presented in Figure 2A. The data collection and statistics are listed in Table 1. egTGR, like mammalian TRs and smTGR, is a homodimer (4, 22). There is one physiological homodimer in an asymmetric unit.
All data (outer shell).
R merge = Σ hkl Σ i |Ii (hkl) − <I(hkl)>|/Σ hkl Σ i (hkl).
Rpim (precision-indicating R merge) = Σ hkl [(1/(N − 1)]1/2Σ i |Ii (hkl) − <I(hkl)>|/Σ hkl Σ i I i (hkl).
R work = Σ hkl ‖F obs| − |F calc‖/Σ hkl |F obs|. R free = Σ Test ‖F obs| − |F calc‖/Σ Test |F obs|, where “Test” is a test set of about 5% of the total reflections randomly chosen and set aside.
The overall fold of the TR domains of egTGR is similar to those of smTGR. Figure 2B presents the comparison between smTGR and egTGR structures. Superimposition of all the Cα atoms of TR domains with those of smTGR resulted in a root mean square deviation (RMSD) of 0.53 Å. The typical TR domains were clearly identified: the discontinuous FAD-binding domain (residues 106–257, 391–460), the NADPH-binding domain (residues 258–390), and the interface domain (residues 461–492).
The dimer interfaces are similar between egTGR and smTGR, consisting of an interacting domain of one antiparallel five-stranded β-sheet, flanked on each side by two α-helices. In addition, the loop of egTGR between residues 184 and 193 forms an antiparallel β-sheet between two monomers. These interactions are missing in smTGR or human TR1. This implies that egTGR has a more stable dimeric structure due to the extra monomer–monomer interactions. In both free egTGR and egTGR-Au complex structures, the C-terminal tail containing the Sec-containing redox center did not show a clear electron density after residue 592, indicating that this portion of the protein would be mobile, as it has been the case for smTGR and mammalian TRs (3 –5, 21, 22).
Although the TR domains of egTGR and smTGR overlaid nicely, their Grx domains (residues 4–105) did not. The Grx of egTGR was found to be rotated 15 degrees toward the C-terminal active site loop (Fig. 2B). This brought the Grx active site 6 Å closer to the Sec-containing C-terminal redox active center. This implies that to accommodate the transfer of electrons from TR to Grx, not only the C-terminal active site loop would undergo a significant conformation change but also the Grx would rotate toward the penultimate Sec596 residue, similar to the Grx domain of mammalian TGR (44, 45). The N-terminal Grx domain had a higher B factor than the TR domains (97.5 and 68.5, respectively), suggesting that it is more mobile than the TR domains.
Interactions of the TR domains with Grx domain and Trx
We observed that a β-strand (residues 44–54) and an α-helix (residues 10–19) of the Grx domain interact with two loops of TR domains (residues 131–134 and 455–460, respectively). Recently, a crystal structure of a complex between human TR1 and thioredoxin was determined (21). The egTGR and TR1 homodimers can be superimposed with RMSD of 0.75 Å. Trp114 of TR1 is critical for thioredoxin binding. Although egTGR does not have the Trp114 counterpart as TR1, there is a Phe210 at the same site, which should be able to form a similar van der Waals contact network with thioredoxin. The C-terminal active site has a similar distance to both thioredoxin and Grx, around 13 Å. This suggests that the flexible C-terminus needs to make a large conformational change and rotate to access the active sites of both substrates.
For TR1 it has been proposed that Trp407, Asn418, and Asn419 in the guiding bar suppress motion of the C-terminal arm (21, 22). On thioredoxin binding, there were no major conformational changes in the C-terminal portion interacting with TR guiding bar for controlled electron transfer. Apparently, egTGR has a more flexible C-terminal tail. This may allow the C-terminal redox center to approach the Grx active site. In both smTGR and egTGR, the Trp407 is missing, and Gln505 in egTGR and Lys506 in smTGR are the counterparts.
NADPH, FAD, and glutathione binding sites in egTGR
The crystal of free egTGR was obtained in the presence of 1 mM NADPH. However, NADPH (or NADP+) is not visible in the electron density. Nevertheless, the lining of the NADPH-binding pocket is the same as in smTGR, including several positively charged arginine residues [260, 317, 322, 413], implying that the NADPH-binding sites of both enzymes are highly comparable. The Cys residues that form part of the active site at the Grx (Cys31-Cys34) and TR domains (Cys156-Cys161) were found to be oxidized.
Each monomer has a cleft, which hosts the cofactor FAD. The FAD is bound in an extended conformation as in eukaryotic pyridine nucleotide thiol–disulfide oxidoreductases. As in unliganded smTGR, egTGR Tyr296 is positioned perpendicularly to the re-side of FAD and, in the van der Waals, contacts with isoalloxazine ring. Tyr296 was suggested to play a role to protect FAD from NADPH access (3). Residues from both subunits interact with FAD through hydrogen bonds, particularly at the interface, similar to smTGR and other TRs (4).
Different glutathione-binding sites, catalytic or regulatory, have been proposed based on different smTGR structures. A covalently bound GSH was observed between Leu397 and Thr404 on the two-turn helix of smTGR (3). In the egTGR structure, Leu397 and Thr404 also adopted a two-turn helix, but the Cys401 is missing, suggesting that the glutathionylation of this residue in smTGR may be lineage specific or a crystallization artifact. A solvent exposed positively charged region present in smTGR and GRs, but absent in TRs, has been proposed to participate in oxidized glutathione (GSSG) binding and reduction (4). The same positively charged region is observed in egTGR (residues Lys126, Lys130, and Arg449) (Supplementary Fig. S1; Supplementary Data are available online at
The glutathione-binding site of the Grx domain resembles that of smTGR (3). When GSH is overlaid into the egTGR Grx pocket, the γ-glutamyl moiety of GSH interacts with Asp84, Ser85, and Gln86, while Gln60 and Lys25 bind the glycine moiety of GSH. These residues are completely conserved among TGRs. A third Cys residue of the Grx domain, Cys88, is only 5 Å away from Cys31 of Grx. When GSH is modeled in the pocket, the Sγ of Cys88 is only 3.3 Å away from the oxygen atom of the Glu residue of GSH. This suggests that Cys88 may have a functional role previously suggested.
AuI compounds are potent inhibitors of TGR
We tested AuI-MPO and AuICl(PPh3) for their capacity to inhibit TR and GR activities of wild-type TGR. Initial time-course experiments indicated that GR inhibition was fast (Supplementary Fig. S2A), while TR inhibition was time dependent (Supplementary Fig. S2B). However, short preincubation of the enzyme with NADPH and gold inhibitors previous to the addition of 5,5′-dithiobis(2-dinitrobenzoic acid) (DTNB) resulted in maximal TR inhibition (Supplementary Fig. S2C). These results would suggest that gold binds to a site close to DTNB-binding site, which is different to the GSSG-binding site.
AuI-MPO inhibited TGR activities slightly better than auranofin, and approximately five times more efficiently than AuICl(PPh3). Inhibition with AuI-MPO was highly efficient: ∼75–80% inhibition of TR and GR activities was achieved at a 1:1 concentration ratio of AuI-MPO:TGR (Fig. 3). However, total inhibition of the enzyme required a large excess of inhibitor (∼50:1). The inhibition data did not fit a tight-binding inhibitor (Morrison equation) and suggest the existence of two gold-binding sites with different affinities.

To further characterize the inhibition, we preincubated NADPH, enzyme, and AuI-MPO mixes at the AuI-MPO:TGR concentration ratios of 10:1 in the enzyme reaction buffer, for 3 min, and then applied the mixes to a PD10 column to remove unbound inhibitor. Inhibition persisted after removal of the excess inhibitor (Fig. 4A). Furthermore, no activity was recovered even after allowing a 1-h equilibration of the TGR eluate (i.e., allowing for inhibitor/enzyme desorption), indicating that AuI-MPO:TGR binding is strong. We then examined whether inhibition was revertible by dithiothreitol (DTT) or GSH. The inhibited enzyme treated with DTT and then desalted was active. However, gold inhibition was not affected by GSH, the most abundant thiol in vivo (Fig. 4B).

Neither egTGR activities nor its inhibition by gold was affected by the [NADPH]/[NADP+] ratios from 10 to 0.1 (data not shown).
The structure of TGR in complex with AuI-MPO
The crystal structure of egTGR in complex with AuI-MPO was determined at 2.88 Å in the presence of 1 mM NADPH. No major conformational change occurred on AuI-MPO binding. The complex structure revealed that only one AuI was ligated to Cys519 and Cys573 (Fig. 5). Similar to the AuI-smTGR complex, the electron density of scaffold of the gold compounds was not observed. Mutation of these Cys residues to Ser resulted in a 75–80% decrease of TR and GR activities (Table 2), mainly due to a decrease in kcat. The activities of these mutants were inhibited by gold, indicative of an additional gold-binding site. In contrast to the AuI-smTGR complex (5), the redox center Cys156-Cys161 was not bound to gold, but forming a disulfide instead.
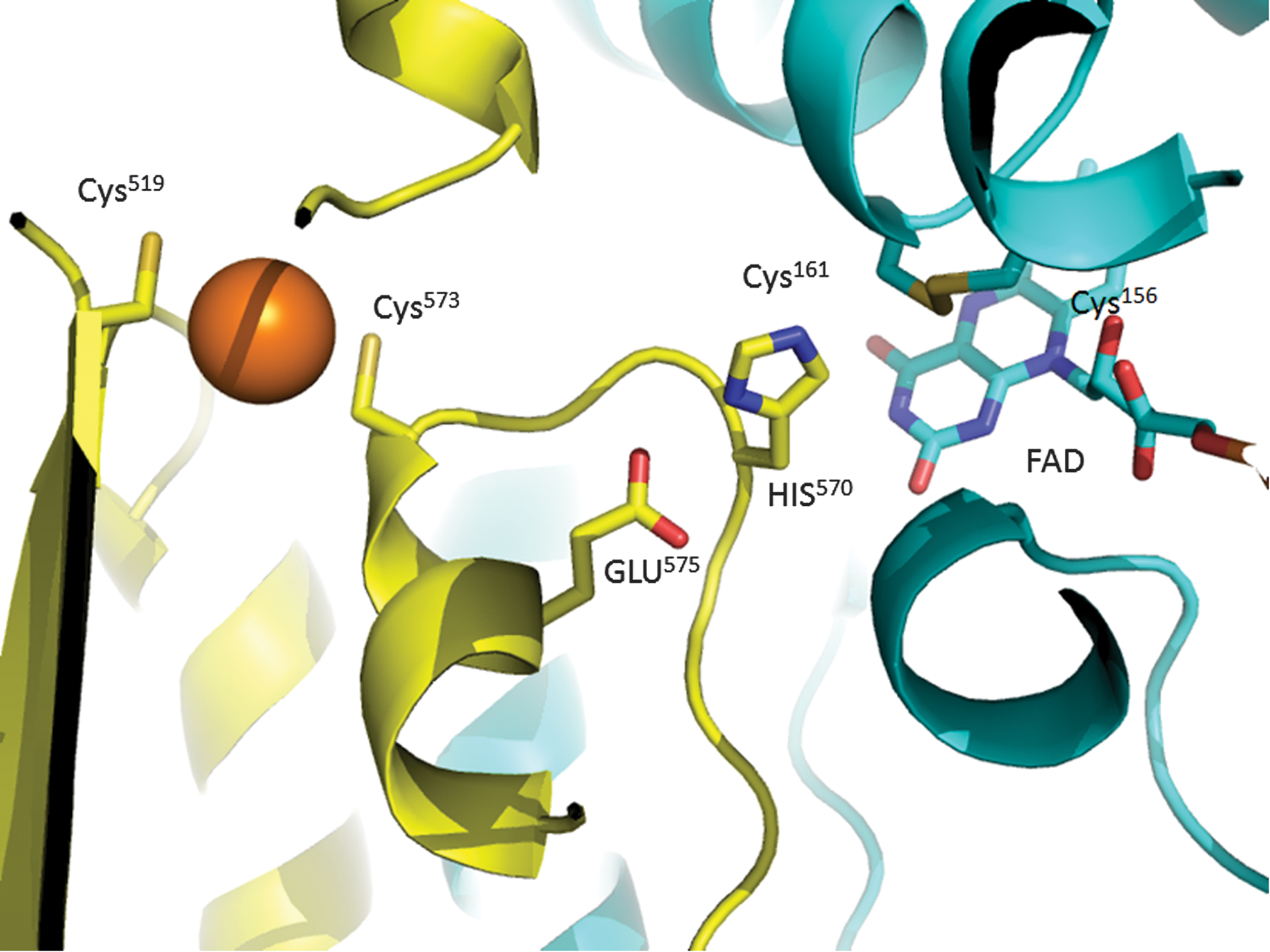
The Fo-Fc electron-density map suggested that the FAD isoalloxazine had an alternative bend butterfly conformation, implying that FAD was partially reduced (32). To explore how Au impacts on the reduction of FAD, spectrophotometric experiments were performed. The visible spectra (300–800 nm) were collected for TGR in the presence and absence of AuI-MPO. No differences were found in the oxidized and reduced spectra at the AuI-MPO:TGR molar ratios ranging from 0.2 to 2 (Supplementary Fig. S3). To gain further information regarding additional gold-binding sites, egTGR was treated with NADPH and AuI-MPO (10:1 concentration ratio) and then analyzed by mass spectrometry (Supplementary Fig. S4). Gold was bound to Cys519 and Cys573 and to a minority proportion of Cys347. The peptide containing the Cys156-Cys161 was oxidized as in the crystal structure. The Sec-containing peptide was not detected either native or bound to gold.
Cys519 and Cys573 could play an important role in the egTGR catalytic cycle. We modeled the C-terminal tail in the position where the GCUG redox center would be reduced. C-terminal tails in TGR structures are mostly disordered. In a reduced smTGR structure, the C-terminal tail was visible (PDB: 2X80C) (3), the GCUG tetrapeptide was stabilized by the long helix of residues 196–218, on the opposite side of the C-terminal active center in the interdomain space of TGR. Based on this smTGR structure, the flexible C-terminal tail starting with residue Ala590 can easily swing into the interdomain groove between the alpha chain Tyr503 to Leu507 and the loop of beta chain His570 to Thr576 without any clashes (Fig. 6A). The residues before Ala590 of the C-terminal loop can still interact with the guiding bar.
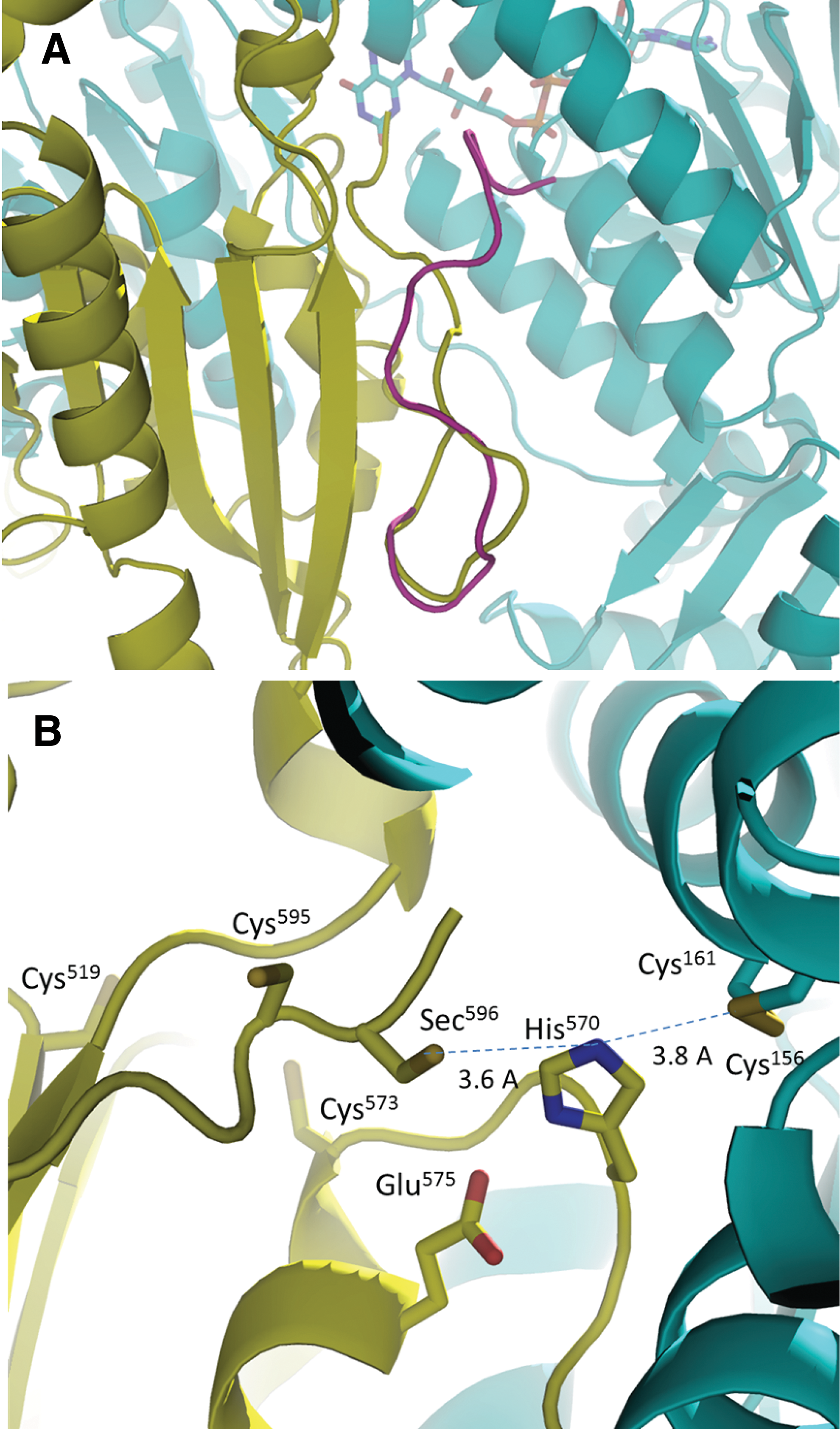
The GCUG tetrapeptide would insert into a “stabilizing cleft” mainly composed of main chain atoms of residues 505–507, Cys573, Pro571, and His570 (Fig. 6). There are no large residues in the cleft causing any clashes and the docking surface would nicely complement the N-terminal active site. During the dithiol–disulfide exchange between N-terminal and C-terminal redox centers, the C-terminal loop needs to be stabilized to perform the reaction. Our model shows that Cys573 can form a potential hydrogen bond or van der Waals contacts with the main chain oxygen of Cys595 (Fig. 6B). This is in agreement with the important role of Cys573 from our data. The impact of Cys517 is secondary. Cys517 could be important in stabilizing both Cys573 and residues of guiding bar loop by hydrogen bonds or van der Waals forces due to the close proximity.
The N-terminal active site disulfide is shield by Ser119, Ile162, Tyr212, Thr441, and His570 (Supplementary Fig. S5). Due to the size and shape of the C-terminal Gly residue, the penultimate Sec596 could not directly access the N-terminal active center. The stabilizing cleft seems to be the only place that can accommodate the GCUG active center and position the Sec596 residue close to the N-terminal active center.
Our model would bring the GCUG tetrapeptide 3.5 Å to the His570, which is the acid-base catalyst for the reduction of the N-terminal active site disulfide. Sec596 is 7 Å away from Cys161, the interchanging thiol of the N-terminal active center. During the reaction, the reduced Cys161 thiolate could directly attack Sec596, which is situated in the stabilizing cleft, while the His570 rotates out of way. We have shown before that His residues often shuffle the positions during the dithiol–disulfide interchange in the catalytic cycles of ferredoxin–thioredoxin reductase (17).
AuI-MPO kills E. granulosus larval worms in vitro
To test whether AuI-MPO was active against parasites, we assessed the effect of AuI-MPO against E. granulosus larval worms (protoscoleces) in vitro. As a control for in vitro studies, we used MPO, an organic parental compound of AuI-MPO, which did not display any TGR inhibition. Incubation with 10 and 20 μM AuI-MPO killed larval worms within 24 and 4 h, respectively, but protoscoleces survived at 2 and 5 μM concentrations of AuI-MPO. Treatment for 24 h at 5 μM AuI-MPO led to an ∼60% decrease in TGR activities, with respect to MPO-treated protoscoleces. These results, summarized in Table 3, indicate a correspondence between TGR inhibition and in vitro killing of larval worms and that the inhibitor was able to cross the protoscolex tegument. Figure 7 shows the effect of AuI-MPO on larval worms at 10 μM AuI-MPO.

In the case of GSSG, the parameters were determined in the presence of GSH to avoid hysteresis. The selenium content of wild-type egTGR and Cys519→Ser and Cys574→Ser mutants was 17%, 15%, and 15%, respectively.
DTNB, 5,5′-dithiobis(2-dinitrobenzoic acid); egTGR, Echinococcus granulosus thioredoxin glutathione reductase; GSSG, oxidized glutathione.
Viability was determined by exclusion of vital dye eosin.
GR, glutathione reductase; TR, thioredoxin reductase.
The disorganization of the parenchyma of the larval worms and the conspicuous loss of the crown of hooks observed on treatment were very similar to those observed with the AuI compound auranofin (11). Interestingly, this phenotype is not observed with oxadiazole N-oxides, which also are TGR inhibitors that kill larval worms (40).
Since TGR inhibition leads to an imbalance of redox homeostasis in flatworm parasites (31, 33), we reasoned that treatment with sublethal concentrations of AuI-MPO would lead to compensatory changes in gene expression and affect its proteome. However, this treatment did not revealed major changes (Supplementary Table S1), suggesting that a deeper coverage is required to detect significant proteome changes on gold treatment.
Discussion
Bacterial yeast and plant NADPH-dependent TRs consist of FAD-binding and NADPH-binding domains (18, 50). In these enzymes, the reducing equivalents are transferred from active site disulfide–dithiol motif directly to thioredoxin. In mammals, TRs are selenoproteins, containing a flexible C-terminal tail to shuffle the electrons to their substrates. Additional complexity is observed in the parasite TGR, which is a fusion protein of TR and Grx enzymes. Its TR domain is similar to the whole mammalian TR. Recently, structural studies of smTGR further elucidated the catalytic cycle of TGRs (3 –5). The structural comparison of egTGR and smTGR with human TRs suggests that they also share the same reaction mechanism. This has also been proposed in previous studies involving mouse TGR (43, 45).
The unique property of TGR, compared to TRs, is that this enzyme uses a flexible C-terminus to transport the electrons to Grx. egTGR structures clarify the GSH and thioredoxin binding sites. Thioredoxin is reduced by the C-terminal active site, and GSH can only be reduced by the Grx domain, which in turn is reduced by the C-terminal active site. Based on our previous and current findings, we suggest that the C-terminal tail, which is stabilized by a guiding bar (21), is more flexible than that of mammalian TR1. This flexibility may be required for direct reduction of Grx.
Thioredoxin and the Grx domain of TGR interact differently with the TR domain of TGR and would be docking on different TR sites (Fig. 2A). This domain arrangement would facilitate NADPH-independent electron transfer between the Grx domain and thioredoxin through the Sec-containing C-terminal tail, independent of NADPH as has been hypothesized by Angelucci et al. (3). To test this hypothesis, we examined whether GSH can reduce Trx via TGR using the insulin reduction assay. Insulin reduction was observed in reaction mixes with NADPH, TGR, and Trx, but not with GSH, TGR, and Trx (Supplementary Fig. S6), suggesting that the Sec-containing redox center and the Grx domain would not function equilibrating the two redox systems.
It is of interest whether TGR possesses additional glutathione-binding sites, glutathionylation sites, and Cys residues involved in catalysis or regulation of TGR activities. The Cys88 residue of egTGR was found in close proximity to the Grx redox active site, and it was glutathionylated at high GSSG concentrations (11), suggesting a physiological role for this residue. In mouse TGR, a third Cys105 residue in the Grx domain is also present (19). Superposition of the Grx domains of mouse and egTGRs showed that these two Cys residues are in the similar location. This implied that the Grx of mammalian TGRs may share the same mechanisms as egTGR. Although a Ser residue is present in smTGR at the homologous position, it is not unusual to observe differences in auxiliary resolving Cys positions among related thiol oxidoreductases. Similarly, Cys401 residue was found to be glutathionylated in one of the smTGR structures (3), but this residue is absent in egTGR. It has also been proposed that a cluster of positively charged residues present in the TR domain of smTGR would be involved in an additional glutathione-binding site and responsible for the marginal GSSG reduction observed in the Sec→stop smTGR mutant (4). egTGR has this basic region, but experiments with the Grx-less TGR revealed that its TR module is devoid of GR activity. Common to all studied flatworm TGRs is the hysteretic behavior (i.e., a lag time before the enzyme is fully active) at high GSSG concentrations (52). In egTGR, this phenomenon is mostly abolished by a Cys34→Ser mutation at the Grx redox active site, suggesting that this Cys residue could be reversibly glutathionylated (10). Collectively, these data suggest that flatworm TGRs are complex enzymes that share common features, but may also exhibit lineage-specific differences.
The crystal structure of smTGR in complex with auranofin showed three Au ions ligated to smTGR. One Au ion coordinated between the two N-terminal active site Cys residues on the si-face of FAD cofactor, a second one coordinated Cys520 and Cys574, and a third one in the putative NADPH binding pocket (5). It has also been observed that an Au+ ion ligated to the active site Cys residues next to FAD in trypanothione reductase and human GR (15, 48). The crystal structure gold-egTGR complex and mass spectrometry of gold-treated TGR revealed that these Cys were forming a disulfide bond. Unlike trypanothione reductase or GR, the active sites of TGR or TR1 are buried in the interface of the homodimers. Furthermore, the binding of Au requires the Cys residues to be reduced, and the reduced state of TGR is transient and short lived and would make TGR inhibition rather inefficient. The AuI-MPO:egTGR crystal was obtained at equimolar concentrations, while the study that described the structure of smTGR with auranofin used an excess of auranofin, which might have led to nonspecific binding. A single Au bound to Cys519 and Cys573 was identified in the gold-egTGR crystal. These Cys residues are conserved in the vast majority of TGRs and mammalian TR1 (Fig. 8). Furthermore, these residues are only 13 Å away from the TGR N-terminal active disulfide and 9 Å away from Glu575 of the catalytic dyad of TGR. Binding of Au may perturb the electron transfer pathway. Cys519 and Cys573 are too distant to form a disulfide bond and have been found to be reduced in smTGR (51). This implies that the thiol form of Cys519 and Cys573 may be an easy target for Au ligation, leading to egTGR inhibition. Cys→Ser mutations of these residues affected electron flow, decreasing TGR activities to ∼20–25%, in agreement with a functional role for these residues. In smTGR, Cys→Ala mutants in these residues also affect DTNB and GSSG reduction although less markedly (28).

It has been shown that the C-terminal loop of hTR1 adopted different conformations to accommodate Trx interaction in different redox states. To bind Trx, the GCUG peptide should flip to the surface. In all these cases, the C-terminal active site is more than 20 Å away from the N-terminal active center. We know that the electrons are transferred from NADPH→FAD→N-terminal redox center→C-terminal redox center. However, how the C-terminal active site is reduced by the N-terminal active center is still unknown. Our data suggest that the catalysis reaction involves Cys573 and Cys519. The reduction of 75–80% activity of Ser mutants precluded their direct participation in the catalytic cycle. Rather, Cys573 and Cys519 must play a crucial role in stabilizing the GCUG tetrapeptide to facilitate the reaction. Our modeling provides a plausible mechanism showing that Cys573 can play an important role in maintaining the position and conformation of the GCUG tetrapeptide in the “stabilizing cleft,” so that Sec596 can be in close proximity with Cys161 and His570, and directly perform the dithiol–disulfide exchange. This explains why Cys573 and Cys519 are an ideal gold inhibitor binding site.
The inhibition of TGR by gold suggested the existence of more than one gold-binding site, with one of them of high sensitivity to gold. Mass spectrometry revealed that in addition to Cys519 and Cys573, Cys347 was marginally bound to gold. This surface-exposed cysteine is distant from both active sites, so this gold binding may be nonspecific. Cys347 was not bound to gold in the structure and it is not conserved. We cannot rule out the possibility of gold binding to the Sec-containing carboxy-terminal peptide, since this peptide was not detected in the structure or by mass spectrometry. Noteworthy, a 75–80% inhibition of the activity is achieved by a nearly 1:1 relationship of active enzyme to gold, implying that the selenium-less protein present in the wild-type egTGR is not binding gold. This would suggest that Sec is involved in the inhibition mechanism.
In this study, we used three different gold compounds, the reference gold compound auranofin, AuI triphenylphosphine, and a novel AuI-triphenylphosphine complexed with the bioactive coligand pyridine-2-thiol N-oxide (AuI-MPO). This latter compound was found to be more potent than its parental form and slightly more potent than auranofin, indicating that the scaffold can be optimized for TGR inhibition. The capacity of AuI-MPO and auranofin to kill E. granulosus protoscoleces in vitro was very similar (11). AuI-MPO was previously noted for its potent antiproliferative effect against trypanosomatids (49). In that study it was shown that DNA was not the main target for the compound (as it is the case of cisplatin). Instead, NADH-fumarate reductase activity was inhibited by AuI-MPO, probably due to the fact that pyridine-2-thiol N-oxide is an effective inhibitor of this enzyme, which is absent in flatworms. However, trypanothione reductase was not assayed in this study as a target for this compound. Treatment of protoscoleces with the AuI-MPO sublethal dose led to a reduction in TGR activities after 24 h, similar to what was observed in S. mansoni (31). A recent work in Taenia crassiceps (a flatworm parasite of the same class as E. granulosus) showed that the addition of auranofin to the larval form of the parasite led to a concentration-dependent decrease of the internal glutathione concentration, formation of glutathione–protein complexes, and export of GSSG to the culture medium (33). Thus, most likely the primary target of gold compounds in flatworms is TGR, although we cannot rule out other targets. A previous report showed that AuI-MPO has low toxicity to mammalian cells (49).
Gold compounds have been used for medical uses since ancient times, and in the last century they became important for rheumatoid arthritis treatment (30). This and other studies indicate that gold compounds can be repurposed for treating flatworm-caused diseases, and contribute to our understanding of their biochemical effects. Overall, our studies revealed insights into the structural basis of the TGR reaction mechanism and inhibition of this enzyme by selected gold compounds. Of particular relevance is the finding that the residues Cys519 and Cys573 constitute a primary site for binding gold and their substitution affects TGR activities. This study also highlights common and specific features of TGRs that may be relevant when considering a rational drug design for flatworms, and that gold compounds are promising prodrugs that can be optimized through scaffold adjustments.
Materials and Methods
Cloning, expression, and purification of E. granulosus TGR
The constructs for expressing wild-type full-length E. granulosus TGR (egTGR), the Sec596→Cys egTGR mutant, the Grx-less TGR mutant, and egThioredoxin were previously generated. Cys519→Ser and Cys573→Ser egTGR mutants were generated by site-directed mutagenesis using the overlap extension method. Recombinant proteins were produced and purified as previously described (11), following protocols optimized for expression of Sec-containing TR (6). The selenium content of wild-type, Cys519→Ser, Cys573→Ser, and Grx-less egTGRs was 17, 15, 15, and 18%, respectively, as determined by atomic absorption using a plasma emission spectrometer (Jarrell-Ash 965 ICP) in Chemical Analysis Laboratory, University of Georgia, and used to correct selenoprotein concentration. For crystallization, the Sec596→Cys mutant was used. Following Ni Sepharose Fast Flow column (GE Healthcare) purification, the eluted protein was concentrated to 2 ml and applied onto a Superdex G200 (GE Healthcare) column equilibrated with buffer consisting of 10 mM Tris-HCl pH 8.0, 100 mM NaCl, and egTGR was collected and concentrated to 1 mg ml−1 for crystallization.
Enzymatic assays
Reduction of DTNB with concomitant NADPH oxidation was determined by the increase in absorbance at 412 nm due to the formation of 5′-thionitrobenzoic acid (TNB) (ɛ = 13,600 M −1 cm−1) (7). The reaction mixtures contained 100 μM NADPH and 5 mM DTNB.
The GR activity was assayed as the NADPH-dependent reduction of GSSG, which is followed by the decrease in absorbance at 340 nm due to NADPH oxidation (ɛ = 6200 M −1 cm−1) (14). The reaction mixtures contained 100 μM NADPH and 1 mM GSSG and 1 mM GSH to avoid conditions of hysteresis (11, 39).
The insulin interchain disulfide reduction was performed as described in an article (24). Assay mixes contained 1.0 mg ml−1 insulin, 10 nM egTGR, 60 μM egThioredoxin (EUB56960.1), and 1 mM GSH or 200 μM NADPH in 100 mM potassium phosphate, pH 7.0. The reaction was followed for 1 h by the increase in absorbance at 620 nm due to the precipitation of free insulin β-chain.
Synthesis of gold compounds
AuI-MPO was synthesized and characterized according to a previously reported procedure (49). Briefly, 0.2 mmol of each AuCl(PPh3) and pyridine-2-thiol N-oxide sodium salt (NaMPO) were heated under reflux in 20 ml methanol for 6 h under N2 using Schlenk techniques. The white grayish solid was filtered off the reaction medium and washed with water, ethanol, and ethyl ether. Elemental analyses, electrospray ionization (ESI)-MS, UV-vis, FTIR, 1H and 31P NMR spectroscopic results agreed with those previously reported (49). Pyridine-2-thiol N-oxide sodium salt and AuICl(PPh3) were commercially available.
Inhibition studies
Stock solutions of AuICl(PPh3) and AuI-MPO were prepared by dissolving the compounds in dimethylsulfoxide at 40°C to a stock solution of 1 mM concentration. The compounds were tested in the nM-μM concentration range. Except otherwise specified, wild-type egTGR was used at a 1.5 nM final concentration in all assays, and NADPH, enzyme, and inhibitor mixes were preincubated for 3 min and the reaction started by the addition of DTNB or GSSG for TR and GR assays, respectively, and followed for 5 min. All assays were performed in duplicate. In every case, a reference progress curve without an enzyme was performed to control for reactions between substrates and inhibitors. The percentage of TR or GR inhibition was calculated as follows: % Inhibition = 100 − (v i/v o) × 100, where v i and v o correspond to the initial velocities of TNB formation or NADPH consumption (μM)/t (s) with and without inhibitor, respectively. To characterize the enzyme-inhibitor binding, NADPH-enzyme-inhibitor mixes were preincubated as described above and then the mixes were applied to a PD10™ column to remove the unbound inhibitor. The TR activity of the eluates was evaluated by adding NADPH and DTNB. Control experiments without either inhibitors or TGR were carried out as control. To test whether inhibition was revertable by low molecular weight thiols, experiments were carried out with mixes containing 2 nM wild-type egTGR, 100 μM NADPH and 200 nM AuI-MPO were preincubated for 3 min, treated with either 100 μM DTT or 100 μM GSH, desalted using a Zeba™ desalting spin column and assayed for GR activity.
Visible spectra of TGR
The 300–800 nm visible spectra were obtained on a Varian Cary 50 Bio spectrophotometer for the oxidized and NADPH-reduced egTGR using 20 μM egTGR in the absence or presence of AuI-MPO at the inhibitor:enzyme ratios from 0.2 to 2.
Crystallization, data collection, and processing
Screening for crystallization conditions took place at 16°C using commercial kits from Hampton Research (Crystal Screen, Crystal Screen 2, Index, PEG Rx1/Rx2, PEG/Ion and PEG/Ion 2). 0.1 μl 10 mg ml−1 protein solution was mixed with an equal amount of reservoir solution and equilibrated against 80 μl reservoir solution. Clusters of needle-shaped crystals appeared after 7 days in Crystal Screen condition at 16°C. Fine-tuning the pH in the range 7.5–9.5 and the PEG 3350 concentration in the range 12–25% produced crystals that were suitable for diffraction. A suitable single crystal for data collection was finally obtained after 10 days in a condition consisting of 0.1 M lithium sulfate, 0.1 M Tris-HCl, pH 7.5, and 18% (w/v) polyethylene glycol 3350 at 16°C. The crystallization condition of the complex is same to the native, and Gold was added to the drop of well.
The harvested crystals were quickly frozen and mounted on the goniometer in a nitrogen stream at −173°C. Data were collected using the Pilatus detector at ID-24c of the Advanced Photon Source, Argonne National Laboratory, at a wavelength of 0.98. The data were indexed, integrated, and scaled with HKL-2000 (37). The data collection statistics is listed in Table 1.
Molecular continuum electrostatic surface potentials
Molecular continuum electrostatic surface potentials were calculated in PDB2PQR (20) and the Adaptive Poisson–Boltzmann Solver (8) with solvent accessible molecular potential isosurface visualization in Jmol (
Modeling of TGR C-terminal loop
The position of TGR C-terminal tail was investigated and manually adjusted on computer modeling program, O (29). There were two criteria for positioning the C-terminal tail. First, the GCUG active center needs to be close to the N-terminal active disulfide without any clashes. Second, the C-terminal loop should have minimal movement from its position in the reduced smTGR (PDB: 2X8C), in which the Sec was 13 Å away from the N-terminal active-site disulfide. The position of GCUG active center was further refined using energy minimization using program CNS (12).
In vitro culture of larval worms
Eighty thousand protoscoleces from E. granulosus sensu stricto (G1 strain, assessed by COX1 amplification) (16), obtained from asceptical punction of a single hydatid cyst from bovine lung, were washed several times with PBS and then incubated at 37°C, 5% CO2, in DMEM supplemented with antibiotics and 20 mM HEPES, pH 7.3. For assessing the effect of the inhibitors on larval worm viability, 2000 protoscoleces were treated with 2, 5, 10, and 20 μM concentrations of AuI-MPO. Protoscoleces were observed under the microscope every 4 h and endpoint viability assessed by exclusion of the vital dye eosin at 24 h (9). The infected bovine viscera were kindly provided by UREXPORT (material normally discarded under the control of the Government Secretary of Agriculture and Livestock).
Proteomic experiments
For proteomic studies, 10,000 protoscoleces were incubated as detailed above, with vehicle (dimethyl sulfoxide) or sublethal concentration of AuI-MPO (2 and 5 μM) for 24 h. Aqueous soluble and insoluble protein extracts were prepared from cultured protoscoleces by resuspension of the lyophilized pellets into the RapiGest™ SF surfactant (Waters), 0.1% diluted in 50 mM ammonium bicarbonate, pH 8.0 (46). The proteins were subsequently reduced by adding DTT (final concentration: 5 mM) for 30 min at 60°C, and alkylated with iodoacetamide (final concentration: 15 mM) for 30 min in the dark at 20°C. The protein samples were then digested with trypsin (Trypsin Gold, Mass Spectrometry Grade; Promega) at the enzyme-to-protein ratio of 1:50 (w/w) overnight at 37°C. After digestion, the RapiGest SF surfactant was removed with trifluoroacetic acid, as previously described (46). The resulting peptides were desalted using an OASIS®HLB Cartridge column (Waters) and analyzed by liquid chromatography separation coupled with tandem mass spectrometry strategy (LC-MS/MS) (34). MS analyses were performed using an ESI quadrupole time-of-flight Ultima API mass spectrometer (Micromass) coupled to a capillary liquid chromatography system (CapLC; Waters). The peptides were separated in a NanoEase C18 (75 μm ID) capillary column by elution with a water/acetonitrile 0.1% formic acid gradient. Data were acquired in a data-dependent mode, and multiple charged peptide ions (+2, +3, and +4) were automatically mass selected and dissociated in MS/MS experiments. LC and ESI conditions included a 200 nL/min flow, a nanoflow capillary voltage of 3.5 kV, a block temperature of 100°C, and a cone voltage of 100 V. Protein identification based on peptide MS/MS data was performed using Mascot software (Matrix Science) with the following parameters: a maximum of one missed cleavage, fixed carbamidomethyl alkylation of cysteines, variable oxidation of methionine, mass tolerance for the monoisotopic peptide window was set to ±0.1 Da, and MS/MS tolerance window was set to ±0.1 Da. All tandem mass spectra were searched against E. granulosus deduced proteome annotation available on
For mass spectrometry analysis of wild-type egTGR treated with AuI-MPO, 2 nM Sec-containing egTGR was treated with 20 nM AuI-MPO and 100 μM NADPH for 1 min and desalted using a Zeba spin desalting column, 40K, 0.5 ml (Thermo). The sample was digested with trypsin and passed through a ZipTip® (Millipore) column and peptides analyzed on an LTQ VELOS nano-ESI (Thermo Scientific). The egTGR preparation contained a [Sec-containing TGR]/[truncated TGR] ratio of 0.17.
Structure coordinates have been deposited in the Protein Data Bank under accession codes: 5W1J for egTGR alone, and 5W1L for egTGR/gold complex.
Footnotes
Acknowledgments
We thank Dr. Martin Fló (Institut Pasteur of Montevideo) and Dr. Beatriz Álvarez (Universidad de la República, Uruguay) for helpful discussions, Dr. Elias Arnér (Karolinska Institutet) for provision of pSUABC, Madelon Portela (Institut Pasteur de Montevideo) and UREXPORT for provision of hydatid material (Lezica 2128, Montevideo, Uruguay). This work was funded by ICGEB (Research Grant URU14-01 to G.S.), the Boettcher Foundation Webb-Waring Biomedical grant to S.D., and NIH ES025797 to S.D., and FOCEM (MERCOSUR Structural Convergence Fund, [COF 03/11]). Proteomic analyses were performed at UNIPROTE-MS, Centro de Biotecnologia, UFRGS, Porto Alegre (Brazil). V.G.V. is a recipient of the FAPERGS (Brazil) DTI1 postdoctoral fellowship. W.G. was a recipient of grant #31070651 from National Natural Science Foundation of China. We thank David Neau for data collection assistance at APS Argonne National Laboratory, Beamline 24 ID-C and Corie Ralston at ALS Lawrence Berkley National Laboratory, Beamline 822.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.