
Abstract
Significance:
Myocardial ischemia/reperfusion (I/R) is an important complication of reperfusion therapy for myocardial infarction (MI). It is a complex process involving metabolic and immunological factors. To date, no effective treatment has been identified.
Recent Advances:
Previous research has focused on the role of innate immune cells in I/R injury. In recent years, increasing evidence has accumulated for an important role for adaptive immune cells, particularly T lymphocytes. Data from ST elevation MI patients have identified prognostic significance for lymphocyte counts, particularly postreperfusion lymphopenia. Dynamic changes in circulating CD4+ T cell subsets occurring early after reperfusion are associated with development of I/R injury in the form of microvascular obstruction. Transcoronary gradients in cell counts suggest sequestration of these cells into the reperfused myocardium. These findings support existing data from mouse models indicating a role for CD4+ T cells in I/R injury. It is clear, however, the effects of lymphocytes in the ischemic myocardium are time and subset specific, with some having protective effects, while others are pathogenic.
Critical Issues:
An understanding of the cellular events that lead to accumulation of lymphocytes in the myocardium, and their actions once there, is key to manipulating this process. Chemokines produced in response to ischemia and cellular injury have an important role, while lymphocyte-derived cytokines are critical in the balance between inflammation and healing.
Future Directions:
Further research into the involvement of lymphocytes in myocardial I/R injury may allow development of targeted therapies, opening a new avenue of considerable therapeutic potential. Antioxid. Redox Signal. 26, 660–675.
Introduction
M
Historically, research into the cellular mechanisms of myocardial ischemic and I/R injury, particularly those involving inflammation, has focused on innate immune cells such as neutrophils and monocytes. Increasingly, however, there is recognition that adaptive immune cells, particularly lymphocytes, have important and potentially diverse rolls in this process (47, 48). This review focuses on the role of these cells in this clinically important phenomenon, and the current understanding of the interactions that affect their accumulation and functions within the reperfused myocardium and microvasculature.
Lymphocyte Subsets and Their Functions
While the role of lymphocytes in myocardial injury is becoming increasingly evident, many cardiovascular researchers may not be well versed in adaptive immunobiology. Consequently, an overview of lymphocyte subsets and their functions is merited. Lymphocytes can be divided into T cells, B cells, and natural killer (NK) cells, each of which has wide-ranging and very different functions.
The main immunological role of B cells is the production of immunoglobulin (antibody). Each B cell is able to produce a single specificity of antibody, which is able to recognize a specific antigen, for example, a molecule on the surface of a pathogen (10, 90). B cells can be activated to proliferate and secrete large quantities of antibody, which has a number of functions in defense against pathogens (87, 88).
NK cells are traditionally considered to be part of the innate immune system. They are not able to recognize a specific antigen, but have a role in destruction of abnormal cells, for instance, tumor cells or those infected with intracellular pathogens (107).
T cells have a wide variety of functions in the adaptive immune system and can be divided into a number of subsets. The characteristic feature of all T cells, however, is the possession of the T cell receptor (TCR). The TCR is a cell surface receptor, able to recognize a specific antigenic peptide, displayed on the surface of another cell in conjunction with a major histocompatibility complex (MHC) molecule. The vast majority of T cells are characterized as either CD4+ or CD8+ T cells, depending on which of these markers they express. These two additional cell surface molecules are coreceptors, essential for the recognition and response to antigen by the TCR.
T cells expressing CD4 on their surface are known as T helper cells. They are so named because following activation, they exert their effects by secretion of cytokines and directly interacting with other cell types, assisting them in their functions. CD4+ T helper cells can be further characterized, depending on the cytokines they secrete and types of responses they facilitate (57, 88).
The first two T helper cell subsets to be discovered are known as T
T
Several other CD4+ T cell subsets have now been identified. These include regulatory T cells (T
CD8
Approximately 4% of human T cells express a different type of TCR, comprising γ and δ polypeptide chains, rather than the more common α and β chain, and are known as γ δ T cells (18). This heterogeneous group of cells comprises several further subsets and has a variety of effector functions, including cytokine production and cytotoxicity (16, 92).
Myocardial I/R Injury
Myocardial I/R injury is thought to contribute up to 50% of the final infarct size in STEMI, impacting subsequent left ventricular ejection fraction and clinical outcome (45). Several diverse pathological processes are recognized and widely considered to collectively contribute to I/R injury. Cardiac arrhythmias can be triggered directly by reperfusion, but are usually self-limiting and harmless (75, 118), while transient impairment of myocardial function, myocardial stunning, occurs and is thought to be related to both oxidative stress and calcium overload (11, 60). These processes, although considered components of myocardial I/R injury, are reversible and do not contribute to final infarct size.
Of more significance, however, are the combined effects of the two other main components of I/R injury, which lead to irreversible myocardial damage. Lethal reperfusion injury is loosely defined as the death of cardiac myocytes viable at the time of reperfusion and is thought to be related to a series of metabolic and biochemical events involving production of reactive oxygen species (ROS) (93), rapid restoration of physiological pH following a period of ischemia-induced acidosis (67), and opening of the mitochondrial permeability transition pore (MPTP), resulting in collapse of the mitochondrial membrane potential (42). A detailed description of these processes is out with the scope of this article, and we would direct readers to other thorough reviews (45, 118).
The final component of I/R injury is microvascular obstruction (MVO), defined as the inability to maintain perfusion of myocardium that has previously been ischemic despite reestablishment of adequate flow in the epicardial coronary vessel supplying the territory. It was first described in animal models involving transient ligation of coronary arteries (61, 62).
Using a canine myocardial I/R model, Kloner et al. demonstrated that after 90 min of ischemia, followed by reperfusion, the uptake of an intravascular tracer in the damaged myocardium was uneven, with areas of absent uptake in the inner half of the infarct (61). Electron microscopy in this and subsequent studies has revealed characteristic findings shedding light on the multifactorial nature of MVO. At the level of the capillaries, there is swelling of endothelial cells, with protrusion of blebs contributing to obstruction of the lumen (61, 95). Plugging of the capillaries with leukocytes, erythrocytes, and platelet thrombi is also typically seen (31, 95). Furthermore, local myocyte swelling and subsarcolemmal blebs contribute further to capillary obstruction by external compression of the vessels (61, 98, 110).
The precise mechanisms and time course of development of MVO are not yet fully understood. Animal studies have suggested that it develops particularly rapidly within the first 2 h following reperfusion (94). In addition to causing mechanical obstruction, accumulated leukocytes can contribute by the release of ROS, which can lead to further damage and impairment of endothelial function, vasoconstriction, and increased accumulation of fibrin/fibrinogen (30). Additional factors, including microembolization of atherosclerotic plaque debris and platelet–fibrin complexes to the distal microvasculature, can also contribute in human patients undergoing PPCI (71).
One important characteristic of MVO as a component of myocardial I/R injury is that unlike lethal reperfusion injury, it can be directly visualized and quantified. It has been shown that 30%–50% of patients with apparently normal flow on angiography following reperfusion have evidence of MVO on further imaging, for instance, by contrast echocardiography or MRI (49, 56). The presence of MVO on myocardial imaging post-PPCI is of great clinical significance, as has been demonstrated by a number of studies showing relationships with adverse outcomes (12, 26, 49, 56, 110, 111). Importantly, in recent years, indirect evidence has emerged for a relationship between lymphocyte kinetics in human patients following PPCI, and myocardial I/R injury in the form of MVO, as will be discussed further below (8, 9).
The Role of Lymphocytes in MI
The vast majority of research investigating the role of leukocytes in MI has focused on innate immune cells, most notably monocytes and neutrophils. These have been extensively reviewed elsewhere (81, 106) and will not be covered in detail in this article. In contrast, there is a comparative paucity of data regarding the role of lymphocytes. However, some solid evidence exists of a key contribution by T
In addition to a role in I/R injury, which will be considered separately in the next section, recent studies have demonstrated an emerging contribution for T cells in the later stages of MI and infarct healing.
Hofman et al., using a model of MI without reperfusion, showed that two types of CD4+ T cell-deficient mice (CD4 knockout [KO] and MHC class II deficient [MHCΔ/Δ]) had reduced collagen deposition and neovascularization in the infarct zone compared with wild-type mice at day 7 (46). Moreover, The CD4 KO mice showed increased left ventricular dilatation, while MHCΔ/Δ mice had excess mortality (46). Both of these groups displayed deranged collagen deposition and scar formation. These findings were associated with elevated numbers of granulocytes and Ly6Chi monocytes in the myocardium at day 7, indicating that CD4+ T cells may be important in limiting inflammation at this stage during the healing process (46). However, in the same study, no differences were seen in the number or composition of innate leukocytes in the myocardium at day 3, suggesting that such effects may be time specific (46).
Several researchers have suggested an important role for T
This is in keeping with two other studies in both rats and mice that have shown attenuated postinfarct myocardial inflammation and reduced adverse remodeling with T
One study has shown a potential pathogenic role for γ δ T cells (115). In a mouse model of MI without reperfusion, mice deficient in these cells were protected from adverse ventricular remodeling and had reduced infarct size at 28 days compared with wild-type mice (115). This protection was associated with reduced sustained infiltration of inflammatory leukocytes, including neutrophils and macrophages, at day 7 post-MI. The mechanism of this appeared to be loss of production of the proinflammatory cytokine IL-17A by γ δ T cells (115). In addition to increased inflammatory cell infiltration, IL-17A also has proapoptotic and profibrotic effects, contributing to adverse cardiac remodeling (115).
Another study, on the other hand, has suggested a protective role during post-MI healing for a further minor T cell subset, called invariant NKT (iNKT) cells (102). Administration of a specific activator of these cells in a mouse model of MI resulted in their increased infiltration into the myocardium at 7 days. This was associated with improved cardiac function and attenuated remodeling at 28 days. There was also increased expression of the anti-inflammatory cytokine IL-10 in the treated group (102).
Finally, one major study has investigated the role of B cells in a mouse model of MI without reperfusion (122). The investigators found that B cell-depleted mice had reduced myocardial monocyte infiltration and inflammatory cytokine expression, as well as improved cardiac function and reduced ventricular remodeling at 14 days post-MI (122). Through a series of reconstitution experiments, they demonstrated that B cell production of the chemokine CCL7 contributed to monocyte infiltration and had an adverse effect on ventricular function post-MI (122). Moreover, the use of mice with leukocytes unable to respond to Toll-like receptor (TLR) signaling suggested that CCL7 production by B cells was, at least in part, dependent on this mechanism of stimulation (122). To date, this is the only study to perform an extensive mechanistic investigation of the role of B cells in MI. However, as with most of the studies discussed in this section, this animal model did not include a reperfusion component, limiting the value in the context of modern treatment in humans.
T Cells and Their Cytokines in I/R Injury
Most of the published research addressing the role of lymphocytes in MI has focused on injury and healing in the absence of reperfusion. However, given the importance if I/R injury with modern reperfusion therapy, further understanding of the involvement of lymphocytes in this process is essential. There is a wealth of evidence implicating T cells, in particular, in I/R injury in various organ systems [reviewed in Refs. (52, 73)]. Surprisingly, myocardial I/R injury has received comparatively little attention in this regard. However, there is robust evidence from mouse models for an important role for CD4+ T cells in this context, particularly from two studies by Yang et al. (116, 117). They used a model involving transient occlusion of the left anterior descending artery, followed by reperfusion. They demonstrated that in wild-type mice, T cells as well as neutrophils accumulated in the previously ischemic myocardium within minutes of reperfusion and that this was associated with a drop in the peripheral blood lymphocyte count (117). Rag 1 KO mice, which lack mature lymphocytes, had reduced myocardial neutrophil accumulation as well as an absence of T cells in the heart following reperfusion (116, 117). Furthermore, these mice were protected from I/R injury, developing smaller infarcts than wild-type mice, and this protection was lost following adoptive transfer of CD4+ T cells (117). Similar levels of protection were achieved with CD4+ T cell depletion in wild-type mice, while CD8+ T cell depletion had no effect (117). Consequently, this provides clear evidence for a critical role for CD4+ T cells in myocardial I/R injury in the mouse, likely due to proinflammatory effects. Moreover, adoptive transfer of CD4+ T cells from IFN-γ KO mice to Rag 1 KO mice failed to abolish the protection from I/R injury, suggesting a critical role for this proinflammatory cytokine produced by T
A number of studies have investigated the role of other T cell subsets in murine myocardial I/R injury. One study assessing the involvement of the T cell-derived cytokine IL-17A found that it was primarily produced by γ δ T cells in the reperfused myocardium (69). Inactivation of IL-17A with a neutralizing antibody, or deletion using IL-17 KO mice, resulted in reduced infarct size and improved ventricular function compared with wild-type mice (69). Moreover, administration of recombinant IL-17A before reperfusion further increased infarct size (69). This cytokine was shown to have proapoptotic effects in cardiomyocytes in murine myocardial I/R as well as increasing neutrophil infiltration through accentuated production of chemokines, including CXCL1, CXCL2, and CXCL6 (69). Furthermore, in a study in rats, the IL-17 pathway was shown to be activated following myocardial I/R, while blockade with an IL-17-neutralizing antibody resulted in reduced cardiomyocyte apoptosis (7). Consequently, there is strong evidence that this cytokine contributes pathologically to myocardial I/R injury and that in this situation it is primarily derived from γ δ T cells.
Another group has investigated the role of iNKT cells in murine myocardial I/R injury (50). They found that administration of an activator of these cells before reperfusion reduced infarct size at 24 h. This was associated with upregulation of the anti-inflammatory cytokine IL-10, although IFN-γ production also increased. Protection from I/R injury was abrogated by neutralization of IL-10, while IFN-γ blockade reduced infarct size (50). This suggests that iNKT cells mediate protective effects through IL-10 production in spite of the concurrent release of harmful IFN-γ.
A recently published article by Xia et al. has addressed the role of T
These studies confirm the importance of T cells in myocardial inflammation and injury following I/R in mice. Moreover, there are clearly opposing subset-specific effects, with IFN-γ-producing T
Recently, we have published a detailed analysis of lymphocyte subset kinetics in the context of PPCI for STEMI in humans (8). We have shown that loss of T cells, and in particular highly differentiated effector T cell subsets, occurs from the peripheral circulation rapidly over the first 90 min following reperfusion. Importantly, there was a strong significant relationship between the extent of the loss of effector CD4+ T cell subsets and myocardial I/R injury in the form of MVO measured on cardiac MRI. Moreover, analysis of transcoronary gradients, determined by blood sampling from the aorta, proximal to, and the coronary sinus, distal to the reperfused myocardium, suggested sequestration of T cells within the myocardium over the early postreperfusion period (8). This work builds on a previous observation of an association between MVO and postreperfusion lymphopenia previously noted by Bodi et al. (9).
Prognostic Relevance of Lymphocytes in MI in Humans
Mechanistic data investigating the role of lymphocytes in MI in humans are limited. However, several observational studies have identified associations between lymphocyte counts and clinical outcome in MI. A number of studies have reported adverse outcomes, including increased mortality, in patients with a high neutrophil-to-lymphocyte ratio (4, 19, 86, 100). The implication from this is of negative prognostic relevance for low lymphocyte counts as well as high neutrophil counts. Our own study is the only one to investigate the prognostic relevance of the minimum lymphocyte count obtained during admission with acute STEMI, in a large cohort of 1377 patients undergoing PPCI (8). We found that lymphocyte counts dropped after reperfusion, and the presence of postreperfusion lymphopenia was an independent predictor for increased mortality at 3 years (82.8% vs. 96.3%, lowest vs. highest tertile; hazard ratio 2.42) (8). The drop in lymphocyte counts was primarily due to postreperfusion depletion of T cells from the circulation. Moreover, our transcoronary gradient data suggested that at least some of the T cells lost from the circulation were sequestered into the reperfusion myocardium (8). This backs up observations from murine studies identifying significant pathological effects of T cells in myocardial I/R injury, as outlined above, suggesting that these roles are likely to remain relevant in the context of human STEMI treated with PPCI.
Lymphocyte Activation in I/R Injury
Lymphocytes are the archetypal adaptive immune system cell, and adaptive immune responses are characteristically antigen specific. Each lymphocyte (T or B cell) expresses a receptor of single antigen specificity, and ligation of these receptors is the initiating event in an adaptive immune response. In the case of T cells, the process is initiated by TCR-mediated recognition of antigen expressed on antigen-presenting cells in conjunction with MHC molecules. Under appropriate circumstances, this leads to activation of the T cell and proliferation, generally within secondary lymphoid tissues such as lymph nodes. These events, however, take time to unfold, and an adaptive immune response generally takes several days to develop following antigen exposure. These lead to the question of how lymphocytes could be activated in the context of acute I/R injury (Fig. 1), including MVO, which is known to develop rapidly within the first few hours of reperfusion (94).
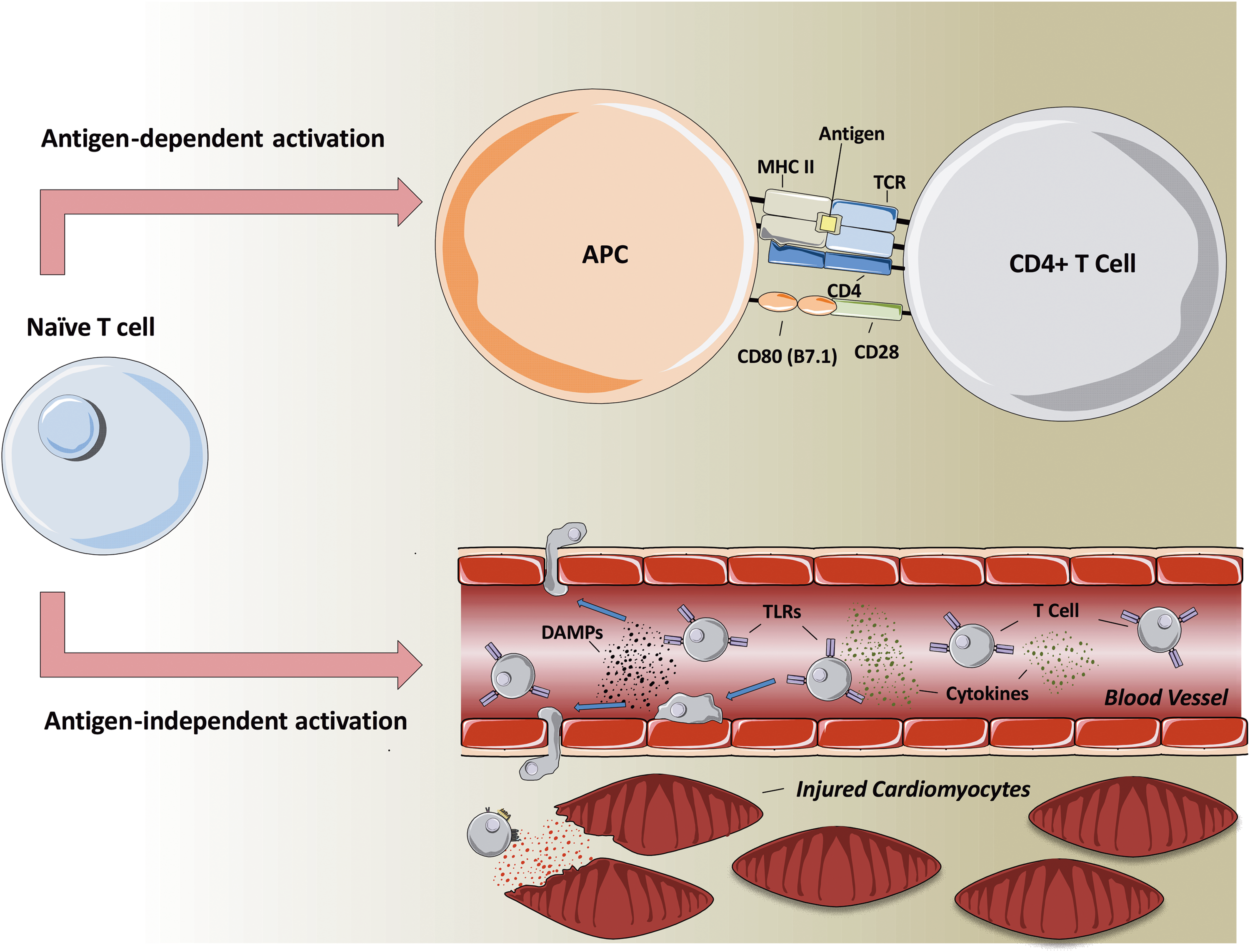
Hofmann et al. have shown evidence of activation and proliferation of T cells in lymph nodes, occurring within days of MI (46). This activation appeared to be dependent on antigen recognition by the TCR as it was reduced in mice with a transgenic TCR for an irrelevant ovalbumin antigen. Moreover, T cell activation in this context appeared to be important in healing following MI, a process that occurs over several days and weeks (46). Consequently, this indicates that antigen-specific T cell activation is likely to be important in the later effects of lymphocytes in MI. It is, however, difficult to envisage how this activation mechanism could be relevant in acute I/R, given the rapid timeframe of injury.
Importantly, it is known that in some circumstances, T cell activation can occur without specific antigen recognition. It has been shown that certain danger-associated molecular patterns (DAMPs), as well as combinations of cytokines, are able to induce proliferation and differentiation of T cells to develop effector functions in vitro without antigen exposure (54, 104). In the case of DAMPs, this occurs through binding to another group of cell surface molecules, pattern recognition receptors (PRRs), including TLRs (see also below) (54). Antigen-independent mechanisms of T cell activation may provide a feasible explanation for their ability to have effects rapidly in I/R injury as cells of a wide variety of specificities could be activated and function without the requirement for a time-consuming period of clonal proliferation of specific cells.
The question of mechanisms of lymphocyte activation in I/R has been addressed to some extent in other organ systems, with varying findings. Kleinschnitz et al. used a murine model of ischemic stroke with reperfusion to investigate the immunological requirements of T cells in mediating cerebral damage (59). They found that mice deficient for all T cells exhibited reduced injury, while reconstitution with T cells abolished protection. Crucially, mice reconstituted with T cells with a single transgenic TCR for an irrelevant antigen, or T cells lacking costimulatory molecules, were similarly susceptible to T cell-mediated damage. This suggests that the deleterious effects of T cells in this setting were independent of their capability for antigen recognition and classical adaptive immune function.
In contrast, however, Kuboki et al. have demonstrated reduced hepatic I/R injury in single TCR transgenic mice compared with wild-type mice, suggesting a requirement for antigen-dependent signaling through the TCR (63). Similar requirements have been found in murine renal I/R injury, with reduced injury in nude mice reconstituted with DO11.10 T cells with limited TCR specificity compared with wild-type mice, although this protection could be abolished by prior immunization to activate the DO11.10 T cells in an antigen-dependent manner (96). Given these contradictory findings, whether T cell activation in acute I/R injury occurs in an antigen-dependent or independent manner remains to be determined. If the former were to be the case, the specific antigens recognized are also unknown, although logically self-antigens made visible to the immune system as a result of tissue damage would seem to be strong candidates in this setting.
Cellular Communication in I/R Injury
Recruitment of leukocytes in MI
To date, few studies have sought to characterize myocardial leukocyte recruitment following MI in humans and most available data come from murine models. One study by Yan et al. used a mouse model of MI with or without reperfusion, quantifying leukocyte subsets within the myocardium using flow cytometry at various time points (114). They found that the predominant leukocytes present were macrophages, although neutrophils, T cells, B cells, and NK cells all show highly significant recruitment compared with mice exposed to a sham procedure (114). In MI without reperfusion, myocardial neutrophil numbers peaked at day 3, while those of macrophages, T cells, B cells, and NK cells all peaked later at day 7 (114). In spite of this late peak, there was highly significant recruitment of most leukocyte subsets, including T cells, by day 1 post-MI (114). In contrast, in MI with reperfusion, the total number of leukocytes recruited was reduced compared with nonreperfused MI, while the lymphocyte peak occurred earlier after only 3 days (114).
In addition to studying myocardial leukocyte numbers, Yan et al. also characterized the cells phenotypically. With regard to the T cells recruited following MI with reperfusion, they found that CD4+ T cells displayed a primarily proinflammatory T
Two further studies have addressed the question of lymphocyte recruitment into the myocardium in mouse models of MI without reperfusion. Zouggari et al. demonstrated infiltration of B cells with a peak at day 5 (122). In the same study, T cells were present when first measured at 0.5 days, before peaking after 1 day and declining thereafter. This represents a significantly earlier peak in T cell infiltration than that found by Yan et al. (114). Another study by Hofman et al. has investigated recruitment of CD4+ T cells. They found these cells to be present in the myocardium by day 3, peaking at day 7 after MI, before their numbers declined (46). Unfortunately, they did not report any findings for any cellular recruitment at earlier time points. Both of these studies, however, involved permanent coronary artery ligation without reperfusion and therefore no I/R injury will have been induced.
Characterization of myocardial leukocyte recruitment following MI in human patients is extremely challenging given the difficulty in obtaining tissue specimens from living patients. Two studies by Abbate et al. have specifically investigated lymphocyte infiltration into the myocardium of patients who had died from cardiovascular disease (2, 3). They demonstrated a T cell infiltrate in infarct regions and remote myocardium in patients with recent MI (1–12 weeks before death) that was not seen in control cases who had died from noncardiac causes (2). At that stage, the infiltrate was greater in patients whose infarct-related artery had remained occluded (2). They subsequently demonstrated less marked myocardial T cell infiltration in patients who had died suddenly, between 0 and 0.25 days after MI onset, as well as in those who had died at a much later stage several months following MI (3).
However, while these studies do point to a role for T cells during and after MI, they are not able to clarify the temporal evolution of lymphocyte recruitment in the early stages of infarction. Moreover, they include patients treated by different strategies, with and without reperfusion, and in the context of concurrent pathologies, including sepsis (2, 3). Consequently, they provide little insight into the involvement of T cells in MI and I/R injury after successful reperfusion therapy.
Overview of mechanisms of leukocyte recruitment
A marked inflammatory response occurs in the myocardium during myocardial ischemia and directly following reperfusion (Fig. 2). This is triggered initially by necrosis of cardiomyocytes, leading to release of intracellular contents (21). Such cellular debris contains a wide variety of DAMPs, which are recognized by other cell types through ligation of PRRs, including TLRs (25). This leads to the release of various inflammatory mediators, triggering inflammatory cell infiltration. Furthermore, production of ROS by ischemic tissue promotes activation of inflammatory cascades and mediator release (64, 85).

Myocardial gene expression studies in a mouse model of MI with reperfusion revealed rapid upregulation of the inflammatory cytokines TNF-α, IL-1β, and IL-6, which then declined from 6 to 24 h postreperfusion (20). DAMPs liberated by necrotic cells and damaged extracellular matrix also cause activation of the complement cascade, resulting in production of anaphylatoxins, which are potent neutrophil chemoattractants, as well as upregulation of adhesion molecules on endothelial cells (78). Exposure to ROS and complement components triggers degranulation of resident mast cells, causing release of preformed mediators, including histamine and TNF-α (38). This contributes to further upregulation of chemokines and adhesion molecules by endothelial cells (21).
The process of leukocyte extravasation from the bloodstream to the tissues occurs in several stages. First, there is an initial phase of rolling along the endothelial surface, which is mediated by adhesion molecules known as selectins (68). Exposure to immobilized chemokines on the endothelium then contributes to activation of another group of adhesion molecules, called integrins, which mediate firm binding and leukocyte arrest (21). Transmigration through the vascular wall is then directed by exposure to chemokine gradients. The particular milieu of chemokines and adhesion molecules present determines the subsets of leukocytes recruited and varies throughout the process of ischemia and reperfusion (39). Once present in the infarct zone, leukocytes help to sustain inflammation through release of mediators and cytokines, although they also have important roles in healing and resolution of inflammation (38, 82).
Role of TLRs in myocardial I/R injury
Myocardial ischemia leads to the release of danger signals such as DAMPs, which can induce the immune system via PRRs, such as TLRs [for review, see Ref. (105)]. Endogenous ligands can also include ROS or heat shock proteins. ROS, for example, has been shown to induce cytokine production through TLR4 (36). In humans, 10 TLRs have been described so far, and 6 in mice. TLRs are transmembrane receptors with an extracellular domain to recognize distinct molecular patterns and an intracytoplasmic tail that resembles the interleukin-1 receptor for inward signaling. TLR signaling occurs via two major pathways, which involve either of the adaptor proteins—MyD88 or TRIF (TIR domain-containing adaptor protein inducing type 1 interferons), ultimately leading to NF-κB activation or induction of type 1 interferons downstream.
Most literature for experimental infarct models involves TLR4, and genetic knockdown of myocardial TLR4 has been proven to reduce myocardial infarct size following I/R (89, 101). Murine cardiomyocytes express several TLRs, including TLR-2, -3, and -4. Ischemia also leads to activation of endothelium, adhesion molecules, and inflammatory chemokines such as CX3CL1 and CXCL10, which might be triggered by TLRs (74). As outlined above, CD4+ T cells have been implicated in I/R injury of several organ systems (heart, liver, gut, lung, kidney, and brain), and CXCR3+CD4+ T cells promote I/R injury in the liver, utilizing the CXCR3 ligand CXCL10 (INF-gamma-inducible protein 10) (119, 120).
Chemokines in leukocyte recruitment
The chemokines are a group of small proteins that play a crucial role in leukocyte migration in health and disease. They do this through interaction with receptors found on the surface of leukocytes, directing movement of the cell toward an increasing chemokine concentration gradient. A total of over 50 chemokines have been identified, which can be divided into four groups based on the number and position of the cysteine residues in the N-terminal region (Fig. 3) (66). The two main groups are CC (e.g., CCL1) chemokines, which have two adjacent N-terminal cysteines, and CXC chemokines (e.g., CXCL1), which have two cysteines separated by another amino acid (66).
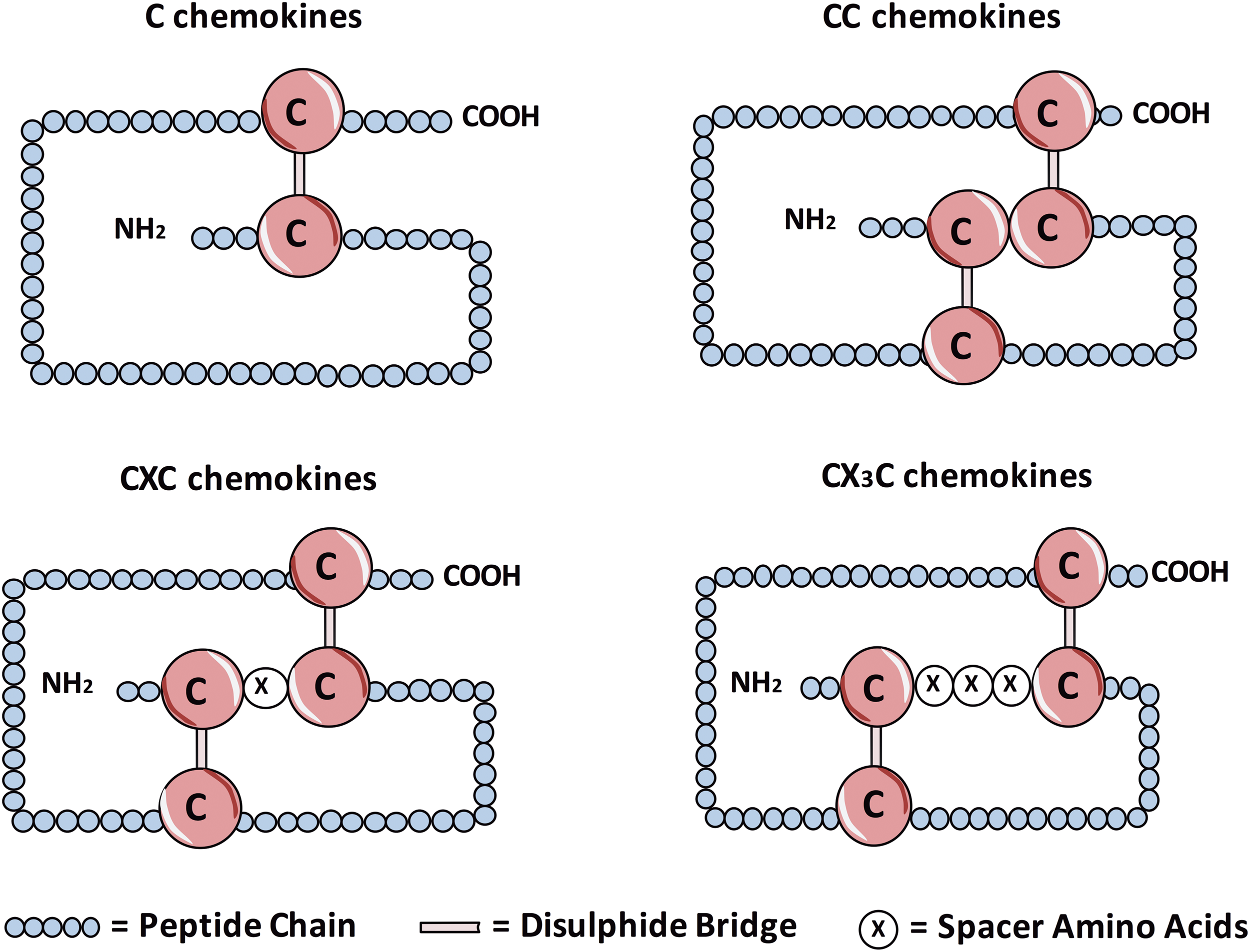
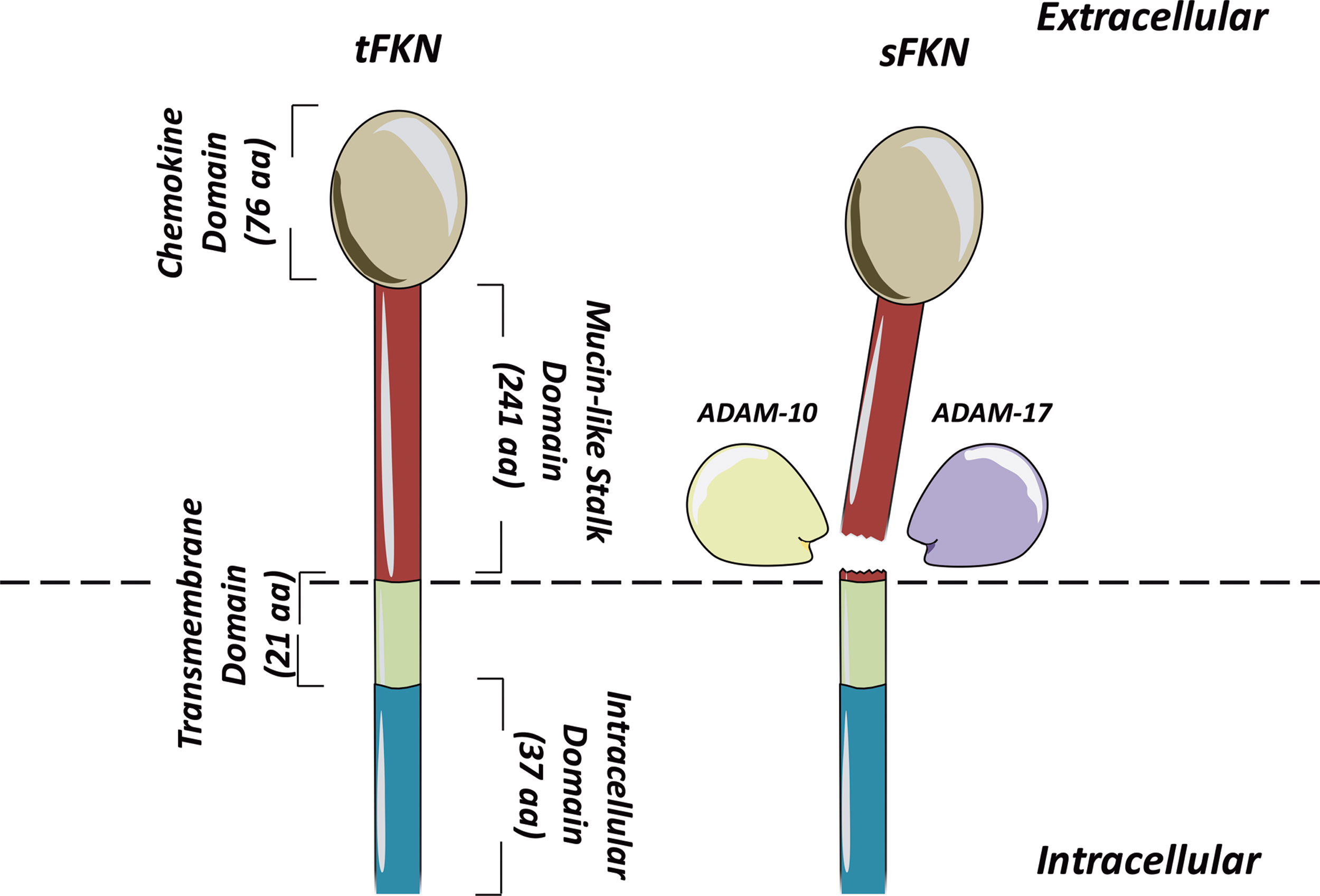
The remaining groups are C chemokines, which uniquely contain only two cysteines in total (one in the N-terminal region, one downstream), and CX3C chemokines, which have two N-terminal cysteines separated by three amino acids. The only known example of the latter group is fractalkine (CX3CL1), which exists both as a secreted and membrane-bound form (Fig. 4) (53). Each chemokine is able to bind to one or more receptor, all of which share a characteristic structure consisting of seven transmembrane domains, with most coupled to a G-protein through which they signal (6, 80). In addition to influencing migration, chemokines can have a number of other effects on cells, including activation and altered gene expression (39). Moreover, as well as their effects on leukocytes, chemokines are able to influence the function of nonimmune cells, including endothelial cells and fibroblasts (39).
The majority of research investigating chemokines in MI and reperfusion has focused on recruitment of innate immune cells, including monocytes and neutrophils. This has been reviewed extensively elsewhere (37, 38, 70). In contrast, relatively little is known about the mechanisms of lymphocyte recruitment in myocardial ischemia and reperfusion (Fig. 5).
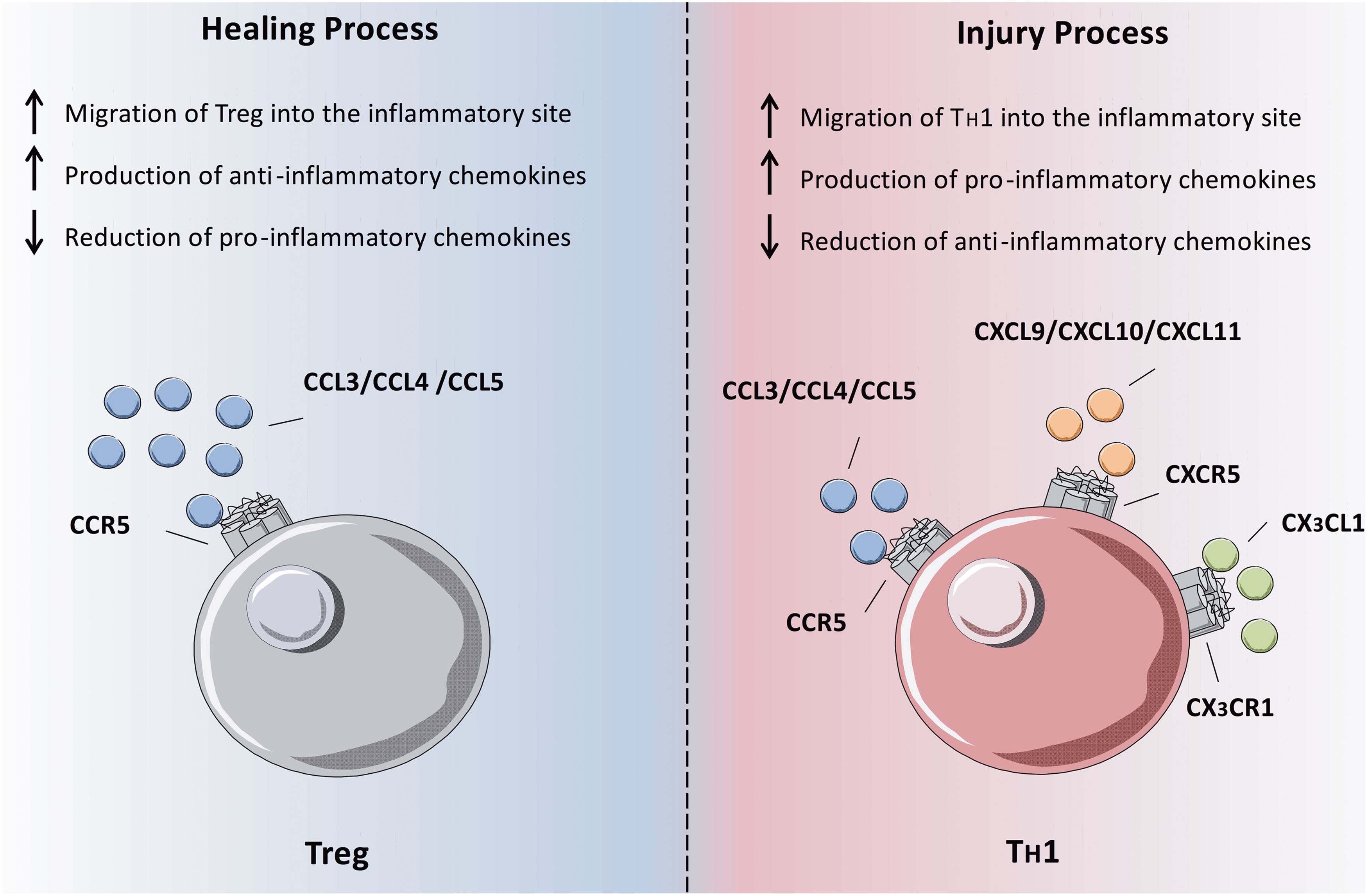
One group of T cells thought to contribute to infarct healing are T
It is worth noting that while CX3CR1 is found on highly differentiated effector T cell subsets in humans, expression on murine T cells is negligible (6, 58), limiting the use of mouse models when studying this receptor in lymphocytes. Our own studies in human STEMI patients undergoing PPCI, however, have shown clear relationship between loss of T cell subsets from the peripheral circulation in the early time period following reperfusion and their expression of CX3CR1 (8). Transcoronary gradient analysis suggested that at least some of these lost cells were sequestered into the reperfused myocardium in the early postreperfusion period. Consequently, we propose that CX3CL1, acting through its receptor CX3CR1, is a key chemokine involved in the recruitment of effector T cells following myocardial reperfusion, where they then contribute to I/R injury.
Role of adenosine signaling in T lymphocytes
One important signaling molecule in the context of I/R injury is the nucleoside adenosine. This molecule is produced through ATP breakdown and its production is dramatically increased following I/R. While its concentration is maintained closely during normal conditions, levels can increase up to 100-fold following cellular injury (14). It exerts a number of important effects through action on a group of four receptors: A1, A2A, A2B, and A3. In general, its actions include vasodilatation, reduced myocardial contractility and heart rate, as well as anti-inflammatory effects (51). It has a variety of effects depending on the concentration and receptors engaged. For example, neutrophils can express all four types of adenosine receptor, and stimulation of A1 receptors enhances neutrophil chemotaxis, inflammatory cytokine release, and ROS production (23), while A2A stimulation reduced ROS production (121) and A2B inhibits chemotaxis (14).
The principal adenosine receptor found on lymphocytes is the A2A receptor (72). One of the main effects of stimulation of these receptors on lymphocytes is the modulation of T cell cytokine production. While T
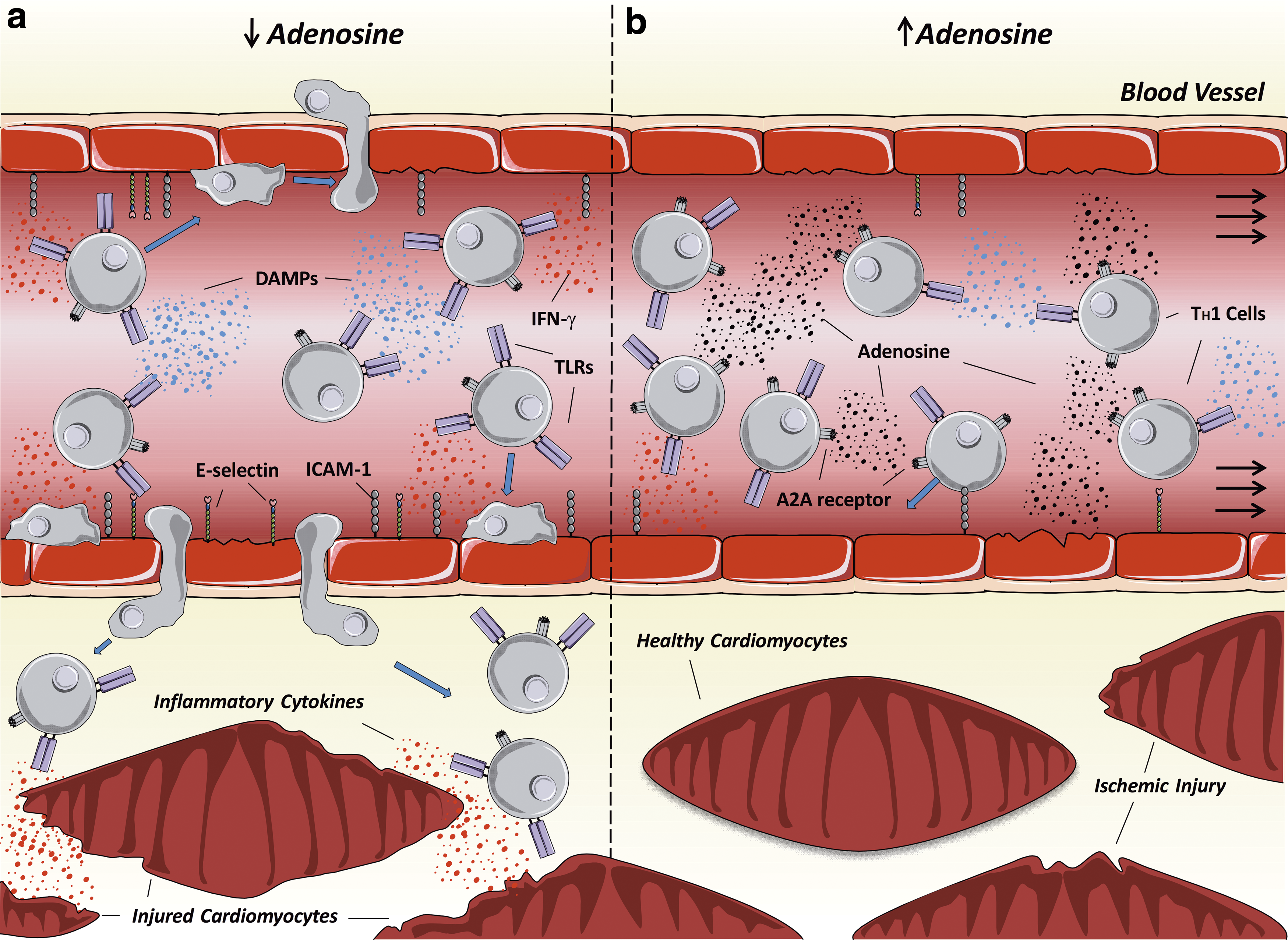
Therapeutic Implications of Lymphocytes in Myocardial I/R Injury
In spite of the considerable potential of myocardial I/R injury as a therapeutic target, successful treatment remains elusive. The failure to date to establish effective treatment strategies is in spite of extensive research and numerous clinical trials. Pharmacological strategies have included the use of anti-inflammatory agents, including pexelizumab, a monoclonal antibody to the C5 complement component (5), and CD11/CD18 integrin receptor blockers (33), as well as antioxidants, aiming to reduce oxidative stress (17, 32, 35). Unfortunately, as has been extensively reviewed elsewhere (28, 45, 99), the outcomes of these treatments have been generally disappointing and none have reached routine clinical use. However, previously attempted strategies to treat myocardial I/R injury have generally not targeted the adaptive immune system, and lymphocytes in particular, which are only recently emerging as candidates for an important role in this process.
One exception to this has been the use of the cyclosporin, which has immunosuppressive effects through inhibition of T cell activation. The original rationale for use of this drug in targeting I/R injury was actually inhibition of opening of the MPTP (91), a key metabolic event in I/R injury, but given its profound immunological effects, this mechanism could also be behind any ultimate outcomes.
An initial small pilot RCT of 58 STEMI patients randomized to receive an intravenous bolus of cyclosporin or normal saline placebo before PPCI found a reduction in infarct size with cyclosporin (91). Since then, a further small trial reported reduced adverse LV remodeling with administration of cyclosporin (77). However, a similar sized study investigating the use of this drug along with thrombolytic therapy failed to find any benefit (41). Unfortunately, a recent large clinical trial involving almost 800 patients has failed to show any improvement in clinical outcome measures at 1 year with cyclosporin treatment before PPCI (24).
While this recent result has been another disappointing development in the search of an effective treatment for myocardial I/R injury, one explanation could be the lack of an adequately targeted therapy. As outlined above, the effects of T cells in the ischemic and reperfused myocardium are complex and highly subset specific. Moreover, our own data suggest a pathological role for highly differentiated T cell subsets in the early postreperfusion period (8), while other studies in mice indicate a protective role for T cells at later stages in myocardial healing, reducing adverse remodeling and subsequent LV dysfunction (46, 103, 108). Consequently, it seems likely that successful therapy would be required to act on the correct cellular subsets at the relevant time, while sparing other T cell subsets to allow them to carry out important protective functions. As such, as our understanding of this important evolving field improves, better-targeted therapies can be developed, which will hopefully have a positive outcome in this area of great therapeutic potential.
Conclusions
The role of lymphocytes in myocardial ischemia and reperfusion is only recently being recognized and explored. In this review, we have summarized current understanding of this complex area, which as yet remains relatively poorly understood. It is clear that different lymphocyte subsets have diverse subset-specific and time-specific effects in these processes. It may well be that the diversity of cellular function in this area explains the lack of success with attempted therapeutic interventions to date. Consequently, further research in this area is essential to develop better targeted therapies able to exploit this area of great untapped therapeutic potential.
Important areas for further study include the detailed mechanisms behind recruitment of the different lymphocyte subsets involved in myocardial injury and healing. Moreover, further translation work in human patients, building on our own recent findings, can place the mechanistic data from animal models in context in the real-world scenario of human STEMI patients treated by PPCI. A better understanding of disease mechanisms may allow development of highly targeted therapies as well as more logical application of existing drugs, utilizing potentially narrow therapeutic windows based on the involvement of particular cellular subsets at specific times. This, therefore, remains an extremely important area of future research.
Footnotes
Author Disclosure Statement
E.A. participates in a postgraduate studentship program at GSK Vaccines, Siena (IT). Other authors have no competing financial interests.