
Abstract
Significance:
Hypertension is the leading risk factor causing mortality and morbidity worldwide. Angiotensin (Ang) II, the most active metabolite of the renin–angiotensin system, plays an outstanding role in the pathogenesis of hypertension and vascular injury. Activation of angiotensin converting enzyme 2 (ACE2) has shown to attenuate devastating effects of Ang II in the cardiovascular system by reducing Ang II degradation and increasing Ang-(1–7) generation leading to Mas receptor activation.
Recent Advances:
Activation of the ACE2/Ang-(1–7)/Mas receptor axis reduces hypertension and improves vascular injury mainly through an increased nitric oxide (NO) bioavailability and decreased reactive oxygen species production. Recent studies reported that shedding of the enzymatically active ectodomain of ACE2 from the cell surface seems to regulate its activity and serves as an interorgan communicator in cardiovascular disease. In addition, collectrin, an ACE2 homolog with no catalytic activity, regulates blood pressure through an NO-dependent mechanism.
Critical Issues:
Large body of experimental data confirmed sustained beneficial effects of ACE2/Ang-(1–7)/Mas receptor axis activation on hypertension and vascular injury. Experimental studies also suggest that activation of collectrin might be beneficial in hypertension and endothelial dysfunction. Their role in clinical hypertension is unclear as selective and reliable activators of both axes are not yet available.
Future Directions:
This review will highlight the results of recent research progress that illustrate the role of both ACE and collectrin in the modulation of NO and oxidative stress in blood pressure homeostasis and vascular injury, providing evidence for the potential therapeutic application of ACE2 and collectrin in hypertension and vascular disease. Antioxid. Redox Signal. 26, 645–659.
Introduction
H
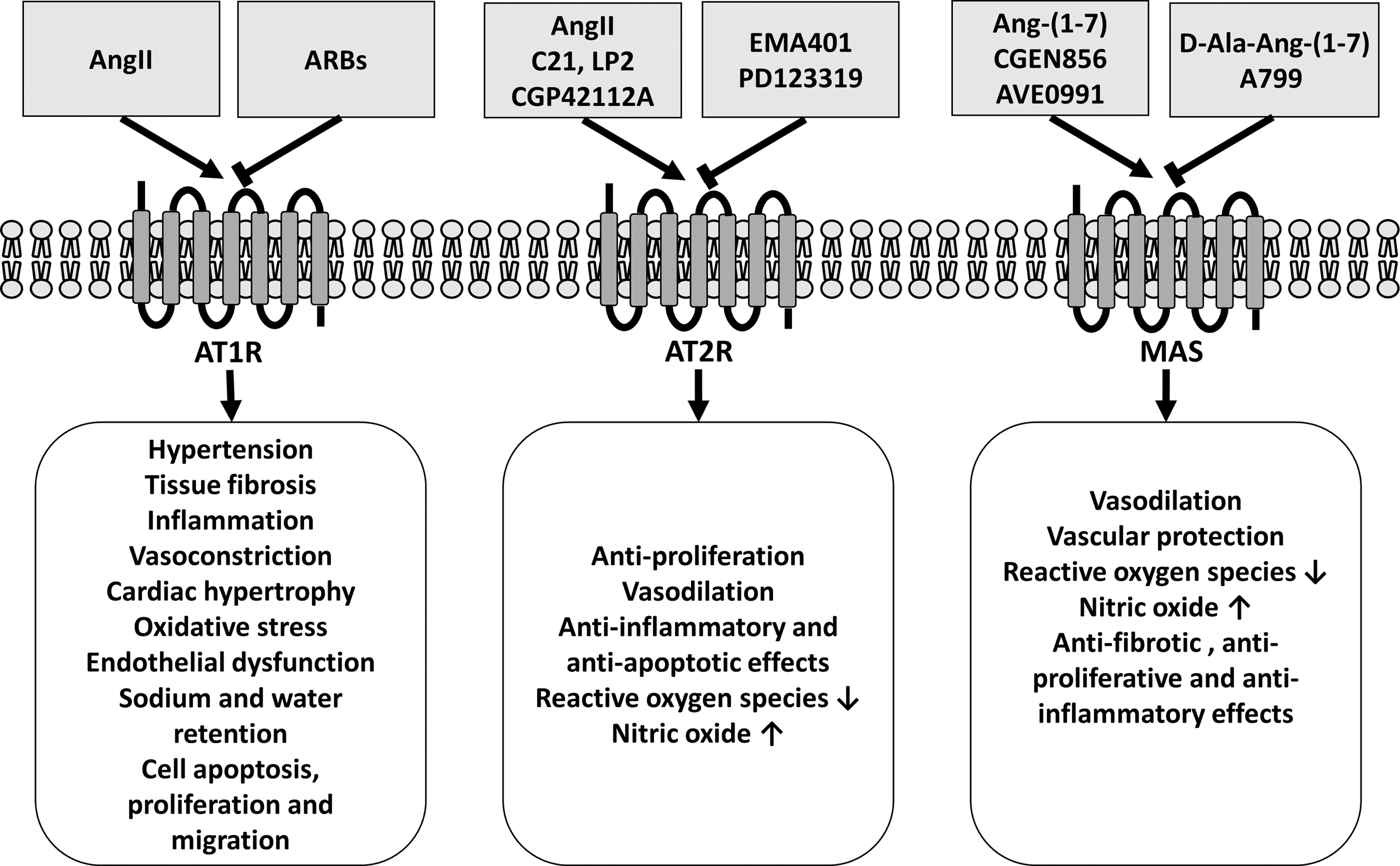
Despite an effective inhibition of this pathway by AT1 receptor blockers or angiotensin converting enzyme (ACE) inhibitors, control rates of hypertension and end organ damage are still not satisfactory (53). One reason for this improvable condition might be explained by a potential downregulation of the alternative RAS in the development of these pathologies. This alternative pathway includes the ACE2/Ang-(1 –7)/Mas receptor axis and activation of the AT2 receptor (Figs. 1 and 2), which have been shown to antagonize the devastating effects of Ang II-mediated AT1 receptor activation.
In the present review, we focus mainly on the ACE2/Ang-(1 –7)/Mas receptor axis and its role on blood pressure homeostasis and vascular function in cardiovascular diseases. Thereby, we will describe the underlying signaling pathways with the main focus on NO bioavailability and oxidative stress. Moreover, we will highlight the more recently discovered role of collectrin, a homolog of ACE2 that shares 50% of its amino acid sequence, in blood pressure and vascular function.
The ACE2/Ang-(1 –7)/Mas Receptor Axis
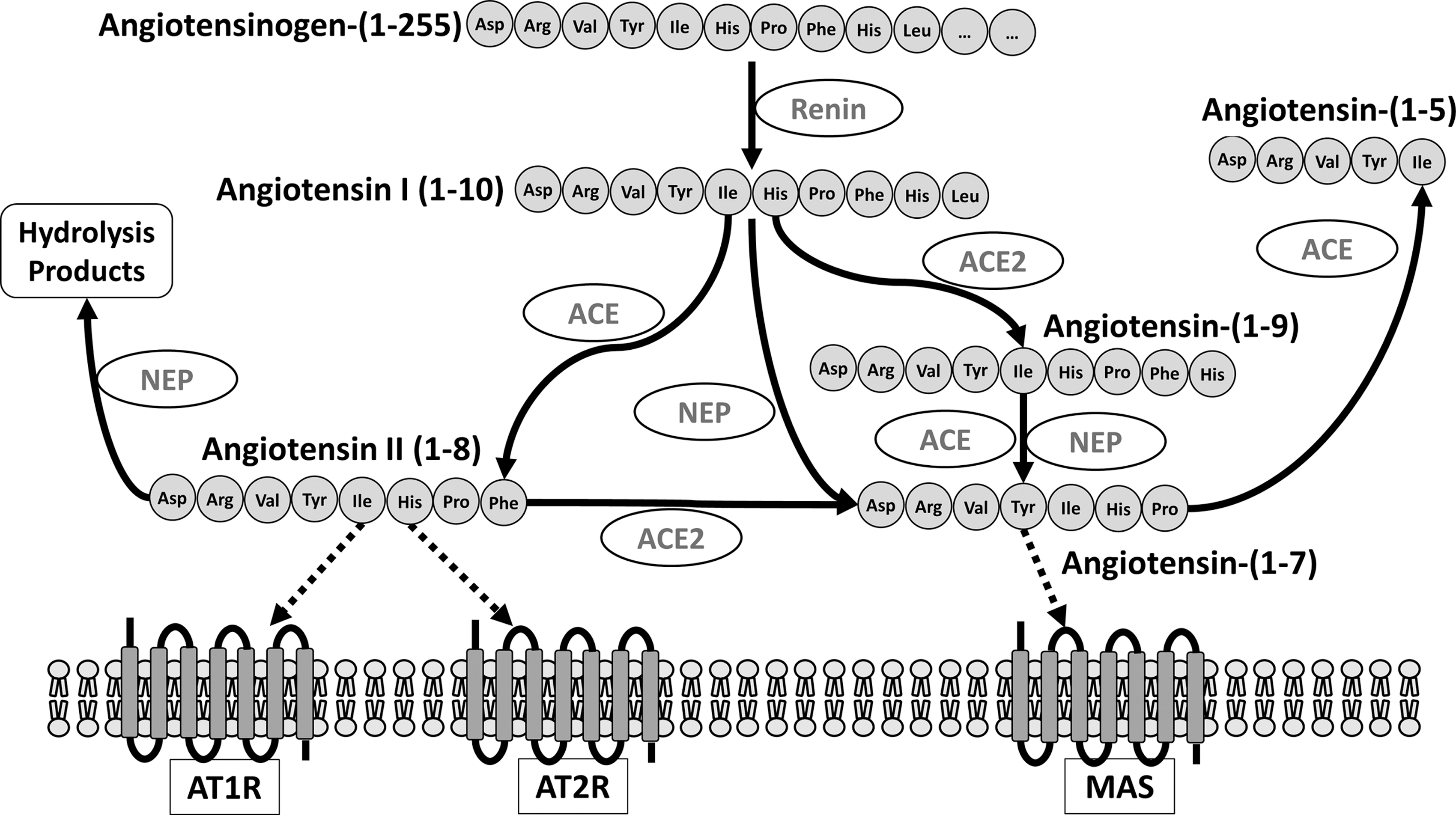
The classical RAS, with Ang II as its most active metabolite, has been studied for more than 100 years. The octapeptide Ang II formed by the removal of two amino acids from the carboxyterminal of Ang I (Fig. 1) activates two different types of receptors, the AT1 and AT2 receptors. Genetic and pharmacological intervention studies have shown that Ang II mediates most of its effects through the activation of the AT1 receptor leading to hypertension, vascular injury, and heart and kidney failure (Fig. 2) (22, 50, 113, 114, 116, 149).
Beside the classical RAS, recent studies provide convincing evidence for an alternative pathway with an additional major impact on cardiovascular physiology and pathophysiology. In this pathway, the mono-carboxypeptidase ACE2, which comprises 42% amino acid homology to the catalytic domain of ACE, is the limiting factor in the degradation rate of Ang I and Ang II. ACE2 metabolizes both Ang I into the nona-peptide Ang-(1 –9) [which is further converted to the hepta-peptide Ang-(1 –7)] and Ang II into the hepta-petide Ang-(1 –7) (Fig. 1) (33, 83, 124). Notably, the efficacy of ACE2 to convert Ang II to Ang-(1 –7) is ∼400 times greater than for Ang I to Ang-(1 –9) (125, 130).
Ang I and Ang II are not the only catalytic substrates for ACE2. For example, ACE2 also cleaves the vasoactive peptide apelin (132). In the kidney, ACE2 is mainly localized in the renal vasculature, the podocytes, and the apical membrane of the epithelial cells in the proximal tubules, where it colocalizes with ACE. Beside its expression in the endothelium and vascular smooth muscle cells of the vasculature, ACE2 is also expressed in the intestine, lung, liver, and central nervous system (CNS) (34, 69, 89).
ACE2 consists of an N-terminal extracellular catalytic domain, a transmembrane segment, and a short C-terminal intracellular tail (33) and is subject to cleavage by several metalloproteinases such as ADAM metallopeptidase domain 17 (ADAM17). ADAM17 cleaves the enzymatically active ectodomain of the extracellular domain from the cell surface to generate a soluble active form of ACE2 (Fig. 3). In the kidney, the ACE2 ectodomain can be detected in the urine. Under high glucose concentrations, urinary levels of the shed ACE2 ectodomain increase and seem to correlate with the progression of diabetic kidney disease (131).

Based on its well-documented capability to cleave Ang I and Ang II, it is not surprising that ACE2 is critically important in regulating Ang II concentration levels in plasma and tissues (47). Indeed, studies in ACE2 knockout (KO) mice have shown more severe Ang II-dependent hypertension, atherosclerosis, abdominal aortic aneurysms, heart failure, and renal kidney damage compared with control mice (2, 21, 46, 88).
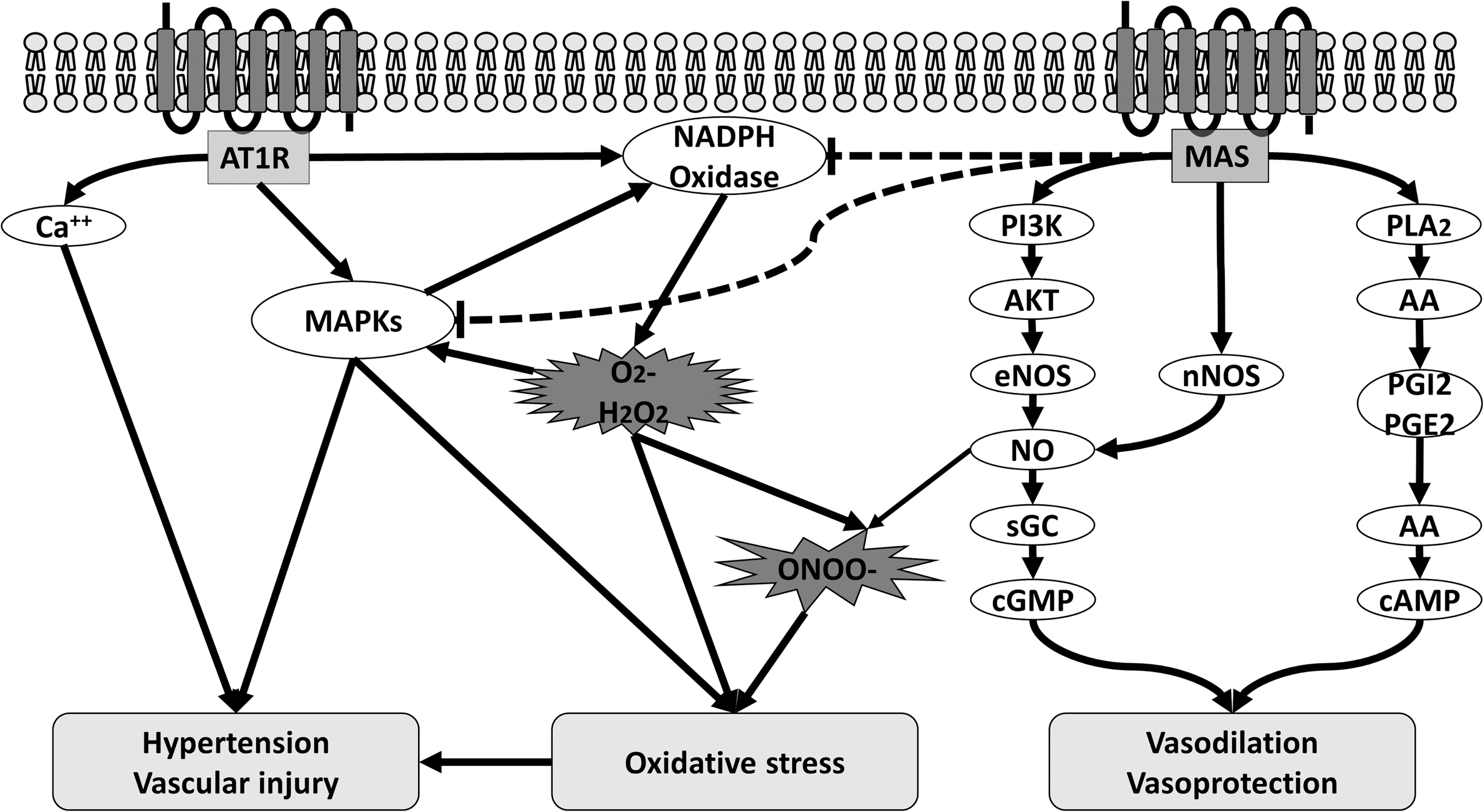
In addition to increased Ang II levels, ACE2 deficiency is also characterized by a reduced production of the hepta-peptide Ang-(1 –7), which mediates its vasoprotective and antioxidative effects via its G protein-coupled receptor Mas (4, 104) (Fig. 2). In this regard, it is not surprising that most of the known signaling pathways of the Mas receptor are involved in the regulation of NO bioavailability, for example, phospholipase A (PLA) generates arachidonic acid (AA) and phosphoinositide 3 kinase (PI3K) and AKT activates endothelial nitric oxide synthase (eNOS) by phosphorylation at serine 1177 and dephosphorylation at threonine 495 (4) (Fig. 4). Thus, Ang-(1 –7)-mediated Mas receptor activation has been shown to ameliorate endothelial dysfunction, atherosclerosis, diabetic nephropathy, and heart failure.
In addition to its function as a mono-carboxypeptidase cleaving Ang I and Ang II to Ang-(1 –9) or Ang-(1 –7), respectively, ACE2 has been shown to serve as a receptor for the SARS-coronavirus (69) and to regulate intestinal immunity by interacting with certain amino acid transporters (26). In this review, we will focus on the role of ACE2 in the RAS and its prominent function as the rate-limiting enzyme in Ang II degradation and Ang-(1 –7)/Mas receptor axis activation.
Ang II, Oxidative Stress, and NO
To understand the protective mechanism of the ACE2/Ang-(1 –7)/Mas axis in the cardiovascular system, it is important to understand the underlying mechanism how Ang II mediates hypertension and vascular injury. Ang II-induced AT1 receptor activation leads to a coupling of G proteins (Gq/11 and/or Gi/o) to the C terminal of the receptor and thereby to stimulation or modification of a variety of different intracellular signaling pathways (56). There is compelling evidence that Ang II-induced hypertension and vascular injury are mediated by decreased NO bioavailability and increased oxidative stress (31, 113, 154).
Oxidative stress reflects a condition of increased reactive oxygen species (ROS) production and reduced antioxidant capacity and is a common result of many Ang II-induced signaling pathways in the vasculature such as phospholipase C, phospholipase A2, Ca2+ channels, RhoA/Rho kinase, mitogen-activated protein kinases (MAPK), and Janus-activated kinase-2-signal transducers and activators of transcription (JAK2-STAT) pathways (67, 91, 107). Vascular ROS are generated in all vascular layers mainly by the NADPH oxidases (NOXs), which generally consist of five subunits (p47phox, p67phox, p40phox, p22phox, and gp91phox) and the small G proteins Rac 1 and Rac 2.
In Ang II-induced hypertension and vascular injury, ROS, generated by activation of NOXs via AT1 receptor, are not only involved in the degradation of NO but also act as intracellular signaling molecules activating the MAPK, protein phosphatases, and tyrosine kinases, which are involved in vasoconstriction, fibrosis, migration, and inflammation (86, 103, 134). In addition to the effects of Ang II on ROS production, we and others have shown that Ang II can also stimulate the cGMP degrading phosphodiesterases 1 and 5 leading to reduced cGMP bioavailability and thereby to vascular dysfunction and injury (85, 117).
While the deleterious effects of Ang II are mediated by activation of AT1 receptor, its beneficial effects seemed to be mediated through the AT2 receptor. Despite similar Ang II ligand binding affinity for both receptors (18), differential gene and protein expression levels of AT1 receptor and AT2 receptor may explain the RAS-dependent cardiovascular diseases (100).
Numerous studies have demonstrated that activation of AT2 receptor can antagonize AT1 receptor-mediated actions, including hypertension, inflammation, and vascular remodeling (Figs. 1 and 2) (61, 65). Genetic deletion of AT2 receptor exaggerated atherosclerosis and nephropathy via heightened oxidative stress (15, 58). Thus, the protective effects of AT2 can be explained by decrease in oxidative stress, suppression of inflammatory responses, downregulation of ACE/ACE2 ratio, increase in vascular NO levels, and reduction in vascular cell proliferation and migration (24, 70, 92, 153). AT2 receptor can upregulate eNOS levels, but endothelial NO also can in turn increase AT2 receptor expression levels in a positive feedback loop (27, 52).
ACE2 in Hypertension and Vascular Injury
In 2002, Crackower et al. first observed that ACE2 mRNA expression levels and protein expression levels were significantly reduced in three different rat models of hypertension (21). ACE2 is highly expressed in the kidney, CNS, vasculature, and heart, suggesting that it might play an important role in the regulation of blood pressure homeostasis, as well as renal and cardiovascular function (33, 130) (Fig. 6).
Under baseline conditions, disruption of ACE2 did not significantly alter plasma Ang II and blood pressure levels in mice. However, during Ang II infusion, plasma and kidney Ang II levels were significantly increased in ACE2-deficient mice and this was accompanied by an accentuated Ang II-dependent hypertension, compared to control mice. In addition, Mas receptor deficiency caused increased blood pressure in mice on an FVB background (140). These observations suggest that blood pressure regulation by ACE2 not only involves its degradation of Ang II but also involves its generation of Ang-(1 –7) leading to the activation of the Mas receptor.
ACE2 in the kidney
During chronic Ang II infusion, ACE2 deficiency is characterized by increased Ang II levels in the kidney. Recent studies have shown that Ang II-mediated AT1 receptor activation within the kidney plays a critical role in the development of hypertension and cardiovascular complications (16, 22, 49, 72, 114). In particular, deletion of the AT1 receptor from renal vascular smooth muscle cells or renal proximal tubular cells but not from principal cells of the collecting duct leads to increased natriuresis and attenuated blood pressure response during chronic Ang II infusion. In this regard, Wysocki et al. provide evidence that ACE2 deficiency leads to an increased renal NOX activity, which can be inhibited by an AT1 receptor blocker (134).
Similarly, in hypercholesterolemic apoE-KO mice, ACE2 deficiency causes an increase of renal superoxide generation, NOX 4 expression, and proinflammatory cytokine production, such as interleukin (IL)-6, tumor necrosis factor (TNF)-α, and IL-1β, leading to a discrete blood pressure elevation and renal impairment compared with apoE-KO mice. Recently, Zhang et al. could demonstrate that IL-1 receptor activation drives sodium retention in the thick ascending limb of Ang II-infused mice by depleting NO (148).
Recombinant ACE2 administration reverses renal superoxide production, inflammation, and blood pressure of apoE/ACE2 double KO mice most likely by reducing Ang II- and increasing Ang-(1 –7) levels leading to Mas receptor activation (59, 75). In addition to the effects seen in ACE2-KO mice, Mas receptor deficiency is characterized by elevated blood pressure levels and increased ROS production. Vice versa, Ang-(1 –7) infusion improves renal endothelial dysfunction by increasing NO-dependent cGMP generation and decreasing hydrogen peroxide production as well as gp91phox and p47phox expression (118).
The mechanism how both ACE2 and the Mas receptor regulate blood pressure by influencing NO generation and ROS production in the kidney is not fully understood. Recently, our group showed that activation of p38 MAPK exaggerates Ang II-dependent vasoconstriction in the kidneys of apoE-KO mice. Ang-(1 –7)-induced Mas receptor activation reduced renal p47phox expression and p38 MAPK activity and thereby the exaggerated renal pressor response to Ang II (95). Moreover, Benter et al. demonstrated a natriuretic and blood pressure decreasing effect of Ang-(1 –7) in spontaneous hypertensive rats (7). A possible explanation for this observation might be the well-documented impact of Ang-(1 –7) on renal NO availability and ROS production affecting sodium handling along the nephron (10, 43, 85) (Fig. 5).
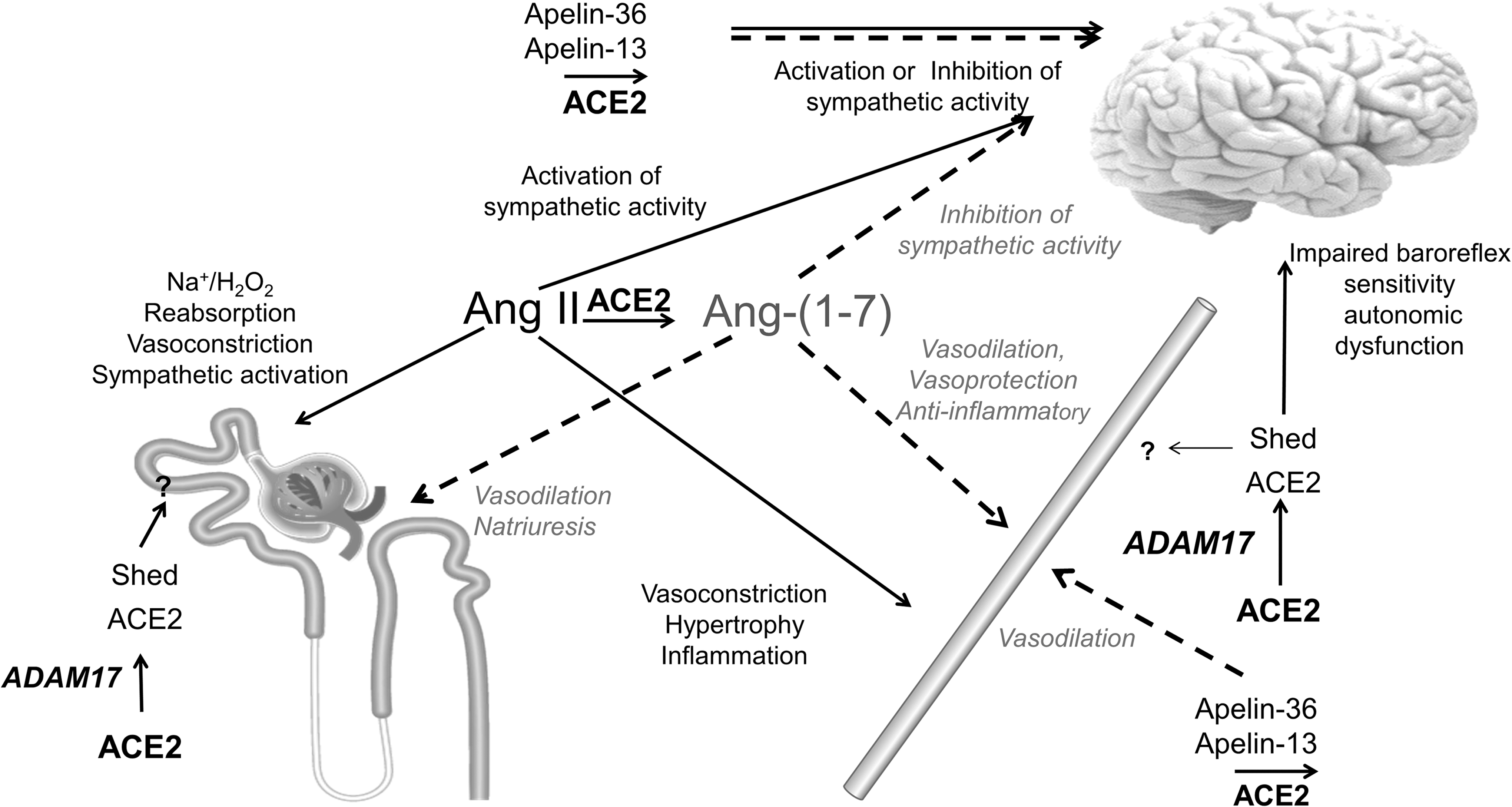
Recent studies report an increased shedding of the active soluble form of ACE2 into the tubular lumen of the nephron under conditions of high glucose, Ang II levels, or increased ADAM17 activity. Ang II-stimulated shedding of the ectodomain ACE2 from renal proximal tubular cells is reduced by ADAM17 inhibition (137). The pathophysiological relevance of ACE2 shedding still remains unknown. One can speculate that the shedding of the ACE2 ectodomain into the proximal lumen of the nephron might regulate the intrarenal RAS in hypertension (137).
ACE2 in the vasculature
Vascular dysfunction is a result of diminished release and function of endothelium-derived NO and increased ROS production. There is large body of evidence that activation of the ACE2/Ang-(1 –7)/Mas receptor axis reduces blood pressure and promotes vasoprotective effects. For example, overexpression of vascular ACE2 in spontaneously hypertensive rats results in increased Ang-(1 –7) levels, a reduction of blood pressure, and restored endothelial function (98). In other studies, both ACE2 and Mas receptor activation improved endothelial dysfunction and slowed the progression of atherosclerosis by several mechanisms in different cardiovascular disease models (41, 122).
First, Ang-(1 –7)-mediated Mas receptor activation has shown to increase NO bioavailability by increasing vascular eNOS expression and activity through a PI3K/AKT pathway (101, 102, 138). In contrast, ACE2 deficiency is characterized by reduced NO bioavailability mediated by decreased aortic eNOS expression (96).
Second, the vasoprotective effects of Ang-(1 –7) are not only related to increased NO production but also to maintaining the NO component of vasodilation. In this regard, Durand et al. have shown that telomerase is essential for the vasoprotective effects of Ang-(1 –7) (35). Telomerase plays not only a critical role in senescence as it regulates the telomere length in the nucleus but also conducts a non-nuclear function where it protects against mitochondrial dysfunction and ultimately improves NO-mediated vasodilation (8, 51). In the respective study, Ang-(1 –7) improved endothelial-dependent vasodilation in microvessels from patients with coronary artery disease. In contrast, the telomerase inhibitor BIBR-1532 abolished the vasoprotective effect of Ang-(1 –7) in those mircovessels, suggesting a critical contribution of telomerase to the vasoprotective effects of Ang-(1 –7).
Third, activation of the ACE2/Ang-(1 –7)/Mas receptor axis reduces ROS production by decreasing the expression level of gp91phox and p47phox, and increasing the catalase activity (95, 118, 139, 140). Vice versa, ACE2 or Mas receptor deficiency caused an increase in lipid peroxidation and NOX activity as well as increased superoxide and peroxynitrite production and reduced superoxide dismutase activity resulting in increased NO degradation and therefore endothelial dysfunction, which was aggravated during Ang II-dependent hypertension (60, 96, 97, 140).
Beside the beneficial effect of the ACE2/Ang-(1 –7)/Mas receptor axis on endothelial function, chronic Ang-(1 –7) treatment also attenuated the Ang II-induced pressor response in apoE-deficient mice through a p47phox/p38 MAKP-mediated pathway. Ang-(1 –7)-mediated inhibition of p38 MAPK reduced not only Ang II-induced vasoconstriction but also attenuated Ang II-induced vascular inflammation and the expression of the adhesion proteins intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion protein-1 (VCAM-1) as well as the proinflammatory cytokines CCL2 and IL-6, hence inhibiting leukocyte recruitment and adhesion to endothelial cells (9, 73).
Thus, Ang-(1 –7) infusion reduces the progression of atherosclerosis by influencing immune cell function and by improving vascular function (122, 144, 151). In addition, ACE2-depleted immune cells aggravate the development of atherosclerosis in LDL receptor-KO mice. Ang-(1 –7) infusion reduces atherosclerosis in ACE2-deficient LDL receptor KO mice, suggesting that ACE2 mediates most of its vasoprotective effects via the ACE2/Ang-(1 –7)/Mas receptor pathway (123).
Beside Ang I and Ang II, there are additional vasoactive active peptides that are catalyzed by the mono-carboxypeptidase ACE2. Thus, ACE2 cleaves the vasoactive polypeptides apelin-36, apelin-17 and apelin-13 on the C-terminal amino acid with a similar catalytic efficiency as Ang II. The cleavage product apelin-12 does not seem to have any cardiovascular effect. Apelin binds to its receptor AJP, a G protein-coupled receptor that displays significant homology with the AT1 receptor (120). Infusion of apelin-13 reduces blood pressure through an NO-mediated mechanism in rats (71, 121) (Fig. 6). Apelin-deficient mice are characterized by heart dysfunction and hypertrophy as well as a downregulation of ACE2. Interestingly, AT1 receptor inhibition or Ang-(1 –7)-induced Mas receptor activation significantly improves heart function in these mice, suggesting a close relationship between the RAS and apelin (105).
ACE2 in the CNS
ACE2 is widely expressed in the CNS, particularly in regions controlling cardiovascular function such as the subfornical organ (SFO), the nucleus tractus solitarii (NTS), the paraventricular nucleus (PVN), nucleus ambiguous, and the rostral ventrolateral medulla (34). In the CNS, several studies have shown that ACE2 expression levels are decreased in genetically modified hypertensive rats or in Ang II and DOCA salt-dependent hypertension (21, 135, 143). Overexpression of ACE2 in one of these regions results in a sustained blood pressure reduction, a decreased release of proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β, as well as reduced oxidative stress (38, 39, 115, 136). The mechanism by which ACE2 in the CNS regulates blood pressure remains unclear.
Recently, Feng et al. have shown that the beneficial effects of ACE2 overexpression in the brain are mediated through the Ang-(1 –7)/Mas axis as D-Ala-Ang-(1 –7), a selective inhibitor of the Mas receptor, abrogates these effects. Interestingly, ACE2 overexpression was associated with an increase in eNOS expression and NO levels, suggesting a role of NO in this pathway (39). Indeed, acute injections of Ang-(1 –7) into the NTS of mice caused a sustained reduction in blood pressure and sympathetic nerve activity via a PI3K/AKT pathway resulting in increased eNOS activity and finally in elevated NO production (133).
In contrast to the kidney, the role of the shed active ectodomain ACE2 on blood pressure regulation in the brain is more definitive. In a recent study, it was shown that DOCA salt-dependent hypertension increases ADAM17 levels resulting in increased levels of shed ACE2. Knockdown of ADAM17 prevented ACE2 shedding and decreased blood pressure reduction in DOCA salt hypertension, suggesting that shed ACE2 impairs the compensatory ACE2 activity in the brain leading to the development of hypertension (135).
Similar to its action in the vasculature, apelin also mediates cardiovascular effects when administrated in the CNS. Depending on the region in the brain, apelin-13 injection induces either blood pressure reduction or blood pressure increase (23, 147). Apelin-13 injected in the SFO decreased blood pressure by influencing the excitability of neurons in this region (Fig. 6). In contrast, apelin-13 administration in the PVN increased blood pressure via sympathetic nerve activation. Interestingly, overexpression of ACE2 in the same region attenuates Ang II-dependent hypertension and decreases the generation of proinflammatory cytokines such as IL-6, IL-1β, and TNF-α (115).
ACE2 in the heart
Hypertensive heart disease is one of the most common end organ damages leading to disability or death. Recent studies highlighted the role of ACE2, which is expressed in cardiomyocytes, fibroblasts, and endothelial cells, in the development of heart failure due to hypertension. Thus, loss of ACE2 resulted in increased cardiac Ang II levels leading to increased cardiac oxidative stress generation, inflammation, and matrix metalloproteinase activation through a MAPK-dependent mechanism (93, 142). Therefore, it is not surprising that ACE2 deficiency caused cardiomyopathy in aged ACE2-deficient mice.
In contrast, overexpression or administration of ACE2 reduced hypertensive heart failure by reducing superoxide production, cardiac fibrosis, and cardiac hypertrophy thereby improving heart function (30, 57, 152). Beside these effects, recent studies have shown that direct Ang-(1 –7)-mediated Mas receptor activation improved or prevented heart failure in experimental hypertension (28, 84). Thus, Mercure et al. could nicely demonstrate that cardiac-specific overexpression of Ang-(1 –7) protected from Ang II-dependent cardiac hypertrophy and fibrosis by reducing p38 MAPK and c-SRC activation as well as increasing SHP-2 protein tyrosine phosphatase activity (84).
Beside the direct influence of ACE2 on cardiac Ang II levels, the underlying pathomechanism how the ACE2/Ang-(1 –7)/Mas receptor axis mediates the beneficial effects in hypertensive heart failure is still not fully understood. Recently, de Almeida et al. have shown that Ang-(1 –7) improved cardiac dysfunction blood pressure independently (28). Application of ACE2 reduced Ang II-mediated oxidative stress and collagen production in cardiomyocytes and cardiofibroblasts through an Ang-(1 –7)-dependent mechanism (152).
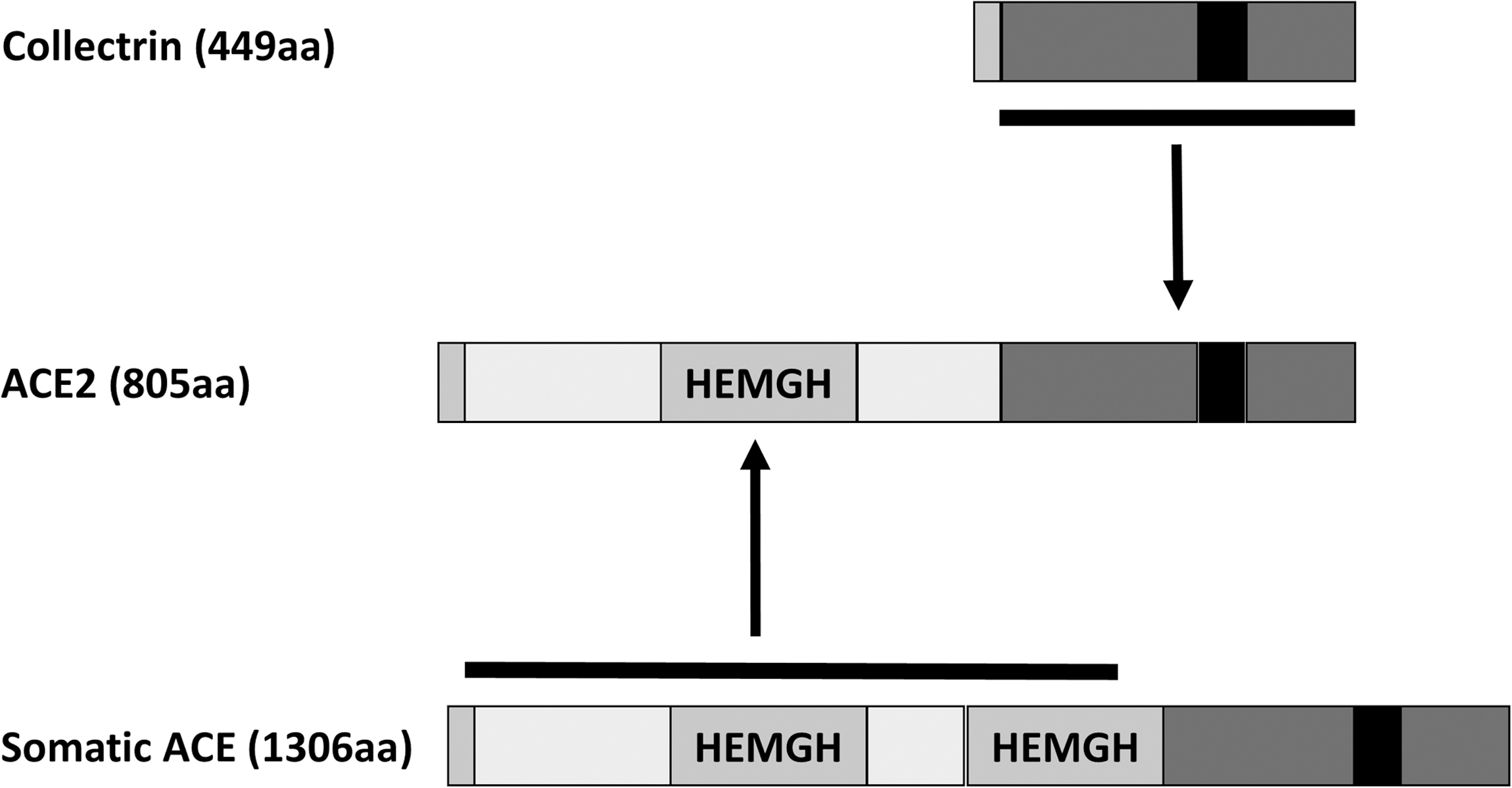
Collectrin, the ACE2 Homolog
The collectrin (Tmem27) gene encodes a 222 amino acid transmembrane glycoprotein, with ∼50% sequence identity to the ACE2 enzyme (Fig. 6). By genomic sequence alignment, ACE2 has been proposed as a chimeric duplication of ACE and collectrin (87). Unlike ACE2, collectrin lacks any catalytic domain (126).
Collectrin is highly conserved among species, sharing >80% identity between mouse, rat, and human (111, 150). It is most abundantly expressed in the kidney, primarily in the proximal tubular and collecting duct epithelia, and is also expressed in the pancreatic β cells, liver, intestinal epithelial cells, retina (1, 26, 80), and endothelial cells (14, 146). The functional role of collectrin may be specific to its tissue localization. Global deletion of collectrin uncovered its central role in amino acid transport in both the renal epithelium and the vascular endothelium. Collectrin-deficient (Tmem27 KO) mice display a generalized urinary amino acid wasting due to reduced expression of neutral and cationic AA transporters in the plasma membrane (PM) of proximal tubule brush border (26, 80).
In endothelial cells, collectrin regulates L-arginine (L-Arg) uptake, likely by controlling the PM expression of the L-Arg AA transporters cationic amino acid transporter (CAT)-1 and y+LAT1 (14). Initially thought to be a chaperone for the trafficking of amino acid transporters in renal epithelial and endothelial cells, collectrin was recently shown to bind B0-like amino acid transporters and is required for both their trafficking and catalytical activity, suggesting that collectrin is an essential subunit of this heteromultimeric secondary active transporter, rather than serving solely as a chaperone (37).
In the pancreas, in vitro and transgenic overexpression studies revealed that collectrin plays a role in islet cell mass (1), and in insulin secretion (42), possibly by facilitating SNARE complex formation by interacting with snapin, a synaptosomal-associated protein (42). Collectrin is a downstream target of HNF1α (1, 42), the mutations of which account for the majority of cases of maturity-onset diabetes of the young (MODY) (108, 141). The deletion of HNF-1α in the mouse leads to Fanconi syndrome that is featured with polyuria, glycosuria, phosphaturia, and aminoaciduria (94). It was thus a surprise that mice globally deficient for collectrin do not display a diabetic phenotype but rather an attenuated insulin resistance with aging that is associated with increased carbohydrate and fat utilization. This condition could be a compensatory mechanism for nutrition loss from aminoaciduria (6, 48, 80).
Collectrin, amino acid transport, and blood pressure regulation
Its homology to ACE2 and its upregulation in the kidney during high-salt feeding and after 5/6 nephrectomy raised the central question whether collectrin might play a role in blood pressure homeostasis. On a permissive and salt-sensitive genetic background in the 129/SvEv mouse strain, deletion of collectrin results in hypertension at baseline and augmented salt sensitivity that is associated with impaired pressure natriuresis (54). These phenotypes are associated with an imbalance of increased superoxide (O2 •−) generation and decreased NO production in the kidney in vivo, possibly involving the uncoupling of NOS and impaired endothelium-dependent relaxation of resistance arteries ex vivo.
Collectrin and the L-Arg-NO pathway
How might the amino acid transport defect be linked to hypertension and salt sensitivity?
The NO signaling pathway is a key second messenger system involved in the regulation of vascular tone and arterial pressure (68). NO is synthesized from the semiessential cationic amino acid L-Arg, a semiessential cationic AA through a reaction catalyzed by NOS in the presence of the cofactor tetrahydrobiopterin (BH4) (120). eNOS and neuronal NOS (nNOS) are the two isoforms constitutively expressed, the third isoform is the inducible NOS (iNOS) that is primarily expressed in activated inflammatory cells. Both eNOS and nNOS are only fully functional in a dimeric form, the stability of which is dependent on the availability of L-Arg substrate and BH4 cofactor, and S-nitrosylation (5). In the absence of L-Arg substrate and/or BH4 cofactor, NOS cannot generate NO but is capable of generating superoxide O2 •−, a phenomenon commonly referred to as “uncoupling” (5, 127) that is associated with endothelial dysfunction.
Collectrin KO mice display lower expression levels of eNOS dimer in the kidney and aorta (54), suggesting a state of NOS uncoupling that favors the generation of O2 •−.
The known functional property of collectrin as a chaperone of amino acid transporters in the proximal tubule raised the possibility that the central defect in blood pressure homeostasis in collectrin KO mice could be due to impaired cellular uptake of L-Arg. Indeed, primary endothelial cells from collectrin KO mice have decreased [3H]L-Arg uptake (54), whereas overexpression of collectrin in primary human coronary endothelial cells results in increased [3H]L-Arg uptake (54). These findings strongly support the hypothesis that collectrin plays a key role in cellular L-Arg uptake.
There are several different intracellular L-Arg sources or pools: (i) PM transport of L-Arg from the extracellular space (19), (ii) intracellular L-citrulline to L-Arg recycling (55), and (iii) lysosomal and proteasomal protein degradation that in turn also generates the L-Arg analog asymmetric dimethyl-L-Arg (ADMA), an endogenous inhibitor of NOS (110). Which L-Arg pool becomes the rate limiting source for NO synthesis may be determined by certain pathological conditions (110). In several cell types, including the endothelium and renal epithelium, de novo import of extracellular L-Arg is required for NO synthesis (3, 32, 62).
Under normal physiologic conditions, intracellular concentration of L-Arg is in far excess (20–760-fold) of the Km for both eNOS and nNOS, and normal plasma L-Arg concentration in humans and animals ranges 5–10-fold the Km for NOS (45, 79). Despite these excess concentrations, exogenous administration of L-Arg to further elevate extracellular L-Arg levels directly enhances endothelial NO production. The dependence of endogenous NO generation on the uptake of extracellular L-Arg has led to the concept of “L-arginine paradox” (45, 79). Thus, impairment in the uptake of extracellular L-Arg may lead to development of hypertension. In this regard, patients with essential hypertension (HTN), as well as in normotensive individuals with a positive family history of essential HTN, have impaired L-Arg transport. How might L-Arg transport defect be linked to hypertension and salt sensitivity?
L-Arg administration in hypertensive humans and animals results in reduced arterial pressure and improved endothelial vasodilator function (17, 109). Renal medullary interstitial infusion of L-Arg to Dahl salt-sensitive rats prevents the development of HTN during high-salt feeding (90). L-Arg supplementation partially lowered blood pressure in collectrin KO mice (54).
L-Arg transport into cells is mediated by different classes of amino acid transporters that are defined by their ion dependency, substrate specificity, and relative affinity (36, 79). In endothelial cells, the system y+, a sodium-independent system, accounts for ∼60% of L-Arg transport, and system y+L, a sodium-dependent system, accounts for ∼40% of L-Arg transport (3, 44, 66). System y+ selectively mediates the cellular transport of cationic amino acids, including L-Arg, whereas system y+L transports both cationic and neutral amino acids (3, 32).
Cloning studies have identified at least 3 cationic transporters in system y+, namely the CAT proteins: CAT1, CAT2A and CAT2B isoforms, and CAT3 (36). System y+L is a glycoprotein-associated amino acid heterodimeric transporter whose heavy chain is the glycoprotein 4F2hc/CD98, and the hydrophobic “light chain,” y+LAT1 or y+LAT2, is responsible for the recognition and operational features of the transport process (128). Both y+ and y+L systems are also expressed in renal endothelial and epithelial cells, and play an important role in influencing L-Arg uptake, NO production, medullary blood flow, and arterial pressure (62, 63, 81, 90).
Analogous amino acid transport systems, encoded by different genes, exist in epithelial cells such as the kidney and intestine. In the kidney, in addition to y+ and the y+L systems, which have been well described in the renal endothelium and inner medullary CD (63, 129), the broad-scope transporters also have been well characterized: B0, system 1, is sodium dependent and is a low-affinity transporter for almost all neutral amino acids (11) and also has preference for the cationic substrates lysine and arginine (25, 29); b0,+, system 2, is sodium independent and is an antiporter that takes up amino acids in exchange for neutral amino acids (11).
These two systems are expressed along the brush border of the proximal tubule (29). Collectrin KO mice display decreased protein expression of B0AT1 (B0 system) and b0,+AT/rBAT heteromeric complex (b0,+ system) (80) as well as y+L system (y+LAT1 and y+LAT2) (54) in kidney PM fractions. Gene knockdown and overexpression studies showed that collectrin directly mediates the PM expression of the y+ and y+L transporter systems in endothelial cells (54).
Compound heterozygosity for two mutations in the SLC7A7 gene that encodes the y+LAT1 transporter was reported to be associated with vascular endothelial dysfunction that was corrected with L-Arg infusion (64). Similarly, the diminished CAT1 (SLC7A1) expression caused by a single-nucleotide polymorphism in the 3′ UTR of SLC7A1 has been linked to hypertension and endothelial dysfunction (145). Thus, collectrin may modulate endothelial function and blood pressure through its central role in chaperoning the proper localization and/or function of amino acid transporters for L-Arg. In addition, collectrin might be a factor that regulates arterial pressure and salt sensitivity, likely by modulating the L-Arg/NO/O2 •− pathway (Fig. 7).

It should also be noted that while decreased L-Arg availability due to impaired uptake may be the cause for decreased eNOS dimerization in collectrin KO mice, it is also possible that collectrin may influence eNOS dimerization independently of its effect on L-Arg uptake by interacting with other proteins that play a role in stabilizing eNOS. For example, caveolae, which are important in determining eNOS intracellular localization and activity (5), contain some members of the SNARE complex (106) that collectrin binds and interacts with (42, 146).
Since ACE2 and collectrin share 50% sequence identity, this raises the question whether ACE2 also influences the expression of amino acid transporters. A study demonstrated that ACE2 in the small intestine regulates the expression of the amino acid transporter B(0)AT1, whereas collectrin regulates its expression in the kidney (13). Furthermore, ACE2 KO mice do not display aminoaciduria (Gurley SB, personal communication, 2010). Whether ACE2 also regulates amino acid transport in endothelial cells remains to be determined. To date, evidence suggests that the effect of ACE2 on blood pressure is primarily through its catalytic effect on Ang II.
Conclusion
This review summarizes the most recent publications in basic research focusing on the role of ACE2 and its homolog collectrin in the regulation of hypertension and vascular injury. Studies have shown that most of the ACE2-induced effects on blood pressure and vascular injury were mediated by changes in Ang II degradation or Ang-(1 –7) generation leading to an imbalance in AT1- or Mas receptor activation, respectively. Thus, Ang-(1 –7)-induced Mas receptor activation within the kidney, vasculature, and CNS increases NO bioavailability and attenuated ROS production affecting natriuresis, vascular function, and sympathetic nerve activity and thereby reduces hypertension and vascular injury.
Shedding of the enzymatically active ectodomain of ACE2 from the cell surface leads to an active soluble form of ACE2 opening up an alternative regulation of the enzyme. Its pathophysiological relevance is still unknown, but it seems likely that the soluble form of ACE2 acts as an interorgan communicator in disease states characterized by an upregulated RAS. In addition to Ang I and Ang II, ACE2 also cleaves apelin, another vasoactive peptide that has been shown to improve cardiovascular function through its own receptor AJP. Although the ACE2 homolog collectrin does not exhibit catalytic activity, it seems to play an important role in blood pressure homeostasis by influencing NO bioavailability. Based on their known beneficial actions, activation or increase in the activity of both ACE2 and collectrin would be promising approaches in the treatment of hypertension and vascular injury.
Footnotes
Acknowledgment
This review was supported by the Deutsche Forschungsgemeinschaft (IRTG1902).