
Abstract
Aims:
Altered redox state has been related to the development of normal pregnancy (NP) and preeclampsia (PE). Endothelial KCa2.3 and KCa3.1 (KCas) play an important role in vasodilation, and KCas levels are affected by oxidative stress. We investigated the mechanisms of oxidative stress-mediated KCas expression modulation during NP and PE.
Results:
Human uterine microvascular endothelial cells were incubated in serum from normal nonpregnant women (n = 13) and women with NP (n = 24) or PE (n = 15), or in vascular endothelial growth factor (VEGF), oxidized low-density lipoprotein (ox-LDL), progesterone, or estradiol-17β (E2)-containing medium for 24 h. NP serum elevated H2O2 levels via reducing catalase and glutathione peroxidase 1 levels, thereby enhancing KCas levels via a H2O2/fyn/extracellular signal-regulated kinase (ERK)-mediated pathway. VEGF enhanced H2O2 and KCas levels and KCa3.1 currents. KCas were upregulated and KCas activation-induced endothelium-dependent relaxation (EDR) was augmented in vessels from pregnant mice and rats. Whereas PE serum, ox-LDL, progesterone, or soluble fms-like tyrosine kinase 1 (sFlt-1) elevated superoxide levels via elevating NADPH oxidase 2 (NOX2) and NOX4 levels and reducing superoxide dismutase (SOD) 1 levels, thereby downregulating KCas. sFlt-1 inhibited EDR. PE serum- or progesterone-induced alterations in levels of KCas were reversed by polyethylene glycol-SOD, NOX inhibition, or E2.
Innovation and Conclusions:
This is the first study of how endothelial KCas levels are modulated during NP and PE. KCas were upregulated by soluble serum factors such as VEGF via H2O2 generation in NP, and were downregulated by serum factors such as progesterone and ox-LDL via superoxide generation in PE, which may contribute to hemodynamic adaptations in NP or to the development of PE.
Endothelial KCa2.3 and KCa3.1 (KCas) induce vasodilation. KCas upregulation may inhibit blood pressure increases, with KCas downregulation having the opposite effect. Oxidative stress, which is increased in pregnancy, might affect KCas levels. We thus examined mechanisms for oxidative stress-induced KCas modulation in normal pregnancy (NP) and preeclampsia (PE). In NP, KCas were upregulated via downregulating H2O2-degrading catalase and glutathione peroxidase 1 and activating a H2O2/fyn/extracellular signal-regulated kinase (ERK) pathway. Whereas KCas were downregulated via upregulating NADPH oxidase 2 (NOX2) and NOX4 and downregulating superoxide dismutase (SOD) 1 in PE. Vascular endothelial growth factor (VEGF), estrogen, progesterone, and oxidized low-density lipoprotein in serum act as modulators of KCas levels.
Introduction
O
Increased oxidative stress leads to endothelial dysfunction, thereby causing increased blood pressure in PE. On the contrary, the proangiogenesis factor vascular endothelial growth factor (VEGF), which induces placental development during pregnancy, also stimulates ROS generation via activation of NADPH oxidases (NOXs) (37). In addition, pregnancy hormones (estrogen, progesterone), which control vascular contractility (9), stimulate ROS generation (5, 26, 38). These results suggest that redox state plays an important role in regulating blood pressure during pregnancy.
Ca2+-activated K+ channels (KCa2.3 and KCa3.1) play important roles in endothelial control of vasorelaxation. KCa2.3 and KCa3.1 evoke endothelium-dependent hyperpolarization and stimulate nitric oxide release by promoting Ca2+ influx through Ca2+ entry channels. Thus, KCa2.3 and KCa3.1 downregulation or deficiency enhances vascular contractility and elevates blood pressure (7, 28), whereas upregulation of these K+ channels promotes endothelium-dependent relaxation (EDR) (10).
Among these K+ channels, KCa3.1 levels and currents are enhanced by H2O2 and reduced by superoxide (11, 12). In addition, KCa3.1 levels are affected by various circulating plasma substances, such as platelet-derived growth factors and ox-LDL. Messenger RNA (mRNA) or protein levels of KCa3.1 are enhanced by platelet-derived growth factors and downregulated by ox-LDL, respectively (6, 11). Platelet-derived growth factors and ox-LDL stimulate ROS generation (18, 31). These results suggest that these plasma substances may regulate KCa3.1 levels via ROS generation. In addition, estrogen and progesterone may control KCa3.1 levels via ROS generation during pregnancy. However, little is known about whether these pregnancy hormones modulate KCa3.1 levels. Furthermore, although KCa2.3 plays an important role in endothelial functions, little is known about KCa2.3 level regulation in NP and PE.
Since redox state is controlled by the superoxide-producing membrane-bound enzymes NOXs and endogenous antioxidant enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPX), altering levels of NOXs and/or antioxidant enzymes might change redox state. Therefore, we hypothesized that an altered redox state caused by altered levels of NOXs and antioxidant enzymes modulates KCa2.3 and KCa3.1 levels in NP and PE. We examined this hypothesis and the roles of plasma substances (VEGF, sFlt-1, ox-LDL, estrogen, and progesterone) in the modulation of these K+ channel levels via altering NOXs and antioxidant enzymes. The results showed that altered levels of these enzymes upregulate KCa2.3 and KCa3.1 by generating H2O2 in NP, whereas KCa2.3 and KCa3.1 are downregulated by superoxide generation in PE.
Results
NP serum upregulates endothelial KCa2.3 and KCa3.1 via VEGF receptor activation
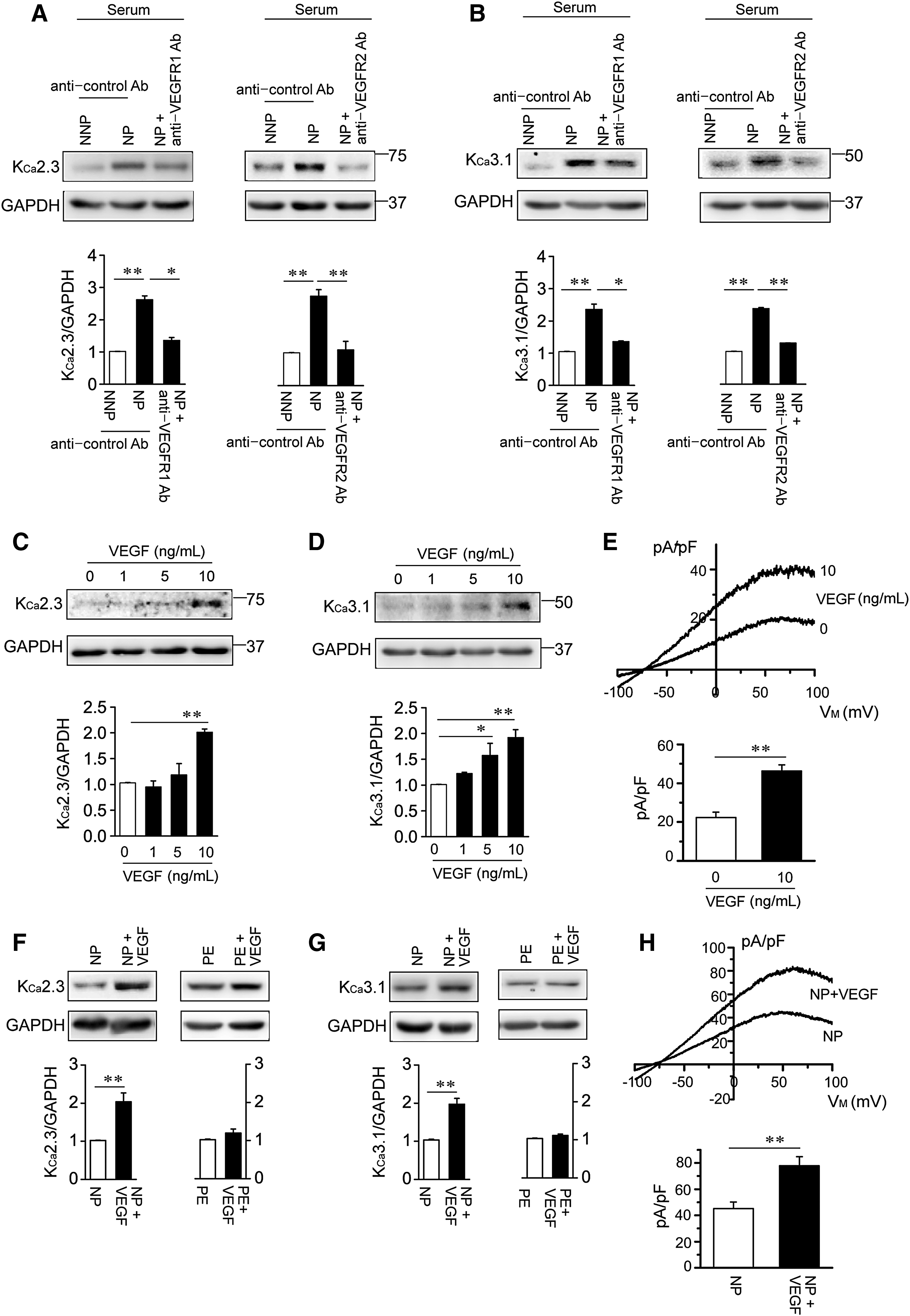
We compared the effects of soluble serum factors in serum from normal nonpregnancy (NNP) and NP women on KCa2.3 and KCa3.1 expression by incubating human uterine microvascular ECs (HUtMECs) in serum from NNP and NP women for 24 h. KCa2.3 (Fig. 1A) and KCa3.1 levels (Fig. 1B) were markedly higher in HUtMECs treated with NP serum than in HUtMECs treated with NNP serum. The NP serum-mediated increase in these K+ channels was reduced by blocking VEGF receptors (VEGFRs) with an anti-VEGFR1 antibody (Ab) or anti-VEGFR2 Ab (Fig. 1A, B), indicating that NP serum increased KCa2.3 and KCa3.1 levels via VEGFR activation. We thus examined the effects of VEGF (10 ng/mL) on KCa2.3 and KCa3.1 levels by incubating HUtMECs in VEGF-containing culture medium for 24 h. VEGF treatment increased protein levels of KCa2.3 and KCa3.1 in a concentration-dependent manner (Fig. 1C, D), and KCa3.1 currents were enhanced from 23.3 ± 2.7 pA/pF of control to 48.3 ± 3.1 pA/pF of VEGF-treated ECs (Fig. 1E). We then examined the effects of VEGF (10 ng/mL) on NP serum- or PE serum-treated ECs. VEGF further enhanced KCa2.3 and KCa3.1 levels (Fig. 1F, G) and KCa3.1 currents from 46.2 ± 5.0 pA/pF of NP serum-treated ECs to 77.8 ± 6.9 pA/pF of (NP serum+VEGF)-treated ECs (Fig. 1H) in NP serum-treated cells. In contrast, VEGF did not affect KCa2.3 (Fig. 1F) and KCa3.1 levels (Fig. 1G) in PE serum-treated cells, indicating the presence of VEGF antagonists in PE serum. These data suggest that KCa2.3 and KCa3.1 are upregulated by soluble serum factors, such as VEGF, during pregnancy.
KCa2.3 and KCa3.1 upregulation enhances EDR during pregnancy
KCa2.3 and KCa3.1 upregulation might promote EDR. We thus examined whether these K+ channels were upregulated to enhance EDR in maternal blood vessels during pregnancy by studying nonpregnant and pregnant mice and rats. Protein levels of KCa2.3 (Fig. 2A) and KCa3.1 (Fig. 2B) were substantially increased in aortic tissues from pregnant mice compared with levels in nonpregnant mice. We then compared the magnitude of EDR of aortic rings from these mice and rings of the first branch and trunk of rat superior mesenteric artery (SMA). The EDR response to acetylcholine (ACh) was significantly increased in aortic rings from pregnant mice (Fig. 2C) and in rings of SMA branch from pregnant rats (Fig. 2D) compared with the response in rings from nonpregnant mice and in SMA rings from nonpregnant rats, respectively. We then inhibited nitric oxide production by pretreatment with N
ω-nitro-
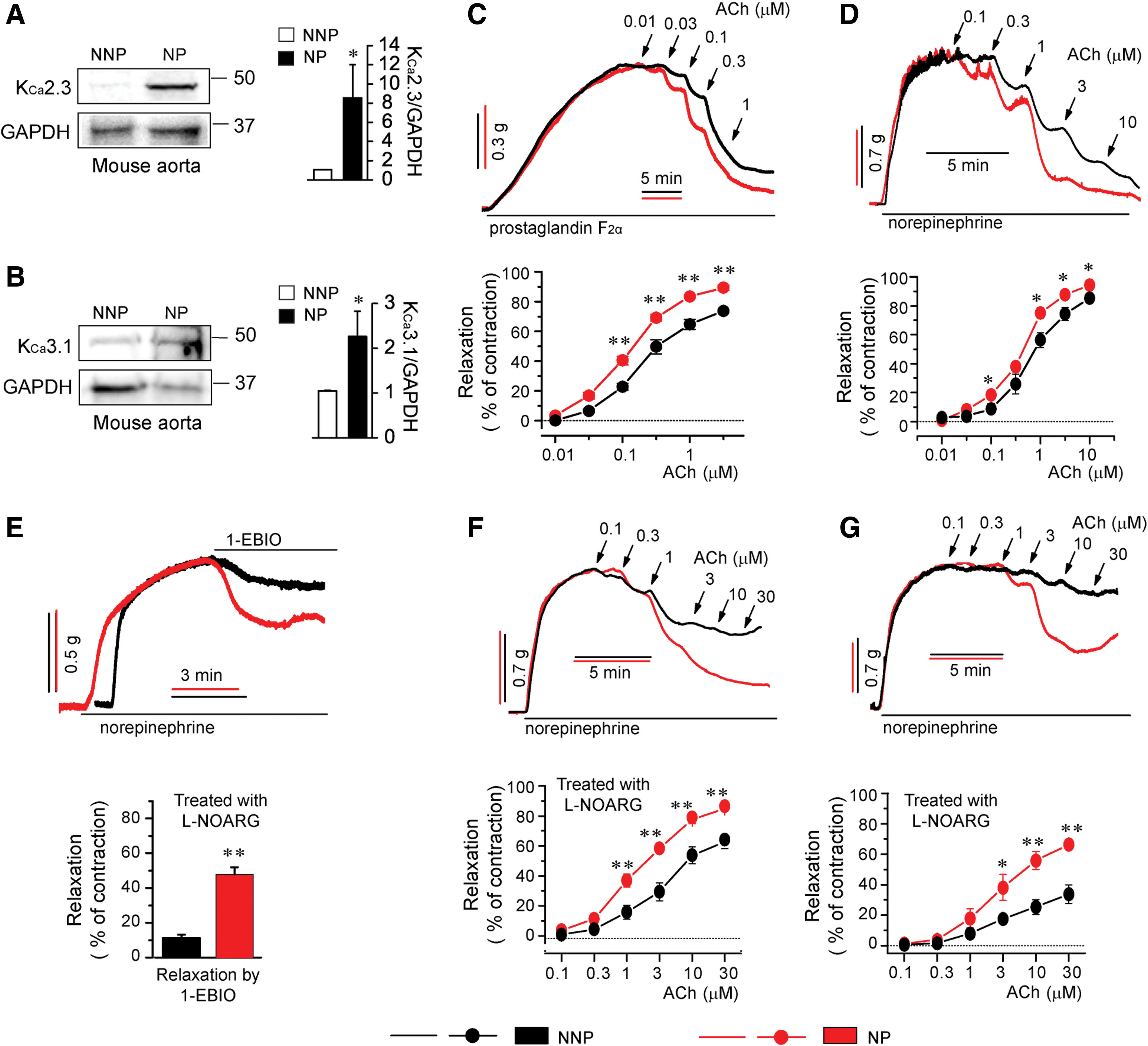
Pregnancy-associated KCa2.3 and KCa3.1 upregulation is attenuated in PE
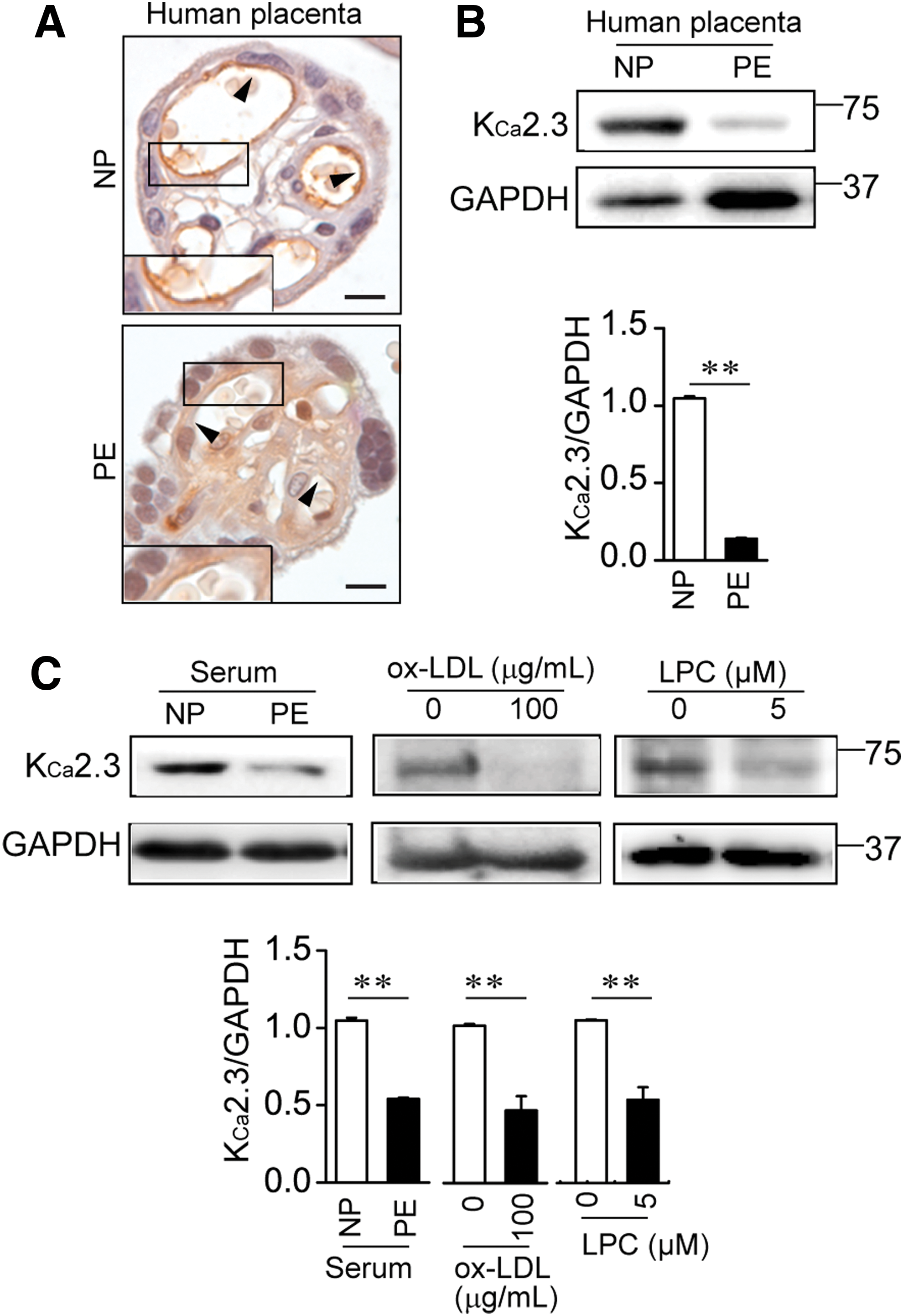
We then examined KCa2.3 expressions in placental tissues from NP and PE using an immunohistochemical technique targeted at the KCa2.3 proteins. Figure 3A shows a cross-sectioned chorionic villus containing blood vessels in placental tissues. KCa2.3 expressions were substantially decreased in the ECs of placental tissues from PE compared with placental tissues from NP (Fig. 3A). The decrease in KCa2.3 expressions in placental tissues from women with PE was confirmed by Western blot analysis. Protein levels of KCa2.3 were markedly decreased in PE placental tissues compared with levels in NP placental tissues (Fig. 3B). In addition, KCa2.3 levels in ECs exposed to PE serum were significantly reduced compared with those in ECs exposed to NP serum (Fig. 3C). Then, HUtMECs were exposed to ox-LDL or lysophosphatidylcholine (LPC). Treatment of HUtMECs with ox-LDL or LPC reduced KCa2.3 levels (Fig. 3C). KCa3.1 levels were also markedly decreased in placental tissues from PE compared with levels in placental tissues from NP (Supplementary Fig. S1; Supplementary Data are available online at
NOX4 and antioxidant enzyme levels in ECs treated with NP and PE serum
We previously reported that endothelial NOX2 is upregulated in umbilical vessels from PE subjects (11). However, levels of NOX1, NOX3, and NOX5 are not altered in vessels from NP and PE (11). We thus compared NOX4 levels, which are highly expressed under normal physiological conditions in ECs and might be constitutively activated (3), between ECs treated with NNP and NP serum, and between ECs treated with NP and PE serum. NOX4 levels were significantly decreased in ECs treated with NP serum versus ECs treated with NNP serum, whereas NOX4 levels were markedly enhanced in ECs treated with PE serum versus ECs treated with NP serum (Fig. 4A). SOD1 levels were significantly increased in ECs treated with NP serum versus ECs treated with NNP serum, whereas SOD1 levels were markedly reduced in ECs treated with PE serum versus ECs treated with NP serum (Fig. 4B). We then compared levels of catalase and GPX1 between ECs treated with NNP and NP serum, and between ECs treated with NP and PE serum (Fig. 4C, D). Catalase and GPX1 levels were significantly reduced in ECs treated with NP serum versus ECs treated with NNP serum, whereas catalase and GPX1 levels were markedly enhanced in ECs treated with PE serum versus ECs treated with NP serum. These results suggest that redox state is altered via alterations in NOXs and antioxidant enzyme levels in NP and PE.

Altered redox state modulates KCa2.3 and KCa3.1 levels in NP and PE
Since catalase and GPX degrade H2O2, catalase and GPX1 downregulation might elevate H2O2 levels. We thus examined whether NP serum-mediated catalase and GPX1 downregulation elevated H2O2 levels by using peroxy-orange 1 in ECs treated with NNP and NP serum. H2O2 levels were markedly increased in ECs treated with NP serum versus ECs treated with NNP serum (Fig. 5A; 156% ± 15% of control). In addition, treating HUtMECs with VEGF (10 ng/mL) elevated H2O2 levels (Fig. 5A; 5 ng/mL: 112% ± 4% of control; 10 ng/mL: 119 ± 6% of control). In contrast, NP serum and VEGF treatment did not elevate superoxide levels in HUtMECs (Supplementary Fig. S2A, B). We then examined whether H2O2 was involved in NP serum-mediated KCa2.3 and KCa3.1 upregulation. NP serum-mediated KCa2.3 and KCa3.1 upregulation was reversed by the membrane-permeable catalase, polyethylene glycol (PEG) catalase (Fig. 5B, C). H2O2 activates the Src family kinase fyn (24). We therefore compared phosphorylated fyn (p-fyn) levels in ECs treated with NNP and NP serum. Treatment with NP serum increased p-fyn levels compared with treatment with NNP serum, and this NP serum-mediated p-fyn increase was reversed by treatment with PEG catalase (Fig. 5D). As H2O2 and fyn play critical roles in extracellular signal-regulated kinase (ERK) activation and KCa3.1 protein is produced via a H2O2/fyn/ERK-dependent pathway (10, 12, 35), we examined whether NP serum elevated phosphorylated ERK (p-ERK) levels in HUtMECs. Treatment with NP serum increased p-ERK levels compared with treatment with NNP serum, and this NP serum-mediated p-ERK increase was reversed by treatment with PEG catalase (Fig. 5E). Since KCa3.1 upregulation by NP serum was reversed by treatment with PEG catalase (Fig. 5C), we examined whether treatment with PEG catalase reduced KCa3.1 currents in ECs treated with VEGF (10 ng/mL). PEG catalase reduced KCa3.1 currents from 46.4 ± 3.8 pA/pF to 30.6 ± 1.3 pA/pF in ECs treated with NP serum (Fig. 5F). These results were consistent with our previous results that KCa2.3 and KCa3.1 are upregulated by catalase and GPX1 knockdown using catalase and GPX1 double knockout mice (10), and suggest that catalase and GPX1 downregulation elevates H2O2 levels, thereby enhancing KCa2.3 and KCa3.1 levels in NP.

Next, we examined whether NOX4 upregulation or SOD downregulation affected endothelial KCa2.3 and KCa3.1 levels via superoxide generation in PE. PE serum markedly elevated superoxide levels in ECs (Supplementary Fig. S2A, B), and KCa3.1 levels were markedly decreased in ECs treated with PE serum versus levels in ECs treated with NP serum (Supplementary Fig. S3A). We then inhibited PE serum-mediated superoxide generation by treatment with small interfering RNA (siRNA) against NOX4 or PEG-SOD. The PE serum-mediated decrease in KCa3.1 levels was recovered by inhibiting NOX4 with siRNA against NOX4 (Supplementary Fig. S3A). In addition, PE serum-mediated KCa2.3 downregulation was reversed by treatment with the membrane-permeable SOD, PEG-SOD (Supplementary Fig. S3B). Previously, we showed that PE plasma reduces endothelial KCa3.1 levels via superoxide generation (11). These results suggest that NOX4 upregulation and SOD downregulation enhance superoxide levels, thereby reducing KCa2.3 and KCa3.1 levels in PE.
Progesterone modulates KCa2.3 and KCa3.1 levels
Progesterone promotes superoxide release via NOX activation and SOD downregulation in blood vessels from progesterone-treated animals (38). Plasma levels of progesterone are increased during pregnancy, and progesterone levels are higher in PE compared with the levels in NP (33). We therefore exposed HUtMECs to progesterone. Progesterone elevated superoxide levels (Supplementary Fig. S2A, B) and reduced H2O2 levels (Supplementary Fig. S2C). The effects of progesterone on KCa3.1 levels varied according to the progesterone concentration. Progesterone increased protein levels of KCa3.1 at low concentrations (≤50 ng/mL), but decreased the K+ channel levels at high concentrations (≥250 ng/mL) (Fig. 6A). In addition, high progesterone concentration (500 ng/mL) reduced protein levels of KCa2.3 (Fig. 6B). As the plasma levels of progesterone range from 8 to 48 ng/mL and from 99 to 342 ng/mL in the first and third trimester, respectively (1), the concentrations of progesterone needed to affect KCa2.3 and KCa3.1 expression are within the ranges observed in NP. We then examined the effects of progesterone on levels of NOX2, NOX4, SOD1, catalase, and GPX1. Treatment of HUtMECs with progesterone increased NOX2 and NOX4 levels (Fig. 6C) and decreased SOD1 levels (Fig. 6D). GPX1 and catalase levels were enhanced by progesterone in a concentration-dependent manner (Fig. 6E). We then examined whether progesterone reduces KCa3.1 levels via superoxide generation. Progesterone (500 pg/mL) decreased KCa3.1 levels, and the progesterone-induced decrease in KCa3.1 levels was prevented by inhibiting superoxide generation using PEG-SOD, the pan-NOX inhibitor VAS2870, or the NOX4 inhibitor GKT137831 (Fig. 6F). These results suggested that progesterone reduced KCa2.3 and KCa3.1 levels by generating superoxide.

Estrogen inhibits progesterone-induced KCa3.1 downregulation
Estrogen inhibits superoxide generation via nitric oxide generation, thereby preventing endothelial dysfunction (2). We thus examined whether estrogen inhibited progesterone-induced changes in the levels of NOX4, antioxidant enzymes, or KCa3.1. HUtMECs were exposed to progesterone or progesterone+estradiol-17β (E2). Progesterone induced NOX4 upregulation and SOD1 downregulation, and these effects were prevented by E2 (Fig. 7A, B). However, catalase upregulation by progesterone was not significantly prevented by E2 (Fig. 7C). In contrast, GPX1 upregulation by progesterone was prevented by E2 (Fig. 7D), and KCa3.1 downregulation by progesterone was prevented by E2 (Fig. 7E). Meanwhile, E2 alone did not increase protein levels of KCa3.1 at low concentrations (≤20 ng/mL), but significantly increased KCa3.1 (Fig. 7F) and KCa2.3 (Supplementary Fig. S4) levels at high concentrations (≥50 ng/mL). In addition, E2 alone did not increase superoxide levels in ECs (Supplementary Fig. S2A, B). Since the plasma levels of E2 range from 188 to 7192 pg/mL during pregnancy (1), the concentrations of E2 needed to significantly increase protein levels of KCa3.1 were above the ranges observed in NP. However, the concentrations of E2 needed to inhibit progesterone were very close to the ranges observed in NP.

sFlt-1 inhibits EDR by elevating superoxide levels
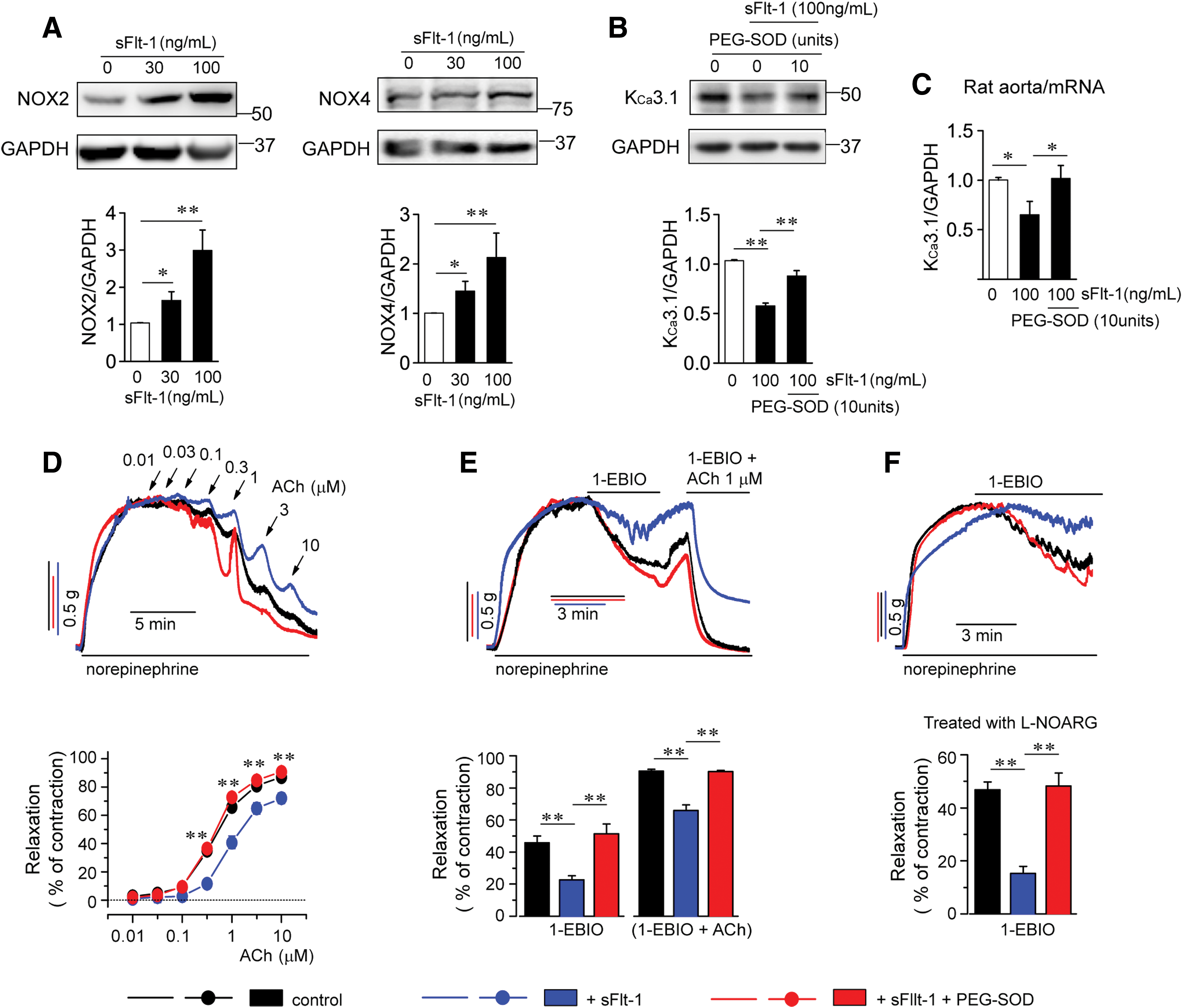
sFlt-1, which acts as an antagonist of VEGF, might contribute to the development of PE (21, 32). We thus examined the effects of sFlt-1 on NOX2, NOX4, superoxide, and KCa3.1 levels. Treatment with sFlt-1 for 24 h markedly elevated NOX2, NOX4 (Fig. 8A), and superoxide levels (Supplementary Fig. S2A, B) and reduced protein levels of KCa3.1 (Fig. 8B) in HUtMECs. Reduced protein levels of KCa3.1 were reversed by PEG-SOD. Then, the effects of sFlt-1 on mRNA levels of KCa3.1 and EDR were examined ex vivo. Rat aortas and SMA trunks were incubated in sFlt-1- or sFlt-1+PEG-SOD-containing culture medium for 24 h. sFlt-1 reduced mRNA levels of KCa3.1 in rat aortas, and reduced mRNA levels of KCa3.1 were reversed by PEG-SOD (Fig. 8C). Treatment with sFlt-1 attenuated EDR to ACh or 1-EBIO in SMA trunks from rats (Fig. 8D, E). In addition,
Discussion
In this study, we observed for the first time that endothelial KCa2.3 and KCa3.1 levels were modulated by altered redox state during pregnancy. During NP, catalase and GPX1 levels were reduced and SOD levels were enhanced, which may increase H2O2 levels and thereby upregulate KCa2.3 and KCa3.1 via a H2O2/fyn/ERK-mediated pathway. Whereas NOX2 and NOX4 levels were enhanced and SOD levels were reduced in PE, which might increase superoxide levels and thereby downregulate KCa2.3 and KCa3.1. Endothelial KCa2.3 and KCa3.1 play important roles in vasodilation. Therefore, KCa2.3 and KCa3.1 upregulation might inhibit blood pressure elevation in NP. Whereas KCa2.3 and KCa3.1 downregulation might contribute to the development of high blood pressure in PE (Fig. 9).

H2O2-induced KCa2.3 and KCa3.1 upregulation in NP and superoxide-induced KCa2.3 and KCa3.1 downregulation in PE suggest that endothelial functions can be modulated by altering redox state during pregnancy. Since SOD upregulation promotes the degradation of superoxide into H2O2, and catalase and GPX downregulation reduces H2O2 degradation, such changes in antioxidant enzymes on treatment with NP serum may increase H2O2 levels and decrease superoxide levels, thereby upregulating KCa2.3 and KCa3.1. Since NOX2 and NOX4 upregulation and SOD downregulation enhance superoxide levels and catalase and GPX upregulation reduces H2O2 levels, such changes in NOXs and antioxidant enzymes by treatment with PE serum may increase superoxide levels and decrease H2O2 levels, thereby downregulating KCa2.3 and KCa3.1. During pregnancy, soluble serum factors such as VEGF, progesterone, estrogen, and ox-LDL might act as modulators of NOXs and antioxidant enzyme levels, thereby regulating redox state. Thus, these factors might affect endothelial functions via modulating KCa2.3 and KCa3.1 levels.
Levels of estrogen and progesterone are elevated during pregnancy (1), and these steroids stimulate VEGF production (17, 27). VEGF, estrogen, and progesterone activate ERK (8, 14, 19, 20, 30). Since KCa3.1 protein synthesis is ERK dependent (34), VEGF and sex steroids might increase protein levels of KCa3.1 by stimulating KCa3.1 protein synthesis, consistent with the finding that VEGF upregulates KCa3.1 mRNA (16). In addition, H2O2, levels of which were increased in NP, activates fyn and ERK (12, 35). Thus, KCa3.1 upregulation that occurs during NP and on treatment with low concentrations of progesterone might be caused by increased production of the K+ channel proteins via activating ERK-dependent pathways. However, little is known about how KCa2.3 proteins are produced in ECs, and further studies are required to clarify regulatory mechanisms behind KCa2.3 protein production.
In contrast, high concentrations (≥100 or 250 pg/mL) of progesterone reduced KCa2.3 and KCa3.1 levels. Since progesterone stimulated superoxide generation via NOX2 and NOX4 upregulation and SOD downregulation, superoxide generated by progesterone might be involved in KCa2.3 and KCa3.1 downregulation by progesterone, which is supported by the finding that progesterone-induced KCa2.3 downregulation was reversed by PEG-SOD. In addition, we previously reported that superoxide induces KCa3.1 downregulation in PE (11). Enhanced NOX4 and GPX1 levels and reduced SOD1 levels by progesterone treatment were reversed by treatment with E2, suggesting that progesterone-induced superoxide production was inhibited by E2. In addition, estrogen has been shown to act as an antioxidant and to inhibit superoxide generation via nitric oxide generation (2, 4, 39). Thereby, estrogen might reverse KCa2.3 and KCa3.1 downregulation by progesterone and recover progesterone-induced endothelial dysfunction.
Progesterone levels in serum are increased during PE compared with NP, and are strongly associated with PE (33). Endothelial KCa2.3 and KCa3.1 downregulation by progesterone at high concentrations might contribute to the development of endothelial dysfunction in PE. Since estrogen inhibited progesterone-induced KCa3.1 downregulation, the progesterone/estrogen ratio might play an important role in controlling KCa2.3 and KCa3.1 levels and endothelial functions as pregnancy progresses. The progesterone/estradiol ratio might correlate positively with mean arterial pressure during pregnancy, and an increased ratio might contribute to the development of PE, as shown in women at high altitude (40).
Other soluble serum factors, such as sFlt-1 and ox-LDL, might also be responsible for the reduced pregnancy-associated KCa2.3 and KCa3.1 upregulation in PE. sFlt-1 elevates superoxide levels and reduces free-circulating levels of proangiogenic factors VEGF and placental growth factor (21), thereby reducing KCa2.3 and KCa3.1 levels. In addition, as reported in our previous studies, PE plasma-induced NOX2 upregulation and KCa3.1 downregulation were inhibited by anti-lectin-like ox-LDL receptor Ab (11), and the major component of ox-LDL, LPC, induces superoxide overload in ECs by reducing SOD1 levels and increasing catalase levels (13). These results suggest that ox-LDL, present in high levels in the sera of women with PE, induces superoxide production, thereby reducing KCa2.3 and KCa3.1 levels.
Endothelial KCa2.3 and KCa3.1 upregulation augments EDR of vascular smooth muscle (10). Increased expression of KCa2.3 and KCa3.1 protein compensated for diminished nitric oxide-induced EDR by enhancing KCa2.3 and KCa3.1 activation-induced relaxation in aged mice (10). The mechanism for KCa3.1 upregulation in aged mice was similar to the mechanism in NP, since downregulation of catalase and GPX1 contributed to KCa3.1 upregulation via H2O2 generation. Estrogen receptor agonists and E2 induce EDR via activating endothelium-dependent hyperpolarization and stimulate EC proliferation (15, 36). Thus, estrogen-induced EDR might be, at least in part, caused by KCa2.3 and KCa3.1 upregulation.
The present study showed that catalase and GPX1 downregulation increased H2O2 levels, thereby enhancing KCa2.3 and KCa3.1 levels in NP. Whereas NOX2 and NOX4 upregulation and SOD downregulation might increase superoxide levels, thereby reducing KCa2.3 and KCa3.1 levels in PE. VEGF, sex steroids, and ox-LDL acted as modulators of these enzymes. Progesterone was involved in the KCa2.3 and KCa3.1 downregulation in PE, and estrogen inhibited progesterone-induced KCa2.3 and KCa3.1 downregulation. Since endothelial KCa2.3 and KCa3.1 play important roles in vasodilation, altered redox state-induced KCa2.3 and KCa3.1 levels may play important roles in controlling blood pressure during pregnancy.
Materials and Methods
Studies involving human subjects were approved by the local ethics committee, the Institutional Review Board of the Ewha Womans University Mokdong Hospital, and Korea University Guro Hospital, and were conducted in accordance with the Declaration of Helsinki. All patients gave their written informed consent before inclusion in the study. Experiments with mice were approved by the local ethics committee, the Institutional Review Board of the Ewha Womans University Mokdong Hospital, and were conducted in accordance with the Declaration of Helsinki, the Animal Care Guidelines of the Ewha Womans University, Medical School, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Human subjects
The study population consisted of Asian women who were not pregnant or had either an NP or PE (Table 1). Pregnancies were considered normal when patients did not have medical and obstetric complications of pregnancy and delivered a newborn at a gestational age of 37–42 weeks. PE was defined by a systolic blood pressure over 140 mmHg and a diastolic blood pressure over 90 mmHg after 20 weeks of gestation in a previously normotensive woman, and the onset of proteinuria exceeding 300 mg of protein during 24 h of urine collection. Nonpregnant women were healthy premenopausal volunteers taking no medications. Preeclamptic patients and normal pregnant women were matched for age (±3 years) and gestational age (±2 weeks), and nonpregnant healthy female volunteers were matched for age (±3 years). Blood samples were obtained from subjects during the third trimester of pregnancy. The study population was monitored at the department of obstetrics and gynecology from the first trimester until their pregnancy was completed without complications. Exclusion criteria included the following: altered renal function, diabetes or chronic diseases, twin pregnancies, recurrent miscarriages, fetal growth retardation, and abruptio placenta. All smokers and women with a history of essential hypertension were also excluded from this study. Gestational age was defined as the interval between the first day of the mother's last menstrual period and the date of delivery.
Blood Pressure and Protein Urea Levels of Study Groups
Values shown are mean ± SEM and exclusively composed of serum donors.
N/S, not specified; SEM, standard error of the mean.
Animals and tissue collection
We studied young C57BL/6 wild-type mice (about 20 weeks old; n = 24) and Sprague-Dawley rats (about 20 weeks old; n = 36). To produce pregnancy, a single male and two to three female mice or rats in proestrous or estrous stage were housed together for about 48 h. Female mice or rats were checked for vaginal plugs each morning and evening. When a vaginal plug was observed, the female mice or rats were separated from the male mice or rats. Gestational day 0 was defined as the day on which a vaginal plug was observed. Pregnant (on gestational days 17—18; n = 12) or age-matched nonpregnant female mice (n = 12) or rats (n = 12) were anesthetized by an intraperitoneal injection of pentobarbital sodium (50 mg/kg body weight) and sacrificed by cervical dislocation. The thoracic aortas were then dissected from mice and rats, and the SMAs (trunk and the first branches) were then dissected from rats.
Cell culture and treatment of cells with serum or reagents
HUtMECs, which were purchased from PromoCell GmbH (Heidelberg, Germany), were maintained in EC growth medium MV2 (PromoCell GmbH). For serum treatment, HUtMECs were plated in six-well plates for 24 h. The concentration of FBS in culture medium was then gradually decreased from 10% to 5%, 2%, and 0% over 30 min, and HUtMECs were incubated in serum-free medium for 30 min. Next, culture medium was substituted with serum from NNP women or women with NP or PE. Cells were incubated in serum for 24 h. For VEGF (751-VE; R&D Systems, Minneapolis, MN), ox-LDL (Intracel, Inc., Frederick, MD), LPC, progesterone (P9776; Sigma-Aldrich, St Louis, MO), E2 (E7879; Sigma-Aldrich), or (E2+progesterone) treatment, cells were incubated in one of these reagents or (E2+progesterone)-containing serum-free medium for 24 h. A pan-NOX inhibitor VAS2870 (SML0273; Sigma-Aldrich), an NOX4 inhibitor GKT137831 (17764; Cayman Chemicals, Ann Arbor, MI), PEG catalase (C4963; Sigma-Aldrich), and PEG-SOD (C4936; Sigma-Aldrich) were pretreated with cells 1 h before reagent application.
Measurement of intracellular ROS
HUtMECs plated on 96-well microplates (for direct quantification) were incubated in the superoxide-sensitive dye dihydroethidine (DHE; 10 μM) or the H2O2-sensitive dye peroxy-orange1 (5 μM) for 20 min. Samples were read directly using a microplate fluorescence reader (model SpectraMax; Molecular Devices, Sunnyvale, CA) or detected by confocal laser microscopy (model LSM 510; Carl Zeiss). Excitation/emission wavelengths used for detecting respective fluorescent dyes in 96-well microplate reader were DHE ex 518 nm/em 605 nm and peroxy-orange 1 ex 540 nm/em 585 nm. To further confirm the superoxide production for each sample tested, Diogenes enhanced superoxide detection kit (National Diagnostics, Atlanta, GA) was used. Cells seeded on 96-well microplates were incubated in serum (from NNP, NP, or PE) or sFlt-1 (AG-40T-0049; AdipoGen, San Diego, CA)-, VEGF-, progesterone- or E2-containing medium for 24 h, and luminescence was measured by using a GloMax 96 luminometer (Promega Corporation, Madison, WI).
Electrophysiology
The patch-clamp technique was used in whole-cell configurations at 20–22°C. Whole-cell currents were measured using ruptured patches and monitored in voltage-clamp modes with an EPC-9 (HEKA Elektronik, Lambrecht, Germany). The holding potential was 0 mV, and currents were monitored by the repetitive application of voltage ramps from −100 to +100 mV with a 10-s interval (sampling interval 0.5, 650 ms duration). The standard external solution contained (in mM) the following: 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4 with NaOH. The pipette solution for whole-cell recording contained (in mM) the following: 40 KCl, 100 K-aspartate, 2 MgCl2, 0.1 EGTA, 4 Na2ATP, 10 HEPES, pH adjusted to 7.2 with KOH. To buffer free Ca2+, the appropriate amount of Ca2+ (calculated using CaBuf software; G. Droogmans, Leuven, Belgium) was added in the presence of 5 mM EGTA.
KCa3.1 currents were activated by loading 1 μM Ca2+ via a patch pipette in whole-cell clamped HUtMECs and adding the KCa2.3 and KCa3.1 activator 1-EBIO (100 μM) to the external solution. KCa3.1 current was normalized to cell capacitance and the selective KCa3.1 blocker TRAM-34-sensitive current was measured as the KCa3.1 current.
Contraction measurement on isolated aortic rings
Thoracic aorta samples from mice and SMA samples from rats were cut into 1.0–2.0 mm rings. A custom myograph was used to record mechanical responses from the ring segments at 37°C. Each aortic or SMA (trunk and the first branch) ring was threaded with two strands of tungsten wire (120 μm diameter) or stainless wire (100 μm diameter for trunk and 40 μm diameter for branches), respectively: one anchored in the organ bath chamber (1 mL) and the other connected to a mechanotransducer (FT03; Natus Neurology, Inc., Middleton, WI). The chamber was perfused at a flow rate of 2.5 mL/min with oxygenated (95% O2 to 5% CO2) Krebs–Ringer bicarbonate solution using a peristaltic pump. The composition (in mM) of the Krebs buffer was 118.3 NaCl, 4.7 KCl, 1.2 MgCl2, 1.22 KH2PO4, 2.5 CaCl2, 25.0 NaHCO3, 11.1 glucose, pH 7.4. Optimal resting tension (0.5–1 g) was applied.
Arterial rings were precontracted with prostaglandin F2α (1 μM) or norepinephrine (1 μM) and EDR was induced by ACh.
Immunoblotting
For immunoblotting, cell lysates were prepared for each type of sample; total protein was measured using a bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). The same amounts of total protein were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis on 7.5–12% gels and transferred to nitrocellulose membranes (Invitrogen, Eugene, OR). Membranes were blocked for 1 h in 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween-20 and incubated overnight at 4°C with primary Abs diluted in blocking buffer. After that, membranes were washed three times with Tris-buffered saline with 0.1% Tween-20 and incubated for 1 h with horseradish peroxidase-conjugated secondary Abs diluted in blocking buffer. The immunoblots were visualized by chemiluminescence reagents obtained from GE Healthcare (Piscataway, NJ). Data processing was performed using a luminescent image analyzer LAS-3000 (Fujifilm, Tokyo, Japan) and IMAGE GAUSE software.
Abs against KCa2.3 (sc-28621), KCa3.1 (sc-32949), p-ERK (sc7383), NOX4 (sc3014), GAPDH (sc-25778), and β-actin (sc-130656) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-ERK Ab (4695S) was purchased from Cell Signaling (Danvers, MA). Abs specific to NOX2 (ab31092), GPX1 (ab22604), and catalase (ab16731) were obtained from Abcam (Cambridge, MA). Anti-fyn (clone EPR5500) and p-fyn (07-909) antibodies were obtained from Millipore (Temecula, CA; MABT208).
Tissue preservation and immunohistochemistry
Placental tissues from NP and PE subjects were fixed by immersion in a periodate-lysine-2% paraformaldehyde solution overnight at 4°C. Tissues were cut transversely into 1–2-mm-thick slices and processed for immunohistochemical studies using a horseradish peroxidase technique. Tissue slices were embedded in paraffin. Four-micrometer sections were deparaffinized with xylene and hydrated in a graded series of ethanol. After rinsing in tap water, sections were incubated with 3% H2O2 for 30 min to eliminate endogenous peroxidase activity. The sections were treated with blocking serum for 30 min and incubated overnight at 4°C in primary Abs (KCa2.3 and KCa3.1). After being washed in phosphate-buffered saline (PBS), the sections were incubated for 2 h with the peroxidase-conjugated donkey anti-rabbit IgG Fab fragment (Jackson ImmunoResearch Laboratories, West Grove, PA) diluted 1:100 in PBS. After being rinsed with Tris-HCl buffer, the sections were exposed to a mixture of 0.05% 3,3′-diaminobenzidine and 0.01% H2O2 for 5 min at room temperature. The sections were dehydrated with graded ethanol and xylene, mounted in Permount, and examined by light microscopy.
Reagents
Reagents were obtained from Sigma-Aldrich and dissolved in autoclaved distilled water unless indicated otherwise. Cells were treated with progesterone (dissolved in ethanol), E2 (dissolved in ethanol), ox-LDL, or LPC (dissolved in chloroform:methanol, 2:1) for 24 h. To neutralize cell surface proteins, cells were pretreated with Abs specific to VEGFR1 (ab32152; Abcam), VEGFR2 (No. 2479; Cell Signaling), or anti-isotype control (rabbit IgG monoclonal) Ab (ab172730; Abcam) for 1 h. 1-EBIO (Tocris Bioscience, Bristol, United Kingdom) was used to activate KCa2.3 and KCa3.1 currents.
The final concentration of dimethyl sulfoxide (DMSO), chloroform, methanol, or ethanol in media was <0.1%, and these solvents did not have any effect on the activities tested in this study (data not shown).
Statistics
Data are mean ± standard error of the mean. To prove the statistical significance between groups, two-tailed Student's t-test or one-way analysis of variance was used. A p value of 0.05 or lower was considered statistically significant. Calculations were performed with SPSS 14.0 for Windows (SPSS, Chicago, IL).
Footnotes
Acknowledgments
This research was supported by the Basic Science Research Program through the Nation Research Foundation of Korea funded by the Ministry of Education, Science and Technology (NRF-2013R1A1A2010851, NRF-2013R1A1A2064543, 2016R1D1A1A09919073, and 2016R1D1A1A09918769) and intramural research promotion grants from Ewha Womans University, School of Medicine.
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.