
Abstract
Significance:
We proposed lysophospholipids (LPLs) and LPL-G-protein-coupled receptors (GPCRs) as conditional danger-associated molecular patterns (DAMPs) and conditional DAMP receptors as a paradigm shift to the widely accepted classical DAMP and DAMP receptor model.
Recent Advances:
The aberrant levels of LPLs and GPCRs activate pro-inflammatory signal transduction pathways, trigger innate immune response, and lead to tissue oxidative and inflammatory injury.
Critical Issues:
Classical DAMP model specifies only the endogenous metabolites that are released from damaged/dying cells as DAMPs, but fails to identify elevated endogenous metabolites secreted from viable/live cells during pathologies as DAMPs. The current classification of DAMPs also fails to clarify the following concerns: (i) Are molecules, which bind to pattern recognition receptors (PRRs), the only DAMPs contributing to inflammation and tissue injury? (ii) Are all DAMPs acting only via classical PRRs during cellular stress? To answer these questions, we reviewed the molecular characteristics and signaling mechanisms of LPLs, a group of endogenous metabolites and their specific receptors and analyzed the significant progress achieved in characterizing oxidative stress mechanisms of LPL mediated tissue injury.
Future Directions:
Further LPLs and LPL-GPCRs may serve as potential therapeutic targets for the treatment of pathologies induced by sterile inflammation. Antioxid. Redox Signal. 28, 973–986.
Introduction
D
Generally, DAMPs act via pattern recognition receptors (PRRs) to drive intracellular signaling pathways, such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), activator protein-1 (AP-1), and initiate innate immune responses, promoting inflammation and tumorigenesis. Canonical PRRs include Toll-like receptors (TLRs), receptor for advanced glycation end product (RAGE), C-type lectin receptors, which are mainly found in the plasma membrane, and scan the extracellular milieu and endosomal compartments for DAMPs. Moreover, NOD-like receptors (NLRs) and retinoic acid inducible gene-I (RIG-I) absent in melanoma-2 (AIM2) are recognized as cytoplasmic PRRs (55).
Although it has been widely studied how DAMPs and PRRs work, the current classification of DAMPs has few limitations as it fails to answer the following concerns: (i) Are molecules which bind to PRRs the only DAMPs contributing to inflammation and tissue injury? (ii) Are all DAMPs acting only via classical PRRs during cellular stress? According to current understanding of how receptors and their ligands work, we pointed out that it is not feasible that all the DAMPs act only via classical PRRs during cellular stress. In addition, classical DAMP model specifies only the endogenous metabolites that are released from damaged/dying cells as DAMPs, but fails to identify elevated endogenous metabolites secreted from viable/live cells during pathologies as DAMPs.
In one of our most recent publications, using endogenous metabolites such as Lysophospholipids (LPLs) and their G-protein-coupled receptors (GPCRs) as a prototype, we argued extensively that LPLs, which mediate homeostatic cellular functions at physiological concentrations, can convert themselves to “conditional DAMPs” at elevated concentrations and initiate sterile inflammation during various pathologies (27, 42, 66). Furthermore, we coined the term “conditional DAMP receptors” for the receptors of such endogenous metabolites, as they would trigger inflammatory signals when the concentration of their ligands reaches pathophysiological levels. In our publication, we recognized LPL-GPCRs as conditional DAMPs.
LPLs, which are a class of bioactive molecules, were initially identified as precursors and metabolites in membrane phospholipid biosynthesis. Later, LPLs were recognized as extracellular signaling molecules, performing their wide range of cellular functions on diverse cell types, including lymphocytes, macrophages, smooth muscle cells, endothelial cells, and neuronal cells.
Prominent among the LPLs are lysophosphatidic acid (LPA), sphingosine 1-phosphate (S1P), sphingosylphosphorylcholine (SPC), lysophosphatidylcholine (LPC), lysophosphatidylethanolamine (LPE), lysophosphatidylserine (LysoPS), lipophosphoglycan (LPG), and lysophosphatidylinositol (LPI). As mentioned above, LPLs bind to groups of specific GPCRs to act as auto- or paracrine regulators of mitogenic effects, cell differentiation, cell survival, cytoskeletal reorganization, process retraction, cell migration, Ca2+ homoeostasis, and Ca2+ dependent functions (71).
Except LysoPS and LPE, which seem to exert anti-inflammatory effects (12, 22), most LPLs are increasingly assumed to represent important disease markers. For example, even though LPC was reported to exert many physiological functions, increased accumulation of LPC was found in ischemic cardiac tissues and plays a significant role as a mediator of arrhythmogenesis following a myocardial infarction (58). Furthermore, others and we have demonstrated increased levels of LPC in atherosclerotic models and implicated LPC as a pro-inflammatory mediator during the progression of the disease (27).
In addition, pathophysiological levels of LPC were shown to induce long-lasting superoxide production, trigger pro-inflammatory actions in leukocytes, endothelial cells, and smooth muscle cells, and further drive cellular damage, thus indicating that it acts as a potential danger signal to the host (42). Meanwhile, studies also revealed that the expression of the GPCRs varies upon the state of immune-cell activation (50). Of note, the binding of LPLs to GPCRs leads to a sequential cascade of transcriptional regulatory events and generation of ROS, which are also commonly associated with conventional DAMPs and PRR mediated signaling (38). Previous publications from our laboratory also demonstrated that LPC significantly induces mitochondrial ROS (mtROS) production and activates an inflammatory cascade in endothelial cells (27 –29).
MtROS have been suggested to play an essential role to innate immunity responses (28), such as inducing production of pro-inflammatory cytokine and reprogramming macrophage, although the mechanisms linking innate immune signaling to mitochondria for mtROS generation remain unclear. Therefore, even LPLs and their GPCRs are not formally considered as DAMPs; based on myriad of evidence published so far, it can be surmised that by modulating its concentration and receptor levels, LPLs play a significant role in regulating wide range of cellular functions in pathological conditions as conditional DAMPs.
The purpose of this review is to broaden the paradigm of DAMPs and consolidate LPLs and their GPCRs can work as conditional DAMPs (66). In addition, we would introduce the significant progress achieved in oxidative stress mechanisms of LPL mediated tissue injury.
Most of the LPLs Act as “Conditional DAMPs”
Lysophosphatidic acid
LPA is a small glycerophospholipid that is present in all eukaryotic tissues at low concentrations. LPA concentrations in human plasma are also very low, but in serum are much higher at several micromolars. Other than the production from activated platelets which accounts for a small percentage of total LPA levels, LPA generation includes multiple derivations from phosphatidic acid (PA) and other LPLs, such as LPC and LPI by the actions of phospholipase A1 (PLA1), phospholipase A2 (PLA2), phospholipase D (PLD), and lysophospholipase D (LysoPD) (2) (Fig. 1). As an important lipid mediator, LPA exerts a series of physiological effects, including platelet aggregation, posthemorrhagic vasoconstriction, vasoregulation, smooth muscle contraction, cytoskeletal reorganization, immune cell chemotaxis, and stimulation of cell proliferation.
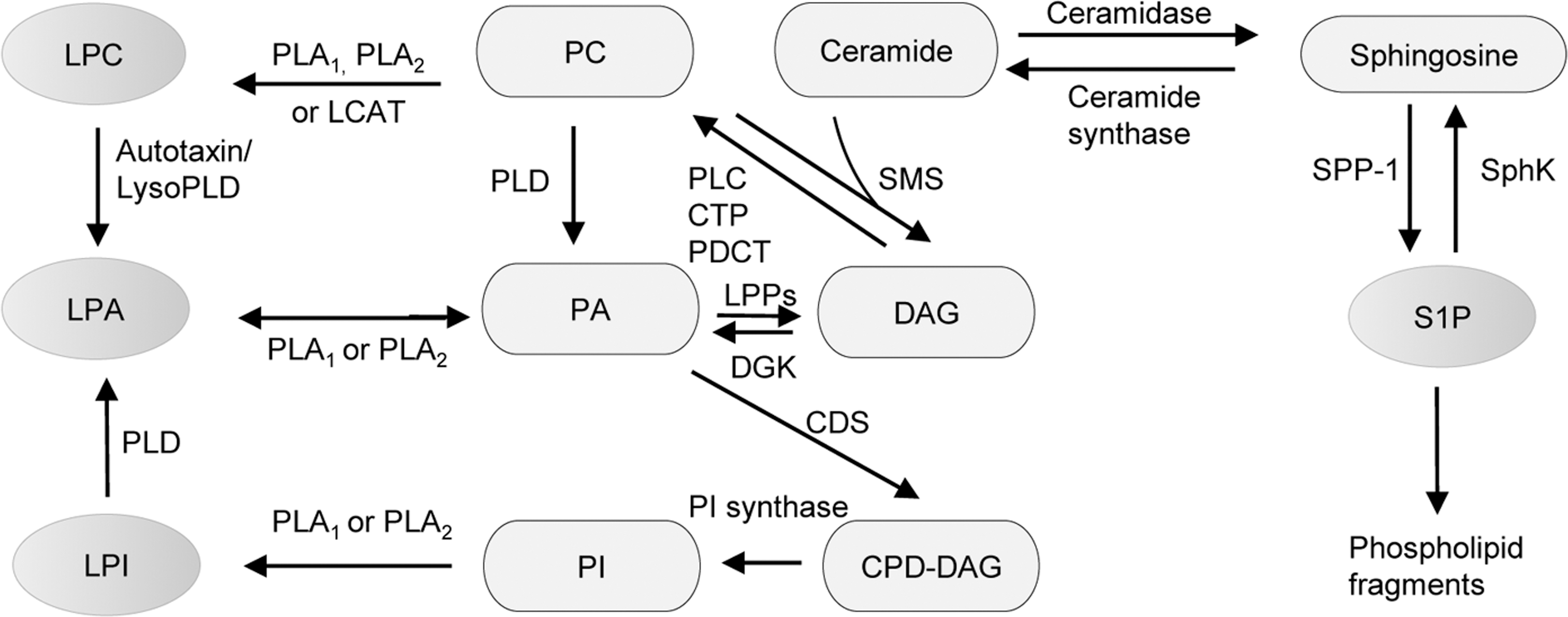
LPA seems to exert its biological activities via a group of high-affinity cell surface GPCRs termed as LPA receptors 1–6 (LPA1–6) (7). LPA1 is the most widely and highly expressed receptor, while LPA2 and LPA3 have more tissue-specific expression patterns, in which LPA2 is most abundant in embryonic brain, kidney, LPA3 in lung, and kidney with low expression in other tissues. Different from LPA1–3, LPA4 expression has a great variance between species. LPA4 has been found in most of the mouse tissues, while human LPA4 mRNA is ubiquitously expressed and specifically abundant in the ovary (26). LPA5 has broad low levels of expression in many tissues, including the embryonic brain, and is enriched in the small intestine (36). P2y5/LPA6 is the most recently identified LPA-GPCR and was implicated in human hair growth.
During development, LPA1–2 mediate LPA induced embryonic vascular formation by regulating endothelial cell survival and migration. Although LPA1 and LPA2 double knockout mice (LPA1−/−/LPA2−/−) have no additional phenotypes relative to LPA1 single knockout mice (LPA1−/−), except for a higher frequency of pups with frontal hematoma, the severe losses of LPA cellular signaling observed in fibroblast cells from LPA1−/−/LPA2−/− indicate that LPA2 does act redundantly with LPA1 to mediate most LPA responses (7).
Aberrant LPA production, receptor expression, and signaling contribute to cancer initiation, progression, and metastasis. LPA released from malignant ovarian epithelial cells or mesothelial cells, acting through LPA4, modulates vascular endothelial growth factor (VEGF) responsiveness by inducing VEGF receptor (VEGFR)-2 expression (48). However, LPA4 also performs a suppressive effect on cell motility; therefore ectopic expression of LPA4 strongly inhibited migration and invasion of human cancer cells (26). LPA5 exerts disparate functions in cancer development in that LPA5 and LPA2 perform an anti-invasive action in melanoma cells in response to LPA, while LPA5 and LPA1 receptors in host tissue microenvironment facilitate the seeding of metastasis.
In addition to significantly elevated levels of LPA in ovarian and other gynecologic cancers, higher levels of LPA were observed in many pathophysiological statuses, such as angiogenesis, inflammation, atherosclerosis, insulin resistance, and obesity (7).
In response to inflammation, LPA diffuses into the extravascular space, where it performs a wide variety of biological activities. Increased LPA stimulates mast cells to proliferate and generate cytokine, which is mediated by LPA1/LPA3 and LPA2, respectively. In addition, LPA acts on LPA5 to induce macrophage inflammatory protein-1 beta (MIP-1 beta) release, which is a potent chemoattractant for, and activator of, monocytes, lymphocytes, and a wide variety of immune cells (36). Monocyte-dependent innate immune responses through LPA receptors for LPA show similar pattern as their activation is through PRRs (such as TLR-2, TLR-4) for bacterial products (PAMP) and complement fragments (DAMP) (62).
Particularly, high levels of LPA play a vital role in development of atherosclerosis. LPA present in the core region of atherosclerotic plaques stimulates LPA1 and LPA3, which are expressed in lesioned endothelial cells, to trigger rapid platelet activation and monocyte adhesion (52). Of note, LPA3 shows a significantly higher affinity for unsaturated LPA than saturated LPA, leading a stimulation of vascular smooth muscle cell (VSMC) phenotypic modulation, which is a hallmark of the development and progression of atherosclerosis (18). Moreover, LPA5 has also been identified involving human platelet activation through the unique ligand selectivity, which provides a basis for future antithrombotic therapies (69). These findings provide clues for understanding that aberrant LPA induces vascular inflammation and VSMC dedifferentiation to promote atherogenesis as conditional DAMP through its GPCRs.
Loss of function studies that were published previously indicated that LPA and its receptors are essential regulators of normal physiological functions. However, previous reports had clearly demonstrated that LPA levels are significantly increased in sterile inflammatory conditions such as cancers, atherosclerosis, vascular injury, and obesity and initiate innate immune response. Therefore, as LPA mediates inflammatory responses at elevated levels and normal biological functions under physiological levels, LPA can be classified as a “conditional DAMP.”
Lysophosphatidylcholine
LPC is a vital plasma lipid component and transports fatty acids and choline to tissues. Unlike other LPLs, the physiological concentration of LPC in body fluids, including blood and ascites, is very high (5–180 μM) (71). Except the generation from hydrolysis of phosphatidylcholine (PC) by PLA2, circulating LPC is majorly produced by the activity of lecithin–cholesterol acyltransferase (LCAT), which transfers the acyl group from PC to lyso-PC instead of to cholesterol. Different concentrations and different forms of LPC present in various cellular and tissue systems may regulate cellular functions differentially (8). G2A, a GPCR expressed predominantly in myeloid and lymphoid cells, had been found to mediate proximal effector of LPC-mediated proatherogenic action. Loss of G2A ameliorates aortic atherosclerosis and provides a candidate for therapeutic inhibition in atherosclerosis (46).
A recent study proposed that circulating levels of LPC are increased in chronic inflammatory diseases with prolonged acute phases as a result of lipoprotein oxidation and the conversion of normal plasma high-density lipoprotein (HDL) into pro-inflammatory “acute-phase HDL” (47). LPC can accumulate in pathological conditions (27), such as ischemia and atherosclerotic aortas, and alter various functions in a number of cell types, including endothelial cells (ECs), smooth muscle cells, monocytes, macrophages, T cells, and B cells.
LPC is considered as a major proatherogenic component of oxidized low density lipoprotein (oxLDL) based on its pro-inflammatory actions in vitro and also an important lipid intermediate that links saturated fatty acids to insulin resistance (16). LPC generated during acute phase response (APR), a physiological alarm response of the body to tissue injury, trauma, or infection, may provide signals of “danger” that are detected by monocyte-derived dendritic cells to potentiate antigen presentation and subsequent adaptive immune responses.
Receptor expression regulated by physiological and pathological conditions would play an important role in controlling LPC-induced effects. G2A not only mediates stimulation of LPC on macrophage and T cell chemotaxis (74) and proapoptotic effects of LPC and therefore promote the death of inflammatory cells within developing lesions (46) but also modulates lipoprotein metabolism, such as limit HDL concentration in plasma and attenuate ApoA1 secretion in hepatocytes. In addition, by implication of specific antagonist, platelet-activating factor (PAF) receptor, a GPCR binding to PAF implicated in diverse pathologic processes, has been identified involving in LPC-induced immune activation (21).
Sphingosine 1-phosphate
S1P, a sphingolipid metabolite, is found in high concentrations in platelets and in blood in forms bound to albumin and lipoproteins. S1P derives from not only hematopoietic system but also from vascular endothelium (64). S1P stimulates members of the Edg family of GPCRs and triggers diverse effects, including cell growth, survival, migration, and morphogenesis. Five S1P receptors have been identified and recently renamed as S1P1 (EDG-1), S1P2 (EDG-5), S1P3 (EDG-3), S1P4 (EDG-6), and S1P5 (EDG-8). Among the S1P receptors, S1P1, S1P2, and S1P3 are widely expressed in various tissues, whereas the expression of S1P4 is confined to lymphoid and hematopoietic tissue and S1P5 to the central nervous system (56).
Similar to LPA1–2, S1P1–3 play a crucial role in blood vessel maturation. S1P1 knockout, S1P1–2 and S1P2–3 double knockout, and S1P1–3 triple knockout mice all exhibit embryonic hemorrhage leading to intrauterine death (24). Conditional S1P1 knockout in endothelial cells indicates that S1P1 signaling in the endothelium is essential to regulate the coverage of vessels by VSMCs. Although showing normal survivor, S1P2 mutant mice have revealed the critical role of this receptor in inner ear physiology, heart and vascular development, vascular remodeling, and vascular tone, permeability, and angiogenesis in vertebrates.
FTY720-P, which is an agonist of S1P1, S1P3, S1P4, and S1P5 receptor at Picomolar to Nanomolar concentrations, is a phosphorylated derivative from immunosuppressor, FTY720 (40). FTY720-P elicits lymphopenia in blood and thoracic duct by sequestering lymphocytes from circulation into the secondary lymphoid organs. Inhibition of lymphocyte recirculation produces clinical immunosuppression preventing transplant rejection, but is associated with transient bradycardia. S1P1-selective agonist does not produce bradycardia, whereas the sustained arrhythmias induced in wild-type mice are diminished in S1P3−/− mice, indicating that S1P1 and S1P3 regulate lymphocyte recirculation and heart rate, respectively.
S1P3 also contributes to lymphoid endothelial cell development and transduces signals that induce important endothelial cell-dependent vascular responses, including eNOS (endothelial cell nitric oxide synthase)-mediated vasodilation (57). S1P4 has been demonstrated involving dendritic cell function and TH17 cell differentiation in a murine model. A number of in vivo models of TH1 (T-helper 1)- or TH2 (T-helper 2)-dominated immune reactions revealed that S1P4 deficiency consistently increased the amplitude of TH2-dominated immune responses, while those depending on TH1-dominated mechanisms were abolished (56).
Moreover, S1P receptors have been found to devote great role in central nervous system. S1P1 and S1P3 are expressed on blood–brain barrier (BBB) endothelial cells and astrocytes that work mutually to coordinate exchanges between the blood and brain. S1P5, which is crucial in cell migration, has been also implicated in regulating BBB function and underlies the expression of specific BBB endothelial characteristics such as tight junctions and permeability (61). Besides, S1P5 could limit neurovascular inflammation and maintain the immunoquiescent state of brain EC with low expression levels of leukocyte adhesion molecules and inflammatory chemokines and cytokines.
In addition to plasma lipoproteins, LDL that is accumulated in the atherosclerotic lesions may release S1P and stimulate S1P production in VSMCs by activating sphingosine kinase activity (60). In injured vessels, activated platelets may also release a large amount of S1P. Elevated level of S1P, in turn, works as danger signal to accelerate development of atherosclerosis. High levels of S1P1 expression in VSMCs result in an increased proliferative and migratory response to S1P, suggesting a role of S1P1 in the pathogenesis of atherosclerosis and restenosis after angioplasty. In addition, S1PR2 has been identified as a novel therapeutic target for vascular disorders due to its role in regulating endothelium proadhesion and pro-inflammatory phenotype (79).
Interestingly, lipopolysaccharides (LPS, a TLR-4 ligand) and S1P cooperate to increase pro-inflammatory molecules in endothelial cells and significantly enhance adhesion of leukocytes under shear stress conditions compared with the effect of either ligand alone (11). This further provides evidence that S1P and its GPCRs not only “work as” but also “work with” DAMPs to alerting the host to drive immune responses.
Lysophosphatidylinositol
LPI/GPR55 system is a new metabolic pathway of clinical relevance. LPI is mainly generated by the calcium-dependent PLA1–2 that catalyzes the hydrolysis of phosphatidylinositol (PI) (39). GPR55 has been identified as a specific and functional receptor for LPI (43). GPR55 is a member of the rhodopsin family of GPCRs and is involved in processes such as migration, proliferation, bone physiology, and inflammatory and neuropathic pain, and in terms of expression it is mainly found in regions of the central nervous system (CNS), adrenal glands, and gastrointestinal tract. In addition, LPI can also act as a potential second messenger in endothelial cells, which allow a receptor-independent fine tuning of Ca2+ sensitive processes within vascular cells and hence essentially contribute to the control of adequate blood flows in various organs and tissues.
Recent finding revealed the potential role of LPI/GPR55 system in human obesity, insulin sensitivity, and atherosclerosis (30, 39). As LPI levels are increased in obese patients, elevated GPR55 levels have been detected in both visceral and subcutaneous adipose tissue of obese subjects and further so in obese patients with type 2 diabetes (39).
Based on ample evidence available so far, it can be concluded that above mentioned LPLs indeed play a vital role as signaling molecules in regulating many biological functions. We have discussed in detail that at elevated levels, LPLs make LPLs become pro-inflammatory mediators, which are very different from their functions at physiological levels. By acting as initiators of inflammation at increased concentrations, many LPLs facilitate the propagation of sterile inflammatory disorders such as cancers and cardiovascular diseases. Thus, based on our previous publication and the evidence provided above, we conclude that LPLs can be categorized as “conditional DAMPs.”
Most interestingly, it is obvious that LPLs use their own LPL-GPCRs to mediate both physiological and inflammatory signaling under different circumstances. Therefore, it can be justified to classify LPL-GPCRs as conditional DAMP receptors. However, it is possible that some endogenous metabolites may have their own receptors to exert biological functions, but may mediate their inflammatory signaling via conventional PRRs. It is evident that LPL-GPCRs and conventional PRRs share the same downstream mediators when propagating inflammation; thus further validating LPLs and LPL-GPCRs are “conditional DAMPs” and “conditional DAMP receptors,” respectively. We have further described the common signaling mediators to LPL-GPCRs and PRRs below.
LPLs Regulate Signaling Pathways as Conditional DAMPs
As we discussed in our previous publication, LPLs act as signaling mediators by GPCRs and either activate or inhibit downstream signaling pathways such as Rho-associated kinase (Rock), diacylglycerol (DAG), inositol 1,4,5-trisphosphate (IP3), mitogen-activated protein kinase (MAPK), Ca2+, adenylate cyclase (AC), and phosphoinositide-3-kinase (PI3K). The specificities of LPL signaling via LPL-GPCRs that has been reviewed in this publication are briefly discussed in Figure 2 (66).

In addition, by analogy, we postulate that LPL specific receptors (GPCRs) share similar functional characteristics with classical PRRs. Once PRRs are activated in response to DAMPs or PAMPs, they trigger activation of transcription factors such as NF-κB, AP-1, CREB (cAMP [cyclic Adenosine mono phosphate] response element binding protein), C/EBP (CCAAT/enhancer binding protein), and IRF (interferon regulatory factor), which are also found in GPCR signaling pathways (13). These transcription factors promote induction of pro-inflammatory gene expression and synthesis of a broad range of molecules, including cytokines, chemokines, cell adhesion molecules, and immunoreceptors, which may initiate positive feedback loops that increases tissue damage.
Furthermore, in response to DAMPs, another subset of PRRs such as NLRs get activated and mediate a cellular reaction cascade that trigger activation of caspase-1, leading to maturation of the cytokines interleukin (IL)-1β and IL-18 (25). Recent findings noted that the implication of GPCRs in activation of NLPR3 (NOD-like receptor pyrin domain containing 3), which is a ligand for NLR, indicates that GPCR pathways could also control caspase-1 activation (51).
Accordingly, in this study we further elaborate how LPLs via LPL-GPCR signaling activate innate immunity and inflammation by sharing the downstream targets of classical PRRs (Fig. 3).
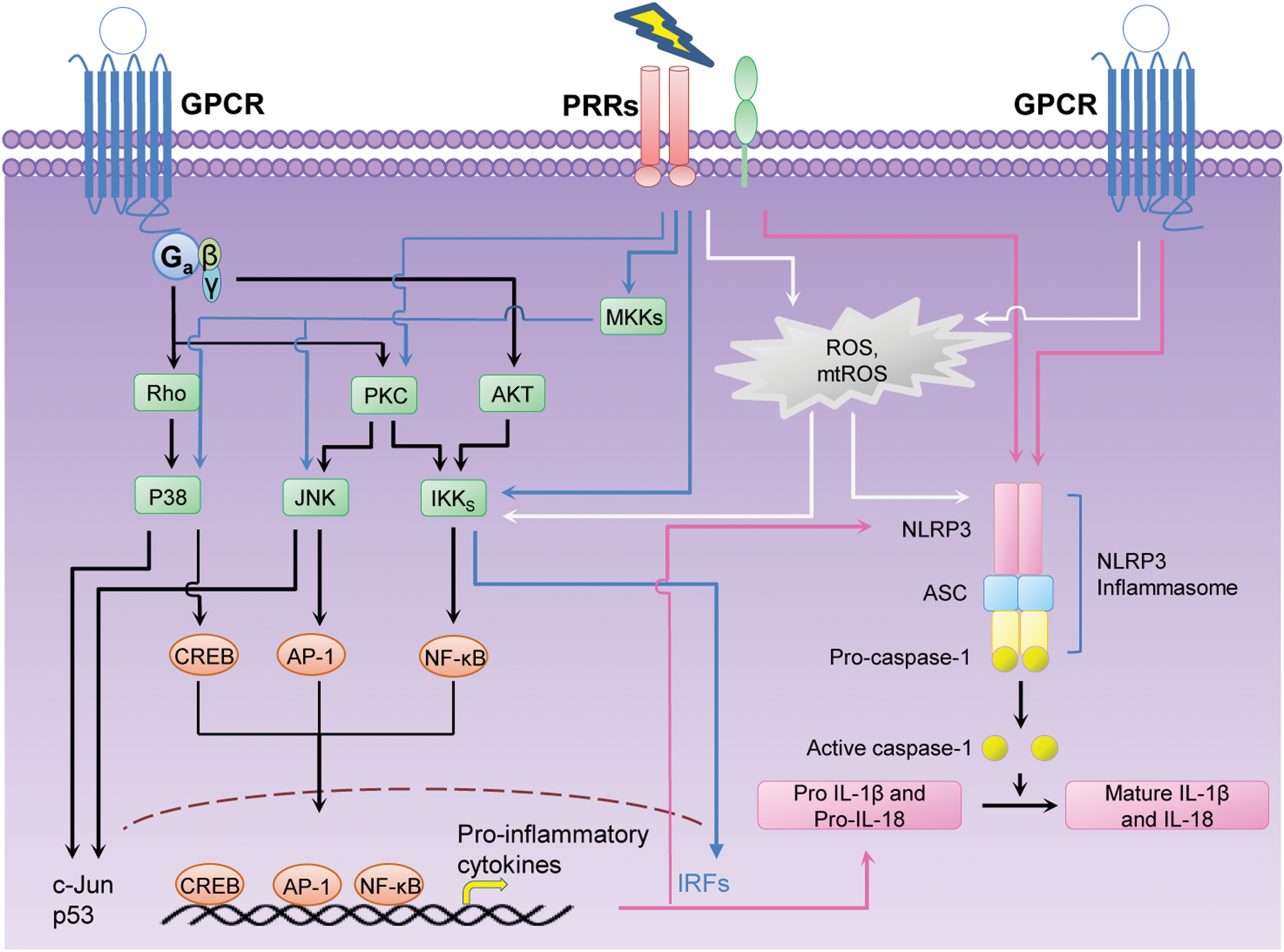
Effects of LPL on transcription factors that mediate inflammation
NF-κB consists of a family of transcription factors that play a critical role in inflammation, immunity, cell proliferation, differentiation, and survival. IκB kinase (IKK) complex is responsible for IκB (inhibitor of kappa B) phosphorylation and is essential for signal transduction to NF-κB (41).
NF-κB pathway has long been considered a prototypical pro-inflammatory signaling pathway. This is largely based on the role of NF-κB in the increased expression of pro-inflammatory genes that lead to production of cytokines, chemokines, and adhesion molecules (20). In turn, inflammatory cytokines and antigen receptor engagement can lead to activation of NF-κB. In addition, opening of calcium channels at the plasma membrane and at intracellular stores is requisite for the basal NF-κB activity. Stimulation of both the calmodulin kinase II and Akt kinase pathways results in the upregulation of the transcriptional potential of the p65 subunit of NF-κB.
Previous publications have shown that LPC exerts its functions via downstream targets such as cAMP and MAPK. Moreover, similar to PRRs, LPC mediated activation of GPCRs and increased the transcription/DNA binding activity of AP-1 and NF-κB in human vascular endothelial cells in a concentration- and time-dependent manner (59). It is necessary to mention that lower concentrations of LPC significantly increased NF-κB activity while higher concentrations inhibited. The time course and the biphasic regulatory actions of LPC on NF-κB might exhibit a unique effect of LPC in arterial walls on the different stages of atherosclerosis, which provide consistent evidence with our hypothesis of “conditional” DAMP.
LPA also can stimulate the same pro-inflammatory transcription factors that are activated by classical PRRs. In epithelial ovarian cancer, LPA contributes to the progression of the disease by promoting cell invasion via NF-κB pathway (9). LPA was also reported to activate NF-κB, AP-1, and EGR-1 in endothelial cells that elevate the mRNA transcripts that encode for encoding E-selectin, intercellular adhesion molecule 1 (ICAM-1), IL-8, and monocyte chemoattractant protein-1 (MCP-1), indicating that it plays a central role in endothelial cell activation, which is the initial step in progression of atherosclerosis. Out of these genes elevated due to increased LPA in endothelial cells, increased expression of E-selectin transcripts was strictly attributed to NF-κB activation (44).
S1P was also implicated as an important regulator of NF-κB activation. It was shown that intracellular S1P, which mediates its actions via GPCR independent pathway, can stimulate NF-κB activation. Similarly, extracellular S1P, which requires GPCRs to exert its effects, is also capable of activating NF-κB (1). This indicates that S1P acts as a mediator in triggering inflammatory, antiapoptotic, and immune response pathways. Moreover, upregulation of sphingosine kinase 1 (SphK1), which is responsible for S1P production and subsequent activation of its S1PR1 receptor, plays an essential role in maintaining persistent activation of NF-κB and STAT3 in a malicious feed-forward amplification loop that leads to chronic inflammation and Colitis-Associated Cancer (CAC) development (32).
In addition, the pro-inflammatory tendency of S1P2 has been revealed in both in vitro and in vivo models. S1P2 deficiency in atherosclerotic ApoE-knockout background (ApoE−/−S1pr2−/− mice) reduced the presence of macrophage-like foam cells in atherosclerotic plaques. Furthermore, S1P2 mediated NF-κB activation has been confirmed to play a crucial role in atherosclerotic disease progression in ApoE−/− aorta. In vitro studies further confirmed that S1P2 mediates a paracrine positive feedback loop that leads to production of tumor necrosis factor (TNF)-α in endothelial cells, which eventually leads to NF-κB activation, and subsequently, increases the expression of adhesion molecules such as in ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1) (80).
Interestingly, LPA can also exert its pathological effects via PRRs similar to classical PRRs. One report indicates that LPA significantly upregulates TLR-4 expression and promotes NF-κB activation, and subsequently, promotes pro-inflammatory cytokine secretion in THP-1 cells, a human monocytic cell line. It is possible that the TLR-4/NF-κB signaling pathway also mediates the atherogenic effect of LPA (73).
Another finding revealed that RAGE was required for LPA-mediated signal transduction in VSMCs and C6 glioma cells and to regulate cell proliferation and migration. Furthermore, inactivation of soluble RAGE or genetic deletion of RAGE reduces LPA-stimulated vascular Akt signaling and LPA-driven Akt phosphorylation in the mammary tissue of transgenic mice vulnerable to carcinogenesis and ovarian tumor implantation and development (49). Meanwhile, similar situation is found in S1P, which regulates TLR trafficking, signaling, and cytokine release (23).
Inflammasome(s)
Besides the activation of the NF-κB, AP-1, and other transcription factors that drive pro-inflammatory cytokine/chemokine production, the effects of LPLs on inflammasome activation platform have drawn widespread attention recently. Inflammasome is a protein complex that is assembled in response to stimulation of DAMPs and PAMPs. An inflammasome is composed of a NLR such as NLRP3 that senses DAMPs, inactive zymogen pro-caspase-1, and apoptosis-associated speck-like protein (ASC), which acts as an adaptor for NLR and pro-caspase-1 (76). An inflammasome assembly is required for pro-caspase-1 to undergo self-cleavage and convert it to active caspase-1, which subsequently induces maturation of pro-inflammatory cytokines such as IL-1β and IL-18 (55, 75 –78).
Caspase-1 processes pro-IL-1β to a mature form that is rapidly secreted from the cell and that may subsequently lead to cell death termed pyroptosis or inflammatory cell death (70). The related enzyme Caspase-11 also promotes activation of caspase-1 by recruiting caspase-1 to the caspase-11 activation platform. Recently, we reported several new findings on caspase-1 pathway, including identification of novel extracellular and nuclear caspase-1 and inflammasomes (65) and identification of caspase-1 regulation on transcriptomes, that is, IL-1β, IL-18, and sirtuin-1 independent (31).
As we mentioned above, several LPLs are implicated in NLRP3 activation through GPCRs. We have demonstrated before that LPC can induce caspase-1 activation in human aortic endothelial cells (HAECs) (34, 76). Endogenous S1P induces the NLRP3-inflammasome-dependent secretion of IL-1β from macrophages similar to traditional DAMPs such as ATP and uric acid crystals (35). Furthermore, studies on S1PR2−/− mice model revealed a diminished expression of caspase-11 in bone marrow-derived macrophages, which suggest that S1P2R is essential for the induction of caspase 11 in macrophages. These findings provide the evidence that S1P exhibits typical behavior of DAMP through specific GPCRs.
Interestingly, a positive autoregulatory loop between SIP2R and TLR-4 exists in macrophages, which stimulate caspase-11 expression. Furthermore, in addition to NF-κB activation, S1PR2 is also required for caspase-1 inflammasome composition and pro-inflammatory IL-1 and IL-18 cytokine release from aortic endothelium in atherosclerotic ApoE−/− mice. Inhibition of this pathway may lead to a novel therapeutic approach to control vascular disorders such as atherosclerosis (15).
Of note, the NF-κB activating pattern and inflammasome activating pattern interact with each other. Studies reveal that expression of NLRP3 itself is tightly controlled by the activity of multiple signaling receptors. NF-κB activators are necessary, but not sufficient for NLRP3 activation, and a second stimulus, such as ATP, is required for NLRP3 activation in macrophage (5). A recent study from our laboratory first revealed that caspase-1 inhibitor could block LPC induced AP-1 binding to AP-1 consensus nucleotides, but could not block LPC induced NF-κB activation in endothelial cells. It proposed a novel signal pathway of Caspase-1-AP-1, which mediates LPC promoted endothelial cell activation as a DAMP (76) (Table 1).
AP-1, activator protein-1; EC, endothelial cell; ERK, extracellular signal-regulated kinase; GPCR, G-protein-coupled receptor; HAEC, human aortic endothelial cell; HUVEC, human umbilical vein endothelial cell; ICAM-1, intercellular adhesion molecule 1; IL, interleukin; LPA, lysophosphatidic acid; LPC, lysophosphatidylcholine; LPLs, lysophospholipids; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemoattractant protein-1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; S1P, sphingosine 1-phosphate; PKC, protein kinase C; THP-1, a human monocytic cell line; TLR, Toll like receptor; TNF-α, tumor necrosis factor alpha; VCAM-1, vascular cell adhesion molecule 1; VSMCs, vascular smooth muscle cells.
LPLs Contribute to Inflammatory Injury by Activating Tissue Oxidative Stress
Oxidative stress refers to the injury caused to cells resulting from imbalance between ROS generation and elimination. The most commonly reported cellular ROS are free radical hydroxyl (OH−), superoxide (O2 −), nitric monoxide (NO), and some other molecules like hydrogen peroxide (H2O2) and peroxynitrite (ONOO−), which are not free radicals but generate free radicals through various chemical reactions in many situations. Toxicity of ROS contributes to proteins and DNA injury, inflammation, tissue damage, and subsequent cellular apoptosis.
Binding of LPLs to GPCR leads to a sequential cascade of transcriptional regulatory events and generation of ROS, which are also common to DAMP stimulated PRR signaling pathways (38). Therefore, LPLs contribute to tissue oxidation and inflammatory injury by multiple mechanisms, including the high-affinity binding of ligands to GPCR family, the effect on specific cellular responses, the generation of a variety of signal mediators, and formation of immune system receptors and sensors.
ROS and inhibitors
It has been wildly accepted that low concentrations of ROS may function as second messengers in intracellular signal transduction pathways. There are multiple sources of ROS within a cell, including mitochondrial electron transport chain, nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), xanthine oxidase, uncoupled nitric oxide synthase, and cytochrome P450 (27). In addition, different environmental stimuli, such as growth factors, inflammatory cytokines, chemical oxidants, chemotherapeutics, and hyperoxia, could induce excessive ROS production, which react with lipids, proteins, and nucleic acids causing oxidative damage to these macromolecules.
LPLs stimulate ROS production followed by activation of cytokine and growth factor signal transduction pathways. The characteristics of ROS have been studied for many years. Several nonenzymatic and enzymatic antioxidants exist as a safeguard against the accumulation of ROS, whose application favors the mechanism study of LPL triggered oxidative injury in different types of cells and tissues.
LPC evokes ROS generation and inhibits nitric oxide (NO) production in a dose-dependent manner in human umbilical vein endothelial cell (HUVEC). This process induces the caspase-3 dependent apoptosis, which is blocked by NADPH oxidase inhibitor (DPI) (19) and superoxide dismutase (SOD) (45). DPI can also prevent LPC-induced proliferation of rat VSMCs with inhibition of ERK1/2 activation that could be involved in the pathogenesis of arteriosclerosis. In bovine aortic endothelial cells, LPC activates NADPH oxidase to enhance ROS generation and, in turn, enhances matrix metalloproteinase (MMPs) induction, which plays a pivotal role in angiogenesis, vascular remodeling after vascular injury, and instability of atherosclerotic plaque (72).
Moreover, LPC can increase O2 − production from isolated rabbit aorta, suggesting that LPC formed during the oxidative modification of LDL might increase O2 − production in vessel wall, which further promotes LDL oxidation and accelerates the development of atherosclerosis. In addition, LPC produced by sPLA2 also plays a role in CNS damage. LPC-induced ROS generation mediates caspase-1 activation in mouse microglial cells that promote secretion of inflammatory cytokines, which are found at elevated levels in the Alzheimer disease brain (68).
Endogenous LPA can be produced by many tumor cells and, in turn, acts as a growth factor and prevents tumor cell apoptosis in a ROS dependent manner. ROS mediates endogenous LPA induced signaling and further stimulates extracellular signal-regulated kinase (ERK) and Akt phosphorylation and NF-κB activities in ovarian cancer cell (54). In PC-3 human prostate cancer cells, LPA1 and LPA3 mediate LPA induced ROS generation by activating protein kinase C (33). When using DPI and antioxidants, such as N-acetyl cysteine, EUK-134, and curcumin, all LPA-dependent NF-κB activity and cell proliferation were blocked. This phenomenon indicates that ROS is an essential mediator in LPA NF-κB pathway.
In addition, LPA stimulated ROS production in HeLa cells. Studies confirmed that ROS are involved in LPA-induced MAPK signaling pathway in HeLa cells. Moreover, inhibition of catalase with aminotriazole enhanced the effect of LPA on induction of MAPK. In contrast, there was one report that demonstrated that elevated ROS from other risk factors can prevent LPA induced biological effects. For an example, in diabetes, ROS stimulated by high glucose block LPA-mediated regression of retinal neovessels, disrupting angiogenic homeostasis and accelerating proliferative diabetic retinopathy (3).
As we mentioned before, S1P is implicated in many biological processes, including cell migration, survival, proliferation, and angiogenesis, as well as immune and allergic responses. A recent finding revealed that the S1P/S1P1 axis regulates progenitor cell egress and mobilization via activation of ROS signaling on both hematopoietic progenitors and bone marrow stromal cells, and release of chemokine stromal cell-derived factor-1 (SDF-1, also termed CXCL12), which is the most powerful chemoattractant of both human and murine hematopoietic stem and progenitor cells (14). In addition, S1P signaling was found to be involved in hyperoxia-mediated ROS generation. In lung endothelium, S1P1/S1P2 signaling axis mediates the hyperoxia-induced ROS generation through p47phox (phagocyte NADPH oxidase protein p47), which is a major subunit that is required for activation of NADPH oxidase system (17).
It has frequently been reported that the small GTPase Rac2 regulates ROS production via its translocation to the plasma membrane and incorporation in the NADPH oxidase complex. LPA activates Rac in response to pertussis toxin in NIH 3T3 fibroblasts and a similar response in VMSC as well. A recent study reported that in pulmonary endothelium and alveolar macrophages, Prdx6-PLA2 modulates NOX2 activation through generation of LPC for conversion to LPA (63). LPA receptor blocker Ki16425 or cellular knockdown of LPA1 decreased oxidant generation and blocked translocation of rac1 to plasma membrane. In addition, LPC also increases vascular radical generation by activating Rac aside its effect on protein kinase C stimulation.
In contrast, S1P does not activate NADPH oxidase and even negatively regulates Rac activity in VSMC (53). Similarly, activated GPR55 mediates LPI to inhibit ROS production and myeloperoxidase release in neutrophils via repression of Rac2 activity (4).
Mitochondrial ROS
ROS are generated in multiple compartments within the cell. Cell membranes, peroxisomes, endoplasmic reticulum (ER), and mitochondria all have “professional” producers of ROS, such as NADPH oxidase (NOX) complexes and various cytosolic enzymes. ROS accumulation in ER stress (62), altered peroxisomal redox homeostasis (14), and modulation of membrane phospholipids in oxidative stress and molecular integrity (48) have been identified as playing causal roles in age-associated diseases, such as atherosclerosis. Although ROS derived from these organelles contribute to the overall oxidative burden, it is believed that the majority of cellular ROS can be traced back to the mitochondria.
mtROS are generated from partial oxygen reduction to form superoxide at complex I and complex III in the mitochondrial electron transport chain (27). mtROS have been suggested to play essential role to innate immunity responses (28), such as inducing production of pro-inflammatory cytokine, reprogramming macrophage, and activating inflammasome, although the mechanisms linking innate immune signaling to mitochondria for mtROS generation remain unclear.
Recent evidence from our laboratory and others revealed that LPLs such as LPC foster mtROS in the redox cell signaling pathways, altering mitochondrial function and leading to ERK activation and adaptation of the pathological stress (27, 67). The late-breaking application of Seahorse Mitochondrial Function Analyzer provides a robust test for analyzing the key parameters of mitochondrial function, including basal respiration, ATP-linked respiration, proton Leak, maximal respiration, spare respiratory capacity, and nonmitochondrial respiration. Our laboratory first announced that ATP synthesis-uncoupled, but proton leak-coupled, mtROS increase mediates LPC-induced EC activation during early atherosclerosis (27, 29).
In healthy mitochondria, a proton gradient is maintained across the membrane and the matrix that facilitate oxidative phosphorylation. Generally, protons from the matrix are pumped out to the mitochondrial intermembrane space at expense of energy and proton gradient that is created by this process called proton motive force. This gradient drives back proton in to the matrix through ATPase (ATP monophosphatase), which result in production of ATP. However, protons can reenter to the mitochondrial matrix independent of ATP production, which usually leads to a decrease in mitochondrial membrane potential (ΔΨm). This is called mitochondrial proton leak (27, 28).
Typically, mitochondrial proton leak and mtROS generation are considered as “an odd couple,” in that proton leak has been shown to decrease mtROS generation, whereas mtROS have been shown to induce proton leak, suggesting the existence of a feedback loop between mtROS and proton leak. Mitochondrial uncoupling proteins (UCPs), which are proton channels or transporters found in mitochondrial inner membrane, mediate proton leak and dissipate the proton gradient in the mitochondria. UCPs have revealed their important roles in cardiovascular diseases rooted in metabolic imbalance and oxidative stress (29).
Unlike in HUVEC we previously mentioned (19), in HAECs, we found that LPC predominantly produces mtROS instead of NADPH oxidase mediated cytosolic ROS. Interestingly, we observed that LPC mediated increase in mtROS production was accompanied with increased proton leak without affecting the ATP production in HAECs. We found that LPC (10 μM) treatment significantly activated HAECs, and treatment of HAECs with MitoTEMPO, which is a mtROS inhibitor, ameliorates the effects of LPC. Therefore, this suggests that LPC increases mtROS production by enhancing proton leak without affecting the viability of the endothelial cells.
The major function of mitochondrial proton leak may be fine-tuning mtROS generation for cell-signaling purpose, such as endothelial activation. This mtROS induction due to LPC treatment triggered ICAM-1 and other EC activation-related gene expression by regulating nuclear binding of AP-1. Combining to Caspase-1-AP-1 pathway proposed by another study of ours, the ATP synthesis-uncoupled, but proton leak-coupled mtROS increase could possibly be an upstream signal to activate caspase-1, which consequently promotes AP1 to trigger endothelial activation in response to LPC stimuli. In early atherosclerosis mouse model, we further demonstrated that blocking mtROS generation by MitoTEMPO could suppress EC activation and monocyte recruitment into the aorta.
The effect of LPLs on mitochondrial function and mitochondrial oxidative stress differs from each other. LPA concentration dependently increases ROS production in porcine coronary arteries and human coronary artery endothelial cells by substantially decreasing mitochondrial membrane potential and ATP content, which lead to an impairment in the endothelium-dependent vasorelaxation (6). Activated autotaxin/LPA signaling axis suppresses brown adipose differentiation by inhibiting UCP1, which contributes to the basal proton leak of brown adipose tissue mitochondria, and promotes diet-induced obesity in mice (10).
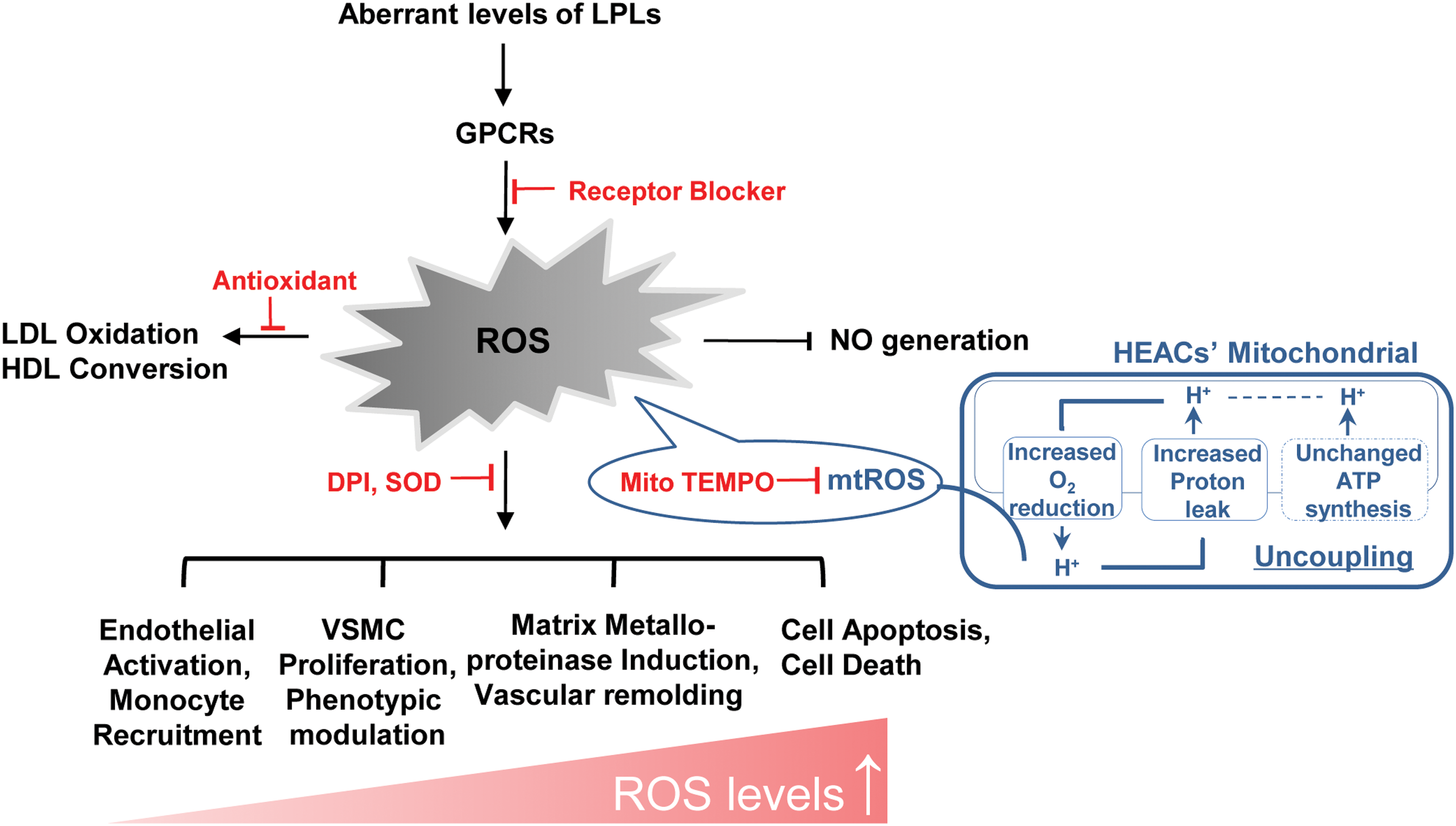
In conclusion, aberrant LPLs, such as LPA, LPC, and S1P, combined with their GPCRs act as conditional DAMPs, initiating innate immune responses, accelerating tissue oxidative stress and inflammation, and leading to atherosclerosis and other cardiovascular diseases (Fig. 4). Attenuating LPL production and blocking LPL-GPCRs can be potential targets for therapeutic intervention in vascular inflammation, tissue oxidative injury, and other inflammatory pathologies such as cancer. Of note, not all LPLs have the potential to become DAMPs. Instead, some LPLs, such as LysoPS and LPE, exist as conditional HAMPs (homeostasis-associated molecular patterns) and perform anti-inflammatory effects, protecting tissue from oxidative injury (66). This indicates that therapies that can enhance LysoPS and LPE also can be novel therapeutic targets for treatment of sterile inflammation.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health Grants to Drs. X.F.Y. and H.W.