
Abstract
Significance:
Hydrogen peroxide (H2O2) is known to act as a messenger in signal transduction. How H2O2 leads to selective and efficient oxidation of specific thiols on specific signaling proteins remains one of the most important open questions in redox biology.
Recent Advances:
Increasing evidence implicates thiol peroxidases as mediators of protein thiol oxidation. Recently, this evidence has been extended to include the peroxiredoxins (Prxs). Prxs are exceptionally sensitive to H2O2, abundantly expressed and capture most of the H2O2 that is generated inside cells.
Critical Issues:
The overall prevalence and importance of Prx-based redox signaling relays are still unknown. The same is true for alternative mechanisms of redox signaling.
Future Directions:
It will be important to clarify the relative contributions of Prx-mediated and direct thiol oxidation to H2O2 signaling. Many questions relating to Prx-based redox relays remain to be answered, including their mechanism, structural organization, and the potential role of adaptor proteins. Antioxid. Redox Signal. 28, 558–573.
The Conceptual Problem of Understanding Hydrogen Peroxide Signaling
H
The first part of the problem is that typical redox-regulated proteins, namely those that are activated or inactivated by thiol oxidation for signaling purposes, including phosphatases, kinases, and transcription factors, have been found to exhibit modest intrinsic H2O2 reactivity (k ≈ 101–102 M −1 s−1) (41). Moreover, many of these proteins are expressed at low levels. It is not obvious how low-reactivity thiols on low-abundance proteins can be oxidized by nanomolar concentrations of H2O2 within time scales appropriate for cell signaling. It is also not clear what mechanisms ensure target specificity of the induced thiol oxidation events. The second part of the problem is that the most prominent group of thiol peroxidases, the peroxiredoxins (Prxs), can be expected to capture nearly all of the H2O2 that is generated inside cells. This is because the intrinsic H2O2 thiol reactivity of Prxs is up to seven orders of magnitude higher (k ≈ 105–108 M −1 s−1) relative to redox-regulated proteins (87). In addition, Prxs are highly expressed proteins. Together, they can amount to ≈1% of the total soluble cellular protein content (7). The concentration of human Prx1 in the cytosol has been estimated as 15–60 μM (49), and that of human Prx2 as 20 μM (84). Arguably, the overall cytosolic Prx concentration is two to three orders of magnitude higher than that of most redox-regulated signaling proteins. Because of the combination of high abundance and high reactivity, Prxs can be expected to easily outcompete all other thiols in the race for H2O2 (85). Given these kinetic considerations, there is currently no overall consensus on how thiols on redox-regulated proteins are actually oxidized.
Within the community of redox signaling researchers, there are two major schools of thought. One school posits that H2O2 directly reacts with thiols on redox-regulated target proteins. It is assumed that H2O2 diffuses from the source to the target protein, where it collides with the target thiol and directly reacts in a bimolecular substitution reaction to form a cysteine sulfenic acid (Cys-SOH) and water as products. Furthermore, it is often assumed or postulated that pK a differences between thiols (as naturally imposed by the local protein environment) can explain the selectivity of thiol oxidation. Prxs (and other peroxidases) are seen as competing H2O2 scavengers that counteract H2O2-mediated oxidation of redox-regulated proteins (60). It is, therefore, reasoned that Prxs need to be inactivated, temporarily and locally, by post-translational modifications. This would facilitate the local accumulation of H2O2 to reach concentrations in the middle to upper micromolar range, which would then allow for the direct oxidation of protein thiols with modest intrinsic H2O2 reactivity (k ≤ 102 M −1 s−1) even in the presence of millimolar glutathione (GSH) concentrations (41). The concept of highly localized H2O2 accumulation is also considered to explain target selectivity, because only proteins localized within or recruited into high-H2O2 microenvironments would be oxidized. The “direct oxidation” concept makes two diagnostic predictions: First, Cys-SOH residues form on the target proteins themselves and should be detectable at these sites. Second, upon experimental deletion of Prxs, H2O2-induced protein thiol oxidation would be predicted to increase.
The second school of thought posits that thiol peroxidases, in particular Prxs, because of their abundance and exceptional intrinsic H2O2 reactivity, would always be the most likely recipients of H2O2, outcompeting all other potential target thiols (58). Indeed, Prxs are known to respond to slight increases in endogenous H2O2 concentration (68) and to become oxidized under signaling conditions, for example, in response to cytokine stimulation (69). It is, therefore, reasoned that redox-regulated target proteins, to become oxidized, must receive oxidizing equivalents from thiol peroxidases. Hence, Prxs (and other thiol peroxidases) are postulated to relay oxidizing equivalents to redox-regulated proteins. They are seen as enablers of protein thiol oxidation, not as competitors (86). If peroxidases act as transmitters of oxidizing equivalents, target and site specificity of thiol oxidation can potentially be explained by protein–protein interactions. These interactions could either be direct or be facilitated by the supramolecular context, that is, by scaffolding proteins that may bring together specific combinations of Prxs and target proteins in appropriate relative orientation. Of note, the “thiol peroxidase redox relay” concept makes predictions that are opposite to those of the “direct oxidation” model: First, Cys-SOH residues should not form on the target proteins themselves, as only thiol peroxidases are prone to react directly with H2O2. Second, upon experimental deletion of Prxs, protein thiol oxidation should be decreased, not increased.
It must be stressed that the two concepts, although making different predictions, should not be perceived as mutually exclusive. H2O2 signaling may occur via different mechanisms in different contexts and situations. Whether H2O2 signals propagate by “direct oxidation” or through a redox relay mechanism may, for example, depend upon the mechanism of H2O2 generation, its duration, and subcellular location. In particular, there may be differences in the mode of H2O2 signal propagation under nonstress and stress signaling conditions. Despite the anticipated plurality of H2O2 signaling mechanisms, it is nevertheless possible that one mechanism is largely predominant under most signaling conditions.
One of the diagnostic predictions for the “peroxidase redox relay model,” as already noted, is that Cys-SOH residues should be found (almost) exclusively in the active sites of thiol peroxidases, but not on the redox-regulated target proteins. On the surface, it could seem that this prediction has already been refuted, by experiments using the SOH reactive compound dimedone (and its derivatives), which suggest that all kinds of proteins form Cys-SOH easily. Thus, this particular observation is discussed in the next section, the specific purpose of which is to point out why dimedone labeling experiments, which have sometimes been portrayed (explicitly or implicitly) as compelling evidence for the direct oxidation of many proteins by H2O2, must be interpreted with caution and should not be used prematurely to draw conclusions about the predominant mechanism of protein thiol oxidation inside living cells. We do not intend to provide a comprehensive overview and discussion of all the observations and arguments that have been put forward in favor of or against the direct oxidation model. Previous reviews have discussed additional aspects not covered here (4, 18, 21, 22, 26, 41, 45, 51, 58, 62).
The Apparent Ubiquity of Protein Sulfenic Acids
1,3-Diketones, in particular dimedone and its derivatives, have been widely used to label Cys-SOH residues, both in lysates and in intact cells (27). Applied to unstimulated and apparently unstressed intact cells, ∼1000 protein thiols in several hundred proteins have been identified as targets of dimedone (91). One interpretation of this finding is that endogenously generated H2O2 molecules spontaneously react with accessible thiols on all kinds of proteins to form Cys-SOH residues, even without the deliberate application of an oxidant or pro-oxidative stimulus. If this interpretation is correct, it would speak in favor of the already mentioned “direct oxidation” model of H2O2 signaling and may even imply that there is no need for peroxidase-based redox relays. Why should the cell make use of a peroxidase as a sensor and transmitter of H2O2-derived oxidizing equivalents if all kinds of thiols on all kinds of proteins are spontaneously and directly oxidized by H2O2? Yet, for this interpretation to be correct, one relies on the correctness of three basic assumptions: first that dimedone is highly specific in forming adducts with Cys-SOH; second that the detected Cys-SOH sites are not created as artifacts of the experimental conditions; and third that the detected Cys-SOH sites are indeed products of the reaction between Cys residues and H2O2.
If we accept all three assumptions as essentially true, how can the direct oxidation of so many protein thiols, inside intact, unstimulated, and unstressed cells, be explained? One possibility is that many thiols in many proteins are surrounded by a local protein environment that makes them sufficiently H2O2 reactive, even at low endogenous H2O2 concentrations. Most commonly it is suggested that a lowering of the thiol pK a value, facilitated by hydrogen bonding and/or electrostatic interactions (64), is sufficient to enable the spontaneous reaction with H2O2 under the given conditions. However, experimental evidence suggests that thiol pK a as such has a limited influence on the overall reaction rate, increasing reactivity by at most one order of magnitude relative to free cysteine (17). This is nowhere near enough to close the five to seven orders of magnitude reactivity gap on peroxidases. To make thiols truly competitive in the race for H2O2, the protein environment would also need to support the execution of the reaction path beyond the initiating nucleophilic attack, namely stabilization of the transition state and promotion of leaving group departure (17). This leads to the question if the thiols detected by dimedone are all located in protein environments harboring such comprehensive catalytic prowess. It has been noted that the majority of dimedone reactive thiols seem to be exposed on protein surfaces (91). Although it is conceivable that solvent-exposed side chains surrounding a surface thiol also interact with the transition state and the leaving group, it seems rather unlikely that they can be structurally organized in a way that would elevate thiol H2O2 reactivity by several orders of magnitude. Alternatively, it may be asked whether the local environment of a thiol could perhaps promote the binding of an ubiquitous small molecule or metal ion capable of catalyzing the direct reaction of the thiol with H2O2. One hypothetical possibility is that zinc ion coordination, usually considered an inert structural feature, under some conditions can promote H2O2 reactivity and thus SOH formation (9, 20). However, it appears to be generally unknown to what extent protein thiol metal interactions contribute to endogenous Cys-SOH generation and thus intracellular dimedone labeling.
Without a known physiological catalyst for the direct reaction between protein thiols and H2O2, it seems inescapable to postulate that the H2O2 concentrations surrounding dimedone reactive thiols must be much higher than the assumed nanomolar range signaling concentrations. Indeed, in an acute signaling context, high local H2O2 concentrations are conceivable. It is frequently assumed that H2O2 emerging from NOX enzymes can reach high local concentrations (79). NOX-generated H2O2 is thought to enter the cytosol most efficiently through the openings of aquaporin channels (2), thus potentially creating high-H2O2 “hot spots” surrounding the aquaporin inlet ports.
The local buildup of high H2O2 concentrations may be facilitated or supported by the local inactivation of Prxs. The first possibility is that the rate of reductive Prx regeneration (or the local availability of reducing equivalents) becomes limiting under high H2O2 flux. Prxs would then accumulate in the disulfide form, and other reactions of H2O2 could occur. The second possibility is Prx inactivation by localized hyperoxidation (89). It is known that Prx hyperoxidation can occur under physiological conditions, for example, hyperoxidation of mitochondrial Prx3 has been observed in corticosterone producing adrenal gland cells (37). However, evidence for the hyperoxidation of cytosolic Prxs under signaling conditions remains to be found (10). The third possibility is that nonoxidative post-translational modifications, specifically phosphorylation, can tune down Prx activity in a localized and transient manner (63). Indeed, there is substantial evidence for kinase-controlled local inactivation of human Prx1 by phosphorylation (40, 59, 88). It should, however, be noted that it remains to be demonstrated that Prx3 or Prx1 inactivation (by hyperoxidation or phosphorylation, respectively) leads to the direct oxidation of redox-regulated proteins by H2O2. It is not obvious that the local inactivation of one Prx isoform is sufficient to allow the direct oxidation of redox-regulated proteins, because other peroxidases and also the highly abundant GSH could still be expected to outcompete redox-regulated proteins. In addition, the inactivation of one Prx family member does not exclude the possibility that target protein oxidation is mediated by one of the other thiol peroxidases (e.g., Prx2 in the case of Prx1 inactivation).
To give one specific example, numerous studies support the conclusion that cysteine-797 on the cytosolic kinase domain of the epidermal growth factor receptor is sulfenylated in response to NOX/dual oxidase activation (30, 50, 78). Indeed, it has been shown that this cysteine, which is exposed on the protein surface, exhibits a lowered pK a (5.5) and elevated H2O2 reactivity (k ≈ 110 M −1 s−1) in vitro (78). Thus, this particular cysteine can be considered a preferential target of H2O2 relative to the majority of “normal” cysteines, which can be expected to be ≈10-fold less reactive. Yet, to rationalize its direct oxidation in vivo, one would need to postulate that there is effectively no competition by peroxidases, and even then, one would probably have to presume local H2O2 concentrations somewhere in the mid-micromolar range, if not in the high micromolar range, depending on the assumed GSH concentration. Despite these uncertainties, it seems conceivable that H2O2 reaches local concentrations that allow cysteines with ≈10-fold increased intrinsic reactivity (k ≈ 102 M −1 s−1) to become preferentially oxidized to Cys-SOH. Perhaps some target proteins are positioned so close to the site of H2O2 generation (or to the point of H2O2 entry into the cytosol) that all potential competitors are spatially excluded. But even in that case, the question remains whether local high-H2O2 hot spots are so common and widely distributed inside untreated and unstressed cells that they can explain the wide-spread dimedone labeling of a very broad variety of proteins (91). It is interesting to note that a recent mathematical modeling study suggests that “direct oxidation” is inefficient even near H2O2 supply sites and even under conditions of Prx inhibition (76).
Given the fact that an explanation for widespread dimedone labeling remains to be found, one is led to the question whether (or to which degree) the three assumptions mentioned at the outset can be justified. Hereunder we briefly consider three theoretical possibilities, namely (i) that some of the detected Cys-SOH sites are created by mechanisms independent of H2O2; (ii) that, to some unknown degree, Cys-SOH formation is caused by the experimental conditions; and (iii) that dimedone is not fully specific and that some unknown degree of dimedone adduction is unrelated to Cys-SOH formation.
(i) If we accept the notion that dimedone adducts specifically indicate Cys-SOH sites and that these are authentic, that is, not caused by the experimental conditions, there is still the question whether all these sites reflect oxidation by H2O2. Indeed, it is known that sulfenic acids can be generated by other reactions. As reviewed recently (27), Cys-SOH can potentially be formed by hydrolytic reactions, including the hydrolysis of nitrosothiols and disulfide bonds. It is, however, uncertain whether these processes play any significant role in living cells. Perhaps more likely, Cys-SOH can also be generated by other oxidants, some of which react more readily with thiols than H2O2, including peroxynitrite. Yet, it is uncertain if peroxynitrite contributes significantly to Cys-SOH formation in intact cells, because redox-regulated proteins would still have to compete with Prxs which are nearly as efficient in reacting with peroxynitrite as they are with H2O2 (77). Another potential source are radical reactions, for example, those caused by the formation and decay of nitrosoperoxycarbonates (13), potentially forming Cys-SOH as one of the oxidation products. But again, the contribution of radical reactions to Cys-SOH formation inside cells is essentially unknown.
Yet another theoretical possibility is that certain enzymes directly sulfenylate thiols in other proteins. In principle, flavin monooxygenases forming a nondiffusible flavin hydroperoxide intermediate could sulfenylate a target thiol on another protein. Indeed, it has been speculated that monooxygenases of the MICAL family use a flavin-bound hydroperoxide to oxidize methionine residues on β-actin (14). However, direct evidence for such a mechanism, for either thioether or thiol oxidation, remains to be found.
Taken together, it seems to be unknown to what extent alternative pathways of Cys-SOH formation, that is, hydrolytic reactions, other kinds of oxidants, radical reactions, or enzymatic sulfenylation reactions, contribute to wide-ranging dimedone labeling in intact cells.
(ii) If we accept the notion that dimedone is highly specific for Cys-SOH, but make no further assumptions, the next question that arises is to what extent the observed adductions truly reflect the unperturbed state of the intact cell. What happens when millimolar concentrations of dimedone or a dimedone derivative act on living cells for up to 2 h (91, 92)? It is conceivable that dimedone inhibits peroxidases and perhaps other enzymes as well (23). It has been suggested, based on whole cell GSH and dichlorofluorescein (DCF) fluorescence measurements, that dimedone does not perturb the “cellular redox state” (50). However, another study observed a strong increase in DCF fluorescence in cells treated with 10 mM dimedone (57). It would seem expedient to address this issue with more refined methods. In the meantime, it seems reasonable not to exclude the possibility that some of the Cys-SOH sites detected by dimedone reagents could have been formed as a cellular reaction to the incubation with the reagent.
(iii) Finally, there is the fundamental question of whether dimedone adduction to a thiol strictly indicates that the trapped thiol was a sulfenic acid before it was trapped by dimedone (Fig. 1; reaction sequence 1 → 2 → 3). It seems difficult to strictly exclude the possibility that there exist other thiol modifications that can also react with dimedone. A case in point may be the observation that 1,3-diketones can react with isothiazolidin-3-ones (i.e., cyclic sulfenyl amides), yielding thioether adducts that are chemically identical to those created from sulfenic acids (20, 65) (Fig. 1; reaction 4 → 3). If cyclic sulfenyl amides can only form from sulfenic acids (Fig. 1; reaction 2 → 4), there is no problem for the interpretation of dimedone experiments, because any dimedone-trapped cyclic sulfenyl amide would faithfully represent a thiol site that had previously been sulfenylated (Fig. 1; reaction sequence 1 → 2 → 4 → 3).
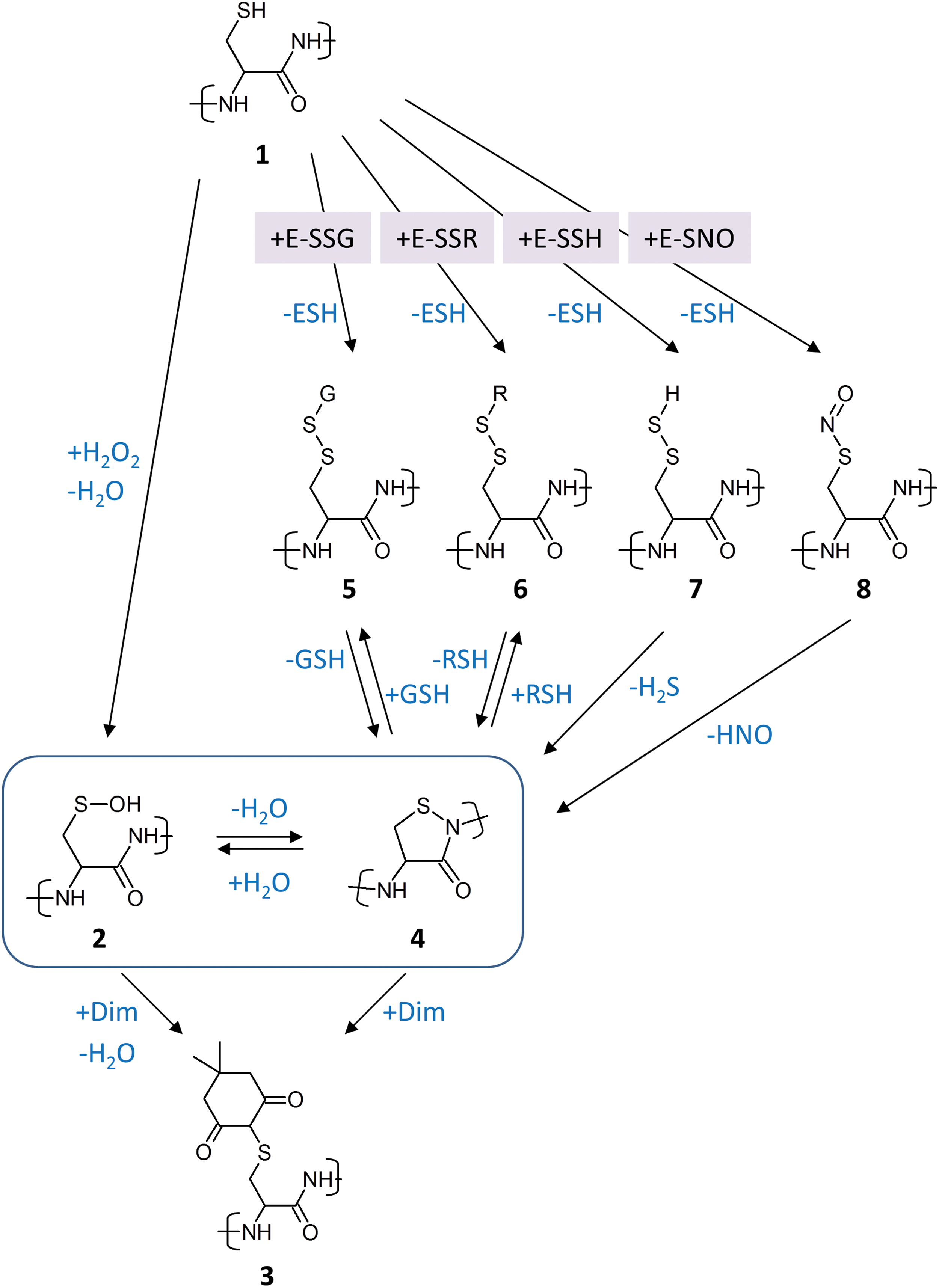
However, the next question that arises is whether cyclic sulfenyl amides could also form from other oxidative thiol modifications, that is, without prior formation of the sulfenic acid on the respective thiol. A recent study using small isothiazolidin-3-one model compounds proposed that glutathionylated thiols can convert into cyclic sulfenyl amides (Fig. 1; reaction 5 → 4) (20). In principle, this seems chemically feasible, as the reverse reaction, that is, ring opening by GSH (Fig. 1; reaction 4 → 5), is known to occur (24). That glutathionylated cysteines can convert into cyclic sulfenyl amides within a protein context, at least in principle, is also suggested by the observation that exposure of phosphatase crystals to glutathione disulfide (GSSG) induced formation of the cyclic sulfenyl amide, as observed by X-ray crystallography (90). Nevertheless, if glutathionylation of protein thiols inside cells would always (or typically) proceed through Cys-SOH condensation with GSH (i.e., Cys-SOH + GSH → Cys-SSG + H2O), then, again, there would be no problem for the interpretation of dimedone experiments, because any cyclic sulfenyl amide generated from a glutathionylated thiol would reflect the prior sulfenylation of the same thiol.
Thus, the next question is whether glutathionylation of reduced thiols by thiol–disulfide exchange reactions is a common event inside living cells. It may be argued that glutathionylation by disulfide exchange is kinetically and/or thermodynamically unfavorable and would require extreme oxidative stress conditions, especially if one is considering GSSG as the glutathionylating agent (31). But there is also the possibility that thiol or selenol peroxidases drive glutathionylation of other proteins by relay mechanisms, that is, by transglutathionylation, either with or without participation of glutaredoxins (20). In this scenario, the sulfenic acid forms and condenses with GSH within the active site of a thiol peroxidase. The peroxidase then glutathionylates proteins in its immediate proximity, in a direct or indirect manner, as will be discussed in detail in the next section. Assuming that peroxidase-driven glutathionylation is efficient, a target thiol would not have to be sulfenylated by itself to become glutathionylated, yet it may then rearrange into a cyclic sulfenyl amide that would potentially be trapped by dimedone, to generate an adduct that would be considered diagnostic for Cys-SOH trapping (Fig. 1; reaction sequence 1 → 5 → 4 → 3).
There does not seem to be a reason to restrict these considerations to glutathionylated thiols. It may be asked whether intra- or intermolecular protein disulfide bonds could also sometimes rearrange into cyclic sulfenyl amides (Fig. 1; reaction 6 → 4). Further along these lines, one may wonder whether yet other kinds of oxidative thiol modifications could do the same, for example, hydropersulfides (Fig. 1; reaction 7 → 4) or nitrosothiols (Fig. 1; reaction 8 → 4). Again, primary thiol oxidation can be expected to take place in the active sites of specialized redox enzymes, which then forward the resulting oxidative modification to other proteins (e.g., thiol peroxidases generating and distributing disulfides, mercaptopyruvate sulfurtransferase and sulfide:ubiquinone oxidoreductase generating and distributing hydropersulfides), as is discussed in the next section. If those redistributed thiol modifications would have some tendency to convert into cyclic sulfenyl amides, many proteins may become susceptible to dimedone adduction without having formed a Cys-SOH residue (Fig. 1; reaction sequences 1→[5, 6, 7, 8]→4 → 3).
Of note, these considerations also imply the theoretical possibility that dimedone, even if it would not react with cyclic sulfenyl amides, could trap Cys-SOH residues which have been formed as hydrolysis products of cyclic sulfenyl amides that did not form from Cys-SOH (Fig. 1; reaction sequence 1→[5–8]→4 → 2 → 3). In this particular scenario, dimedone would be detecting Cys-SOH as intended, yet dimedone adduction would not be indicative of direct Cys oxidation by H2O2. In other words, the very fact that sulfenic acids and sulfenyl amides can interconvert (27) implies the possibility that any thiol oxidation pathway that gives rise to cyclic sulfenyl amides will also give rise to Cys-SOH and, therefore, to dimedone adduction. Indeed, dimedone may be expected to shift the overall sulfenic acid/sulfenyl amide equilibrium by trapping Cys-SOH (Le Chatelier's principle).
The plausibility of any of the mentioned chemical rearrangements to occur inside living cells to any significant extent is certainly debatable, and the actual prevalence of cyclic sulfenyl amides inside cells is essentially unknown. Nonetheless, it seems reasonable to conclude that at present it remains uncertain to which extent dimedone labeling is truly specific for or indicative of Cys-SOH residues. Even under the assumption of absolute Cys-SOH specificity, it remains to be clarified whether dimedone labeling identifies sites of primary thiol oxidation (i.e., sites of direct peroxide–thiol reactions) or rather the hydrolysis products of “secondary” thiol modifications, that is, modifications transferred to the site by redox enzymes via thiol–disulfide exchange, transglutathionylation, transpersulfidation, transpolysulfidation, transnitrosylation, or other transfer mechanisms. Hence, the problem of how to interpret the outcome of dimedone labeling experiments may be considered largely unsolved.
The General Concept of the Thiol Peroxidase-Based Redox Relay
The idea that thiol peroxidases relay H2O2 signals to redox-regulated proteins evades the problems that haunt the idea of direct thiol oxidation, namely low intrinsic thiol reactivity and kinetic competition, as discussed in the previous sections. In the direct oxidation model, thiol peroxidases are only seen as H2O2 scavengers, and thus as competitors of protein thiol oxidation. In the redox relay model, they are also recognized as the cell's most sensitive and abundant collectors and forwarders of peroxide-derived oxidizing equivalents, and thus as highly efficient enablers of protein thiol oxidation. Hence, what is a problem for direct thiol oxidation can be a solution for mediated thiol oxidation. Another conceptual advantage of the relay model is that the selectivity of protein thiol oxidation in redox signaling is easier to rationalize, because protein–protein interactions should allow for more specific recognition of target sites than the diffusional collision of small H2O2 molecules with protein surfaces. Nevertheless, the redox relay concept also raises several difficult questions. Hereunder we first try to define the concept and scope of the redox relay concept, before we look at the existing experimental evidence and discuss open questions.
Thiol peroxidases may participate in redox signaling in several conceptually different ways. One possibility is that the reduced and oxidized forms of the peroxidase (e.g., reduced noncovalent Prx decamers vs. oxidized covalent Prx dimers) differ in how they interact with other proteins, thus influencing downstream events in a manner that depends on the peroxidase redox state, yet without actually involving a redox reaction between the peroxidase and the interacting protein. This is a valid concept, but it does not bear on the problem of how redox-regulated proteins become oxidized and thus will not be discussed here.
The other possibility is that the thiol peroxidase engages in a redox reaction with another protein, thus changing the redox state of that protein. Here we use the term “peroxidase redox relay” to indicate that the peroxidase relays oxidizing equivalents to another protein. The term “signal peroxidase” has previously been used to denote the same concept (35). However, the latter term could be understood to indicate any process by which a peroxidase contributes to signaling, including interactions that do not change the redox state of the interacting target protein, as mentioned in the previous paragraph. Therefore, we here refer more specifically to peroxidase redox relays.
In principle, transmission of oxidizing equivalents from a thiol peroxidase to a redox-regulated target protein can be either indirect or direct. In the case of indirect transmission, representing a two-step redox relay (Fig. 2a), the peroxidase first transfers oxidizing equivalents to a thiol–disulfide oxidoreductase, as it normally does as part of its H2O2 scavenging cycle. The oxidoreductase then oxidizes the target protein. In the case of direct transmission, representing a one-step redox relay (Fig. 2b), the peroxidase oxidizes the target protein directly, by transferring oxidizing equivalents across a common protein–protein interface.
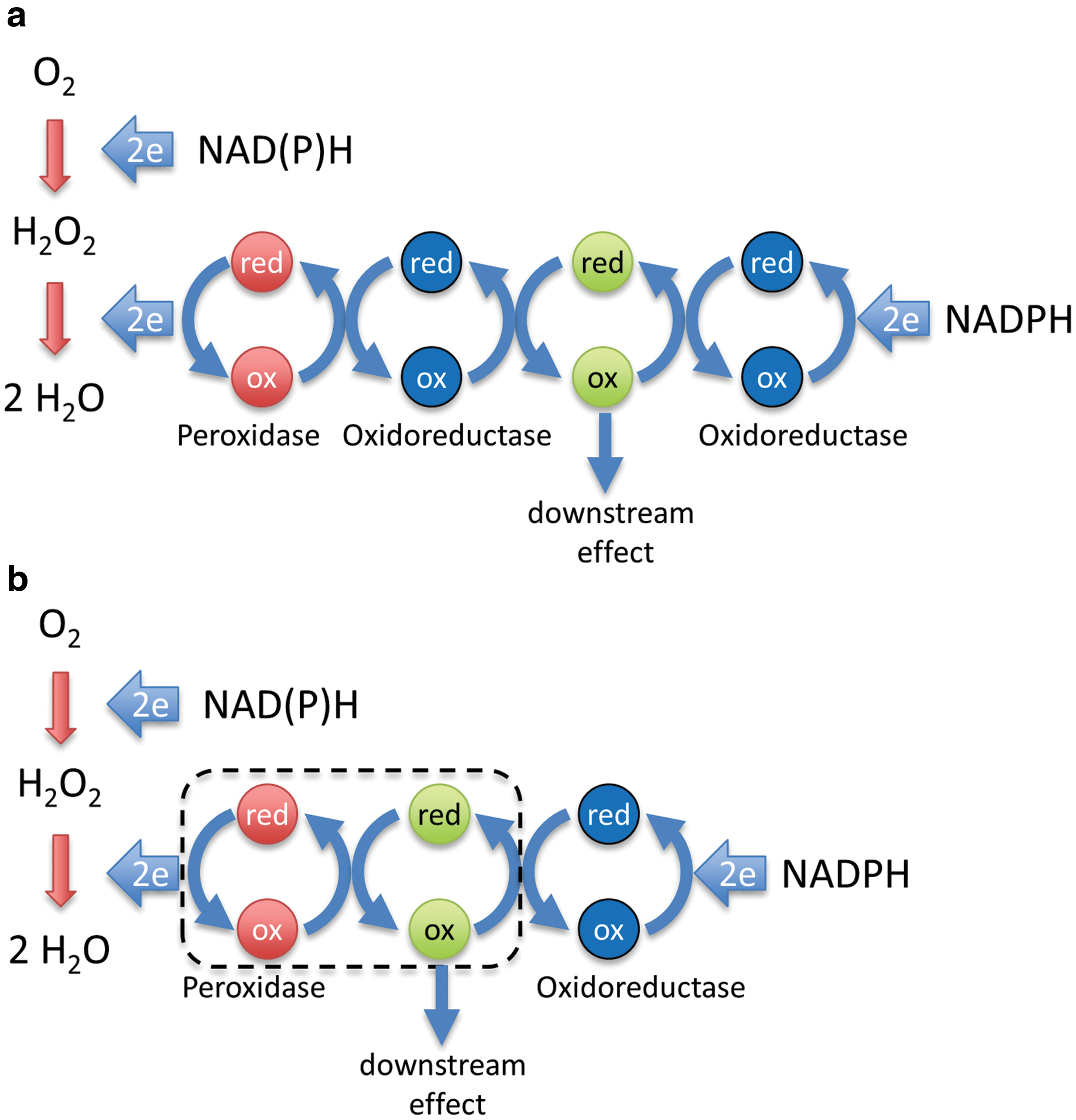
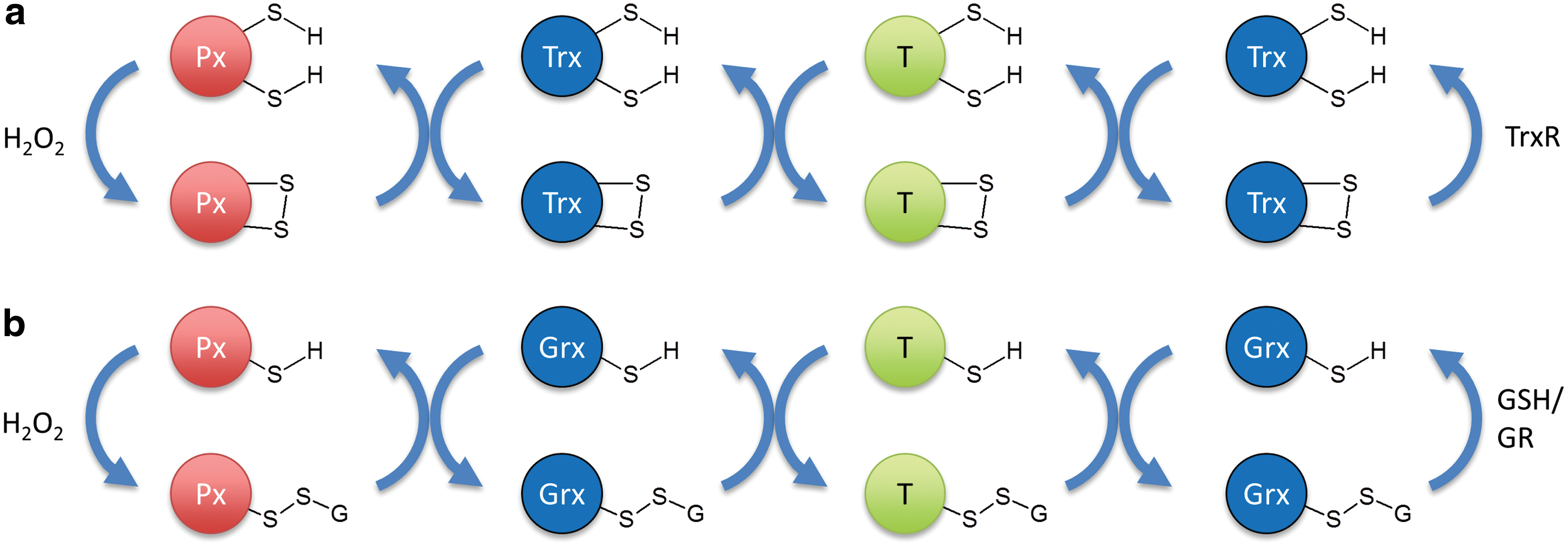
What are possible mechanisms for two-step relays? Prxs, as well as certain members of the glutathione peroxidase (Gpx) family, are very efficient oxidizers of thioredoxin (Trx) (45). If peroxidase-mediated Trx oxidation is faster than Trx reduction, Trx would accumulate in the oxidized state and this may cause a role reversal for Trx, meaning that it would introduce disulfide bonds into target proteins, rather than reducing them. It is quite well documented that Trx can act as a protein thiol oxidase in oxidizing environments or when reducing equivalents are limiting (25, 70). Trx would then oxidize the redox-regulated target protein, and would also reverse the process once H2O2 levels subside and Trx switches back to its reducing function (Fig. 3a). Another possibility for a two-step redox relay would be a transglutathionylation cascade, in which a glutathionylated peroxidase is deglutathionylated by a glutaredoxin, which, in turn, is deglutathionylated by a target protein (Fig. 3b). The latter possibility may also apply to Prxs, which can be glutathionylated at the peroxidatic cysteine and then deglutathionylated by glutaredoxin (53). That glutathionylated glutaredoxins can efficiently forward oxidizing equivalents to proximal proteins is not just a theoretical possibility, but experimentally demonstrated by the intracellular behavior of the rxYFP-Grx1p and Grx1-roGFP2 fusion proteins (3, 28, 42, 66).
What are the expected mechanisms for one-step relays? Several molecular mechanisms can be envisaged: For any thiol (selenol) peroxidase, the primary oxidation product is Cys-SOH or cysteine selenenic acid (Cys-SeOH). This sulfenic (selenenic) acid can directly condense with a target protein thiol. Yet, for this to happen, the incoming target thiol must be brought into proximity. Otherwise it would not be able to compete with the thiol that would normally condense with Cys-SOH (Cys-SeOH), which is typically provided by a resolving cysteine on the peroxidase (Fig. 4a) or by an incoming GSH molecule (Fig. 4b). Alternatively, if the thiol peroxidase already formed a protein disulfide bond, oxidation could be transferred to the target protein by a thiol–disulfide exchange reaction. For this to happen, the target thiol must be positioned in proximity to successfully compete with the canonical disulfide reductant, typically Trx (Fig. 4c). Correspondingly, if the peroxidase is already glutathionylated, the proximal target protein may be oxidized by transglutathionylation or through an intermediary protein–protein disulfide bond. Again, the target thiol must be able to successfully compete with the canonical reductant, typically GSH (Fig. 4d). In principle, there are several other mechanisms that could transfer oxidizing equivalents from a thiol peroxidase to a target protein. For example, the Cys-SOH on the thiol peroxidase may react with H2S to form a hydropersulfide (Fig. 4b), which may then be transferred to proximal recipient proteins by transpersulfidation (Fig. 4e). Thus, the key event of sulfenic (selenenic) acid formation in the active site of peroxidases can be the entry point for the generation and transfer of several kinds of thiol modifications.
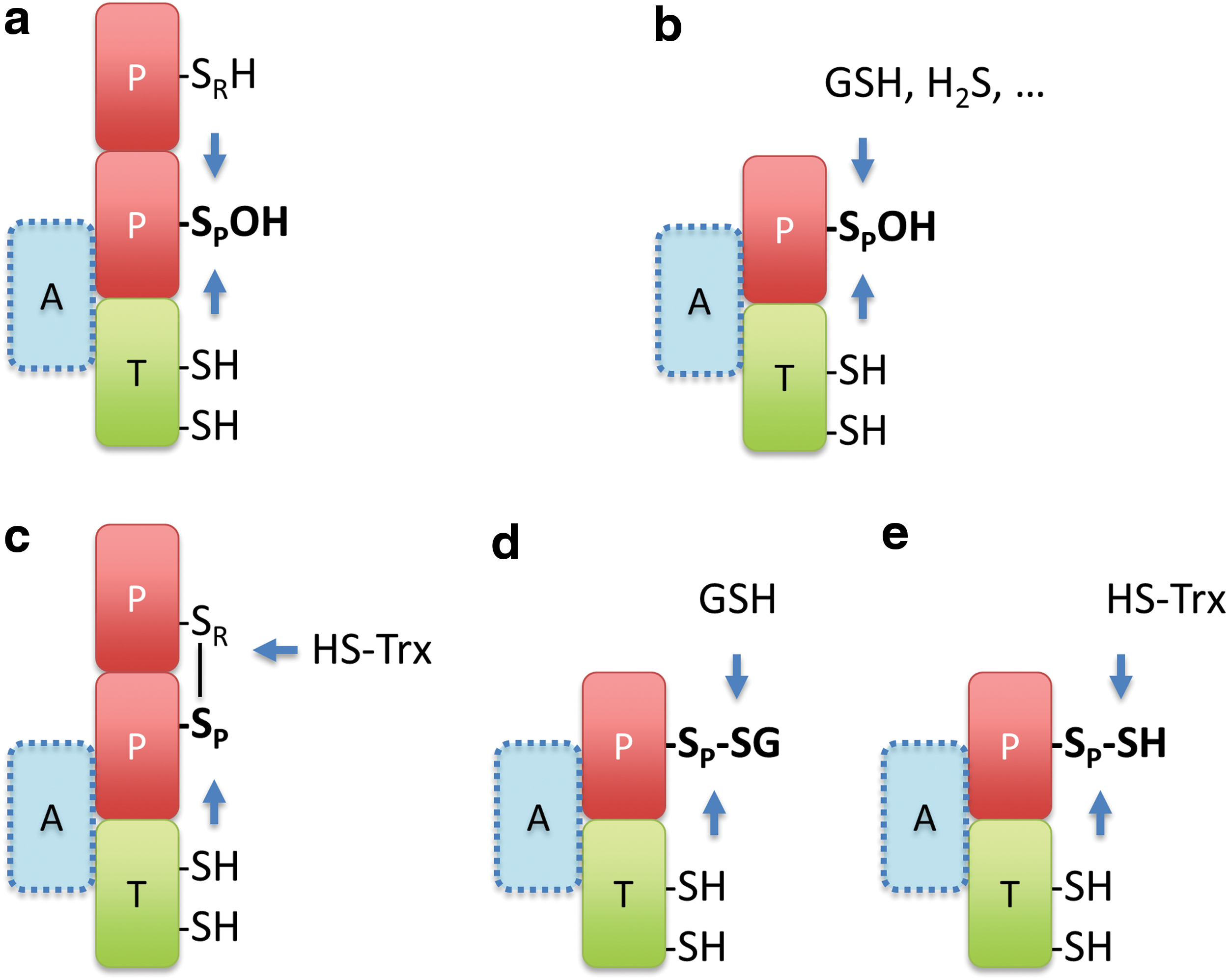
Based on these considerations, it may be postulated that practically all kinds of thiol peroxidases have the potential to form redox relays with other proteins as long as the target protein thiol can be recruited into the immediate proximity of the oxidized peroxidatic cysteine. This allows the client thiol to compete with those thiols that by default reduce the peroxidase and thus tap into the prime cellular source of kinetically accessible oxidizing equivalents. That different families and subfamilies of thiol peroxidases are indeed capable of relaying oxidizing equivalents to other proteins is, for example, suggested by the observation that all eight yeast thiol peroxidases (including members of the Gpx, 1-Cys Prx, typical, and atypical 2-Cys Prx families) do oxidize redox-sensitive green fluorescent protein 2 (roGFP2) when brought into proximity by translational fusion (43).
In summary, we would like to define the general notion of “thiol peroxidase-based redox relays” to include all molecular arrangements and mechanisms, whether direct or indirect, that move an oxidizing equivalent that is initially embodied in a peroxidase sulfenic (or selenenic) acid to a redox-regulated protein, no matter how the oxidizing equivalent is transformed, transmitted, and finally epitomized on the target protein (including disulfide bonding, glutathionylation, persulfidation, and potentially other modifications as well). The only common and defining property of all thiol peroxidase redox relays is that the oxidizing equivalents ending up on redox-regulated proteins must originate from a sulfenic (or selenenic) acid that was first created on a peroxidatic cysteine (or selenocysteine) in the active site of a Gpx or Prx family member.
So far, we have assumed that the oxidizing equivalent always translocates completely from the peroxidase to the target protein and that it is the oxidation of the target protein per se that expresses the relevant functional change. However, there is another formal possibility of how a protein could be functionally redox regulated by a thiol peroxidase: the oxidizing equivalent, instead of being completely transferred, could just be temporarily shared between the peroxidase and the target protein and then either “fall back” onto the peroxidase or be taken away by another reductant. Sharing of an oxidative equivalent is taking place when a peroxidase is disulfide bonded to a target protein. The target protein is half-way oxidized, as the formal oxidation number of its thiol changes from −2 (thiol) to −1 (disulfide). Correspondingly, the peroxidase is half-way reduced as its peroxidatic thiol has changed in formal oxidation number from 0 (SOH) to −1 (disulfide). The disulfide-linked conjugate per se could serve to cause a functional change within the target protein, rather than being an intermediate en route to target protein oxidation. In this scenario, the disulfide bonding would be reversed to release the target protein from the Prx conjugate in the fully reduced state, for example, by the resolving cysteine of the peroxidase or by an oxidoreductase. This conceptual possibility would still fall under the mentioned definition of a peroxidase redox relay, because the oxidizing equivalent that caused a change in the behavior of the target protein, although never completely transferred, originated as a sulfenic (selenenic) acid on a thiol peroxidase.
Evidence for Prx-Based Redox Relays
Having considered the definition and scope of the thiol peroxidase redox relay concept, what is the actual experimental evidence that such relays exist in nature?
The very first thiol peroxidase recognized to form a redox signaling relay was Orp1 in yeast, a member of the Gpx family, also known as Gpx3. This discovery is almost 15 years old (12) and is recognized as the archetypical example of its kind. Orp1 relays H2O2 signals to the transcription factor Yap1, to activate its transcriptional activity. Its peroxidatic cysteine reacts with H2O2 to form a sulfenic acid, which then condenses with a Yap1 cysteine. A thiol–disulfide exchange reaction completes the transfer of the oxidizing equivalent to the transcription factor. The formation of the relay depends on an adaptor protein (Ybp1) that brings Orp1 and Yap1 into proximity (81). The Ybp1-mediated proximity relationship between Orp1 and Yap1 thus closely corresponds to the situation depicted in Figure 4a (except for the fact that Orp1 is a monomer). After the seminal discovery of the Orp1-Yap1 relay, Gpx family members from other organisms have also been found to oxidize other proteins, although not necessarily for signaling purposes. For example, human Gpx4 was found to introduce structural disulfide bonds into sperm protamine (8). The endoplasmic reticulum (ER)-resident mammalian Gpx family members, Gpx7 and Gpx8, were found to forward oxidative equivalents to protein disulfide isomerase, to make use of Ero1-generated H2O2 in oxidative protein folding (47, 82). Gpx7 was also found to oxidize the ER chaperone BiP, presumably to enhance its activity under stress conditions (83). Last but not least, all three yeast Gpx family members (Gpx1–3) as well as human Gpx4 were shown to efficiently transmit oxidation to roGFP (29, 43).
Given the evidence that Gpx family members can and do relay oxidation to other proteins, the question arises whether the same is also true for the Prxs. Numerous Prx-interacting proteins, many of them involved in signal transduction, have been identified over the years (48, 61). It can be noticed that several of these studies report observations that are compatible with or suggestive of a redox relay interpretation.
First of all, there are studies reporting Prx interactions that are triggered by elevated H2O2 levels and/or Prx interactions that depend on the presence of the peroxidatic cysteine. These observations indicate that the interaction depends on the Prx redox state, but as already mentioned, this does not necessarily imply the transfer of oxidizing equivalents to the target protein. For example, the atypical 2-Cys Prx5 was found to interact with the lipoamide acyltransferase component of the mitochondrial branched-chain alpha-keto acid dehydrogenase complex (1). The interaction, as analyzed by coimmunoprecipitation, depended on the presence of the peroxidatic cysteine of Prx5. This finding is compatible with a relay interpretation, but without further evidence it remains unclear whether Prx5 actually donates oxidizing equivalents to the acyltransferase. Another recent example is the finding that an increase in H2O2 levels triggers an interaction between Prx2 and the protein deglycase DJ-1, coinciding with the formation of disulfide-linked DJ-1 dimers (16). Again, this finding is compatible with a relay interpretation, but remains to be substantiated by direct evidence.
Then there are studies that have found disulfide-linked conjugates between Prxs and other proteins. These findings are highly suggestive of Prx-mediated target protein oxidation, but, again, in the absence of additional evidence, these observations must be interpreted with caution. A notable example is the observation that various forkhead box O (FOXO) transcription factors engage in disulfide interactions with various cytosolic Prxs (54 –56). Although suggestive, it has not been substantiated that Prxs actually oxidize FOXO transcription factors.
Finally, some studies have deleted or depleted Prxs, alone or in combinations. An important example is the demonstration that yeast cells lacking all eight thiol peroxidases are unable to alter gene expression in response to H2O2 (19). This remarkable result effectively showed that all sensing of H2O2 for the purpose of gene regulation is mediated by thiol peroxidases. Thus, at least in yeast, thiol peroxidases, including Prxs, are the primary sensors for H2O2, and direct oxidation of redox-regulated proteins does not seem to play a significant role in transcriptional regulation. Nevertheless, these results do not prove that all peroxidase-dependent signaling is through relay mechanisms. Notably, this study also suggests some degree of redundancy among the thiol peroxidases in relation to H2O2 signaling, which may also exist and complicate experimentation in other organisms.
It is evident from the mentioned examples that several pieces of evidence need to be combined. First of all, it is important to be able to monitor the oxidation state of the implicated redox-regulated target protein. Then it seems important to demonstrate that the target protein is as sensitively oxidized as the implicated Prx, inside living cells. Then it should be shown that target protein oxidation fully or largely depends on the presence of the Prx. There is, however, the possibility of redundancy. Depending on the organism or compartment, perhaps more than one Prx has to be deleted to expose a relay mechanism. The relay case is substantially strengthened if a transient mixed disulfide intermediate can be demonstrated. Again, there is a caveat: as already discussed, some anticipated types of relays are not expected to involve disulfide-linked intermediates (e.g., those based on transglutathionylation or trans-persulfidation) and a two-step relay involving an oxidoreductase would also not produce a peroxidase-target disulfide intermediate.
There are relatively few studies in which several lines of evidence have been brought together with the intention to make a case for Prx-based redox relays. Three examples come from budding yeast. First, Tsa1, the major typical 2-Cys Prx in yeast, was reported to take over the role of Orp1 in oxidizing Yap1 when the Ybp1 adaptor protein is nonfunctional or missing, that is, when Orp1 cannot be recruited to oxidize Yap1. In a Ybp1 deletion strain Yap1 oxidation and activity depended on Tsa1, and disulfide-linked Tsa1-Yap1 disulfide conjugates were detected (73). Second, Tsa1 was proposed to form a redox relay with pyruvate kinase (Pyk1) (33). Tsa1 and Pyk1 formed a disulfide-linked conjugate in response to H2O2, depending on the presence of the peroxidatic cysteine. Tsa1 facilitated an adaptive decrease in the activity of Pyk1. Unfortunately, the study did not investigate the formation of Pyk1 oxidation products, making a mechanistic interpretation difficult. Third, the atypical 2-Cys Prx Ahp1 was shown to facilitate the oxidation of the transcription factor Cad1. Ahp1 was required for Cad1-dependent transcriptional activity, and disulfide-linked intermediates were detected (34).
Further examples come from fission yeast. Here, the typical 2-Cys Prx Tpx1 is required for peroxide-induced oxidation and activation of the protein kinase Sty1. Tpx1 deletion suppressed Sty1 activation, whereas Tpx1 overexpression enhanced Sty1 activation. Moreover, a disulfide-linked conjugate between Tpx1 and Sty1 was identified (80). Later it was also shown that the Sty1-dependent transcriptional response to H2O2 requires the formation of an intramolecular disulfide bond within Sty1 (11). However, it has not been directly demonstrated that the Tpx1-Sty1 disulfide conjugate found in the first study leads to the formation of the Sty1 intramolecular disulfide bond found in the second study.
Tpx1 is also needed for the oxidation and activation of the transcription factor Pap1. A direct one-step redox relay mechanism was suggested by the identification of a disulfide-linked Tpx1-Pap1 conjugate (6). Another study, published side-by-side, concluded that Pap1 oxidation can only happen when Tpx1 oxidizes a particular member of the Trx family, Txl1, which normally keeps Pap1 in the reduced state (5). The latter study shows that Tpx1 acts as an inhibitor of the reductive function of Txl1, but it does not address or answer the question of how Pap1 is actually oxidized. Thus, the two studies do not necessarily contradict each other. It could well be that Tpx1 directly oxidizes Pap1 and that Pap1 oxidation will only be maintained and detectable when the Pap1 reducing Trx family members are also oxidized (i.e., inactivated) by Tpx1. Thus, it may be the combination of two effects: Tpx1 directly mediating Pap1 oxidation plus Tpx1 indirectly inhibiting Pap1 reduction, together allowing functionally relevant steady-state Pap1 oxidation. Obviously, the redox state of reversibly oxidized proteins is a function of both oxidation and reduction activities.
Further evidence for Prx-based redox relays comes from human cells. Human Prx4, the only ER resident Prx, oxidizes members of the PDI family to support oxidative protein folding (74, 93). Other examples relate to redox signaling: Prx1 forms a redox relay with the stress kinase apoptosis signaling kinase 1 (ASK1), as depletion of Prx1 abolished peroxide-induced ASK1 oxidation and activation (35). The same study also indicated a disulfide-linked Prx1-ASK1 conjugate. Prx2 was found to form a redox relay with the transcription factor STAT3. Oxidation of STAT3 was shown to depend on the presence of Prx2 and several STAT3 cysteines formed mixed disulfide intermediates with Prx2 (69). More recently, a redox relay between Prx1 and the nuclear DNA repair enzyme APE1 has been proposed. Prx1 depletion affected APE1 location and function. Indications for a covalent intermediate between Prx1 and APE1 were reported. Unfortunately, the influence of Prx1 on the oxidation state of APE1 was not investigated (44).
In summary, there is limited yet increasing evidence that not only Gpxs but also Prxs facilitate protein thiol oxidation, in organisms ranging from yeast to humans. Lastly, the principal prowess of Prxs in relaying oxidation to proximal proteins is also demonstrated by the successful use of roGFP2 Prx fusion proteins as in vivo real-time H2O2 probes (43).
Open Questions Regarding Prx-Based Redox Signaling Relays
As discussed in the previous section, direct evidence for Prx-based redox relays does exist, but so far remains restricted to a few case studies. Thus, a key question is how common (and important) are Prx-based relays for protein thiol oxidation in general? Even if Prx-based relays are indeed much more common than hitherto recognized, uncovering the whole extent of Prx relay activity will likely remain a challenging task. Many redox-regulated proteins may assemble with Prxs in a highly dynamic and transient manner, perhaps in response to other (nonredox) signals, which can be short-lived and localized. Such fleeting relay assemblies may be hard to detect and characterize, also because only a small subpopulation of the redox-regulated protein may form part of the specific relay, and likewise, only a tiny fraction of the total Prx pool would be involved in any specific relay. However, some target proteins may be associated with Prxs more continuously and more stably. In such complexes, the two partners would be prepositioned and poised to act in a redox relay, just waiting for the arrival of H2O2 molecules. Preassembled relays may be more abundant and easier to detect, and it is conceivable that existing Prx interaction databases (e.g., from co-IP studies) already include redox relay partners that remain to be recognized as such.
These considerations lead to the question of what makes Prxs assemble with certain proteins and not with others. In other words, where does the target specificity of redox relay signal transmission come from? One possibility is that target proteins directly bind to Prxs. In this regard, it is interesting to note that many Prxs exhibit chaperone-like properties under various conditions (61). For example, a reduced 2-Cys Prx from Leishmania was observed to bind an unfolding client protein in the center of its decameric ring structure (75). It may be speculated that proteins with disordered domains or stretches can interact with the center of Prx rings, thus being recruited into proximity of peroxidatic cysteines. Alternatively, the pairing of thiol peroxidases and target proteins may be facilitated by the supramolecular scaffolding context of larger signaling complexes, potentially involving dedicated adaptor proteins, as it is the case for the prototypical Orp1-Yap1 redox relay (81). Indeed, mammalian Prxs have often been copurified as components of larger signaling complexes (61).
A related question is how individual Prxs can switch between an antioxidative scavenging mode (relaying oxidative equivalents to Trx) and a pro-oxidative signaling mode (relaying oxidative equivalents to redox-regulated proteins). This may simply depend on local interactions. Conformational changes taking place within multiprotein assemblies may reposition associated Prx oligomers relative to other proteins. These rearrangements would then determine whether incoming oxidizing equivalents are delivered toward Trx for disposal or are relayed to a neighboring redox-regulated recipient protein. Alternatively, the switching between Prx scavenging and signaling modes may be regulated by more general mechanisms. For example, one interesting possibility is that the canonical transfer of oxidizing equivalents from Prx to Trx to TrxR is redox regulated on its own terms. It has been reported that Prx1 can oxidize Trx1 at its nonactive site cysteines 62 and 67 (15). The resulting C62-C69 disulfide bond was found to inactivate the transmission of oxidizing equivalents along the Prx-Trx-TrxR axis, thus potentially allowing Prx1 to deliver incoming oxidizing equivalents to other targets. The local and transient inactivation of Trx is expected to have a dual effect: not only would it promote the rerouting of oxidative equivalents from Prx to other recipients, it would also ensure that those proteins oxidized by Prx will stay oxidized, namely as long as Trx remains oxidatively inactivated. Perhaps it is a general principle of redox signaling that the local activation of oxidative transfer to client proteins is accompanied by the local inactivation of the reduction of the same clients. In this scenario, Trx is not just a redox relay bridge between Prx and TrxR, but also a redox-regulated target protein of Prx.
Another open question is the potential redundancy between different Prxs in terms of their redox relay activities. If certain Prxs can substitute for each other, it may not be sufficient to delete or deplete just one Prx to determine the impact on the redox state of a target protein. For the cytosolic-nuclear environment of mammalian cells, this question predominantly relates to Prx1 and Prx2. Both are typical 2-Cys Prxs and highly homologous. Are Prx1 and Prx2 both able to form redox relays? To what extent do they target different proteins? Do they cooperate in redox signaling? Prx1 and Prx2 are known to differ in hyperoxidation sensitivity and chaperone-like activity (38), suggesting that they also differ in conformational dynamics and preferred oligomeric state. This may be relevant for how they interact with target proteins.
Thus, the question arises how redox relays can be envisioned from a structural point of view. Here we consider this issue specifically for the typical 2-Cys Prxs. As discussed, the most direct reaction path for a relay is the condensation of the peroxidatic Cys-SOH with the thiol on the target protein. The existence of this mechanism is indicated by the fact that both Tsa2ΔCR and Prx2ΔCR can oxidize roGFP2 (43), and also by the observation that Prx2ΔCR can oxidize STAT3 (69). How can the transfer of oxidation be envisaged in structural detail? Based on the well-established reaction cycle of typical 2-Cys Prxs (51), the reaction with H2O2 takes place in the fully folded (FF) state. The resulting sulfenylation of the peroxidatic cysteine shifts the conformational equilibrium toward the locally unfolded (LU) state. During the transition into the LU state, the sulfenylated peroxidatic cysteine moves from the center to the surface of the subunit (Fig. 5a), and the C-terminal tail of the opposing subunit, covering that surface in the FF state, swings away in a concerted manner (Fig. 5b) (52). With the C-terminal tail displaced, the sulfenic acid is exposed and remains accessible on the Prx surface until the C-terminus happens to fold back in a manner that allows the resolving cysteine to condense with the sulfenic acid. It is thus conceivable that there is a window of opportunity for another protein to come in, to bind to the surface and to bring its own thiol into the proximity of the exposed sulfenic acid (Fig. 6a). Thus, the incoming client thiol effectively takes the role and position of the resolving cysteine. The resulting condensation reaction creates a mixed disulfide bond that covalently links the target protein to the Prx. This disulfide may then be resolved by a second cysteine in the target protein. If there is no other cysteine in the target protein, the complex would be arrested as a conjugate, but perhaps the CR or GSH would have a resolving function in that case.
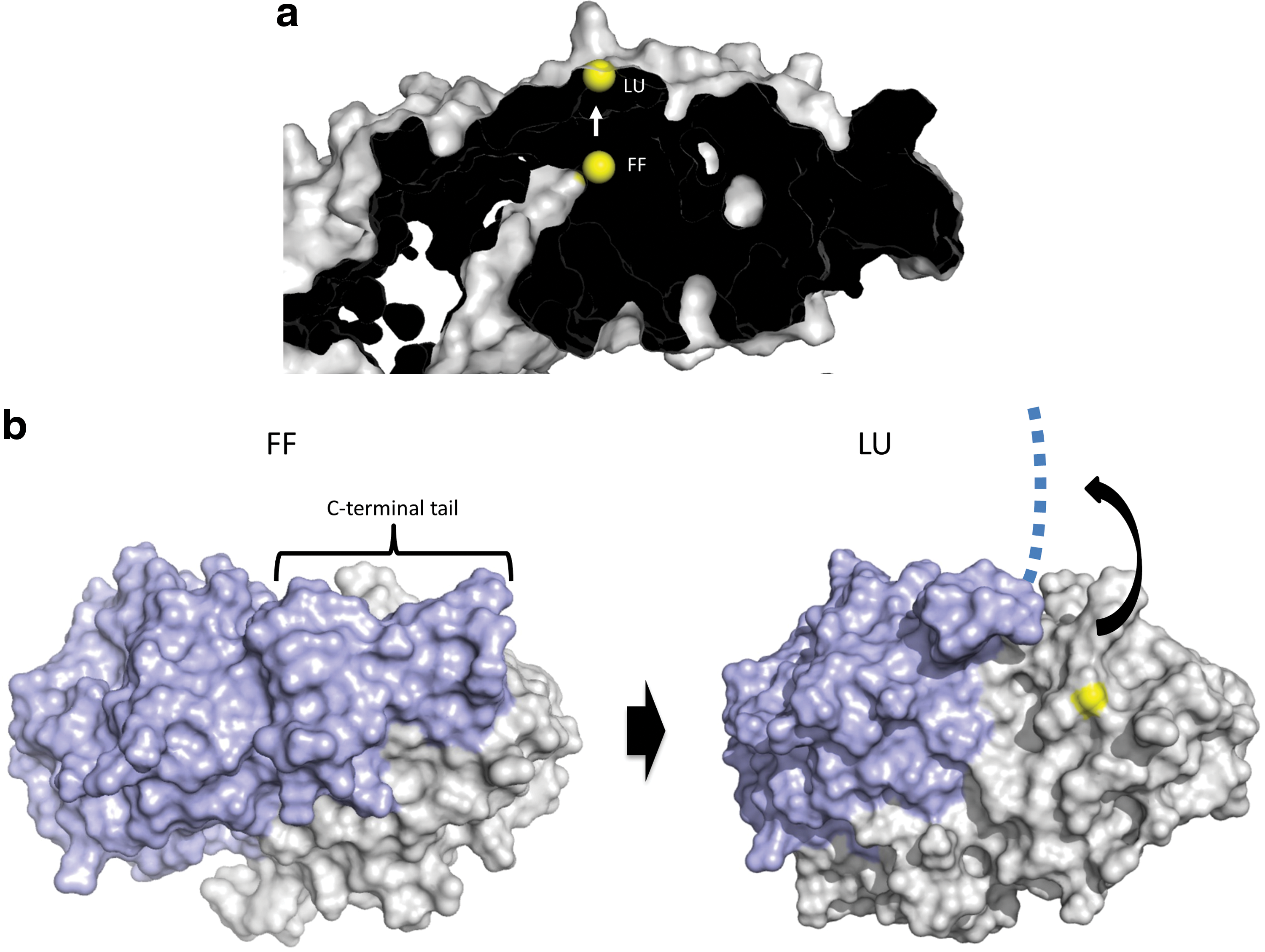
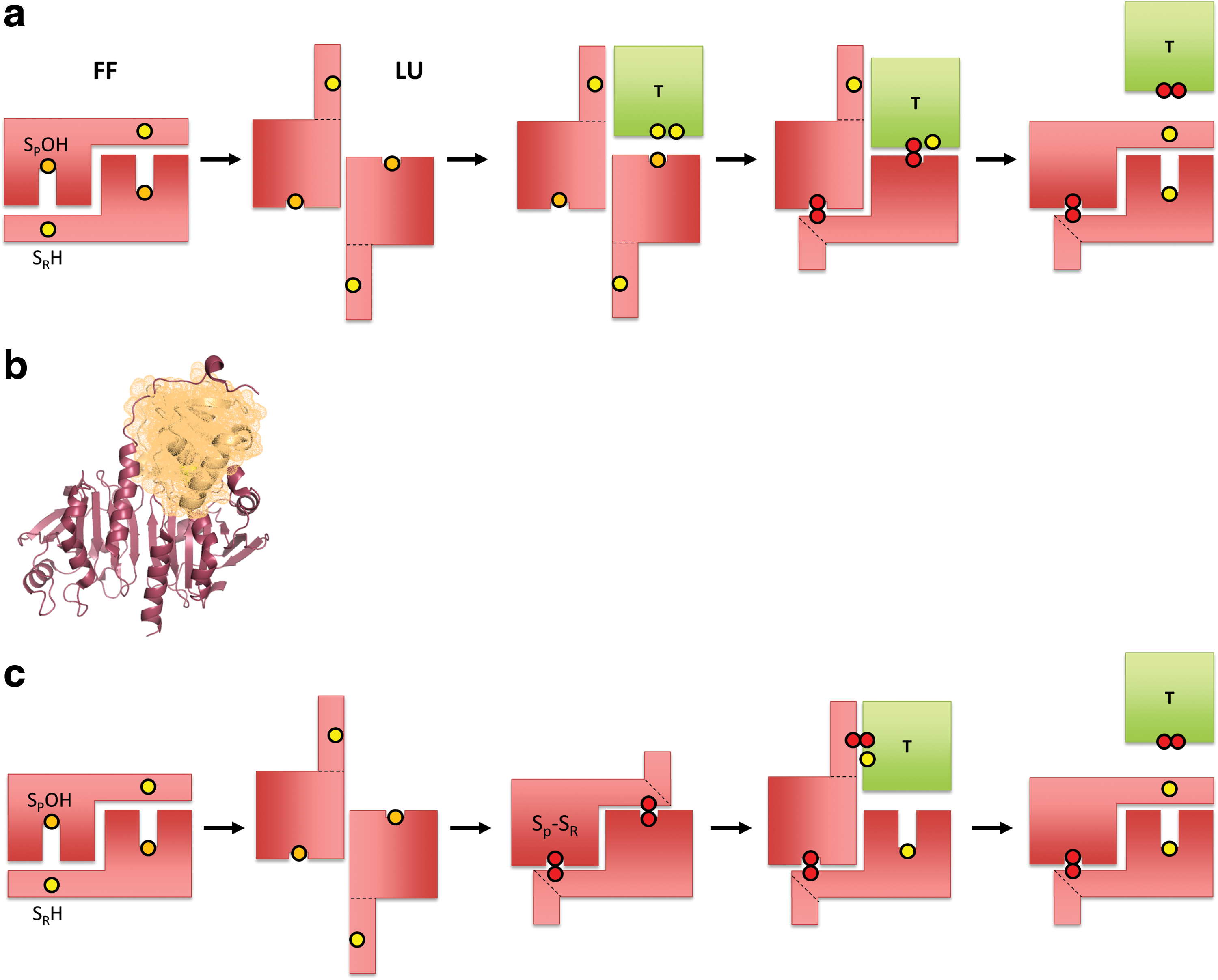
Of note, the positioning of a redox-regulated client protein on the surface of LU state Prxs may be mimicked by the known organization of the Prx-sulfiredoxin (Srx) complex (36) (Fig. 6b). Here the C-terminal tail of the opposing subunit makes way for the Srx to position its active site thiol close to the surface-exposed peroxidatic cysteine. Although the function and chemistry of this particular complex are unrelated to a relay mechanism, its structural organization shows that the previously postulated protein–protein interactions are possible and precedented.
The alternative scenario, based on the assumption that the Prx forms the disulfide first, would be analogous to the reduction of Prx by Trx (Fig. 6c). The structural arrangement and predicted interaction surface would be different from the first model. Trx preferentially attacks the CP-CR disulfide at the sulfur that belongs to the resolving cysteine, as suggested by previous studies (39), and thus is expected to form a transient mixed disulfide intermediate with the C-terminal tail before it resolves itself. Unfortunately, there is no crystal structure of a complex formed between a typical 2-Cys Prx and a Trx to provide further structural insight into this arrangement.
Outlook
Which specific experiments may help to resolve the conundrum of H2O2 signaling? How do we clarify whether direct or Prx-mediated oxidation is more prevalent inside cells? Importantly, the two concepts (direct vs. mediated oxidation) make different predictions, which should be testable by experiment. For example, the two cytosolic Prxs (Prx1+Prx2) are estimated to consume >99% of H2O2 inside the cytosol of mammalian cells (85). Hence, it is interesting to ask what happens to protein thiol oxidation in the cytosolic compartment (under a range of conditions) when both cytosolic Prxs are deleted or depleted. This experiment would require a general readout of cytosolic protein thiol oxidation (e.g., disulfide formation), as potentially provided by redox proteomics or kinetic trapping strategies. If protein thiol oxidation is predominantly direct (rather than Prx mediated), then the simultaneous deletion of the two cytosolic Prxs should increase overall cytosolic protein thiol oxidation. However, if thiol oxidation is predominantly mediated by Prxs, the opposite outcome can be expected, namely a decrease in protein thiol oxidation.
In the end, there is little doubt that H2O2 and other peroxides are inherently unspecific oxidants and that intracellular signal transduction requires precise spatiotemporal control (4). All the well-known nonoxidative post-translational protein modifications are catalyzed in both directions and hence controlled by the location and activity of enzymes that introduce and remove the respective modification. It is not obvious why reversible protein thiol oxidation should be an exception and how it could play a role in signaling without bidirectional enzymatic control (14, 18, 46). Thiol peroxidase-based redox relays, in particular those based on Prxs, may serve as the major mechanism for the specific and efficient transmission of oxidative signals under physiological conditions, that is, under conditions of nanomolar-range H2O2 signaling. It is, however, not unlikely that alternative and/or complementary mechanisms exist, depending on the strength and subcellular distribution of the H2O2 signal. Untangling the various kinds of redox signaling mechanisms and their contributions remains a central goal for the field of redox biology.
Footnotes
Acknowledgments
We wish to thank Marcel Deponte and Bruce Morgan for critical comments on the article and Peter Nagy for the discussion of chemical aspects. Our work on redox signaling is supported by the Deutsche Forschungsgemeinschaft (DFG) through collaborative research program SFB 1036 and priority program SPP 1710.