
Abstract
Significance:
Hydrogen peroxide (H2O2) is produced on stimulation of many cell surface receptors and serves as an intracellular messenger in the regulation of diverse physiological events, mostly by oxidizing cysteine residues of effector proteins. Mammalian cells express multiple H2O2-eliminating enzymes, including catalase, glutathione peroxidase (GPx), and peroxiredoxin (Prx). A conserved cysteine in Prx family members is the site of oxidation by H2O2. Peroxiredoxins possess a high-affinity binding site for H2O2 that is lacking in catalase and GPx and which renders the catalytic cysteine highly susceptible to oxidation, with a rate constant several orders of magnitude greater than that for oxidation of cysteine in most H2O2 effector proteins. Moreover, Prxs are abundant and present in all subcellular compartments. The cysteines of most H2O2 effectors are therefore at a competitive disadvantage for reaction with H2O2.
Recent Advances:
Here we review intracellular sources of H2O2 as well as H2O2 target proteins classified according to biochemical and cellular function. We then highlight two strategies implemented by cells to overcome the kinetic disadvantage of most target proteins with regard to H2O2-mediated oxidation: transient inactivation of local Prx molecules via phosphorylation, and indirect oxidation of target cysteines via oxidized Prx.
Critical Issues and Future Directions:
Recent studies suggest that only a small fraction of the total pools of Prxs and H2O2 effector proteins localized in specific subcellular compartments participates in H2O2 signaling. Development of sensitive tools to selectively detect phosphorylated Prxs and oxidized effector proteins is needed to provide further insight into H2O2 signaling. Antioxid. Redox Signal. 28, 537–557.
Introduction
S
H2O2 oxidizes the cysteine thiol (–SH) to sulfenic acid (–SOH), which then either forms an intramolecular or intermolecular disulfide bond, a mixed disulfide with glutathione (glutathionylation), or sulfenylamide (–S–NR–CO–) or is further oxidized to sulfinic (–SO2H) and sulfonic (–SO3H) acids (Fig. 1). Oxidation to sulfenic acid, disulfide, or sulfenylamide can be reversed by various cellular thiols, whereas that to sulfonic acid is irreversible. Oxidation to sulfinic acid appears to be irreversible for most proteins, with one exception being the active-site cysteine of the family of antioxidant enzymes known as peroxiredoxins (Prxs). Changes in cysteine oxidation state can affect the susceptibility of a protein to other posttranslational modifications, influence protein turnover or enzymatic activity, regulate protein–protein interactions, and alter subcellular protein trafficking.
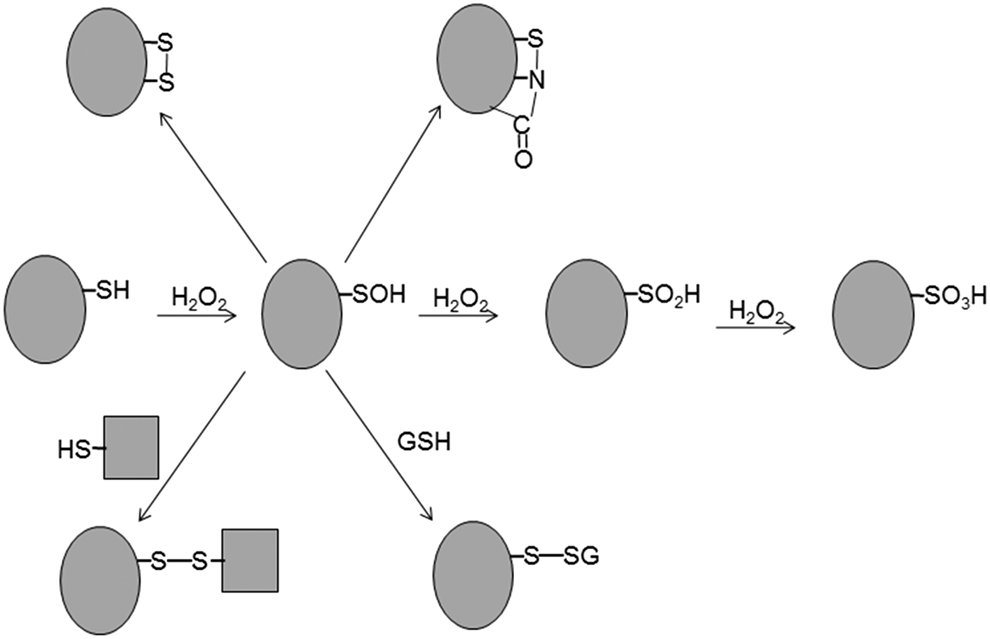
Many proteinaceous cysteines have now been identified as primary sites of reaction with H2O2 (see section on Protein Whose Function Is Regulated via Reversible Cysteine Oxidation). However, the mechanism by which H2O2 transmits its signal by oxidizing the specific cysteine residues of its target proteins in the presence of vigilant antioxidant enzymes is not well understood. H2O2 molecules are eliminated by several such enzymes, including catalase, glutathione peroxidase (GPx), and Prx. Whereas catalase is present mostly in peroxisomes, GPx and Prx exist in multiple isoforms that are found in a variety of subcellular compartments. The primary sites of reaction with H2O2 in catalase, GPxs, and Prxs are heme, selenocysteine, and cysteine, respectively.
Kinetic and structural analyses have revealed that all Prxs possess an active-site pocket that gives rise to a high-affinity (submicromolar) peroxide binding site that is lacking in catalase and GPxs (123, 135, 147). In addition, this pocket of Prxs is arranged so as to stabilize the transition state intermediates that aid breaking of the O-O bond of H2O2, thus eliciting rapid oxidation of the catalytic cysteine by H2O2, with the second-order rate constant for the oxidation of Cys–SH to Cys–SOH being several orders of magnitude greater than that for the oxidation of cysteine in well-characterized H2O2 target proteins (135, 188). Given that Prxs are abundant proteins and are present in various compartments of the cell, cysteine residues of most target proteins are at a competitive disadvantage for reaction with H2O2.
It has recently become increasingly apparent that Prxs are more than simple peroxide-eliminating enzymes and that they actively participate in H2O2 signaling by assisting H2O2 effector molecules to overcome their kinetic disadvantage with regard to cysteine oxidation. Although the role of Prxs in H2O2 signaling remains largely uncharacterized, a full understanding of this role will require identification both of the relevant sources of H2O2 and of the effector proteins. In this review, we have compiled various known sources of H2O2 production as well as classified proteins known to be modulated by H2O2 through cysteine oxidation according to their biochemical and cellular functions.
We then describe how some of these H2O2 target proteins are able to overcome their kinetic disadvantage with regard to cysteine oxidation by H2O2. One way that this can be achieved is through temporary inactivation of Prx so as to protect H2O2 from destruction. Prx I is reversibly inactivated either through phosphorylation by Src family kinases at the plasma membrane in response to growth factor receptor or immune receptor activation (190) or through phosphorylation by cyclin B-dependent kinase activity at the centrosome during mitosis (27, 101). Such localized inactivation of Prx allows H2O2 to accumulate in specific regions of the cell without global redox disturbance.
Another way in which oxidation of H2O2 target proteins can be achieved is through oxidation of Prx I or Prx II by H2O2 and subsequent transfer of their oxidation state to the target protein. Such target proteins include apoptosis signal regulating kinase 1 (ASK1), the transcription factor STAT3 (signal transducer and activator of transcription 3) (72, 126, 168), the redox-sensitive chaperone DJ-1 (48), and APE1/Ref-1 (apurinic/apyrimidinic endonuclease 1/redox effector factor-1), a reductive activator of many transcription factors (125).
Sources of Intracellular H2O2
The complete reduction of one oxygen molecule (O2) to water requires four electrons, with incomplete reduction by one electron producing the superoxide anion (O2 −•), which is spontaneously or enzymatically (by superoxide dismutase) converted to H2O2. Incomplete reduction of molecular oxygen by two electrons generates H2O2 directly. Enzymes that generate H2O2 directly or indirectly in mammalian cells include NADPH oxidase (Nox) (5, 92), the mitochondrial electron transport chain (ETC) complexes (5, 152, 164), cytochrome P450 (CYP) (5, 55, 60), arachidonic acid-metabolizing enzymes such as lipoxygenase and cyclooxygenase (32), xanthine oxidase (78, 128), and monoamine oxidase (MAO) (46, 109) (Fig. 2). Among these enzymes, Nox is the only one whose primary function is to generate superoxide or H2O2, with the others producing these oxidants as a by-product of their main catalytic function.

Members of the Nox family of enzymes exist as membrane-bound complexes that face the extracellular space when located at the plasma membrane. Nox generates superoxide by transferring electrons from intracellular NADPH across the membrane to molecular oxygen. The Nox family consists of seven members—Nox1 to Nox5 as well as two dual oxidases designated Duox1 and Duox2—that are differentially expressed and regulated in various tissues and which have different subcellular localizations (20). Nox1 is present in lipid rafts at the plasma membrane as well as in endosomes; Nox2 in phagosomes, endosomes, and at the leading edge of lamellipodia; Nox3 at the plasma membrane; Nox4 in focal adhesions, the nucleus, and endoplasmic reticulum (ER); and Nox5, Duox1, and Duox2 at the plasma membrane.
The signaling targets of H2O2 molecules produced by Nox enzymes are located in close proximity to the site of H2O2 generation (65, 129, 182). Early studies revealed that growth factors such as platelet-derived growth factor (PDGF) and epidermal growth factor (EGF) induce the production of H2O2 and that this H2O2 is required for the mitogenic action of these proteins (4, 173). Nox enzymes were subsequently identified as the source of H2O2 for such proliferation signaling: Nox1 for PDGF- or EGF-induced mitogenesis in nonphagocytic cells (114, 171) and for angiotensin II-induced proliferation of vascular smooth muscle cells (93), and Nox4 for proliferation of pulmonary artery smooth muscle cells induced by transforming growth factor-β (170).
The first proteins identified as targets of H2O2 produced by Nox were members of the PTP family. H2O2 generated in response to EGF, PDGF, or insulin signaling was thus found to inactivate PTPs by oxidizing the active-site cysteine of these enzymes and thereby to enhance growth factor-induced protein tyrosine phosphorylation (96, 106, 113). Many other signaling proteins, including the transcription factor AP1 (activator protein 1) and Src protein kinase, were later shown to be regulated via cysteine oxidation [reviewed in Refs. (31, 71, 148)].
Mitochondria generate ATP by transferring electrons derived from the tricarboxylic acid cycle to the ETC, which ultimately delivers them to molecular oxygen. The ETC comprises four protein complexes (complexes I–IV) that are located at the mitochondrial inner membrane. Electron flow through these complexes requires a diverse set of cofactors that include quinones, flavin groups, heme moieties, and iron/sulfur centers. Electrons can escape from the flavin groups or iron/sulfur centers of complexes I, II, and III in the ETC and can then be captured by O2 to generate superoxide, which then undergoes dismutation to yield H2O2 (152, 164). Many other mitochondrial proteins, including those that participate in the tricarboxylic acid cycle, contain flavin groups or iron/sulfur centers. As with the ETC complexes, electrons can leak from the cofactors of these proteins and react monovalently with O2 to form superoxide (152, 164).
The production of H2O2 by mitochondria was long viewed as a wasteful side reaction of ATP production. However, substantial evidence now indicates that such mitochondrially generated H2O2 serves an important signaling function in the regulation of diverse biological processes such as cell proliferation, differentiation, autophagy, immunity, the response to hypoxia, and, most recently, circadian rhythm (37, 59, 80, 81, 134, 152, 164). Some of the H2O2 produced in mitochondria is released into the cytosol, where it serves as a key regulator of various signaling pathways. However, mitochondria are equipped with several H2O2-eliminating enzymes, with Prx III being an especially abundant and efficient such enzyme in mitochondria of most cell types. As the production of H2O2 increases in mitochondria, Prx III undergoes reversible inactivation through hyperoxidation of its catalytic Cys–SH to Cys–SO2H, likely resulting in the release of mitochondrial H2O2 into the cytosol [reviewed in Ref. (151)].
Mitochondria are highly dynamic organelles that frequently undergo fission, fusion, and translocation along the cytoskeleton. The overall morphology of mitochondria is therefore highly variable, ranging from numerous fragmented individual organelles to a single large interconnected tubular network depending on environmental conditions and cell type. Recent studies have revealed that the mitochondria of HEK293 cells are arranged in elongated tubules that undergo fragmentation during mitosis (74, 75). Given that cell division represents a major commitment to energy expenditure, mitochondrial H2O2 likely plays a major role in coordinating the cell cycle with metabolism.
The CYP family comprises many enzymes that catalyze the monooxygenation of hydrophobic organic molecules. CYPs bind and activate oxygen via a heme group in their active site, and they use one atom of an oxygen molecule to hydroxylate their substrate and the other to produce H2O. The two electrons required for the conversion of an oxygen atom to H2O are provided by NADPH. During the catalytic cycle, a substantial portion of the heme-bound (activated) oxygen is released from the catalytic intermediate and forms superoxide or H2O2 (55, 60, 199). CYPs are abundant in the ER of liver cells as well as in mitochondria of steroidogenic cells. The production of H2O2 by CYPs is generally considered to be a side reaction with accompanying adverse effects. However, H2O2 produced by a CYP in mitochondria of the mouse adrenal gland was recently shown to contribute to a negative feedback mechanism that is critical for the circadian rhythm of corticosterone production (80).
Arachidonic acid metabolism by lipoxygenase and cyclooxygenase enzymes also plays a role in the generation of reactive oxygen species (ROS) in various cell types. Arachidonic acid is released from glycerophospholipids in the nuclear envelope and plasma membrane through the activity of cytosolic phospholipase A2, and it can then be oxidized to a variety of bioactive eicosanoids by lipoxygenase and cyclooxygenase enzymes. Oxidation of arachidonic acid by these two groups of iron-containing dioxygenases is accompanied by the side production of ROS, including H2O2 (32). Certain arachidonic acid metabolites have also been found to increase H2O2 levels by activating Nox enzymes (32).
Xanthine oxidase is a large (270 kDa) protein that contains two molybdopterin and two flavin groups as well as eight iron atoms and which catalyzes the oxidation of hypoxanthine to xanthine as well as the further oxidation of xanthine to uric acid (78, 128). Substrate-derived electrons are first transfered to the molybdenum cofactor and then, via several intermediates, to the flavin center, where they can reduce O2 either univalently to yield the superoxide anion or divalently to yield H2O2 (78, 128).
MAO enzymes are flavoproteins and catalyze the oxidative deamination of neurotransmitters (such as dopamine, serotonin, and norepinephrine) and food-derived biogenic amines (such as tyramine and phenethylamine). Substrate-derived electrons reduce the flavin groups, which then react with O2 stochiometrically to form H2O2 (46). Two forms of MAO are found in mammals, with MAOA being located at the cytosolic face and MAOB at the intermembrane face of the mitochondrial outer membrane (46). Although a signaling role has been demonstrated for CYP-derived H2O2 in steroidogenic cells, H2O2 produced by CYP, lipoxygenase, cyclooxygenase, xanthine oxidase, and MAO enzymes has been studied mainly in connection with pathological conditions such as hepatotoxicity, vascular inflammation, and neurodegeneration.
Proteins Whose Function Is Regulated via Reversible Cysteine Oxidation
Many proteins are susceptible to modification by cysteine oxidation, with their subcellular localization and sensitivity to H2O2-mediated oxidation varying widely. The regulation of cellular processes by H2O2 thus depends on the concentration of the oxidant, the duration of exposure of the target protein, and cell type. Taking cell cycle progression as an example, low levels of H2O2 generated by Nox enzymes promote the transition from G0 to G1 phase by increasing the expression of cyclin D, whereas prolonged exposure to higher levels of H2O2 induces adaptive cellular responses by activating the tumor suppressor protein p53, which triggers either growth arrest at the G1-S or G2-M transitions of the cell cycle or apoptosis depending on the extent of such exposure [reviewed in Refs. (22, 31)].
The conversion of H2O2 to hydroxyl radicals and consequent induction of DNA damage result in recruitment of the protein kinase ATM (ataxia telangiectasia mutated) to the damage sites, where it phosphorylates and activates p53. H2O2 can also activate ATM directly in the absence of DNA damage by inducing the formation of a disulfide-linked dimer of the kinase (57). Low levels of H2O2 produced by Nox4 and Duox2 in response to stimulation of normal human fibroblasts with PDGF were also found to promote cell cycle entry not by increasing cyclin D expression but by downregulating the p53-dependent checkpoint (156). This attenuation of p53 function is likely achieved through a reduction in p53 concentration or through oxidation of cysteine residues of p53 and consequent inhibition of its DNA binding activity (156, 172).
Given that many regulatory proteins participate in multiple signaling pathways, it has also become clear that the outcome of activation of a specific regulator depends on contextual factors such as cell type and the activation state of related signaling pathways. For example, activation of the three types of mitogen-activated protein kinase (MAPK)—extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38—all of which are sensitive to H2O2, has been implicated in cellular outcomes as diverse as proliferation, survival, differentiation, and death (94). Activation of ERK and p38 thus contributes not only to promotion of the G0-G1 transition via the AP1–cyclin D pathway but also to growth arrest at the G1-S or G2-M transitions through phosphorylation of p53 and consequent inhibition of p53 degradation (66, 165, 178).
In the following sections, we will group proteins whose function is regulated via H2O2-mediated cysteine oxidation on the basis of their biochemical and cellular functions (Fig. 2).
PTPs as H2O2 targets
It has been well established that H2O2 inactivates PTPs via oxidation of their catalytic cysteine residue. The PTP family of proteins comprises four subfamilies: classical (phosphotyrosine-specific) PTPs that include PTP1B, SH2 domain-containing PTPs (SHPs), T cell PTP (TCPTP), and receptor PTPα (RPTPα); dual-specific (phosphotyrosine- and either phosphoserine- or phosphothreonine-specific) PTPs that include Cdc25, Cdc14B, and MAPK phosphatase (MKP); phospholipid-specific phosphatases that include phosphatase and tensin homolog (PTEN); and low-molecular-weight PTP (LMW-PTP) (179). All PTPs share a CX5R signature motif (where X is any amino acid), in which the invariant cysteine residue functions as a nucleophile in catalysis. The catalytic cysteine is oxidized by H2O2 to Cys–SOH, which can then either react with another cysteine residue to form an intramolecular disulfide (as in PTEN, Cdc25, and Cdc14B) (90, 97, 101, 159, 169), with glutathione (GSH), or with a main-chain amide nitrogen to form a sulfenylamide bond (–S–N–) or undergo irreversible oxidation to Cys–SO2H or Cys–SO3H [reviewed in Ref. (176)].
Reversible oxidation of a PTP was first demonstrated for PTP1B in cells stimulated with EGF or insulin (96, 107). The colocalization of PTP1B and Nox4 attests to the importance of a confined microdomain containing the H2O2 source and the target protein for this reaction (28). The crystal structure of oxidized PTP1B revealed that Cys215–SOH at the active site forms a sulfenylamide bond with the nitrogen of Ser216, with the product being readily reduced back to Cys–SH (155, 184).
Glutathionylation of PTP1B was also detected in vitro (9). Reversible oxidation of SHP-1, SHP-2, or TCPTP has also been observed in cells stimulated with PDGF, insulin, or collagen or via the T cell receptor (TCR) (33, 70, 91, 112, 113). RPTPα contains a single transmembrane domain and two conserved intracellular PTP domains (domains 1 and 2) arranged in tandem, with domain 1 being responsible for most of the catalytic activity and domain 2 functioning predominantly as a regulatory domain. The conserved cysteine (Cys723) of domain 2 was found to be more susceptible to oxidation than that (Cys433) of domain 1 (15, 137).
H2O2 produced in response to cell stimulation with EGF, PDGF, or vascular endothelial growth factor (VEGF) was found to potentiate signaling by the protein kinase Akt through oxidation of the catalytic Cys124 residue of PTEN and consequent formation of an intramolecular disulfide with Cys71 (37, 90, 97). Both Nox and mitochondria were implicated as the source of H2O2 for PTEN oxidation.
Exposure of cells to H2O2 also results in the formation of an intramolecular disulfide between the conserved catalytic cysteine (Cys377 and Cys314, respectively) and a nonconserved cysteine (Cys330 and Cys228, respectively) in Cdc25C and Cdc14B (101, 159). However, Cdc25C and Cdc25B showed a much lower sensitivity to H2O2 compared with Cdc14B and PTEN in H2O2-treated cells (101). Given that Cdc25C activity is essential for activation of the cyclin B–Cdk1 (cyclin-dependent kinase 1) complex and that oxidized Cdc25C undergoes degradation in H2O2-treated cells, cysteine oxidation of Cdc25C has been proposed as a mechanism for induction of cell cycle arrest (159). It is interesting to note that, although Cdc25 enzymes contain the PTP signature motif, they bear no structural similarity to other PTPs, instead manifesting an architecture similar to that of rhodanese (47).
Several lines of indirect evidence suggest that MKPs are inhibited through reversible oxidation of their active-site cysteine to sustain JNK activation in cells stimulated with tumor necrosis factor-α (73). Direct evidence of MKP oxidation was obtained for MKP3 (a phosphatase specific for ERK), which was shown to form disulfide-linked high-molecular-weight aggregates with unidentified proteins via its catalytic Cys293 residue in H2O2-treated cells (73).
LMW-PTP is an important regulator of cell proliferation that counteracts phosphorylation by the PDGF receptor (PDGFR) (29). Oxidation of LMW-PTP, which has been observed in H2O2-treated cells as well as in NIH3T3 cells stimulated with PDGF, results in the formation of an intramolecular disulfide between Cys12 and Cys17, both of which are present in the active site (24, 30).
Protein tyrosine kinases as H2O2 targets
Increasing evidence suggests that H2O2 enhances protein tyrosine phosphorylation through cysteine-mediated activation of protein tyrosine kinases (PTKs) as well as through inhibition of PTPs (124). Lck was the first member of the Src family of PTKs (Src, Fyn, Yes, Lck, Hck, Blk, Fgr, Lyn, and Yrk) found to exhibit H2O2-dependent activation (122). Src itself was subsequently shown to be activated when its two cysteine residues (Cys245 and Cys487) were oxidized by H2O2 produced in response to cell attachment to the extracellular matrix (52). Activation of Src via H2O2-mediated cysteine oxidation was later demonstrated in cells stimulated with insulin-like growth factor 1 or VEGF (95, 193).
It was initially thought that Cys245 and Cys487 form an intramolecular disulfide in oxidized Src (53), but analysis of Lyn, which becomes oxidized and activated to initiate signaling pathways underlying wound healing in zebrafish neutrophils, indicated that oxidation of Cys466 (corresponding to Cys487 of mammalian Src) to sulfenic acid was sufficient for this role, with oxidation of Cys224 (corresponding to Cys245 of Src) not being necessary (198). An essential role for Cys487 oxidation, but not for Cys245 oxidation, was confirmed for the activation of Src by hypoxia-induced mitochondrial H2O2, which leads to activation of the transcription factor NF-κB (nuclear factor-κB) (104).
The EGF receptor (EGFR) provides another clear example of activation of a PTK via cysteine sulfenylation in vivo. In A431 cells stimulated with EGF, EGFR associates with Nox2 and its Cys797 residue located in the ATP binding pocket undergoes oxidation to sulfenic acid, resulting in kinase activation. The sulfenylated form of activated EGFR mutant proteins has also been detected in both lung and breast tumors (181). The Cys797 residue of EGFR is conserved among nine additional receptors, including human EGFR 2 (HER2) and HER4 but not in Src family members. In contrast to Src- or EGFR-related proteins, in which a critical cysteine is oxidized to a nondisulfide form to play a positive role in kinase activation, VEGF receptor 2 (VEGFR2) forms an intramolecular disulfide bond between Cys1199 and Cys1206 in vascular endothelial cells stimulated with VEGF, which renders the receptor resistant to activation by VEGF binding (76).
Adding another level of complexity to PTK regulation via cysteine oxidation, both inactivation and activation of Src were found to occur in H2O2-treated cells in a manner dependent on the exposure time and subcellular localization of Src: Src localized to focal adhesions and other regions of the plasma membrane was rapidly inactivated by low levels of H2O2 at short exposure times, whereas cytoplasmic Src was activated gradually over longer exposure times (175). Such inactivation of Src was subsequently shown to result from homodimerization mediated by oxidation of Cys277 located in the glycine loop of the catalytic site (79).
Although five cysteine residues—Cys238, Cys245, Cys400, Cys487, and Cys498—are conserved among all Src family members, Cys277 is conserved in only two other members of the family (Yes and Fgr) as well as among all members of the fibroblast growth factor receptor (FGFR) family of PTKs. FGFR1 was also found to undergo reversible inhibition as a result of homodimerization mediated by formation of a disulfide bond by the cysteine residue in its glycine loop (79). The regulation of Src through cysteine oxidation therefore represents a good example of spatially and temporally dependent signaling by H2O2. In cells exposed to ultraviolet radiation, the proto-oncogenic receptor PTK c-Ret undergoes homodimerization and consequent activation as a result of the formation of a disulfide bond, likely between Cys376 of each monomer (77). The Cys376 residue of c-Ret is conserved among several receptor and nonreceptor PTKs and corresponds to Cys498 of Src, the nitrosylation of which has been found to enhance Src activity (143).
MAPK pathways as H2O2 targets
MAPK pathways constitute major signaling hubs and have been shown to assess the intracellular level of H2O2 and to dictate cell fate accordingly. In general, low levels of H2O2 promote cell proliferation, whereas high levels drive cell cycle arrest. MAPK pathways consist of a series of serine/threonine kinases—an MAPK kinase kinase (MAPKKK), an MAPK kinase (MAPKK), and an MAPK—that are sequentially activated by phosphorylation. The three types of MAPK (ERK, p38, and JNK) are preferentially activated by different MAPKK and MAPKKK enzymes and are inactivated by different phosphatases (MKPs). H2O2 acts at several levels to activate MAPK signaling. Whereas MKP inactivation appears to be a common means to increase MAPK activity, MKPs show different sensitivities to H2O2 depending on the oxidation state of their binding partner Prx I (94).
ASK1 is an MAPKKK (MAPKKK5) that is important for activation of the two stress-related kinases p38 and JNK. H2O2 oxidizes thioredoxin (Trx) and thereby induces its dissociation from ASK1 and elicits ASK1 oligomerization, resulting in the autophosphorylation and activation of ASK1 (56, 111, 154). Alternatively, H2O2 induces the oligomerization and activation of ASK1 in a manner dependent on formation of an intermolecular disulfide bond between unidentified cysteine residues of the kinase (118, 119), with this disulfide-dependent oligomerization having been shown to be mediated by Prx I (72) (see section on Sensor and Transducer Function of Prx).
In addition, two conserved cysteine residues of MAPKs—for example, Cys38 and Cys214 of ERK1 or ERK2—have been found to be directly oxidized to sulfinic and sulfonic acid in mitochondria of cells exposed to H2O2 (49). This mitochondrial oxidation increases the interaction of MAPKs with their cognate upstream kinases (MAPKKs), resulting in the phosphorylation (activation) of the MAPKs and their translocation to the nucleus.
Cysteine oxidation can also promote MAPK inhibition. Like ASK1, MAPKKK1 (also known as MAPK or ERK kinase kinase 1 or MEKK1) is an upstream kinase kinase of p38 and JNK. Strikingly, however, oxidative stress induced by menadione was found to inhibit MAPKKK1 through glutathionylation of its Cys1238 residue while at the same time activating ASK1 (39). MAPKK6, an upstream kinase of p38, also undergoes oxidation-induced inactivation through formation of a specific intramolecular disulfide bond between Cys109 and Cys196, which blocks the binding of ATP to its kinase domain (44). Consistent with the fact that these two cysteines are conserved in all seven members of the MAPKK family, other MAPKKs have also been found to be sensitive to oxidation (44).
The final outcome of the regulation of MAPK pathways via cysteine oxidation has thus been shown to be dependent on cellular context, with factors such as the subcellular location of the target protein, the level of H2O2 around the target, and the timing of target oxidation playing a key role.
AGC protein kinases (PKA, PKG, and PKC) as H2O2 targets
The AGC protein kinase family includes serine/threonine kinases such as cAMP-dependent protein kinase (PKA), cGMP-dependent protein kinase (PKG), protein kinase C (PKC), and Akt (protein kinase B). The optimal activity of PKA is dependent on phosphorylation of Thr197 located in the activation loop of the catalytic subunit. Cys199 in the activation loop readily undergoes oxidation either to form an intramolecular disulfide with Cys343 or to become glutathionylated. PKA oxidized at Cys199 is preferentially dephosphorylated at Thr197, resulting in a loss of activity (67).
Akt is also regulated both positively and negatively by H2O2. Akt isozymes (Akt1 to Akt3) are activated by phosphorylation of threonine and serine residues (Thr308 and Ser473 for Akt1) mediated by phosphoinositide-dependent protein kinase (PDK). The inactivation of PTEN by H2O2 and the consequent increase in the abundance of its substrate phosphatidylinositol 3,4,5-trisphosphate therefore promote Akt phosphorylation and activation, as observed in cells exposed to H2O2 or to the growth factors EGF or PDGF (90).
Conversely, direct oxidation of cysteine residues of Akt by H2O2 results in the downregulation of Akt activity (116, 174, 186). At H2O2 concentrations that induce apoptosis in H9c2 cells, Akt2 was found to undergo phosphorylation, likely promoted by PTEN inactivation, followed by dephosphorylation (116). Under the same stressful condition, Akt1 was gradually oxidized to form an intramolecular disulfide between Cys297 and Cys311, resulting in an increase in the affinity of the kinase for protein phosphatase 2A (PP2A) and thus an increased susceptibility to dephosphorylation and consequent inactivation by the phosphatase.
Inhibition of Akt2 through cysteine oxidation has also been observed but does not require stressful levels of H2O2, having been detected in PDGF-stimulated NIH3T3 cells (186). This inhibition of Akt2 was attributed to the oxidation of its Cys124 residue to sulfenic acid (186). Both Cys297 and Cys311 of Akt1 are conserved in the other two Akt isoforms, whereas Cys124 is found only in Akt2, allowing isoform-specific downregulation of Akt2 activity in response to the intracellular accumulation of H2O2 (186). The phosphorylation-dependent activation of Akt2 by upstream kinases and its Cys124 oxidation-dependent inhibition by H2O2 were found to occur in parallel in PDGF-treated cells.
PKC has long been known to be activated by H2O2 (84, 102). The PKC family of enzymes comprises at least 11 isoforms that are divided into three groups: conventional, novel, and atypical. Conventional PKCs (α, β1, β2, and γ) are activated by both diacylglycerol (DAG) and Ca2+ ions; novel PKCs are Ca2+ independent but are still activated by DAG; and atypical PKCs are independent of both Ca2+ and DAG. DAG binds to conventional PKCs at the C1 domain, which contains six conserved cysteine and two conserved histidine residues that are tetrahedrically coordinated by two Zn2+ ions. DAG binding to the C1 domain activates conventional PKCs by anchoring them to the plasma membrane at basal intracellular Ca2+ levels.
In cells exposed to exogenous H2O2, cysteine residues in the C1 domain are oxidized to form a disulfide bond (or bonds), resulting in release of the Zn2+ ions, and the oxidized PKC translocates from the cytosol to the plasma membrane, where it is activated independently of DAG (84, 102).
Mammals express three isoforms of PKG: the alternatively spliced α and β isoforms of type 1 PKG (PKGIα and PKGIβ) as well as type 2 PKG (PKGII). All PKG isozymes exist as dimers of two identical subunits. PKGIα forms a disulfide linking Cys42 of each subunit of the dimer in cells exposed to H2O2 (21), with this cysteine residue being unique to the Iα isoform. This oxidation of Cys42 was shown to directly activate the kinase in vitro as well as in rat cells and tissues, providing an alternative activation mechanism in addition to the classical mechanism mediated by nitric oxide and cGMP. Oxidation of PKGIα at this cysteine residue was also observed in cells stimulated with insulin, consistent with the ability of insulin to generate H2O2 (21).
Other kinases as H2O2 targets
ATM is a serine/threonine kinase that is important not only for DNA damage responses such as cell cycle arrest and apoptosis but also for maintenance of cellular redox homeostasis, a function mediated by multiple pathways, including feedback inhibition of mammalian target of rapamycin (45). ATM is directly activated by H2O2 in vitro as well as in various cell types exposed to H2O2 in the absence of DNA damage (45), with this activation having been attributed to homodimerization mediated by formation of a disulfide bond between Cys2991 of each subunit (45).
The use of a sulfenic acid-specific chemical probe revealed that a cysteine residue in the catalytic site of phosphatidylinositol 3-kinase (PI3K) was oxidized to sulfenic acid in HeLa cells exposed to H2O2, although the functional consequence of this oxidative modification remains unclear (98).
Transcription factors as H2O2 targets
The expression of many genes is sensitive to changes in intracellular redox state (161). The regulation of transcription factor function is achieved through multiple mechanisms, including changes in transcription of the transcription factor gene itself, in turnover of the encoded protein, in phosphorylation of the transcription factor, and in its interactions with other transcriptional regulators that can either promote or interfere with its activity. All of these mechanisms are susceptible to modulation by changes in the intracellular level of H2O2 and in the activity of signaling networks that include H2O2 target proteins.
Signaling pathways mediated by PTKs or serine/threonine kinases, which are interconnected at various levels by cross talk, lead to the activation of transcription factors, including AP1 (7), NF-κB (64), STAT3 (19, 99), specificity protein 1 (Sp1) (120), cAMP response element-binding protein (CREB) (120), ternary complex factor (TCF) (166), and c-Myc (117).
Alternatively, transcription factors, including AP1, NF-κB, the estrogen receptor, STAT3, and p53, can be regulated via oxidation of their own cysteine residues. Under conditions of oxidative stress, the AP1 subunit c-Jun, the p50 and p65 subunits of NF-κB, and STAT3 are glutathionylated at a cysteine residue located in the DNA binding site, resulting in repression of cyclin D gene expression (82, 141, 194). The estrogen receptor, which coactivates gene expression with AP1, contains two Zn2+ ions tetrahedrically bound to eight cysteine residues in its DNA binding domain (89, 187). Similar to the case of conventional PKCs, oxidative stress triggers formation of an intramolecular disulfide bond (or bonds) by these cysteine residues that prevent coordination of the two zinc-finger motifs for DNA binding (187).
The tumor suppressor protein p53 mediates the responses of cycling cells to oxidants, with these responses including cell cycle arrest, DNA repair, or cell death. The DNA binding domain of p53 contains four conserved cysteine residues, two of which (Cys275 and Cys277) undergo oxidation in vitro (108, 144). The better known mechanisms for regulation of the transcription factor function of p53 include posttranslational modifications such as phosphorylation, acetylation, and ubiquitylation. The proposed redox regulation of transcription factors via cysteine oxidation is mainly based on studies performed in vitro or with cells exposed to oxidants. Recently, however, stimulation of HEK293 cells with oncostatin M or interleukin-6 was shown to induce the oxidation of STAT3 at cysteine and consequent suppression of its transcriptional activity (168). As described below, this oxidation of STAT3 resulted in the formation of disulfide-linked oligomers mediated by Prx II (168).
Ion channels as H2O2 targets
The cellular levels of H2O2 and metal ions are tightly connected and control each other through multiple pathways. Calcium ions play a prominent role in such interplay (17, 54). For example, Ca2+ increases H2O2 levels by activating Nox5, Duox1, and Duox2 as well as by stimulating several mitochondrial dehydrogenases, resulting in increased mitochondrial respiration, whereas H2O2 activates various Ca2+ channels, including the inositol 1,4,5-trisphosphate receptor (IP3R), the ryanodine receptor (RyR), and STIM-ORAI channels by oxidizing their cysteine residues. Some K+ channels are also activated via targeting of their H2O2-sensitive cysteine residues.
Both IP3R and RyR channels are located in the membrane of the ER or sarcoplasmic reticulum, and they release Ca2+ from intracellular stores contained in these organelles in response to a wide array of hormones or during excitation/contraction coupling. Three different isoforms each of IP3R and RyR are expressed in mammalian cells. Although the site of modification appears to be isoform specific, all of these proteins harbor multiple redox-sensitive cysteine residues (2, 8). For example, RyR1 can undergo redox modification (nitrosylation, glutathionylation, or disulfide formation) at 12 cysteine residues (Cys36, Cys253, Cys315, Cys811, Cys906, Cys1040, Cys1303, Cys1591, Cys2326, Cys2363, Cys3193, and Cys3635) (2).
Depletion of Ca2+ in the ER store triggers Ca2+ entry from outside the cell to refill the store and maintain long-term Ca2+ signaling processes. This store-operated Ca2+entry is mediated by the cooperation of two protein families—designated STIM (stromal interaction molecule) and ORAI—that are localized in the ER membrane and the plasma membrane, respectively. STIM proteins convey the message that the Ca2+ store in the ER is depleted to the plasma membrane by translocating to contact points of the two membrane systems and physically interacting with the Ca2+-selective ORAI channels in the plasma membrane to trigger refilling of the store (12).
Two STIM isoforms (STIM1 and STIM2) and three ORAI isoforms (ORAI1, ORAI2, and ORAI3) have been identified in humans. Exposure of cells to low levels of H2O2 promotes STIM1 translocation and activation of store-operated Ca2+ entry even in cells in which the ER store is not depleted (63). Two cysteine residues (Cys49 and Cys56) located in the luminal domain of STIM1 have been identified as redox sensors, with Cys56 undergoing glutathionylation in the presence of H2O2 (63). A cysteine residue (Cys195) in the extracellular loop of ORAI1 has also been implicated in sensing of extracellular H2O2 (12). Interestingly, after ORAI1 forms a complex with an STIM protein in response to store depletion, its Cys195 residue becomes inaccessible to exogenous H2O2 (16).
Members of the transient receptor potential (TRP) family of proteins contain six transmembrane domains and assemble into tetramers to form nonselective cation channels that function as biosensors (86). TRP channels are activated by diverse stimuli from the extracellular or intracellular milieu that include receptor ligands, heat, osmotic pressure, mechanical stress, and environmental irritants.
Mammalian TRP channels are divided into six subfamilies: canonical (C), vanilloid (V), melastatin (M), polycystic kidney disease (P), mucolipin (ML), and ankyrin (A). Many TRP channels—including TRPV1, TRPV3, TRPV4, TRPC1, TRPC4, TRPC5, and TRPA1—sense alterations in the redox environment through oxidative modification of their cysteine residues (86). TRPV channels are highly selective for Ca2+. A detailed study of human TRPV1 recently revealed that an intersubunit disulfide bond is formed between Cys258 and Cys742 of two subunits and that the C258S mutant is completely insensitive to H2O2-mediated activation (130).
Voltage-gated K+ channels constitute a large family of K+-selective ion channels in which the channel pore is formed by four α subunits (153). The largest subfamily of these channels is designated Kv and is further subdivided into Kv1 to Kv12 channels. Potassium channels activated by both voltage and Ca2+ (SLO1–SLO3) have also been identified. Numerous studies have described redox sensitivity of K+ channels (153).
Although the ROS target in K+ channels was initially identified as methionine (36, 157), low levels of H2O2 were later shown to increase the current mediated by Kv7 channels (also known as M channels) by acting on three cysteine residues corresponding to Cys112, Cys175, and Cys519 of Kv7.4 (50). H2O2 was also shown to inhibit the activity of Kv10 (also known as EAG), Kv11 (ERG), and SLO channels by oxidizing cysteine residues. The major sites of oxidation were identified as Cys532 and Cys562 of Kv10.1, Cys723 of Kv11.1, and Cys911 of SLO1 (83, 153, 200).
Uncoupling proteins (UCPs) are mitochondrial transporters that mediate proton leakage across the inner mitochondrial membrane, thus uncoupling oxidative phosphorylation from ATP synthesis. Acutely activated thermogenesis in brown adipose tissue was recently found to be accompanied by a substantial increase in mitochondrial ROS levels and the consequent oxidation of Cys253 of UCP1 to sulfenic acid. This oxidation was required for the adrenergic activation and uncoupling function of UCP1 (35). This cysteine residue of UCP1 is conserved in UCP2 and UCP3.
Intracellular Messenger Function of H2O2 and Its Regulation by Prx
The Prx family of peroxidases reduces H2O2 or alkyl peroxide (ROOH) with a conserved cysteine residue (the peroxidatic Cys or CP–SH) serving as the site of oxidation by peroxides (147, 149). In addition to the CP–SH located in the NH2-terminal region of the molecule, most Prx enzymes contain an additional conserved cysteine (the resolving Cys or CR–SH) in the COOH-terminal region. On the basis of the location or absence of the CR residue, Prxs are classified into 2-Cys, atypical 2-Cys, and 1-Cys subfamilies. Mammalian cells express six isoforms of Prx: four 2-Cys Prxs (Prx I to IV), one atypical 2-Cys Prx (Prx V), and one 1-Cys Prx (Prx VI).
These isoforms vary in subcellular localization, with Prx I, II, and VI being localized mainly in the cytosol; Prx III being restricted to mitochondria; Prx IV being found predominantly in the ER; and Prx V being present in the cytosol, mitochondria, and peroxisomes. Peroxiredoxins are abundant proteins, with Prx I and Prx II together constituting a total of 0.2%–1% of soluble protein in cultured mammalian cells (25).
During the catalytic cycle of Prxs, the CP–SH is first oxidized to sulfenic acid (CP–SOH) by H2O2, which promotes a local conformational change that allows the reaction of CP–SOH with CR–SH to form a disulfide that is subsequently reduced by an appropriate electron donor to complete the cycle (136, 150). The CP–SOH of 2-Cys Prxs is occasionally further oxidized to sulfinic acid (CP–SO2H) before the disulfide can be formed, resulting in inactivation of peroxidase function (197). This hyperoxidation is reversed by the ATP-dependent enzyme sulfiredoxin, which restores peroxidase activity (14, 189). The reversible hyperoxidation confers a chaperone function on 2-Cys Prxs in cells subject to severe oxidative stress (69). It has also been shown that the hyperoxidation allows accumulation of H2O2 and so H2O2-mediated signaling (80).
Kinetic analysis revealed that peroxides bind to Prx with submicromolar affinity (110, 132, 180). A specific binding site for peroxide was confirmed by determination of the crystal structure of Prx with bound peroxide or peroxide-mimicking molecules (58, 123, 136). Structural analysis also yielded a model for the transition state of the peroxidase reaction. In this model, all Prxs have an active-site structure with a nearly universal sequence, PXXXTXXC (where X is any amino acid), as well as a conserved arginine residue that is distant in sequence but located nearby in the three-dimensional fold. The transition state is characterized by an extensive hydrogen bond network formed by the CP thiolate anion (CP–S−), peroxide, the threonine and proline residues located within the universal PXXXTXXC motif, and the conserved arginine residue. The importance of the conserved arginine was also revealed by mutational study (121).
This network of hydrogen bonds not only provides a binding site for peroxide but also properly aligns the peroxide for attack by the CP sulfur atom as well as lowers the activation energy for both breaking of the O–O bond of peroxide and formation of the S–O bond of CP–SOH by stabilizing the transition state intermediate. In accord with this model, the second-order rate constants for the oxidation of CP–SH to CP–SOH measured in several laboratories are very high, lying in the range of 1 × 106–1 × 108 M −1 s−1 (38, 110, 131, 132, 135, 139, 180).
H2O2 is distinct from other messenger molecules in that it acts not by binding to effectors but by oxidizing cysteine residues of effector proteins. The mechanism by which H2O2 selects its target proteins and oxidizes specific cysteine residues thereof is not well understood, however. It has been generally accepted that such selective oxidation is attributable to the fact that thiol oxidation is affected by pK a (where K a is the acid dissociation constant). The pKa of a typical proteinaceous cysteine thiol is ∼8.4, but characteristics of the microenvironment centered on the cysteine residue, such as proximity to a positively charged basic residue, can lower this value and thereby increase the susceptibility of the thiol group to deprotonation (oxidation) at physiological pH.
The active-site cysteines of members of the PTP family thus have low pKa values (4.6–5.5) (105, 169), and the rates of Cys–SH oxidation to Cys–SOH by H2O2 for PTPs (PTP1B, Cdc25, leukocyte antigen-related, and vaccinia H1 related) have been kinetically measured as 9–160 M −1 s−1, values that are 10–180 times that (0.87 M −1 s−1) for oxidation of the glutathione thiol (pKa of 8.4) (42, 169, 188). Although the effect of a low pK a on thiol reactivity is clear, the target thiol of PTPs still reacts with H2O2 at a rate that is several orders of magnitude slower than that for the catalytic thiol (CP–SH) of Prxs. Given that Prxs are abundant proteins that are present in various compartments of the cell, cysteine residues of H2O2 target proteins such as PTPs are at a competitive disadvantage for reaction with H2O2.
Recent studies have revealed that this disadvantage can be overcome by transient inactivation of neighboring Prx molecules so as to allow the target cysteines to react with H2O2 (see below). Furthermore, in some instances, redox-regulated proteins are not directly oxidized by H2O2, their oxidation instead being mediated by Prxs. In such a scenario, Prx is first oxidized by H2O2 and then transfers its oxidation state to the redox-regulated protein, thus serving as both a sensor and transducer of H2O2 signaling (also see below).
Localized Regulation of H2O2 Concentration at the Plasma Membrane
Light was shed on the mechanism by which H2O2 produced in response to receptor engagement is able to propagate its signal by the finding that Prx I localized in the vicinity of the receptor can be reversibly inactivated. Prx I becomes phosphorylated on Tyr194 by an Src family kinase in several cell types stimulated via various receptors, including the PDGFR, EGFR, TCR, and the B cell receptor (190).
In vitro analysis revealed that such tyrosine phosphorylation of Prx I impairs its peroxidase function. Importantly, tyrosine-phosphorylated Prx I, which accounts for about 0.2%–0.4% of total Prx I in PDGF-stimulated NIH3T3 cells, was found to be present exclusively in the low-density, detergent-insoluble fraction of these cells. This fraction is derived mainly from lipid rafts, which are membrane microdomains enriched in glycosphingolipid-cholesterol and are similar in their lipid composition and resistance to detergent solubilization to other biochemically well-defined membrane structures known as caveolae.
Lipid rafts and caveolae compartmentalize cellular processes by serving as organizing centers for the assembly of signaling molecules. Growth factor and immune receptors, Nox enzymes, and Src family kinases are all membrane-associated proteins that are highly localized to lipid rafts (3, 85, 133). Nox1 appears to be the major source of H2O2 in cells stimulated with PDGF or EGF (190), with this Nox1-derived H2O2 being critical for the propagation of growth factor signaling, including protein tyrosine phosphorylation, Akt and MAPK activation, and target gene transcription (4, 90, 146, 173).
These various findings indicate that localized H2O2 production by Nox1 might not be sufficient to support the messenger function of this molecule in growth factor-stimulated cells, with inactivation of Prx I by phosphorylation on Tyr194 being necessary to protect the pool of H2O2 signaling molecules from destruction by this abundant enzyme so as to allow the oxidation of target molecules such as PTPs, Src family kinases, and PTEN (Fig. 3). PTPs such as SHP-1 and SHP-2 associate with tyrosine-phosphorylated (activated) receptors via their SH2 domains. PTEN is present mostly in the cytosol, but it transiently associates with lipid rafts to antagonize PI3K signaling (87).
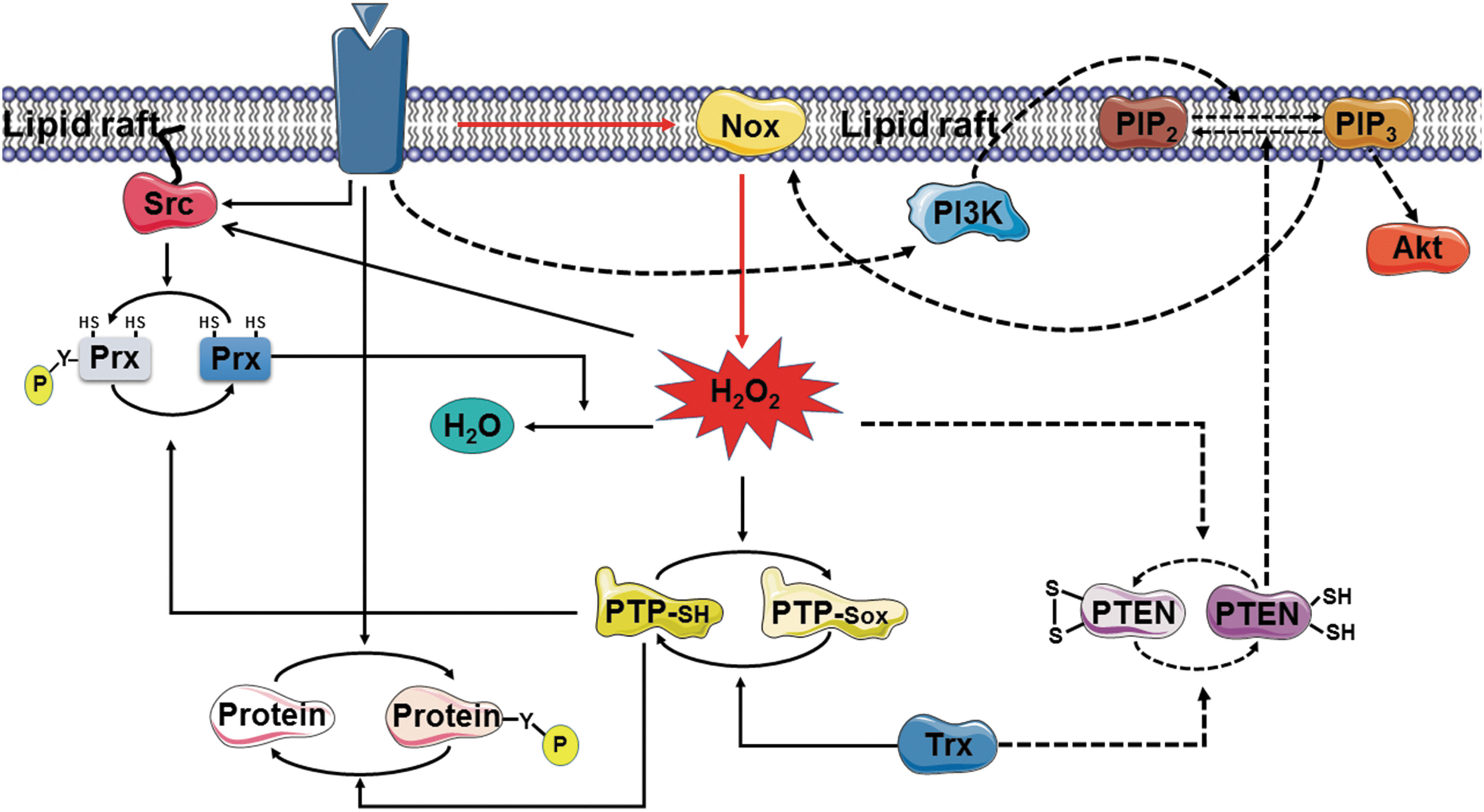
The accumulation of H2O2 around lipid rafts would be expected to promote further phosphorylation and inactivation of Prx I through both activation of Src family kinases and inactivation of PTPs (Fig. 3). These two positive feedback loops likely sustain the H2O2 levels necessary for the oxidation of target cysteines. The oxidative inhibition of PTPs by H2O2 is critical for growth factor signaling because activation of receptor PTKs alone is not sufficient to achieve the levels of protein tyrosine phosphorylation necessary for such signaling, with the concurrent inhibition of PTPs by H2O2 preventing the futile cycle of phosphorylation and dephosphorylation.
For a similar reason, oxidative inactivation of PTEN by H2O2 is necessary for the accumulation of PI(3,4,5)P3 that is generated by PI3K and needed for Akt signaling (90). PI(3,4,5)P3 was also found to be required for PDGF-induced Nox activation (6), thus revealing the operation of another positive feedback loop (Fig. 3). A similar scenario likely applies to H2O2 signaling in cells stimulated via the B cell receptor or TCR.
Whereas both Prx I and Prx II are present in the low-density, detergent-insoluble membrane fraction, only Prx I is phosphorylated by PTKs. Given that the accumulation of a small amount of H2O2 at the plasma membrane can trigger rapid signal amplification through the action of multiple positive feedback loops (Fig. 3), this presence of Prx II in addition to Prx I might serve as a safeguard to prevent inappropriate activation of signaling cascades by basal production of H2O2 at the cell membrane. Consistent with this notion, cells derived from Prx II-deficient mice show enhanced tyrosine phosphorylation of PDGFR when stimulated with PDGF (34).
A similar protective role for Prx II at specific microdomains was also demonstrated in human aortic vascular endothelial cells stimulated with VEGF (76). A fraction of Prx II, but not of Prx I, was thus detected in caveolae of these cells. Furthermore, depletion of Prx II rendered VEGFR2 insensitive to VEGF stimulation as a result of the formation of an intramolecular disulfide between Cys1199 and Cys1206 in its COOH-terminal tail, suggesting that Prx II protects the PTK activity of VEGFR2 from oxidative inactivation (76). Whereas VEGFRs are located in both caveolae and other regions of the plasma membrane, the protective function of Prx II was found to be restricted to caveolae, with the oxidized form of VEGFR2 being detected only in this fraction of the endothelial cells after Prx II knockdown. Deficiency of Prx II was also shown to impair VEGFR2 activation and angiogenesis in mice (76).
Functional differences between Prx I and Prx II are also apparent in the phenotypes of the corresponding knockout mice. Mice lacking Prx I thus manifest an increased susceptibility to cancer, whereas those deficient in Prx II do not (127), suggesting that Prx I may function as a tumor suppressor. Prx I directly binds to PTEN and thereby protects its lipid phosphatase function from inactivation by removing H2O2, whereas Prx II does not bind to PTEN (23).
Whether PTEN located at lipid rafts is also protected by Prx I binding and whether such PTEN-bound Prx I becomes inactivated through phosphorylation are questions that remain unanswered, however. Nevertheless, tumor suppression by Prx I is likely due, at least in part, to its ability to remove H2O2 from the region surrounding growth factor receptors and its ability to protect PTEN by binding to the lipid phosphatase. As described in the following section, the tumor suppressor function of Prx I is also likely related to its role as a regulator of H2O2 concentration around the centrosome.
Localized Regulation of H2O2 Concentration Around the Centrosome via Cdk1-Dependent Phosphorylation of Prx I
Another example of spatially confined inactivation of Prx is provided by Prx I phosphorylation at Thr90 in mitotic cells. Mammalian 2-Cys Prx enzymes (Prx I–IV) contain a consensus sequence, (S/T)PX(K/R), for phosphorylation by Cdks. Among 2-Cys Prxs, Prx I is phosphorylated most efficiently by the purified Cdk1–cyclin B complex, with the susceptibility to such phosphorylation then decreasing in the order Prx II, Prx III, and Prx IV, and such phosphorylation inactivates peroxidase function (27).
Threonine phosphorylation of Prx I occurs during mitosis, being virtually undetectable in interphase. The amount of threonine-phosphorylated Prx I in mitotic HeLa cells was estimated to be ∼0.4% of total Prx I, suggesting that phosphorylation of Prx I at Thr90 is likely a localized event, as in the case of its phosphorylation at Tyr194. Indeed, Prx I was found to be present at the centrosome, and such centrosome-associated Prx I, but not cytosolic Prx I, was enriched in the Thr90-phosphorylated form of the protein. Furthermore, threonine-phosphorylated Prx I was detected at the centrosome of HeLa cells during the early stages of mitosis (prophase, prometaphase, and metaphase) but not during interphase or late mitotic stages (anaphase, telophase, and cytokinesis) (101).
In addition to being the major microtubule-organizing center of mammalian cells, the centrosome acts as a centralizing platform that coordinates the complex sequence of events that lead to the activation of Cdk1 and the consequent entry of cells into mitosis (18). Many proteins that function in the mitotic entry network—such as cyclin B, the phosphatases Cdc25B and Cdc25C, and the kinases Polo-like kinase 1 (Plk1) and Aurora A—are specifically recruited to the centrosome in G2 phase of the cell cycle, resulting in the generation of high local concentrations of these mitotic activators (18).
Activation of the Cdk1–cyclin B complex occurs first at the centrosome during prophase, and its amplification through multiple feedback loops involving cyclin B, Plk1, and Aurora A also occurs at this organelle (103). Exit of cells from mitosis also requires that these regulators (cyclin B, Plk1, and Aurora A) be degraded in a timely manner. Such degradation is mediated by the 26S proteasome and is triggered by ubiquitylation of the regulators by the multisubunit ubiquitin ligase APC/C (anaphase-promoting complex/cyclosome), which requires CDC20 homolog 1 (Cdh1) as an activator protein (138, 140, 192).
Cdh1 is prevented from interacting efficiently with APC/C as a result of its phosphorylation by Cdks during S and G2 phases, but it is able to activate APC/C after its dephosphorylation by Cdc14B (138, 192). A fraction of each of APC/C, Cdh1, and Cdc14B is present at the centrosome (140, 191), as are Cdk1, cyclin B, Plk1, and Aurora A. Activation of APC/C also occurs at the centrosome (18, 103). At the onset of mitosis, cyclin B, Plk1, and Aurora A need to be protected from APC/C–Cdh1-dependent ubiquitylation and degradation to ensure the robust and irreversible activation of Cdk1–cyclin B. Such protection appears to be achieved through suppression of the Cdc14B-mediated dephosphorylation of Cdk1-phosphorylated Cdh1.
In the absence of such suppression of centrosomal Cdc14B activity, further activation of Cdk1 would not be expected to occur because of the premature degradation of cyclin B, Plk1, and Aurora A. The catalytic Cys314 residue of Cdc14B readily forms an intramolecular disulfide with Cys228 in cells exposed to H2O2, with Cdc14B appearing to be more sensitive to such oxidation than is PTEN.
The intracellular concentration of ROS—as measured with the chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate, an irreversible fluorescent probe sensitive to various oxidants—has been found to change during cell cycle progression (62). Analysis with a genetically encoded fluorescent probe (HyPer) that reacts reversibly and specifically with H2O2 (10) revealed that the intracellular level of H2O2 increased steadily from G1 to S phase and was maximal at G2-M (101). Whereas HyPer fluorescence reflects global changes in H2O2 concentration during cell cycle progression, however, the production of H2O2 and its elimination occur via highly localized and regulated processes. The measured global changes in H2O2 levels may thus not be representative of local changes.
The accumulation of H2O2 in S and G2-M phases might be due to increased energy metabolism, which is accompanied by mitochondrial H2O2 production. H2O2 derived from Nox4 has also been shown to be required for G2-M progression (195). Indeed, the Nox inhibitor diphenyleneiodonium attenuates mitotic entry in HeLa cells (101). In addition, inhibition of cytosolic phospholipase A2 activity—which produces arachidonic acid for H2O2-generating lipoxygenases—as well as inhibition of lipoxygenase activity each delayed mitotic entry, suggesting that G2-M progression is affected by H2O2 produced from multiple sources (101).
Mitochondrially produced H2O2 might also be directed to the centrosome, as appears to be the case in T cells. Ligation of the TCR thus induces H2O2 production that is required for the activation of various signaling molecules, including ERK (43). This H2O2 might be produced in T cells by different sources, including Nox2 and mitochondria (68, 162). Mitochondria accumulate in a region close to the immunological synapse formed between T cells and antigen-presenting cells and trigger localized Ca2+ entry that is critical for T cell activation and proliferation (88, 142, 160).
TCR activation also induces translocation of the centrosome to the vicinity of the synapse (1). Changes in mitochondrial morphology and positioning might therefore determine the site of H2O2 release from these organelles and thus the proteins targeted for oxidation by the released H2O2. The immediate targets of H2O2 in the region close to the synapse include two Src family kinases (Lck and Fyn) that are associated with the TCR.
Taken together, these many observations indicate that, during interphase, the centrosome is shielded from the effects of H2O2 by Prx I that is physically associated with the organelle (Fig. 4A). During early mitosis, however, centrosomal Prx I is inactivated as a consequence of phosphorylation by Cdk1, resulting in exposure of the centrosome to the high tide of H2O2 and consequent inactivation of Cdc14B (Fig. 4A). This inactivation of Cdc14B is critical to prevent the premature dephosphorylation of Cdh1 and the consequent untimely ubiquitylation of cyclin B, Plk1, and Aurora A by APC/C–Cdh1 (Fig. 4B). Consistent with this scenario, downregulation of the pericentrosomal H2O2 level through forced expression of a centrosome-targeted form of catalase delayed mitotic entry (101).
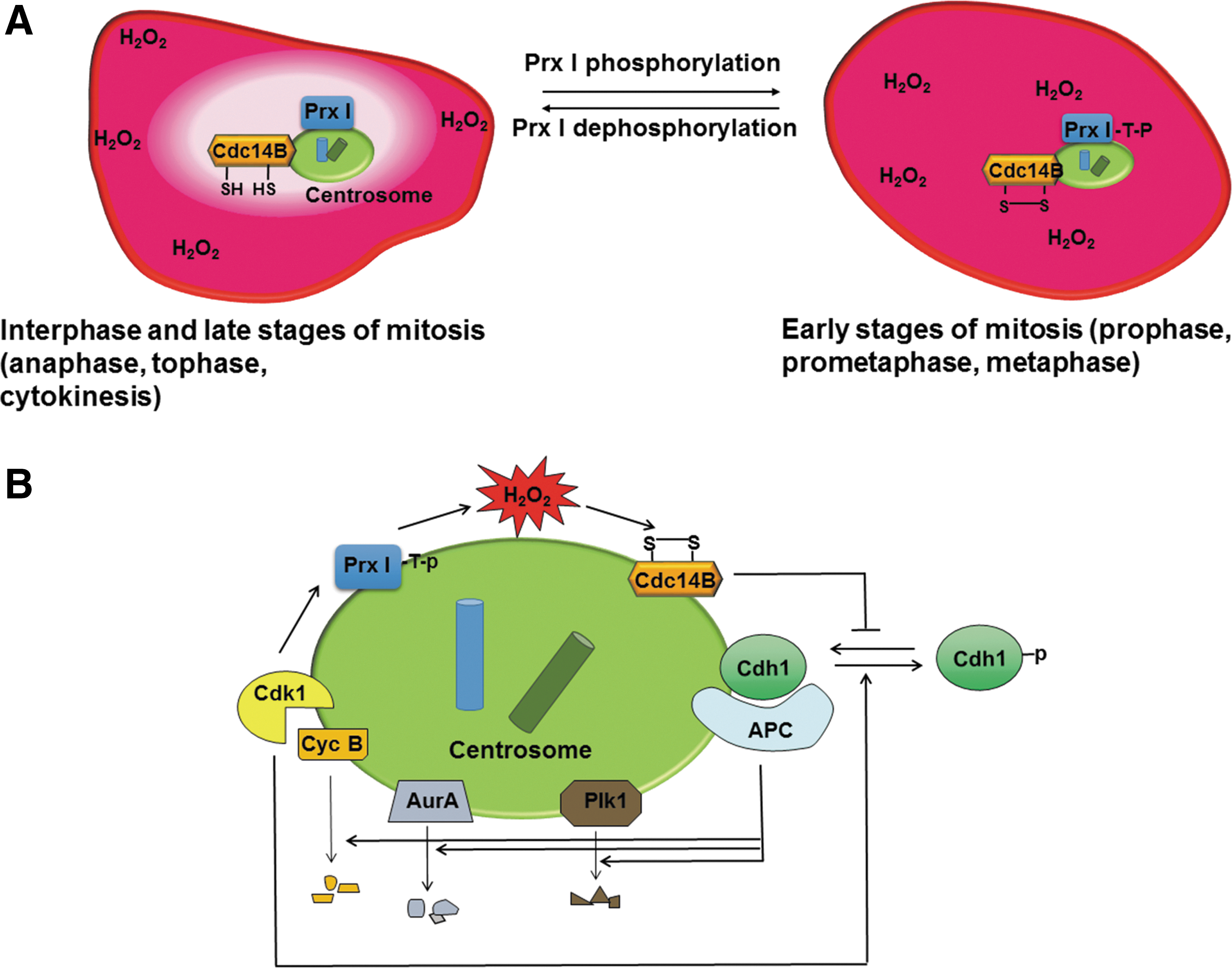
Later during mitosis, Prx I is dephosphorylated by protein phosphatase 1 (PP1) and PP2A, which are also concentrated at the centrosome (192), and the centrosome is consequently again shielded from H2O2, allowing Cdc14B activation, Cdh1 dephosphorylation, and activation of APC/C–Cdh1 (Fig. 4).
The phosphatases Cdc25B and Cdc25C, which function during the entry of cells into mitosis, are not sensitive to oxidation by H2O2 under the same conditions as those supportive of the oxidation of PTEN and Cdc14B. The accumulation of H2O2 at the centrosome during the early stages of mitosis as the result of Prx I inactivation therefore does not lead to the inactivation of centrosome-associated Cdc25B and Cdc25C, with the role of these phosphatases in the feedback loops underpinning activation of Cdk1–cyclin B thus being maintained. The oxidative inactivation of Cdc25C has been suggested to occur at higher levels of H2O2 as a means to induce cell cycle arrest (159).
The regulation of APC/C–Cdh1 by endogenous ROS (H2O2) was first demonstrated by the observation that antioxidant treatment of synchronized cells induced cell cycle arrest in late G1 phase (62). Although cyclin A expression is induced at the transcriptional level in early G1 phase, cyclin A protein does not accumulate until late during the G1-S transition as a result of its ubiquitylation by APC/C–Cdh1. The activity of cyclin A-associated Cdk2 (or Cdk1) is required to initiate DNA synthesis, but endogenous H2O2 is required to inactivate APC/C–Cdh1 activity and thereby to allow cyclin A accumulation at late G1 phase (62).
The mechanism of APC/C–Cdh1 inactivation by H2O2 at late G1 phase is not known, but our knowledge of the control of pericentrosomal H2O2 levels (101) suggests that it is also achieved through a combination of Cdh1 phosphorylation by Cdks and Cdc14B inactivation by H2O2. Given that Prx I associated with the centrosome remains active (not phosphorylated) during all of interphase, however, the centrosome is not likely the site of Cdc14B inactivation by H2O2 at this time.
In quiescent cells, APC/C–Cdh1 must remain active to ensure that certain positive regulators of S and M phases do not accumulate prematurely. Given that mitosis persists in the absence of Cdk1 activity when phosphatase activity is suppressed (167) and that H2O2 can inactivate phosphatases and APC/C–Cdh1 at the centrosome, the presence of Prx I at the centrosome might serve as a safeguard to prevent unscheduled mitotic entry as a result of an accidental increase in H2O2 abundance such as might occur in response to ultraviolet radiation or in association with inflammation. Consistent with this notion, Cdc14B, Cdh1, and Prx I have all been suggested to function as tumor suppressors (51, 127, 191).
Sensor and Transducer Function of Prx
In addition to being associated with subcellular compartments such as lipid rafts and the centrosome and its regulation of the susceptibility of proteins present in these compartments to oxidation by H2O2, Prx is able to catalyze the oxidation of H2O2 effector molecules. In this process, the catalytic cysteine sulfhydryl group (CP–SH) of Prx is first oxidized to sulfenic acid or disulfide intermediates, either of which can then transfer its oxidation state to an effector protein to transduce the H2O2 signal (Fig. 5). Binding of the effector protein to Prx can theoretically occur at any stage of and persist throughout the Prx catalytic cycle. It is also possible that Prx and the effector protein are recruited for transient disulfide formation by a scaffold protein.
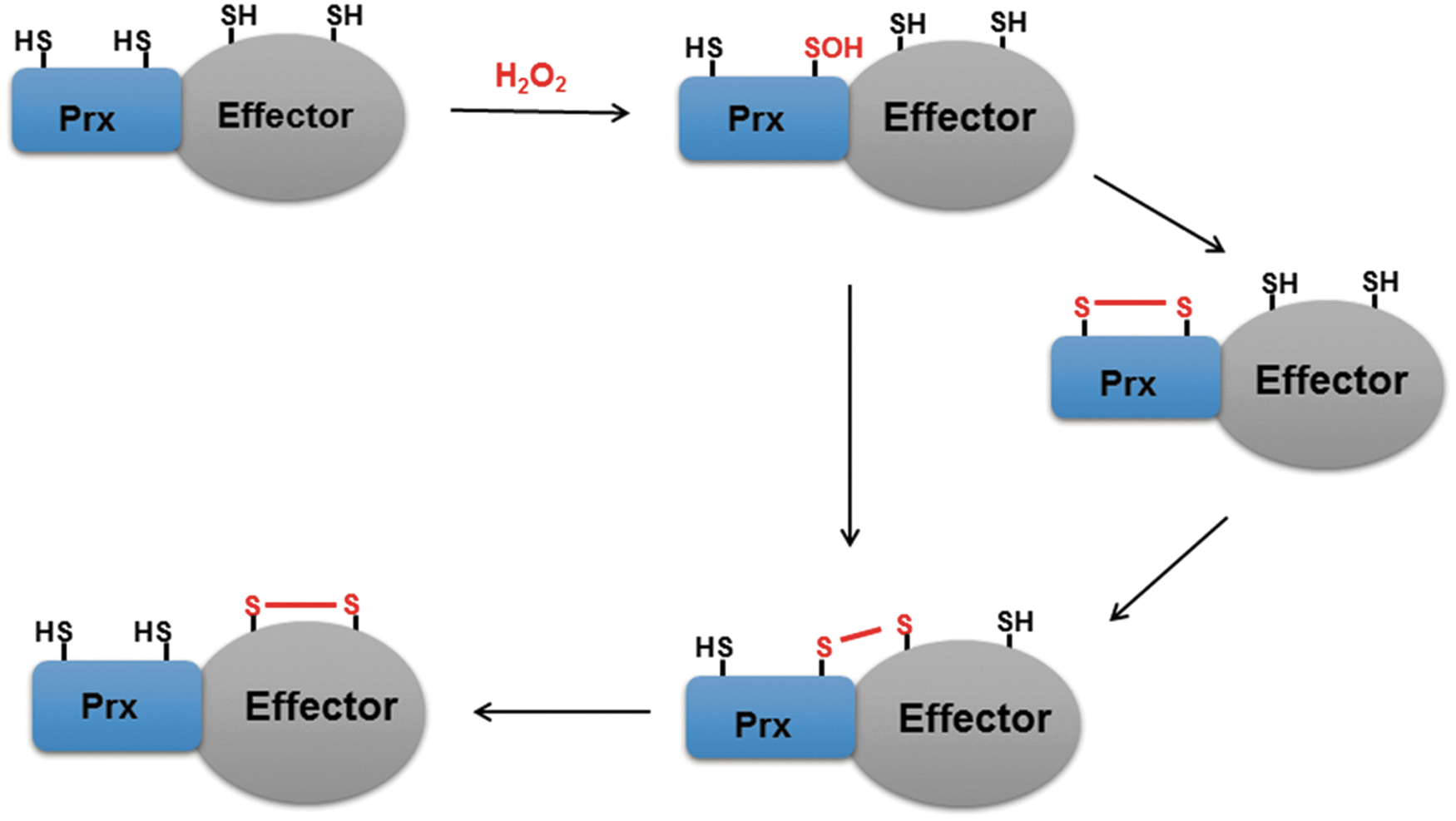
Peroxidase-catalyzed protein thiol oxidation by H2O2 was first demonstrated in yeast, in which the AP1-like transcription factor Yap1 was shown to be oxidized indirectly through the action of a thiol-dependent peroxidase (41, 185).
It was later shown that H2O2 produced by ER membrane-associated oxidoreductin 1 (Ero1) in mammalian ER can be used to oxidize Prx IV, the ER-specific isoform of Prx, and that the oxidized state of Prx IV can then be transferred to the thiols of protein disulfide isomerase (PDI). The resulting formation of an intramolecular disulfide in PDI then allows PDI to promote protein folding in the ER by transfer of the disulfide to a target protein (177, 201). The physical interaction of Prx IV with PDI was revealed by determination of the crystal structure of oxidized Prx IV bound to ERp44 (an ER-specific PDI) (196). It was thus suggested that Prx IV functions as a sensor of H2O2 in PDI-mediated protein folding.
ASK1 is activated subsequent to oligomerization mediated by the formation of intermolecular disulfide bonds in cells exposed to H2O2 (118). This disulfide-dependent oligomerization is catalyzed by Prx I, which forms a transient disulfide-linked intermediate with ASK1 in H2O2-treated cells (72). Knockdown of Prx I thus resulted in inhibition of both ASK1 oxidation and phosphorylation of the downstream kinase p38, suggesting that Prx I can function as a sensor of H2O2 and transduce its signal to ASK1. This ASK1-activating function was found to be specific to Prx I, with Prx II exhibiting a negative effect on ASK1 oligomerization, probably because it lowers cytosolic H2O2 levels.
Prx I was also shown to play a role in the oxidation of APE1/Ref-1, a multifunctional protein that contributes to both DNA repair and transcriptional regulation, with Prx I-catalyzed APE1/Ref-1 oxidation having been proposed as a mechanism to prevent NF-κB activation and upregulation of interleukin-8 expression (125).
On the contrary, Prx II is thought to play a sensor and transducer role in the formation of disulfide-linked oligomers of STAT3 or DJ-1 (48, 168). Dimerization and tetramerization of STAT3 were initially observed in cells exposed to H2O2 (100), and this oligomerization was shown to be mediated by the oxidation of several cysteine residues located in the DNA binding and COOH-terminal transactivation domains of STAT3 and to result in diminished STAT3 binding to serum-inducible elements in cells treated with H2O2 or interleukin-6 (100). It was then shown that Prx II catalyzes both the dimerization and tetramerization of STAT3 in a reaction involving the formation of disulfide-linked Prx II–STAT3 intermediates.
Prx I did not form such disulfide-linked conjugates with STAT3. Two cysteine residues (Cys712 and Cys718) in the COOH-terminal transactivation domain and two cysteines (Cys418 and Cys426) in the DNA binding domain of STAT3 were found to be critical for dimerization and tetramerization, respectively.
Importantly, Prx II-catalyzed STAT3 oligomerization was observed in cells stimulated with interleukin-6 or oncostatin M (168). In contrast to the case of Prx IV and PDI, direct evidence for the physical association of Prx II and STAT3 has not been obtained to date. An observed dominant negative effect of a catalytically inactive form of Prx II (in which both CP and CR are substituted by serine) on STAT3 oligomerization, however, suggests that such physical association occurs before the formation of disulfide-linked conjugates, providing specificity for the relay of oxidative equivalents from H2O2 to Prx II and then to STAT3 (168). Prx II also interacts with DJ-1 and promotes its disulfide-linked homodimerization in cardiomyocytes exposed to H2O2 (48).
The catalytic cysteine (CP) of Prx enzymes is highly sensitive to oxidation by H2O2, whereas direct oxidation of the cysteines of H2O2 target proteins is generally very slow. It is also likely in the interest of cell survival that the concentration of the potentially toxic oxidant H2O2 not remain elevated for the time required for direct oxidation of kinetically inefficient targets. The Prx-catalyzed oxidation of target proteins is thus likely a biologically selected mechanism of H2O2 signaling.
The few examples of such regulation identified to date likely reflect difficulties in capturing the disulfide-linked Prx-target intermediate, which is formed transiently and in specific compartments. It is important to note, however, that Prx isoforms are known to interact with many other proteins (11, 150). Moreover, Prx I and Prx III each transiently form a number of disulfide-linked higher molecular weight intermediates in H2O2-treated cells, suggesting that cysteine residues of multiple proteins are targets for Prx-catalyzed oxidation (72).
Perspective
As our understanding of signaling networks has become more advanced, the importance of strict temporal and spatial regulation of signaling events has become evident. This is especially so for events related to signaling by the precarious molecule H2O2. The outcome of oxidative modification of an H2O2 effector molecule depends on contextual factors such as cell type as well as the subcellular domain and stage of the cell cycle in which the oxidation occurs. Oxidative modification and signaling events observed in cells exposed to exogenous H2O2, even at physiologically relevant concentrations, can thus be very different from those that occur in response to a specific physiological signal.
Several challenges remain to achieving a full understanding of H2O2 signaling. First, it is important that changes in H2O2 levels be monitored at discrete locations during cell cycle progression. Although sensitive probes for such monitoring are not yet available, substantial progress has recently been made toward this goal by taking advantage of the high sensitivity of Prx enzymes to H2O2 and their capacity to transduce the H2O2 oxidation state to target proteins. Genetically encoded fluorescent probes for H2O2 that are based on the fusion of a Prx isoform to green or yellow fluorescent protein and in which Prx catalyzes the formation of an intramolecular disulfide within the probe and thereby changes its fluorescence properties have been designed (115, 183).
Second, given that Prxs are localized to various subcellular compartments and regulate local H2O2 levels, unraveling of Prx regulation at specific locations—as exemplified by Prx I regulation via tyrosine phosphorylation at lipid rafts and via threonine phosphorylation at the centrosome—will be required. Prx enzymes also undergo additional posttranslational modifications such as lysine acetylation (163) as well as cysteine glutathionylation (26) and nitrosylation (145), although the in vivo relevance of these additional modifications remains to be established.
Third, given that Prx-catalyzed oxidation appears to be an economic means by which to convey the H2O2 message with spatiotemporal precision and target specificity, cysteine residues of many signaling proteins are likely to be oxidized indirectly via the disulfide transferase activity of Prx. Identification of such target proteins will be difficult, however, because only a small fraction of the total pools of a given Prx and target protein is likely to form a disulfide-linked intermediate and such an intermediate is likely to be transient.
Fourth, tools to selectively detect the oxidized form of H2O2 target proteins are needed. Again, given that Prx-catalyzed oxidation is likely restricted to a small subpopulation of target molecules localized in a specific subcellular compartment, the development of such tools for various target proteins will be a long-term goal. However, the successful development of phage antibodies that specifically recognize the conformation of oxidized PTP1B but not that of the reduced form (61) may provide a guide for the accomplishment of this goal.
Footnotes
Acknowledgments
This study was supported by grants from the National Research Foundation of Korea (NRF-2015R1D1A1A09057154 and NRF-2015R1D1A1A01059571) and from the Ministry of Food and Drug Safety in 2016 (16173MFDS009).