
Abstract
Significance:
Essential metals such as copper, iron, manganese, and zinc play a role as cofactors in the activity of a wide range of processes involved in cellular homeostasis and survival, as well as during organ and tissue development. Throughout our life span, humans are also exposed to xenobiotic metals from natural and anthropogenic sources, including aluminum, arsenic, cadmium, lead, and mercury. It is well recognized that alterations in the homeostasis of essential metals and an increased environmental/occupational exposure to xenobiotic metals are linked to several neurological disorders, including neurodegeneration and neurodevelopmental alterations.
Recent Advances:
The redox activity of essential metals is key for neuronal homeostasis and brain function. Alterations in redox homeostasis and signaling are central to the pathological consequences of dysfunctional metal ion homeostasis and increased exposure to xenobiotic metals. Both redox-active and redox-inactive metals trigger oxidative stress and damage in the central nervous system, and the exact mechanisms involved are starting to become delineated.
Critical Issues:
In this review, we aim to appraise the role of essential metals in determining the redox balance in the brain and the mechanisms by which alterations in the homeostasis of essential metals and exposure to xenobiotic metals disturb the cellular redox balance and signaling. We focus on recent literature regarding their transport, metabolism, and mechanisms of toxicity in neural systems.
Future Directions:
Delineating the specific mechanisms by which metals alter redox homeostasis is key to understand the pathological processes that convey chronic neuronal dysfunction in neurodegenerative and neurodevelopmental disorders. Antioxid. Redox Signal. 28, 1669–1703.
Introduction
M
Prospective epidemiological studies have associated cognitive, motor, and behavioral alterations to environmental exposure to metals and metalloids (153, 158, 250, 371), effects that are exacerbated when environmental exposures occur chronically and during development (153, 158, 250). Long-term effects of either environmental metal exposure or alterations in metal homeostasis in the central nervous system (CNS) and peripheral nervous system (PNS) have been proposed to play a role in neurodegenerative disorders (379). Importantly, alterations in the cellular redox environment of the cell are central to the toxic effects of metals.
Previous reviews address the general role of metals in neurodegeneration, or the mechanisms by which metals produce oxidative stress or neurotoxicity (85, 86, 89, 163, 355). In this work, we present an integrated review on recent advances in (a) the metabolism of both essential and xenobiotic metals; (b) the mechanisms by which distinct metals determine or modify the cellular redox homeostasis; (c) the link between metal redox activity and function in neural systems; and (d) how alterations in metal homeostasis or intracellular/extracellular levels participate in neurotoxicity and neurodegeneration.
Overview of Oxidative Stress and Redox Homeostasis
Reactive species is a term used to describe compounds that can receive or provide a couple of electrons or one electron participating in nucleophilic, electrophilic, or redox metabolic reactions, respectively. Reactive oxygen species (ROS) are molecules derived from O2, an obligate component of aerobic organisms. The reduction of O2 is one of the primary reactions that sustain aerobic life, yet it is also the main source for ROS. ROS include free (•) and nonfree radical species such as hydroxyl radicals (•OH), superoxide anion radicals (O2 •−), and hydrogen peroxide (H2O2). Reactive nitrogen species (RNS) contain both nitrogen (N) and O (oxygen atom), and thus can be categorized as ROS. RNS include nitric oxide (NO•), nitrogen dioxide radical (NO2), and peroxynitrite (OONO−) (120, 252).
A major source of intracellular ROS production are the mitochondrial electron transport complexes, primarily the one-electron reduction of O2 to O2 •− by complex I (ubiquinone: NADH oxidoreductase), by the semiquinone of ubiquinone (coenzyme Q), and by complex III (cytochrome bc1 complex or CoQH2-cytochrome c reductase). O2 •− undergoes rapid dismutation into H2O2 through the action of superoxide dismutases (SODs). Three types of SODs exist in mammalian cells that use an essential metal as cofactor. Cu/Zn-dependent SOD1 and 3 are localized in the cytosol (SOD1), the extracellular space (SOD3) and to a lesser extent, in the inner membrane space of the mitochondria (SOD1). MnSOD (SOD2) is solely localized in the mitochondrial matrix. The transition metal (Cu or Mn) in the active site of SODs is required for the breakdown of O2 •− by catalyzing both the one-electron oxidation and one-electron reduction of separate O2 •− to give the overall disproportionation reaction that produces O2 and H2O2. Binding of Zn to SOD1 or 3 is not essential for O2 •− dismutation reaction but confers higher thermal stability to the proteins (64).
One to 2% of the total mitochondrial O2 consumed is leaked and contributes to the formation of ROS. Usually, this occurs at a slow rate and can be counteracted by mitochondrial antioxidant systems, but in damaged or aged mitochondria, increased ROS formation occurs. O2 •− and H2O2 fuel •OH formation through Fenton/Haber–Weiss reactions, where H2O2 oxidizes a redox-active metal (Fe or Cu) leading to the formation of •OH. Then, the oxidized metal is reduced back by O2 •− or other cellular reductants promoting metal-catalyzed free radical chain reactions (100, 120, 252).
Other sources of ROS are the nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidases (NOX), enzymes whose principal function is to generate O2 •− or H2O2. The formation of RNS begins with the synthesis of NO•, catalyzed by nitric oxide synthases (NOSs), which are Fe dependent. Zn is an important structural element of NOS enzymes and is also known to inhibit their activity. NO• reacts with O2 •− to produce OONO−, which is a strong oxidizing agent. Microsomes and peroxisomes are important sources of ROS due to the presence of NOX and NOS (302). In addition, ROS production can be mediated by the activity of enzymes such as xanthine oxidase (that contains an Fe-sulfur [S] cluster), the heme proteins cyclooxygenases, cytochrome P450 enzymes, lipoxygenases, and myeloperoxidases, as well as the protein folding machinery in the endoplasmic reticulum (ER) (96, 120, 252).
ROS/RNS act as signaling molecules affecting the stability, expression, function, and activity of a multiplicity of proteins controlling almost all cellular functions, including proliferation, cell survival, metabolism, and signaling. An adequate balance between the formation and elimination of ROS/RNS facilitates the signaling role of these reactive species. However, an imbalance between an increase in the steady-state levels of ROS/RNS and the ability of the cell to metabolize/detoxify them leads to a nonhomeostatic state referred to as oxidative stress. Oxidative stress results in the irreversible oxidative modification of biomolecules with the concomitant loss of function of proteins, damage to cellular organelles, and eventual cell death (96, 100, 252, 282). Polyunsaturated fatty acids are one of the preferred oxidation targets for ROS; particularly, free radicals that are potent initiators of lipid peroxidative chain reactions. Lipid peroxidation products, including malondialdehyde (MDA) and 4-hydroxy-2-nonenal (HNE), can react further with DNA bases and proteins (83). DNA bases are also susceptible to direct oxidation by free radicals that can cause mutations as well as deletions in both nuclear and mitochondrial DNA (328).
Protein oxidation can be a reversible or irreversible phenomenon depending on the type of modification, ROS involved, and the extent of oxidation. Tyrosine (Tyr) nitration, protein carbonylation, and protein crosslinkage generated by adduct formation between oxidized proteins, lipid peroxides, or glycative products are irreversible modifications that promote a loss of function, aggregation, and degradation of the targeted protein, and in some cases, the formation of toxic by-products (71). In contrast, reversible oxidative modifications in the sulfur-containing amino acids methionine (Met) and cysteine (Cys) act as sensors and transducers of ROS/RNS-mediated signaling. Thiol-based oxidoreductases thioredoxins (Trxs), glutaredoxins (Grxs), and Met sulfoxide reductases reduce such modifications acting as the OFF-switch for redox signaling processes (101, 160). On the contary, both Trxs and peroxiredoxins (Prxs) have been proposed to act as redox sensors, buffers, and relays for H2O2- and NO•-mediated signal transduction (46, 195, 268, 323). Cys are also targeted by electrophiles generating irreversible modifications, which are thought to be a primary mechanism of toxicity by xenobiotics (193).
Cells are equipped with enzymatic and nonenzymatic antioxidant systems to counteract the toxic effects of ROS/RNS and maintain redox homeostasis (96, 100, 120, 252). The reducing power of glutathione (GSH) is essential for the detoxification of peroxides by GSH peroxidases (GPX) with the resultant conversion of GSH to GSH disulfide (GSSG). GSSG is reduced back by GSH reductase (GR) in an NADPH-dependent manner (105). Catalases also detoxify peroxides but their localization is primarily restricted to peroxisomes. Other endogenous nonenzymatic antioxidants include uric acid, lipoic acid, and ubiquinol (or reduced coenzyme Q), and those obtained from the diet, such as vitamins and flavonoids (96, 100, 120, 252).
The CNS is particularly sensitive to oxidative damage, from which neurons and oligodendrocytes seem to be more susceptible than astrocytes and microglia. The basis for this increased sensitivity is linked to the high levels of O2 consumption (and electron leakage as a consequence), the low levels of antioxidant defenses when compared to other cells, and the abundance of lipids or fatty acids (262, 295).
Higher levels of endogenous antioxidants and antioxidant systems in astrocytes are explained by the activation of the nuclear factor erythroid-2-related factor 2 (Nrf2) transcription factor (314). Nrf2 recognizes antioxidant response elements to trigger the transcription of antioxidant systems. The ubiquitin ligase Kelch-like ECH-associated protein 1 (Keap1) negatively regulates Nrf2 signaling by inducing its ubiquitination and degradation. Upon modification of specific Cys residues within Keap1 by oxidants or electrophiles (including metals), Nrf2 is released from Keap1 and translocates to the nucleus to induce gene expression dependent on antioxidant response elements (ARE). Nrf2 signaling in neurons has been reported to be epigenetically silenced (27), and induction of the Nrf2 pathway does not seem to be able to promote antioxidant protection (148). Astrocytes also have higher levels of NADPH and glucose 6-phosphate dehydrogenase (G6PD) (109). In contrast, antioxidant genes in neurons seem to be transcriptionally regulated, independent from Nrf2 by synaptic activity, through the triggering of the activating transcription factor 4 (ATF4) and the activator protein 1 (AP-1) (23, 177). Furthermore, while both neurons and astrocytes can synthesize GSH, neurons depend on the supply of GSH precursors via GSH efflux (13, 26).
Neurotoxicity of Metals and Metalloids
Neurotoxicity is defined as a damaging effect on the nervous system caused by a biological or chemical agent. The neurotoxic effects of chemicals are the result of a series of events that include the following: the entry and/or changes in the distribution of a chemical into the brain, interactions with specific cellular targets (neurons and glia), and the initiation of biochemical changes, resulting in structural and functional changes of the nervous system (270). Environmental neurotoxicants include organic and inorganic chemical compounds, such as heavy metals, organic solvents, and cytotoxic substances that can also contain heavy metal mixtures (e.g., pesticides, cigarette smoke, diesel exhaust particles,). The neurotoxic effect of environmental agents is determined by their chemical composition, metabolic function, and pathologican consequence, differing widely according to the brain region targeted and the mechanism(s) of action (270).
Essential Metals
Micronutrients are defined by their essentiality and very limited quantity in humans, where their deficiency results in the impairment of biological functions (103, 205). Some metals are essential for the maintenance of cellular homeostasis. Essential metals display important roles as signaling agents or cofactors and, in particular, as activators or redox system components (Supplementary Table S1) (103, 205). Traditionally, cellular osmotic balance and signaling (including synaptic communication and excitability) are associated with nontransition metal ions, such as Na+, K+, and Ca2+, which form complexes with proteins using low-affinity binding sites, are found at high concentrations, and move quickly across cellular compartments. On the contrary, transition metal ions are known as catalytic cofactors or structural elements in enzymes. Transition metals are present in lower concentration (“trace elements”) and are usually coordinated to proteins at high-affinity binding sites. In the last decade, a role for Zn2+ ion as a second messenger has been recognized; however, the role of redox-active metals, such as Cu, Fe, and Mn, in cellular signaling is less explored.
Cu, Fe, and Mn are cofactors of many enzymes that catalyze redox reactions. Although the high reactivity of these metals is essential for life, they can also be involved in uncontrolled redox reactions associated with oxidative stress and cellular damage. Hence, a highly conserved network of proteins strictly regulates the homeostasis of redox-active metal ions, by controlling their uptake, intracellular distribution, storage, and export (205). In the following sections, we describe how brain homeostasis of redox reactive metal ions requires close communication between the blood-brain barrier (BBB), neurons, astrocytes, oligodendrocytes, and microglia. The cases where metal trafficking is tightly linked to the cellular redox environment are highlighted, while the potential role of metal redox cycling in redox signaling is also discussed. Finally, for each essential metal ion, we review how the disruption of its homeostasis may cause two major features associated with neurodegenerative diseases: dysfunction of metalloproteins and aberrant metal-protein interactions that can lead to protein aggregation and uncontrolled ROS production.
Copper
Cu is present in biological systems as Cu+ (cuprous ion) and Cu2+ (cupric ion) (Supplementary Table S1). Cu is a redox-active metal and a cofactor of many enzymes involved in cellular respiration, radical detoxification, as well as biosynthesis of neurotransmitters, neuropeptides, and hormones. For example, Cu is required as cofactor of several important enzymes in the brain, such as peptidylglycine monooxygenase (PHM), dopamine β-monooxygenase (DBM), tyrosinase (TYR), and cytochrome C oxidase (COX). Cu can activate O2 for reduction and although its high reactivity with O2 is essential for life, if uncontrolled it can promote oxidative stress and cellular damage. Cu+ can react with H2O2 to produce highly reactive •OH. Cu also induces microglial activation and mitochondrial ROS formation (137).
The control of Cu homeostasis in the brain requires a close interrelationship between the BBB, neurons, and astrocytes (300) (Fig. 1). Astrocytes regulate the properties of the BBB, which is the entry point for Cu into the brain from the blood stream, where Cu is bound to albumin or ceruloplasmin (Cp) (58) (Fig. 1a). At the same time, neurons require Cu as a cofactor and neuromodulator, while astrocytes are key players in synaptic transmission and Cu homeostasis (58). The Cu trafficking machineries of the BBB endothelial cells, neurons, and astrocytes resemble those of other extensively studied mammalian cells (Fig. 1a–c). Extracellular Cu is primarily transported into cells as Cu+ via the Cu transporter 1 (CTR1) (58). Cu2+ reduction to Cu+, and Cu uptake from Cp via CTR1, has been proposed to involve a reduction step but no Cu2+ reductase has been identified (281). CTR1-independent mechanisms have also been proposed. The divalent metal transporter 1 (DMT1) seems to play a compensatory role for Cu uptake under certain conditions such as in the absence of CTR1 or under low Fe conditions (147, 231). Interestingly, DMT1 loss promotes brain Cu accumulation and oxidative stress (122). Other potential candidates recently proposed to mediate Cu uptake are the Zrt (Zn-regulated transporter)- or Irt (Fe-regulated transporter)-like protein 4 (ZIP4) (11, 29).

Intracellular Cu distribution depends on the relative concentration and metal affinity of chaperones or chelators (18) (Fig. 1b, c). The antioxidant protein 1 (Atox1), the Cu chaperone for superoxide dismutase 1 (CCS1), and GSH have been proposed to take Cu+ from CTR1 (197). Chaperones not only bind Cu but they also deliver it to specific targets. CCS1 transfers Cu to SOD1, where its reactivity with O2 is required for SOD1 maturation via the formation of a disulfide bridge (17). Copper chaperones COX19 and COX17 deliver Cu to the COX assembly proteins (SCO1 and SCO2) and COX11. Finally, Atox1 transports Cu to the ATPase copper transporting alpha (ATP7A) and beta (ATP7B) in the secretory pathway, where cuproenzymes such as SOD3 are metalized (Fig. 1b, c). Upon an excess in cytosolic Cu levels, vesicles in the secretory pathway are loaded with Cu and trafficked to the plasma membrane, where Cu is released into the extracellular space (196, 197).
Strikingly, although GSH has a lower affinity for Cu, compared to Atox1 and CCS1, the rate of Cu entry into the cell via CTR1 is affected by GSH, but not by Atox1 or CCS1 depletion, likely due to the higher concentration of GSH (208). Thus, GSH is the most important cytosolic first acceptor of Cu from CTR1, providing a tight link between cellular Cu uptake and cellular redox homeostasis. GSH in turn is known to transfer Cu ions to metallothioneins (MTs), small Cys-rich proteins that play a major role as scavengers for metal ions (93) (Fig. 1b, c). Three distinct isoforms of MTs are expressed in the human brain. MT-I, MT-II, and MT-III are found in astrocytes, while MT-III is the main isoform expressed in neurons. MTs can be secreted, playing a crucial role in modulating Cu homeostasis and protecting the cell from oxidative damage (299).
Chaperones use Cys residues to coordinate Cu+ in their reduced state. Thus, Cys oxidation affects Cu dynamics. Atox1 coordinates Cu using a CysXXCys motif that can form a disulfide bond, which can be reduced directly by GSH or by Grx1 in a GSH-dependent manner (Fig. 1d) (41, 127). During neuronal differentiation, the GSH/GSSG ratio increases promoting a more reductive environment, which in turn reduces Cys residues at the Cu binding site of Atox1. These events enable Cu transport from Atox1 to ATP7A/B and enhance Cu availability to load the active sites of newly synthetized cuproenzymes (128) (Fig. 1d). After neuronal differentiation, both Cu and MT-III levels increase (249).
The recent development of fluorescent sensors has revealed new important roles of intracellular Cu in neuronal activity (78); for example, in the spine neck of hippocampal neurons, Cu is essential for the control of the dendritic actin cytoskeleton (269). Cu export by ATP7A has been reported to be triggered by the activation of glutamate (Glu)/N-methyl-

Cu trafficking at the synapse is complex and involves several Cu binding proteins, such as the membrane-bound PrPC, the amyloid precursor protein (APP), and the amyloid beta (Aβ) peptide from neurons, or the neurokinin B (NKB) peptide from astrocytes (Fig. 2). PrPC has several Cu binding sites and it might be involved in Cu sensing and transport into neurons (Fig. 3a). During synaptic transmission, the extracellular Cu concentration may reach ∼100 μM. Cu binding to PrPC induces its endocytosis, possibly contributing to Cu delivering into the cytosol (263). APP has been reported to regulate Cu efflux, as the APP knockout mice display higher levels of Cu in the brain and in neurons, while APP overexpression leads to decreased intracellular Cu concentrations (28, 330, 375). In contrast, Aβ peptides produced from the proteolytic cleavage of APP have been proposed to act as Cu scavengers (264), particularly those cleaved to yield 4–40 peptides that have higher affinity for Cu and make up to 50% of the Aβ in plaques (374). NKB has been suggested to compete for Cu from PrPC and transport it into astrocytes by endocytosis (Fig. 2c) (308). PrPC and Aβ contain intrinsically disordered regions with Cu binding sites that have the capacity to adopt different Cu coordination modes, some of which have been proposed to activate O2 and produce ROS (Fig. 3a) (184, 350).
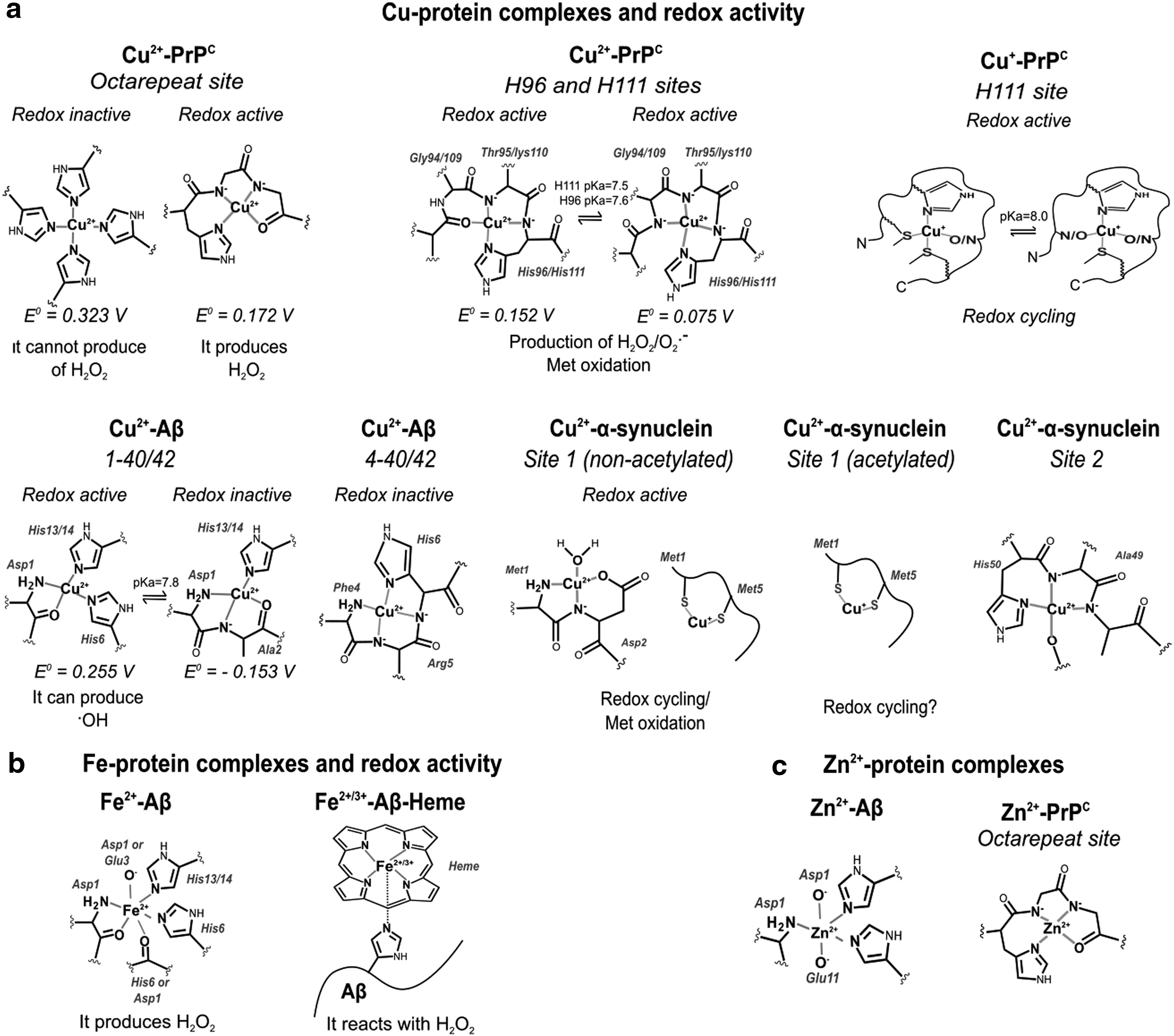
A dysfunction in Cu homeostasis is reported to alter neuronal function and lead to disease progression, including neurodegeneration (Supplementary Table S1). Menkes disease and Wilson disease are caused by mutations or partial deletions in ATP7A and ATP7B, respectively. These Cu transporters have different patterns of expression in the CNS, explaining the distinct pathological features of each disease. ATP7B is found in the visual cortex, anterior cingulate cortex, caudate, putamen, substantia nigra (SN), and cerebellum. ATP7A is detected in astrocytes and neurons from the hippocampus and cerebellum, the BBB and choroid plexus, and during neural development (338). Wilson patients display parkinsonism, underscoring the importance of Cu homeostasis in the motor controlling systems. On the contrary, the ubiquitous expression of ATP7A has challenged mechanistic investigations in Menkes disease. However, a recent study shows that depleting ATP7A in neurons and glia does not lead to neurodegeneration, but to an increased susceptibility to NMDA seizures (132), underscoring its neuroprotective role as described above (Fig. 2a,b). Although neurodegeneration is clearly linked to alterations in Cu homeostasis in Menkes and Wilson diseases, the mechanisms are still unknown.
Neurodegenerative disorders such as amyotrophic lateral sclerosis (ALS), Huntington's disease (HD), Parkinson's disease (PD), and Alzheimer's disease (AD) have been associated with alterations in Cu homeostasis, but a link to specific genetic alterations in Cu transport or handling is missing (Supplementary Table S1). These neurodegenerative diseases are associated with the formation of amyloid aggregates composed of proteins that are either a Cu-dependent antioxidant enzyme, such as SOD1 in ALS, or Cu-binding proteins, such as Aβ and α-synuclein in AD and PD, respectively.
ALS is characterized by the degeneration of motor neurons, and mouse models show increased intracellular Cu levels and the formation of protein aggregates composed of SOD1 and Cu transport proteins such as Ctr1, CCS, Atox1, and Cox17 (344, 346). SOD1 aggregation has been associated with an alteration in protein stability, which is impacted by metallation and Cu-dependent dimerization. Although SOD1 plays an important role in clearing O2 •−, studies have demonstrated that the mechanism is likely other than the alteration in the antioxidant capacity of SOD1 (312). Increased intracellular Cu levels have been reported in ALS models (346). Consistently, Cu chelators or MT-I overexpression extend the life span and slow disease progression of the SOD1 (G93A) ALS mouse model (345).
HD is an autosomal dominant genetic disorder caused by polyglutamine (polyQ) repeat expansions near the N-terminus of the huntingtin (Htt) protein. HD is characterized by movement dysfunction, psychiatric and cognitive alterations linked to the degeneration of striatal spiny neurons. Cu accumulation in the striatum of HD transgenic mice has been reported (102). Mutant Htt forms toxic aggregates, but the mechanisms of toxicity are still unclear. A putative Cu binding site in Htt involving Met8 and His82 has been identified, and this interaction promotes polyQ aggregate formation (102, 381), which is reduced by MT-III overexpression (123).
PD is characterized by the degeneration of dopaminergic neurons in the SN, which are rich in Cu. PD patients show decreased Cu content in the SN (70) without changes in Cu levels in serum, plasma, and cerebrospinal fluid (CSF) (203). Accordingly, decreased levels of CTR1 (70) have been reported in the SN, while MT-I and MT-II levels in active astrocytes are increased, reflecting a glial response to the loss of Cu homeostasis in PD (226). In contrast, occupational exposure to Cu has been linked to an increased risk to develop PD (115).
The accumulation of intracellular protein inclusions (Lewy bodies), where α-synuclein is the main protein component, is another hallmark of PD (362). α-Synuclein is a small intrinsically disordered protein (IDP) enriched in presynaptic terminals and nucleus that can interact with cytoskeleton components and lipid membranes (170). IDPs are proteins that can adopt different conformations, and thus respond to changes in their biochemical environment. This property allows them to engage in interactions with multiple protein targets (23). Cu2+ and Cu+ ions are capable of binding to α-synuclein at three different sites (20, 33, 228, 361) (Fig. 3a). Interestingly, the H50Q SNCA/α-synuclein mutation linked to hereditary PD abolishes one Cu binding site altering Cu-induced α-synuclein aggregation (361). Moreover, Cu binding to the high-affinity Cu-binding site at the N-terminal region of α-synuclein accelerates its amyloid aggregation in vitro (20, 33); although this effect is abolished in acetylated α-synuclein (234), which is found in Lewy bodies (10).
While several studies have suggested that Cu-induced aggregation of α-synuclein is directly linked to its neurotoxicity, recent studies suggest a lack of correlation between protein aggregation and cytotoxicity (361); in fact, it has been demonstrated that α-synuclein potentiates the toxicity of Cu in dopaminergic cells in the absence of enhanced accumulation of protein aggregates (8). Clearly, further investigations are needed to completely understand the structural impact of Cu-α-synuclein interactions and their role in PD. On the contrary, acetylation and Cu+ binding to α-synuclein are two synergistic events that turn the intrinsically disordered N-terminal region into an α-helix conformation (228) (Fig. 2d), which displays higher affinity for membranes (75). These observations suggest a potential link between Cu+-α-synuclein interactions and the proposed function of α-synuclein in vesicle trafficking; a link that might be perturbed in PD.
Different redox modifications of α-synuclein are found in Lewy bodies, including Met oxidation, Tyr nitration (67), and formation of di-Tyr-linked α-synuclein dimers. Cu+-α-synuclein complexes have been implicated in these modifications, as they are capable of activating O2, leading to Met oxidation and di-Tyr bond formation (5, 227) (Fig. 3a). In contrast, a recent report suggests that Cu+ complexes with oligomeric or fibrillar α-synuclein reduce metal-catalyzed ROS formation (264). Cu has also been shown to potentiate oxidative damage induced by the dopamine (DA) analog 6-hydroxydopamine (63). Cu also interacts with the early-onset PD recessive genes (protein) PARK7 (DJ-1) and PARK2(Parkin). Cu binding to DJ-1 protects against metal toxicity, possibly acting as a chaperone for SOD1 (35, 114); while Parkin mutations have been reported to increase the cytotoxic effect of heavy metals, including Cu (1).
AD is a neurodegenerative disease associated with the degeneration of hippocampal and cortical neurons and eventual loss of memory and progressive dementia (326). Decreased levels of Cu are found in AD brains (42), while the total Cu content and labile nonprotein-bound Cu fraction are increased in the plasma of AD patients (325). Interestingly, polymorphisms in ATP7B have been linked to AD (326). AD is associated with the formation of extracellular amyloid plaques composed of Cu bound to Aβ peptides 1–40 and 1–42 fragments produced by cleavage of the APP, as well as N-truncated forms 4–40 and 4–42, and to a lesser extent 11–40/42 (210, 273, 364). The Cu binding features of Aβ 4–40/42 and 11–40/42 are different from those of 1–40/42, leading to the formation of Cu-Aβ complexes with distinct redox properties (Fig. 3a) (229). Different aggregation properties have also been described, as illustrated by the faster fiber assembly rate of Aβ 11–40/42 when compared with 1–40/42 (21). Substoichiometric Cu2+ concentrations trigger Aβ aggregation through a different pathway that involves the formation of oligomers more neurotoxic than those generated by the peptide alone (25, 204). On the contrary, the redox activity of Cu-Aβ complexes has also been proposed to lead to the generation of ROS (52, 214), but contradictory results exist as well (264). Interestingly, interaction of Cu with N-truncated 4–40 and 4–42 peptides yields redox-inactive Cu-Aβ complexes (229, 350). A dysfunction in Cu homeostasis in AD is also evidenced by decreased levels of MT-III (390). MTs are capable of exchanging Cu2+ with Aβ1–40/42, reducing Cu and stabilizing it. This Cu exchange by MTs has been proposed as a redox-silencing mechanism that prevents ROS formation by Cu-Aβ complexes (223).
The impact of dysfunctional Cu homeostasis in AD might go beyond the neurotoxicity of Cu-Aβ complexes. APP, whose mutations are associated with AD, is a type 1 transmembrane protein that displays three Cu binding sites in its extracellular domain (22, 140). One Cu-binding site is located in the growth factor-like domain, and has been implicated in Cu-induced dimerization of APP, a process that would be important in cell adhesion and signaling (22). Cu was found to induce APP phosphorylation at Thr668 promoting its localization to the axonal membrane, suggesting an important link between Cu and APP functions at the synapse that might be perturbed in AD. Aβ peptides can also interfere with Cu-PrPC interactions implicated in the regulation of NMDAR activity (Fig. 2b) (389). In addition, Cu has been reported to promote the degradation of the low-density lipoprotein receptor-related protein 1 (LRP1) via Tyr nitration and proteasomal degradation, which was linked to a decrease of Aβ clearance and its resultant accumulation in brain vasculature (319). Clearly, Cu plays important roles in neuromodulation and signaling processes, which would be perturbed in AD.
Iron
Fe is found in biological systems primarily as ferrous (2+) and ferric (3+) ions (Supplementary Table S1). Fe is a redox-active metal involved in several redox reactions that catalyze the formation of ROS. Fe is tightly bound to Fe storage and transport proteins, while <5% is present as labile redox-active Fe bound to low-affinity molecules. Fe is required as cofactor of several important enzymes for respiration and synthesis of neurotransmitters, including tryptophan hydroxylase (serotonin) and Tyr hydroxylase (norepinephrine and DA), cholesterol, and fatty acids; the latter particularly important for nerve myelination (161, 342). Electron transfer in many Fe enzymes, including the mitochondrial respiratory complexes, is facilitated by heme and Fe/S clusters (291). Fe is also important as cofactor for peroxidases and catalases, which are important for cellular redox homeostasis (6).
Fe is heterogeneously distributed in the brain; it is highly concentrated in the SN, hippocampus, striatum, interpeduncular nuclei (125), and myelin (329) (Supplementary Table S1). Fe homeostasis is regulated by communication between the BBB and astrocytes. In the blood stream, Fe3+ is found coordinated to transferrin (Tf) or ferritin (Ft), and as heme. Fe uptake into the BBB can occur by two pathways: (a) by direct transport of Fe2+ into the cytosol via DMT1; or (b) by endocytosis of Tf-bound Fe3+ via the Tf receptor (TfR), where the low pH of the endosome causes the release of Fe3+ from Tf. In both cases, Fe3+ is reduced to Fe2+ by the duodenal cytochrome b (Dcytb) or by the six transmembrane epithelial antigen of the prostate 2 (Steap2) ferrireductases and then transported by DMT1 (Fig. 4a) (215, 218).
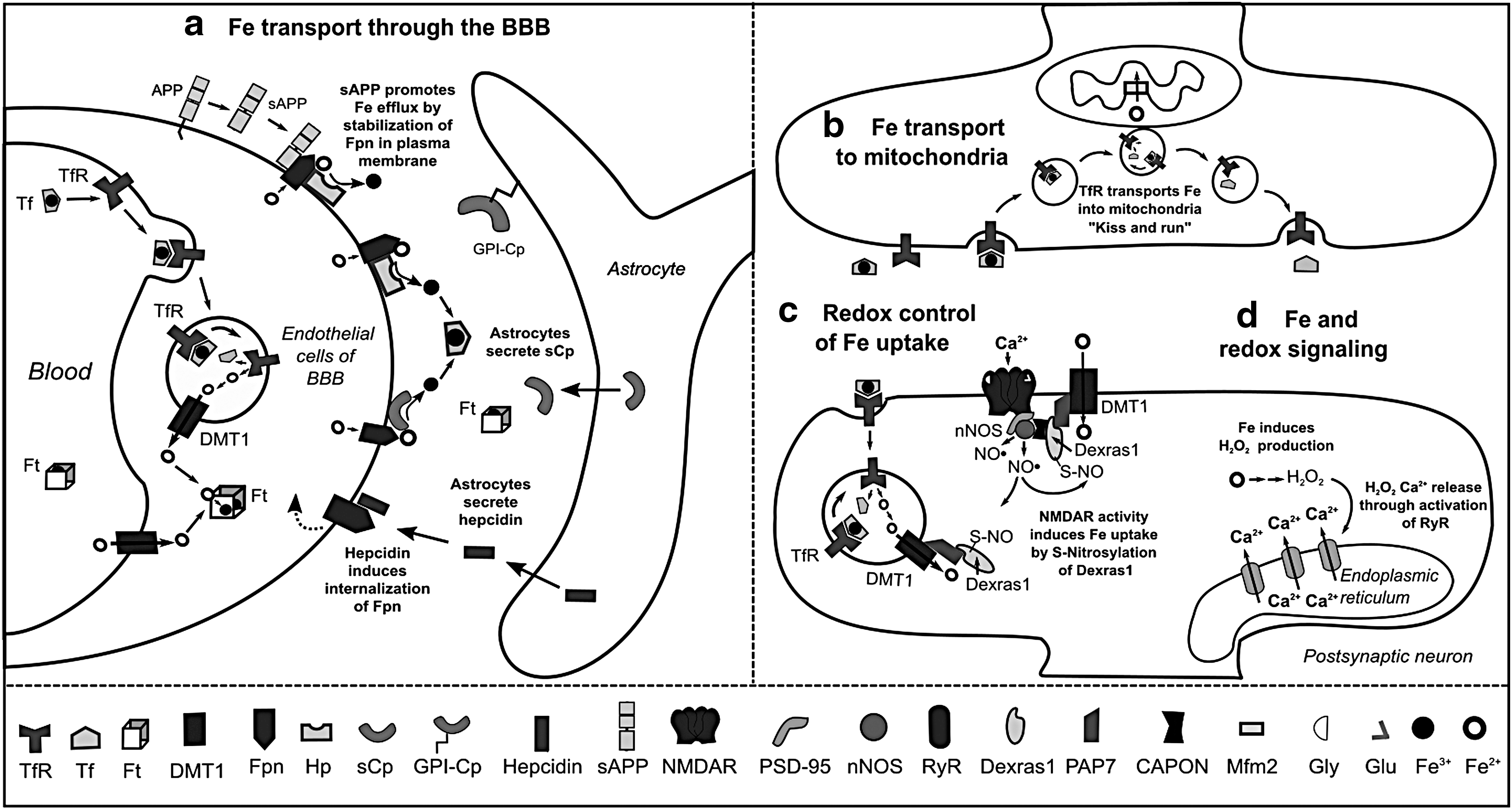
Fe efflux from endothelial cells to the interstitial space occurs via the coordinated activity of the Fe2+ transporter ferroportin (Fpn) and the Cu-dependent ferroxidases hephaestin (Hp) and soluble ceruloplasmin (sCp), which oxidize Fe2+ to Fe3+. Astrocytes regulate the release of Fe from BBB by either secretion of sCp, which stimulates Fe release, or by production of hepcidin, a peptide that induces internalization and ubiquitination of Fpn and thus, decreased Fe efflux (216). Astrocytes also express a glycosylphosphatidylinositol (GPI)-anchored form of Cp, which interacts with Fpn and participates in Fe efflux (Fig. 4a) (144). In general, oligodendrocytes, astrocytes, microglia, and neurons have the same machinery for Fe efflux, involving the concerted action of Fpn with Cp or Hp (60).
The mechanisms of Fe uptake differ between brain cell types. Fe, Tf, and Ft are primarily found in oligodendrocytes (61). Most Tf in the brain is synthesized and secreted by oligodendrocytes as Fe-free Tf or apo-Tf and is required for Fe mobilization within the interstitial fluid brain (87). Although oligodendrocytes and astrocytes can accumulate high levels of Fe, they do not express TfR. Fe uptake into oligodendrocytes has been recently proposed to involve the internalization of H-Ft by a mucin-domain containing protein (Tim-2) (343). In astrocytes, ascorbate-dependent Fe2+ uptake is mediated by DMT1 (167) and transient-receptor potential channels (265).
In neurons, the mechanisms involved in Fe uptake are still unclear. DMT1 is found in human neurons and its expression levels are negatively regulated by Fe exposure via ubiquitination and degradation (134). However, DMT1 seems to colocalize in cytoplasmic vesicles with TfR and to primarily contribute to Tf-bound Fe uptake (232, 266). The ZIP8 and ZIP14 members of the ZIP family of metal transporters have also been demonstrated to mediate Fe2+ uptake (188) and to be expressed in the brain (113). A recent report demonstrates that ZIP8 at the plasma membrane is the primary transporter involved in non-Tf-bound Fe into neurons (145).
In brain cells, the ferrireductases Dcytb and stromal cell-derived receptor, SDR2, are expressed in astrocytes (192, 351), while SDR2 and Steap2 are found in neurons (145). In all cases, it is important to note that Fe uptake and efflux always require redox cycling between Fe2+ and Fe3+ oxidation states.
Cytosolic Fe is stored by Ft, which is a protein complex formed by 24 subunits of heavy-ferritin (H-Ft) and light-ferritin (L-Ft) chains. Ft genes are regulated by Nrf2 (272). While H-subunits have ferroxidase activity and participate in Fe uptake, L-subunits are involved in Fe mineralization and long-term storage. Ft can store ∼4500 Fe ions in its core. However, the mechanism by which Fe is released from Ft remains unclear (47). Although Fe is mainly stored in the cytosol, mitochondria are the organelles with the highest Fe demand, as they require Fe-S clusters and heme groups for electron transfer during respiration. Similarly, mitochondria are also considered the main sources of ROS under physiological conditions. Therefore, mitochondrial Fe homeostasis must be tightly regulated to prevent uncontrolled ROS production.
The mechanism(s) involved in mitochondrial Fe uptake are still unclear. Fe is delivered by a direct “kiss and run” interaction between the endosomes containing Tf-bound Fe and mitochondria (69). Subsequently, the metal crosses the inner membrane via the Fe importer mitoferrin-2 (Mfrn2) (Fig. 4b). In the mitochondrial matrix, Fe is either used for the biogenesis of prosthetic groups, Fe-S clusters, or heme, or it is stored by mitochondrial ferritin (FtMt), which is homologous to H-Ft, and it displays the same Fe uptake efficiency but lower ferroxidase activity. Fe distribution between mitochondria and cytosol depends on FtMt expression and the export of Fe prosthetic groups (106, 176).
Heme is a component of globins, a superfamily of heme-containing proteins involved in binding and/or transporting O2, and a cofactor for cytochromes, catalase, NOX, NOS, and myeloperoxidase, which is also found in microglia (59, 151, 198). Hemoglobin (Hb) is involved in O2, NO, and carbon dioxide (CO2) transport in cells of erythroid lineage, but Hbα and β transcripts have been found in dopaminergic neurons, oligodendrocytes, and cortical or hippocampal astrocytes as well (32). Brain Hb levels are altered during neurodegeneration (94), but their functional consequences are unclear. Neuroglobin is another globin expressed in the CNS and PNS and found in both neurons and astrocytes. Neuroglobin has a higher affinity for O2 than Hb and exerts a protective effect against oxidative and ischemic insults (7, 43, 81, 179, 331, 368). Oxidative stress has the potential to release heme from Hb, and labile heme can induce oxidative damage via Fenton reactions or NOX activation (165, 239). Heme oxygenases (HOs) catalyze heme degradation. HO-1 transcription is induced by oxidative stress, inflammation, hypoxia, and metal exposure, while HO-2 is constitutively expressed. HO activity has both antioxidant and pro-oxidant effects that relate to the ability of the cell to detoxify labile Fe released from heme (286, 369).
Cellular Fe trafficking is controlled by the iron regulatory proteins (IRP) IRP1 and IRP2 which regulate translation of proteins involved in Fe storage (H-Ft and L-Ft), Fe uptake (TfR), and Fe efflux (Fpn). Under conditions of Fe depletion, IRPs can bind to messenger RNAs (mRNAs), decreasing the levels of Ft and Fpn and promoting the translation of TfR via the stabilization of its mRNA. Conversely, Fe overload prevents mRNA binding to IRPs, promoting Ft and Fpn translation while reducing TfR levels. The ability of IRP1 and IRP2 to bind mRNAs exerts an important redox control via two distinct mechanisms. IRP1 has two conformations: (a) a closed one, triggered via an Fe-S cluster that prevents mRNA binding and has aconitase activity; and (b) an open conformation that is favored when NO•, O2, and H2O2 cause dissociation of the Fe-S cluster. In contrast, the ability of IRP2 to bind mRNAs is controlled by proteasomal degradation, involving the ubiquitin ligase F-box/LRR-repeat protein (FBXL5), which is also an Fe and O2 sensing protein. FBXL5 has a binuclear nonheme Fe site, and it is stabilized on Fe and O2 binding, promoting IRP2 degradation. Together, these two mechanisms illustrate the redox control of cellular Fe trafficking (164).
In neurons, Fe uptake is induced by a redox signaling cascade that starts with the activation of the NMDAR (Fig. 4c). Increased intracellular Ca2+ induces NO• production by the neuronal (n) NOS, leading to S-nitros(yl)ation of the small GTPase Dexras1 (Ras-related dexamethasone induced 1), which in turn induces Fe uptake through DMT1 and TfR (51). In hippocampal neurons, NMDAR activation increases intracellular Fe and H2O2 production, which activates the redox-sensitive ryanodine receptor (RyR) and promotes Ca2+ release from the ER (237) (Fig. 4d). Thus, Fe uptake is clearly part of a redox signaling mechanism that might be important for neuronal plasticity.
Cell death induced by an increase in the labile Fe pool within cells has been defined as a specific entity named ferroptosis. Ferroptotic cell death is a necrotic-like cell death characterized by Fe-dependent lipid peroxidation due to either the formation of •OH and H2O2 via Fenton-like reactions or the activation of lipoxygenases. As such, ferroptosis is counteracted by Fe-chelators and the GSH/GPX4 system. GPX4 knockout in neurons induces motor neuron degeneration, paralysis (53), and cognitive impairment (121). During ferroptosis, Tf-dependent Fe uptake and release of Fe from lysosomal compartments have been shown to act as important sources for Fe. Interestingly, lysosomal permeabilization is a common phenomenon observed in a number of neurodegenerative disorders, including PD (40, 73). However, during pathological conditions such as neurodegeneration and hemorrhagic stroke, cell death is likely to involve a combination of different pathways and a complex balance between them, including apoptosis, necrosis, autophagic cell death, and ferroptosis as well (77, 143, 357, 398).
During aging, accumulation of Fe in the frontal lobes and striatum is associated with motor dysfunction, loss of myelin sheaths, and memory decline (2, 327). Inflammation, a common hallmark of many brain disorders, increases the expression of DMT1 and hepcidin (354).
Abnormal accumulation of Fe is a common feature of many neurodegenerative diseases, including a group of twelve diseases known as neurodegeneration with brain iron accumulation (NBIA) (225). The most common clinical features of NBIA are movement disorders, such as ataxia, parkinsonism, and dystonia. Although in NBIA Fe is usually accumulated in the globus pallidus, and in some cases in the SN and cerebellum, only two types of NBIA involve the dysfunction of a protein that participates in Fe trafficking: aceruloplasminemia (lack of Cp) and neuroferritinopathy (loss of function mutations in L-Ft) (65, 261, 290, 360). Another neurodegenerative disease associated with disturbed Fe homeostasis is Friederich ataxia, which is linked to mutations in frataxin, an Fe-chaperone involved in Fe-S cluster biogenesis (55).
Altered Fe homeostasis and mitochondrial dysfunction are also hallmarks of PD. While no significant differences in Fe levels have been found in the blood, serum, and CSF (203), Fe is increased in the SN of PD patients (365). Neuromelanin is an Fe-rich pigment found in the dopaminergic neurons targeted in PD (A9) and it has been suggested that its presence makes this neuronal population vulnerable to oxidative damage (91). Decreased levels of serum Cp or its oxidation has also been proposed to exacerbate Fe accumulation in PD (149, 251). DMT1 is found increased in the SN of PD brains (294). Interestingly, Parkin regulates DMT1 expression levels via ubiquitination and proteasomal degradation (289).
Fe accumulation in SN might be associated with a dysfunctional delivery of Fe to mitochondria through the “kiss and run” interaction mentioned above (Fig. 4b) (69). Indeed, a Tf/TfR2-dependent mechanism for Fe transport into the mitochondria of dopaminergic neurons has been described (211), while a role for Tf and TfR2 genes in Fe accumulation and mitochondrial dysfunction in PD has been implicated as well (285).
Mitochondrial dysfunction in PD might lead to decreased synthesis of Fe-S clusters, which in turn would activate IRP1 binding to mRNAs, resulting in augmented Fe accumulation (142). Accordingly, knockdown of mitochondrial Grx2 impairs Fe-S cluster biogenesis in dopaminergic cells, decreases the activity of Complex I and aconitase, and increases the activation of IRP1 (171). Loss of PINK1/PARK6, another autosomal early-onset PD-related gene, has also been reported to inactivate Fe-S clusters via O2 •− formation. As such, overexpression of FtMt exerts a protective effect on mitochondrial dysfunction and oxidative stress induced by loss of PINK1 (88).
HO-1 protects against neuronal cell death induced by PD-related mitochondrial toxins and α-synuclein, suggesting a role of heme in dopaminergic cell loss. However, HO-1 becomes toxic at high levels, but this effect is counteracted by FtMt (391). Interestingly, the pathogenic PINK1 mutation G309D impairs the induction of HO-1 on oxidative stress (56).
Fe2+ can interact with the negatively charged C-terminal region of α-synuclein, accelerating its amyloid aggregation (34). Conversely, α-synuclein overexpression also enables Fe accumulation (254). In PD, oxidative stress has been linked to intracellular Fe levels due to the redox activity of Fe-DA and Fe-neuromelanin complexes (400). Furthermore, overexpression of FtMt protects against neuronal cell death induced by 6-hydroxydopamine (313). Interestingly, a recent report demonstrates that depletion of Fpn has no consequence on the survival of dopaminergic neurons. In contrast, loss of TfR causes Fe deficiency and a PD-like neurodegeneration in mice (212). Previous findings have also suggested a link between Fe deficiency and predisposition to PD, while Fe overload seems to be protective (157, 190, 271). Thus, the exact role of Fe homeostasis in PD is still far from being understood.
A role of Fe in AD has also been proposed based on observations showing that Fe levels are decreased in the serum of AD patients (324), and increased in AD-related brain areas such as the hippocampus, neocortex, and basal ganglia (168). Mitochondrial dysfunction is also observed in AD, which might be related to a disruption in Fe homeostasis. Accordingly, overexpression of FtMt protects against the toxicity of Aβ (380). Fe is accumulated in amyloid plaques in the AD brain (38, 194), consistent with the observation that Aβ is able to bind Fe2+ in vitro (Fig. 3b) (39). Fe bound to Aβ plaques catalyzes H2O2 formation (321).
A novel mechanism for controlling Fe efflux from the BBB was recently discovered, involving the soluble fragment of APP (sAPP), which interacts with Fpn, stabilizing its membrane location (219) and counteracting the effects of hepcidin (216). APP expression is also negatively regulated by IRP1 (57), and production of sAPP and Aβ is increased in AD. Thus, the proposed role of sAPP in Fe movement from BBB into the brain is consistent with the accumulation of Fe in AD brains and the observation of higher levels of Aβ plaques surrounding the brain blood vessels. Indeed, BBB damage is a common feature in AD (217).
Heme metabolism also contributes to AD. APP binds and inhibits HO-1 activity and this effect is enhanced by pathogenic APP mutations (334). In addition, Aβ can form a complex with heme that displays peroxidase activity (16, 112). Conversely, neuroglobin has been proposed to protect against the toxicity of 1–42 Aβ peptides (180). Overall, disruption of Fe homeostasis in AD is likely linked to oxidative stress, and it may also impair NMDAR- and Fe- dependent redox signaling mechanisms (Fig. 4d) impacting neuronal plasticity and memory.
Manganese
Mn is an essential metal required for the activity of a plethora of enzymes, including hydrolases, isomerases, ligases, lyases, oxidoreductases, and transferases, involved in diverse metabolic functions such as amino acid (arginase and glutamine synthetase [GS]), lipid, protein, and carbohydrate metabolism (phosphoenolpyruvate decarboxylase), as well as protein glycosylation, energy production, and redox homeostasis (SOD2). The main source for Mn intake is food, but occupational/environmental exposures also occur associated with mining, smelting, welding, alloy, battery, pesticide, and electrical industries. Ingestion and inhalation are the primary routes of Mn exposure. Importantly, inhalation can transfer Mn directly to the brain. The brain is a major target for chronic Mn intoxication (manganism) where Mn is accumulated in nonheme Fe-rich regions. Manganism is defined as a parkinsonism that results in dystonia, hypokinesia, and rigidity as a consequence of impaired neurotransmitter function (133, 352).
Mn can be transported via Tfr and DMT1 as it competes with Fe for its binding sites (333). Other proposed Mn transporters include the Zn carriers ZIP-8 and ZIP-14, voltage-regulated, store-operated, and ionotropic Glu receptor Ca2+ channels, and the Mn-citrate complex shuttle (Fig. 5a) (352). Mn is efficiently detoxified by Fpn at the plasma membrane. In addition, Mn is also detoxified by sequestration in the Golgi via the solute carrier family 30 member 10 or human Zn transporter 1 (SLC30A10/hZnT1) transporter whose mutations are directly associated with manganism (178). Alternatively, the Ca2+/Mn2+ ATPases SPCA1 and SPCA2 also detoxify Mn via the secretory pathway (356). Finally, the autosomal recessive early-onset PD-related gene ATP13A2/PARK9 mediates sequestration of Mn in lysosomes (Fig. 5b) (336). Recently, direct comparison of different detoxification proteins demonstrated that hZnT1 and SPCA1, but not ATP13A2, are involved in Mn detoxification and resistance (246).

Mn2+ is the predominant species found in cells that can be oxidized to the more reactive and toxic species Mn3+. Neither Mn2+ nor Mn3+ can generate free radicals via Fenton-type reactions. However, it has been proposed that Mn enhances ROS generation via the Mn-catalyzed autoxidation of DA that involves the redox cycling of Mn2+ and Mn3+ and the generation of ROS and DA-o-quinone (Fig. 5c) (79, 89). Mn accumulates in the mitochondria via the mitochondrial Ca2+ uniporter (MCU) (111) and increases the accumulation of labile Fe. Both mitochondrial dysfunction and Fe lead to ROS formation and oxidative damage (Fig. 5d) (54, 206). Mn specifically generates H2O2 but not O2 •− in the mitochondria via complex II (92, 186, 322). Furthermore, Mn impairs oxidative phosphorylation and ATP production.
Astrocytes seem to have a high capacity to accumulate Mn (14). Mn-induced neurotoxicity has been linked to a decrease in Glu uptake by astrocytes (156) leading to excitotoxicity in neurons, as well as the induction of inflammation and increased activity of NOS (Fig. 5e) (95, 185).
Zinc
Zn is a redox-inactive transition metal ion with an oxidation state of +2. The majority of intracellular Zn is bound to proteins and is distributed in the cytoplasm (∼50%) and nucleus (∼40%) (304) (Supplementary Table S1). The function of Zn as enzyme cofactor is limited to structural roles (e.g., SOD1) (312), or as a Lewis acid that activates substrates for nucleophilic attack (e.g., carbonic anhydrase, where Zn2+ catalyzes the hydration of CO2 to form bicarbonate [HCO3 −]) (138). Enzymes with Zn-dependent catalytic activity control many cellular processes, including DNA synthesis and brain development. Zn also plays an important role in cell signaling associated with development and learning. In the brain, Zn is highly concentrated in the hippocampus and cortex (304).
The key players in Zn homeostasis are the ZIP transporters that mediate Zn uptake into the cytosol, the zinc transporters (ZnT) that participate in Zn efflux, and MTs involved in Zn chelation. ZIP1 is expressed in astrocytes and microglia (303). In neurons Zn uptake is mediated by, voltage-gated Ca2+ channels, ZIP1 and 3 transporters, as well as Ca2+ and the Zn2+ permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (AMPAR) (146, 278, 335). Interestingly, Zn has been reported to be transported into postsynaptic neurons through a complex formed by PrPC, which is evolutionary linked to ZIP proteins, and the AMPAR (Fig. 6b) (373). ZnT1 is expressed in astrocytes, microglia, and oligodendrocytes, and its expression levels are directly modulated by Zn (247). Hypoxia decreases the levels of ZnT1 in astrocytes inducing the accumulation of cytosolic Zn (257). At the postsynaptic density, the ZnT1 transporter interacts with NMDAR and this complex is regulated during synaptic plasticity (Fig. 6c) (222, 373).
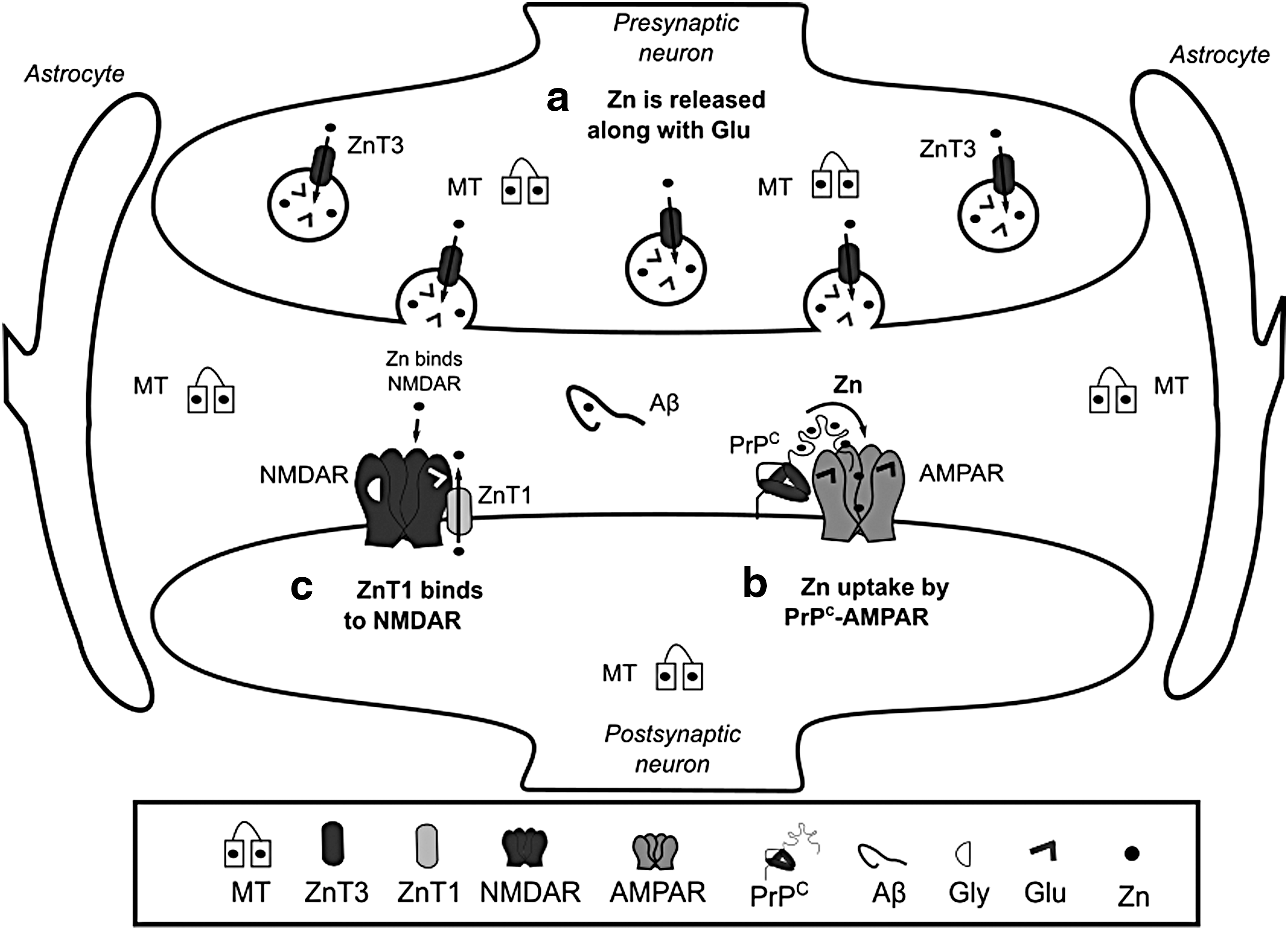
Cytosolic Zn is distributed to different organelles, including synaptic vesicles, Golgi, ER, and mitochondria. Similarly, Zn uptake by these organelles is performed by ZnTs, while ZIPs participate in Zn efflux into the cytosol (155). Zn modulates cellular signaling pathways and acts as a neuromodulator. ZnT3 participates in the transport and accumulation of Zn within synaptic vesicles of glutamatergic terminals (Fig. 6a) (125). During synaptic transmission, Zn is released with Glu, where it inhibits the activity of NMDAR and AMPAR (9, 154), modulating neuronal excitability and long-term synaptic plasticity (long-term potentiation [LTP] and long-term depression). In addition, Zn regulates the activation of the tropomyosin kinase receptor B (TrkB) (304). A metabotropic Zn-sensing receptor (mZnR) has also been reported (31). At the synapse, Zn can also bind to MTs, Aβ, and PrPC (Figs. 3c and 6).
Although Zn2+ is a nonredox-active metal ion, it participates in redox signaling through several mechanisms. MTs bind about a fifth of the intracellular Zn with a stoichiometry of 1:7. MTs and proteins containing Zn-finger domains use Cys residues to bind to Zn2+ ions. Zn coordination stabilizes the reduced state of Cys thiol groups preventing their oxidation and subsequent formation of disulfide bonds. Transcription of MTs is induced by oxidative stress via Nrf2, and by heavy metal exposure via metal-response elements (MREs). MREs are recognized by the Zn-finger domain containing protein metal-responsive transcription factor-1 (MTF1, also known as MRE-binding transcription factor-1 or metal regulatory transcription factor-1). MTF1 senses Zn levels. Furthermore, via Zn displacement from MTs or Cys oxidation, MTF1 also senses heavy metal toxicity (Cd) and oxidative stress. In addition to MTs, MTF1 regulates the transcription of a number of genes involved in redox homeostasis and metal ion detoxification, including Zn transporters, Fr, Fpn, ATP7(A/B), Trx, selenoproteins, and γ-glutamate-cysteine ligase (GCL), a rate-limiting enzyme in GSH synthesis (119). The role of MTF1 in brain function and redox homeostasis is unclear. MTF1 has been shown to regulate the expression of β-synuclein (220), which is thought to act as a negative regulator of its homologue α-synuclein (126). Interestingly, deletion of MTF1 induces lethality in Parkin-deficient flies (Drosophila melanogaster) (293).
Altered Zn levels have been reported to promote neuronal injury. Upon Cys oxidation, Zn is released from MTs (255). Cellular acidification also releases intracellular Zn in neurons (159). In a recent report, AMPA-induced oligodendrocyte cell death was shown to be linked to Zn mobilization from mitochondria and protein-bound pools that were mediated by cytosolic acidification, independently from ROS (213). Exposure of mitochondria to Zn promotes increased ROS formation (305). While no specific mitochondrial Zn transporter(s) has been identified, potential candidates include the MCU, ZIP8, and Znt2 transporters (30, 199, 307). Zn also increases NOX-derived ROS formation and NOS activity (162). High extracellular Zn enhances microglia activation and ROS formation (130). Zn deficiency also induces oxidative stress via a reduction in the activity of SOD1 (378), and an impairment in the transcriptional regulation of GCL by Nrf2 (253).
Alterations in Zn homeostasis are associated to neurodegenerative diseases. Serum levels of Zn are decreased in AD patients (358), while Zn is enriched in Aβ plaques (364). Furthermore, a decrease in the levels of Znt3 and MT-III (390) is found in AD. Importantly, the predominant localization of Aβ plaques in Zn-containing glutamatergic synapses might explain why they are primarily found in the neocortex (304). Zn promotes a rapid, but reversible, aggregation of Aβ that is different to the aggregation of Aβ or Aβ-Cu complexes (25). Zn also reduces the toxicity of Cu-induced Aβ aggregates (214). ZnT3 knockout increases soluble Aβ in transgenic APP mice corroborating the role of extracellular Zn in plaque formation (172).
Elevated Zn levels have been found in the SN of PD brains (74), while reduced levels of Zn in serum and plasma have been linked to an increased risk for PD (80). Zn has been shown to potentiate the toxicity of DA as well (189). Furthermore, Zn chelation reduces the toxicity of mitochondrial PD-related toxins (310). Recently, ATP13A2 was identified as a Zn transporter localized to multivesicular bodies. Loss of function mutations of ATP13A2 induces alterations in Zn homeostasis and mitochondrial dysfunction (259).
Xenobiotic Metals
Measurable concentrations of xenobiotic metals with no physiological functions are present in humans (Supplementary Table S1) (103). In addition, environmental or occupational exposures to xenobiotic metals may take place by inhalation, ingestion, or skin penetration and are often linked to the development of toxicity and pathological conditions (Supplementary Table S1). Metals can reach the CNS from the vascular lumen affecting neuronal and glial function. Metals hijack transport systems of essential metals to pass through the BBB and enter neuronal tissues (molecular mimicry). Metal toxicity is largely attributable to their physicochemical properties, which mediate their interference with cellular biochemical systems, including redox-related processes (205, 355).
Environmental or occupational exposure to xenobiotic metals has been reported to contribute to neuronal dysfunction (cognitive, motor, and behavioral) and in some cases, neurodegeneration. However, the mechanisms involved are largely unclear. We next review the sources and routes of exposure to xenobiotic metals; the metabolic pathways involved in their transport and activation, and the mechanisms by which they alter cellular redox balance to promote neurotoxicity.
Arsenic
As is naturally present in air, water, and soil and is the 20th most abundant element in the earth's crust and 12th in the human body. This metal is named inorganic As (iAs) when found combined with other elements such as O2, chlorine (Cl), and sulfur (S). Combined with carbon (C) and hydrogen (H) is referred to as organic As. In the environment and within the human body, iAs predominantly exists in two oxidation states: arsenite +3 (or AsIII, found as arsenic trioxide [As2O3], sodium arsenite [NaAsO2], and arsenic trichloride [AsCl3]), and arsenate +5 (or AsV found as arsenic pentoxide [As2O5], arsenic acid [H3AsO4], and arsenates [PbHAsO4, Ca3(AsO4)2]).
iAs has been widely used as a therapeutic agent to treat leukemia. Currently, iAs compounds are predominantly used in pesticides, herbicides, cotton desiccants, wood preservatives, alloys for batteries, and in semiconductors and light-emitting diodes. Millions of individuals are currently exposed to iAs across the world due to natural groundwater contamination. Fish and crustaceans contain very high levels of organic arsenobetaine but no toxicity has been reported in vivo (382). The concentration of iAs in natural surface and groundwater is generally about 1 parts per billion (ppb) of water but it may exceed 1000 ppb in contaminated areas or where iAs soil levels are high (118, 200) (Supplementary Table S1).
While As is considered a carcinogen, in the brain, acute exposure to iAs can induce encephalopathy, with symptoms such as confusion, hallucinations, reduced memory, and emotional lability (exaggerated changes in mood or affect). Long-term exposure to lower levels of iAs can lead to the development of peripheral neuropathies.
There are reports of neurobehavioral alterations (cognitive function, verbal abilities, long-term memory, and motor skills) in children exposed to As concentrations ranging from 5 to 50 ppb in water, in Bangladesh (260), Mexico (45, 287), and in the United States (370). Although scientific understanding of the developmental neurotoxicity of As is still evolving, epidemiological and toxicological studies clearly show that As is a developmental neurotoxicant that affects intellectual function. Moreover, exposures even below current safety guidelines are associated with decrements in full-scale intelligence quotient (IQ) and memory (90, 347). Evidence in experimental models, including mice, rats, Caenorhabditis elegans (worm), and Danio rerio (zebrafish), has replicated many of the observations in humans supporting the notion that As can lead to cognitive, locomotor, and neurological impairment (85). Gestational exposure to NaAsO2 leads to a significant iAs accumulation in the mice offspring's brain (280). As neurotoxicity has been linked to changes in neurotransmitter metabolism and synaptic transmission (85, 276, 280). However, the mechanisms involved remain unclear.
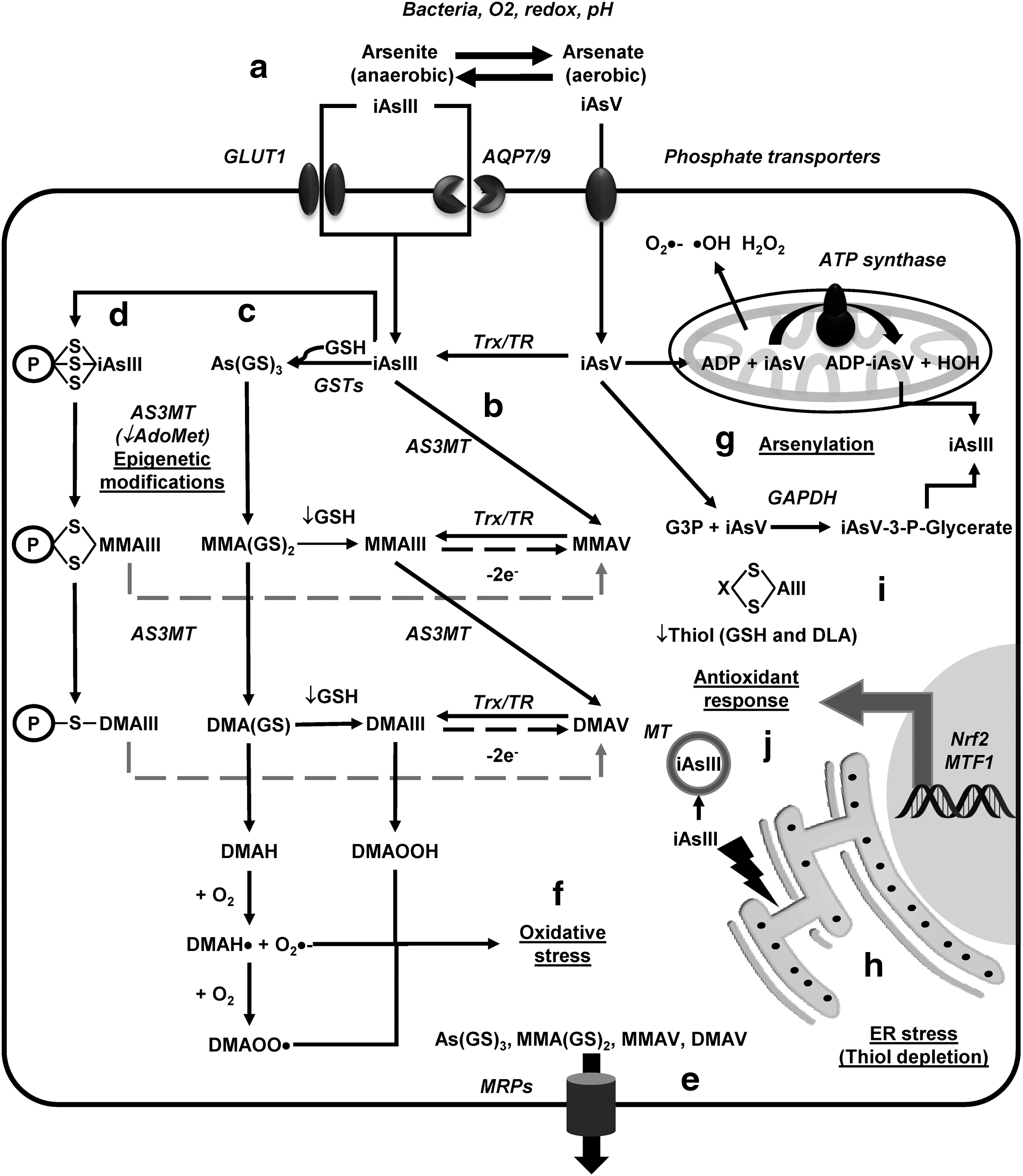
In the environment, oxygenated water contains iAsV species, while in reducing environments iAsIII species are prevalent. iAsV enters cells through phosphate transporters to be subsequently reduced to iAsIII, while iAsIII is transported via aqua(glycerol)porins (AQP), organic anion transporters, and glucose transporters (GLUT) (Fig. 7a) (44, 187, 348). Once in the cytoplasm, iAsIII is methylated by different mechanisms. Oxidative methylation (Fig. 7b) is mediated by arsenite methyltransferase (AS3MT) that uses S-adenosylmethionine (AdoMet) as a cosubstrate. AS3MT methylates iAsIII to monomethylarsonic acid or arsonate (MMAV) that is reduced to monomethylarsonous acid (MMAIII) before being methylated again to dimethylarsinic acid (DMAV) by AS3MT (353, 372). Finally, DMAV is reduced generating dimethylarsinous acid (DMAIII). The reduction of pentavalent arsenicals (iAsV, MMAV, and DMAV) in this pathway is now well recognized to be mediated by the Trx/TR system, but GSH seems to increase the methylation rates by an unknown mechanism (76).
Developmental exposure to As alters the methylation patterns of genes involved in neuroplasticity likely due to changes in AdoMet, but its long-term implications are unclear (207). Recent in vivo studies demonstrated that the alterations in synaptic plasticity (LTP), memory, and learning induced by gestational exposure to iAs were associated with an increase in extracellular Glu levels and downregulation of AMPAR subunits (244).
As methylation via the GSH conjugation mechanism is based on the formation of GSH complexes with iAsIII resulting in arsenic triglutathione [As(SG)3] (Fig. 7c). Conjugation of iAsIII with GSH has been proposed to occur nonenzymatically, but enzymatically as well by the activity of glutathione-S transferases (GST isoforms GSTO1, GSTM1, or GSTP1) (173, 372). As(GS)3 is subsequently methylated by AS3MT to form monomethylarsinic diglutathione [MMA(GS)2] and then again to generate dimethylarsinic GSH [DMA(GS)]. At low GSH levels, As(GS) conjugates are hydrolyzed and then oxidized to generate MMAV and DMAV (372). A third mechanism for iAsIII methylation has been recently proposed, where instead of As(GS) conjugate formation, iAsIII binds to protein-Cys (thiol) and is methylated while still being conjugated to proteins (Fig. 7d). This hypothesis is supported by the preferential binding of iAsIII to protein-Cys when compared to GSH (284).
Methylated (and maybe unmethylated) As metabolites are exported through the multidrug resistance proteins (MRP1, MRP2, or MRP4) (173, 317, 388) (Fig. 7e). AS3MT is ubiquitously expressed in all brain regions, and animal studies have shown that the iAs that crosses the BBB is methylated and accumulated across the brain, with the highest accumulation observed in the pituitary gland (297). Interestingly, knockout mouse for P-glycoprotein accumulates more As in the brain (183). Endothelial cells and astrocytes feet surrounding capillaries are the first barrier of detoxification of xenobiotics entering from the circulation. We (unpublished data) and others have observed that the resistance of astrocytes to iAsIII is mediated by MRPs (332).
iAs generates ROS and dimethylarsenic or peroxyl radicals that in turn lead to lipid peroxidation and the accumulation of oxidized by-products (MDA and HNE) (Fig. 7f). Importantly, MMAIII and DMAIII are proposed to be more potent toxicants than iAsIII due to their increased ability to generate radicals (392). Oxidative stress has been reported in brain regions of different animal models and in neurons and glial cell cultures exposed to As compounds (48, 107, 108, 243, 394).
Mitochondria have been proposed to be a primary source for ROS formation by iAs (Fig. 7g) (97, 150). Chronic iAs exposure generates mitochondrial oxidative stress in the rat brain by impairment of mitochondrial complexes I, II, and IV activities followed by increased ROS generation, lipid peroxidation, and protein carbonylation (275). Mitochondrial pyruvate dehydrogenase is also directly inhibited by iAs (136). In addition, iAs reduces the levels of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), downstream targets Nrf1 and Nrf2, and the mitochondrial transcription factor A (TFAM) decreasing mitochondrial biogenesis (274). ER stress has also been shown to contribute to iAs toxicity, but the mechanisms involved remain unclear (Fig. 7h) (182).
iAs toxicity has also been attributed to the ability of AsV to replace phosphate in several metabolic pathways (arsenylation) where the end product is the reduction of AsV to AsIII, because the arsenylated by-product is more readily reduced than AsV itself (Fig. 7g). AsV uncouples oxidative phosphorylation and ATP formation in the mitochondria by binding to ADP via ATP synthase. Replacement of phosphate in glycolysis also impairs carbon flux and ATP production. Reaction of AsV with glucose generates glucose 6-arsenate, an analog of glucose 6-phosphate that is suggested to act as an inhibitor of hexokinase. AsV is also arsenylated by glyceraldehyde 3-phosphate dehydrogenase (GAPDH) to produce the unstable product 1-arsenato-3-phospho-D-glycerate (iAsV-3-P-glycerate). Purine nucleoside phosphorylase, glycogen phosphorylase, and mitochondrial ornithine carbamoyl transferase (OCT) have also been shown to arsenylate AsV (139, 245, 340). Thus, energy failure, alterations in central carbon metabolism, and mitochondrial dysfunction are consequences of AsV toxicity.
AsIII binds to thiol containing molecules (coenzyme A, GSH, and dihydrolipoamide also known as dihydrolipoic acid [DLA]) and protein-Cys thiols inactivating enzyme function (Fig. 7d, i). AsIII has higher affinity for dithiols than monothiols as demonstrated by the transfer of AsIII from the GSH-adduct to 2,3-dimercaptosuccinic acid (DMSA) a sulfhydryl-containing metal chelator used to treat heavy metal toxicity. In addition, AsIII conjugated with GSH has the ability to bind protein thiols, which highlights the importance of detoxification of GSH-As adducts from the cell (230). Dithiol molecules such as the cofactor DLA and dithiol oxidoreductases Trxs, Trx reductase (TrxR), Prxs (except for monothiol Prx6), Grx, and GR, as well as proteins with adjacent Cys (MT), have been reported to avidly bind AsIII (Fig. 7d, j) (50, 279, 311, 393, 397). In addition to binding AsIII, Zn finger domains have been shown to be oxidized upon As binding (396). Binding of AsIII to DLA (Fig. 7i) is expected to interfere with the TCA cycle and energy production as DLA is a cofactor for the pyruvate dehydrogenase and 2-oxoglutarate dehydrogenase complexes that catalyze the synthesis of acetyl-CoA and succinyl-CoA, respectively. DLA reverses protein oxidation and loss of protein-SHs in the brains of rats exposed to high levels of iAs (82, 296, 315, 316).
As activates the cystine/Glu exchanger system (xCT) in microglia to increase extracellular Glu levels (320), while in astrocytes it decreases the expression levels and activity of GS and Glu transporters (GLAST/excitatory amino acid transporter (EAAT) 1 and GLT-1/EAAT2) (48, 395). Importantly, these effects were linked to an increase in GSH levels and Nrf2 activity (Fig. 7i), but not oxidative stress (48). Accordingly, activation of Nrf2 by As seems to involve a noncanonical pathway where inhibition of autophagy leads to the accumulation of the ubiquitin-binding protein/adaptor p62 that sequesters Keap1 (169). iAsIII toxicity is also counteracted by the transcriptional regulation of MT via MTF1 (Fig. 7j) (129).
Lead
Inorganic Pb remains one of the most studied toxic elements due to several reasons. To begin with, human contact with Pb started very early in human civilization, and its toxic effects were also known since then. However, more importantly, Pb is neurotoxic leading to lower IQ even at lower doses than those recommended by the World Health Organization (10 ppb in drinking water). Human exposure to Pb not only occurs occupationally but also environmentally. The presence of Pb in the environment has multiple sources such as gasoline, industrial processes, paint, water pipes, and solder in canned food. It is present in air, household dust, soil, water, and food (Supplementary Table S1). Environmental Pb levels have fortunately decreased, especially in those countries where the Pb addition to gasoline and paints was banned. This prohibition was enforced after several studies associated the presence of high blood levels of Pb with impaired or diminished cognitive functions. Epidemiological studies have clearly shown that exposure to Pb in early stages of development is associated with significant deficits in neurobehavioral performance, including lower IQ, attention deficits, and aggressiveness later in life. Despite all the enforced restrictions, Pb contamination is still a major public health concern. For example, in November 2000 in Washington DC, there was a “lead drinking water crisis” triggered by a change in the disinfectant used to clean the water, this contamination affected hundreds of kids for 3 years. The health consequences of the recent crisis of Pb-contaminated water in Flint Michigan (United States, 2015) are still to be revealed in the future (68, 84, 124, 298).
Inhalation and ingestion of Pb and Pb-containing particles or products are the main routes of Pb entry into the body. Young children are especially vulnerable because they show higher gastrointestinal absorption than adults. Inhaled Pb particles are quickly absorbed in alveoli and distributed to other organs through the circulation. Thus, blood lead levels (BLL) are reliable biomarkers of exposure and risk. However, BLL do not reflect the total Pb body burden because Pb is absorbed in bones where it can be stored for several years (68). Currently, the acceptable BLL for children is lower than 10 μg/dl (0.48 μM) (49, 376), but due to the devastating effects that might occur later in life, there is a consensus to recommend efficient surveillance methods for children protection to reduce BLL to the lowest possible level (141). Pb binds with high affinity to erythrocytes' δ-aminolevulinic acid dehydratase (ALAD) that catalyzes the second step in the porphyrin and heme biosynthetic pathway, causing the accumulation of aminolevulinic acid (ALA) in both plasma and urine, which is used as a biomarker of exposure (248).
Pb can cross the BBB and cell membrane because of its ability to mimic Ca2+ and Fe2+ ions (Fig. 8a) (205, 298). In children, due to a more permeable BBB and a lower bone storage capacity for Pb, the amount of Pb passing into the nervous system is higher than in adults. The highest accumulation of Pb has been reported in the hippocampus, amygdala (116), and choroids plexus (201). The PNS may accumulate considerably more Pb than the CNS. Animals chronically exposed to Pb had impaired dendritic spines and synapse formation (306). Developmental exposure to Pb impacts the prefrontal cerebral cortex, hippocampus, and cerebellum regions, which can lead to neurological disorders, mental retardation, behavioral problems, and nerve damage (242). Early life exposure to Pb has also been linked to neurodegenerative diseases such as AD and PD (62, 209).
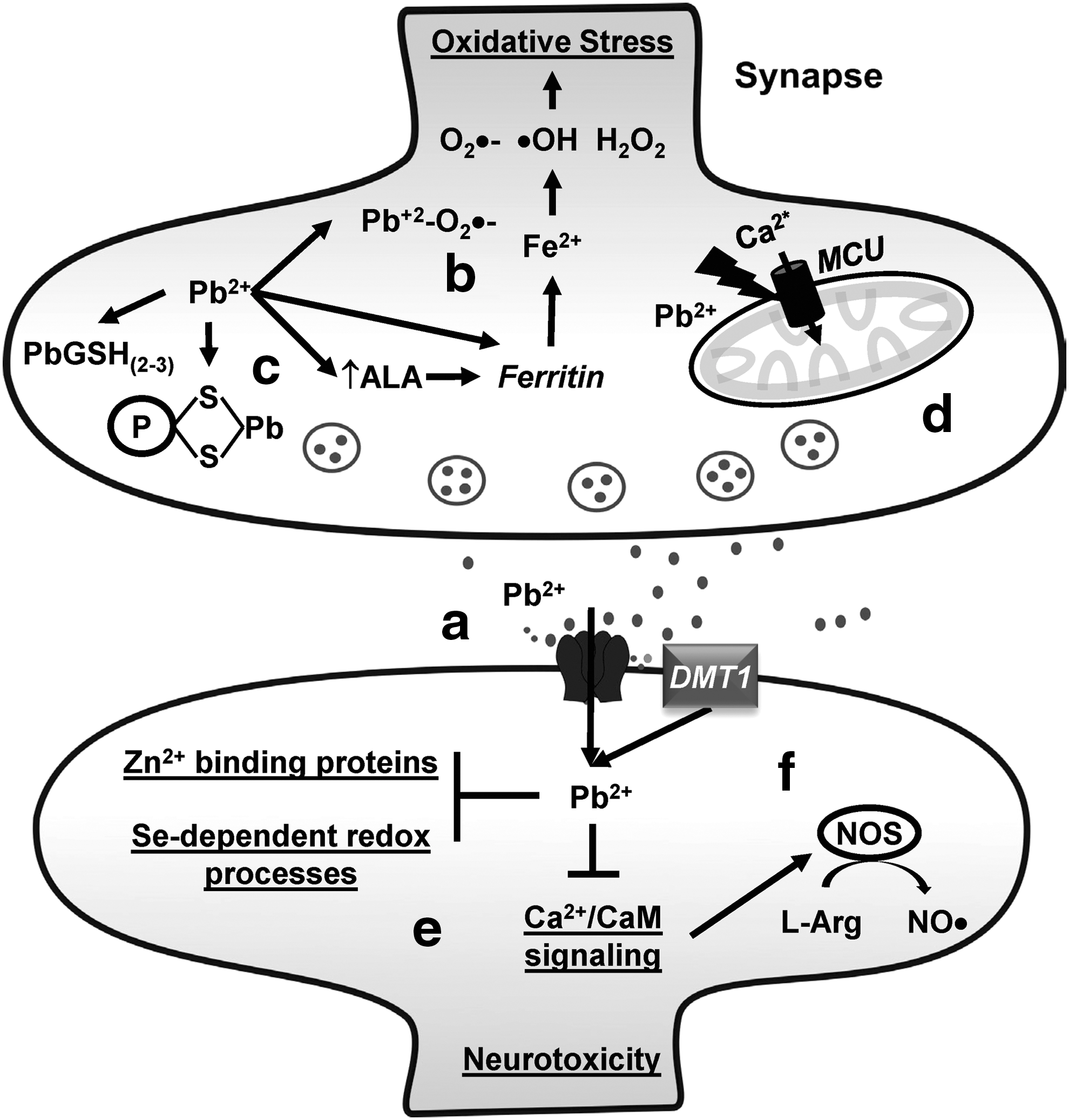
The mechanisms of Pb toxicity include the ability of Pb to bind SH groups of proteins Cys and to mimic or compete with Ca2+, Fe2+, and Zn2+ (Fig. 8b, c, e, f) (99, 283). Zn deficiency increases the toxicity of Pb (4). The generation of oxidative damage by Pb in vitro and in vivo suggests that ROS also participate in Pb toxicity. For example, Pb acetate induces the opening of the mitochondrial permeability transition pore in human neuroblastoma SH-SY5Y cells via ROS (384). Pb can form a Pb2+–O2 •− complex with higher oxidizing capacity than O2 •− (3). In addition, accumulated δ-ALA by Pb-induced ALAD inhibition can be subsequently oxidized to generate O2 •−, •OH, and H2O2. Pb per se has been reported to stimulate Fe2+-initiated lipid peroxidation (Fig. 8b) (337). Early postnatal exposure of rats to Pb leads to a higher accumulation of oxidative DNA damage in the cerebral cortical tissue when compared with aged controls or aged mice exposed acutely to Pb (36).
Perinatal exposure to Pb acetate inhibits the activity of brain acid and alkaline phosphatases, catalase, acetylcholinesterase, and ATPases (12). Similar observations have been made for the activities/levels of SOD1, GPX1, and GPX4 in the hippocampus, and for mitochondrial SOD2 and GSH, both in the cortex and hippocampus (19). Antioxidant nutrients such as vitamin E, vitamin C, vitamin B6, β-carotene, and DLA, as well as metal chelators such as DMSA, or replenishment of displaced metals has been shown to be beneficial against Pb-induced oxidative stress in the brain (98, 236, 256, 258, 277, 367). Diet supplementation with Zn and Se, which participates in the regulation of the GSH and Trx antioxidant systems, can effectively outcompete Pb binding to Zn- and Se-binding sites (Fig. 8e) (135).
Pb interferes with and disrupts Ca2+ signaling and homeostasis leading to excitotoxicity. In addition, Glu potentiates Pb-induced cell death in PC12 cells (267). Recently, oxidative stress induced by Pb has been shown to be linked to changes in the levels of MCU (Fig. 8d) (383). Other important intracellular targets of Pb in the brain are both neural NOS and endothelial NOS due to an impairment in their Ca2+/calmodulin (CaM)-dependent activation (Fig. 8f) (241). Importantly, Pb amplifies Glu-induced oxidative stress in a Ca2+-independent manner, but neither Ca2+ nor ROS seem to be essential for the enhanced cytotoxicity of combined exposure to Glu and Pb (191, 238).
Mercury
Hg is a transition metal that exists as elemental, inorganic, and organic Hg (Fig. 9a). Hg is ubiquitously found in the environment as sulfide compounds generated from volcanic activity and erosion, or released by anthropogenic sources such as fuel combustion, waste disposal, and industrial activities (Supplementary Table S1). Elemental or metallic Hg (Hg0) used in thermometers and amalgams is primarily absorbed via inhalation, while inorganic mercury (Hg1+ or 2+) used in medicine and everyday life products is partially absorbed through the gut. Organic Hg (ethylmercury [EtHg or C2H5Hg] and methylmercury [MeHg or CH3Hg]) is originated from atmospheric sources that are deposited in water body surfaces to be biomethylated and magnified in the food chain (Fig. 9a). Around 95% of MeHg is absorbed by the gastrointestinal tract making it the most toxic Hg species. Neurotoxic signs of Hg intoxication are vast and include ataxia, dizziness, insomnia, speech impairment, arthralgia, cognitive and behavioral changes, seizures, fatigue, and sensory disruption. While there has been an association between Hg exposure and neurodegeneration or autism, the neurological effects of chronic exposure to Hg are largely unclear. However, research has clearly demonstrated that Hg impairs neuronal development, communication, and myelination (89).
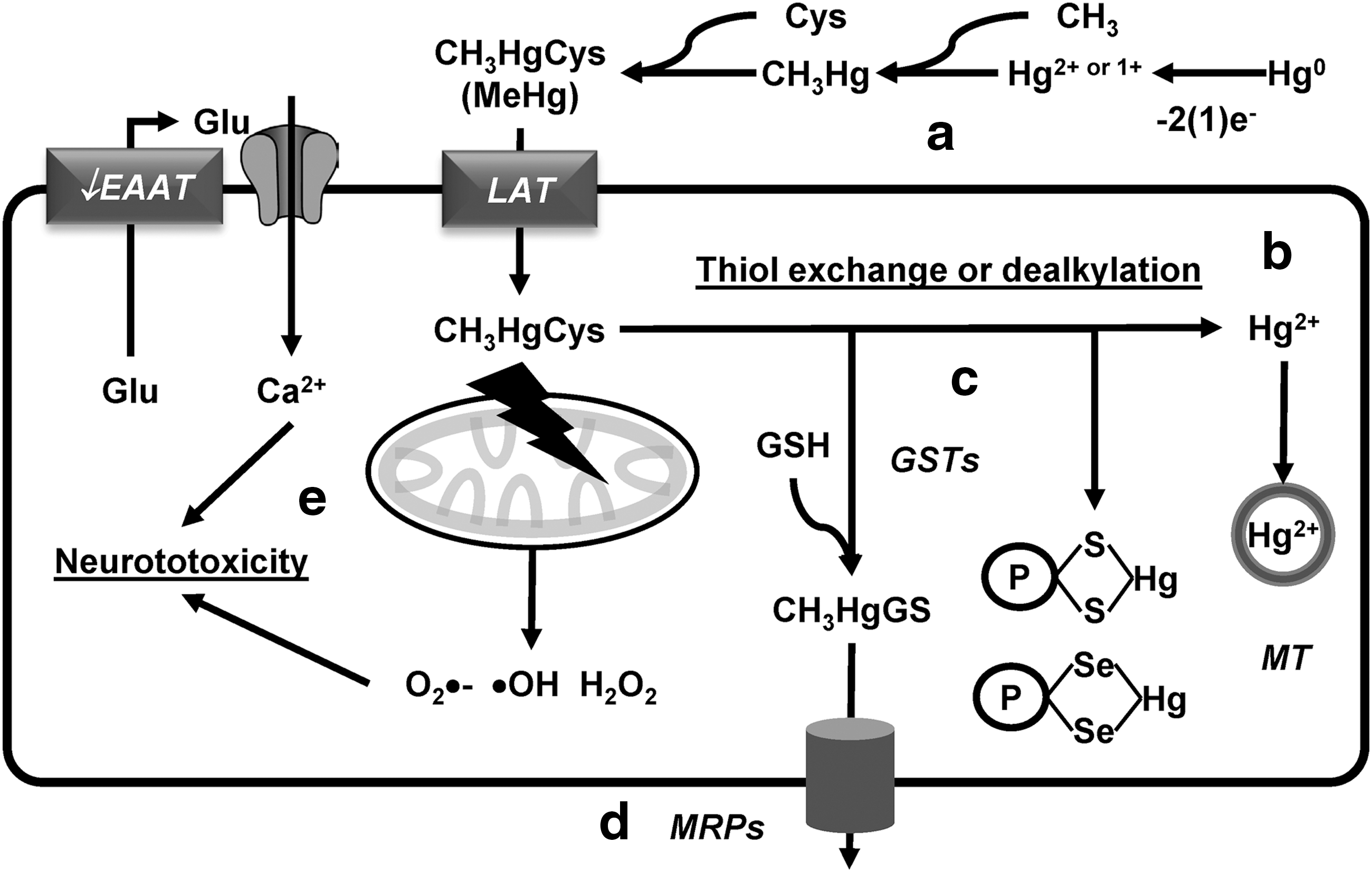
Most of the studies regarding the mechanisms involved in Hg neurotoxicity have been done using MeHg. MeHg and EtHg are potent electrophiles that form a complex with Cys (CH3HgCys or C2H5HgCys), and then transported across the BBB and into neuronal cells via L-type neutral amino acid transporters (LAT1 and 2) (Fig. 9a) (318, 385, 399).
A high percentage of Hg in individuals intoxicated with MeHg is found as Hg2+, suggesting that dealkylation of MeHg is an important mechanism for the high persistence of Hg in the brain (Fig. 9b) (72). Thiol exchange from CH3HgCys to low-molecular-weight thiols (GSH) and protein thiols has been proposed to be central mechanisms by which MeHg induces GSH depletion, inhibition of thiol-dependent antioxidant systems, and alters the activity or function of proteins with redox-sensitive Cys (signaling proteins, metabolic enzymes, neurotransmitter receptors, and transporters) (Fig. 9c) (89). MeHg also has a stronger affinity for selenol groups (selenohydryl groups in selenocysteines) compared with thiol groups. As such, selenoproteins are important targets for direct electrophilic attack of MeHg or transfer from thiol adducts (CH3HgCys, CH3HgGS, or CH3HgPS [protein-Cys adduct]) (Fig. 9c) (104, 221). GSTs have been proposed to mediate the formation of CH3HgGS adducts, which are detoxified by MRP1-mediated transport (Fig. 9d). GSH synthesis, GST, and MRP1 levels are regulated transcriptionally by the Nrf2 antioxidant system (152, 292, 349).
MeHg induces mitochondrial ROS and energy failure (175, 233). Neurotoxicity induced by MeHg has also been ascribed to its inhibitory effect on Glu uptake by astrocytes, triggering neuronal excitotoxicity (15, 235) (Fig. 9e).
Hg0 absorbed through the respiratory tract is oxidized to inorganic mercurous (Hg1+) and mercuric ions (Hg2+) (Fig. 9a). While inorganic Hg ions have limited access to the CNS, they induce profound neurotoxic alterations that seem to be mediated as well by their binding to thiol groups (89). Accordingly, MTs exert protective effects against Hg0-induced neurotoxicity (387) (Fig. 9b).
Other xenobiotic metals
Aluminum
Al is one of the most abundant metals in the earth's crust (8.1%). Al has a plethora of uses in industry and manufacturing, as well as in food additives. As such, human exposure is primarily originated from food and drinking water. Importantly, pharmaceuticals have higher levels of Al compared to food. Occupational exposures to Al are related to mining, processing, and welding (359) (Supplementary Table S1). While Al is poorly absorbed in the gut, inhalation mediates direct transfer to the brain via the olfactory system (341). Importantly, ∼85% of Al in blood is bound to Tf, which is considered to mediate its transport across the BBB (Fig. 10a) (288), but Tf-independent Al transport also exists. Interestingly, monocarboxylate and xCT transporters have also been proposed to mediate the transport of Al-citrate complexes (240, 386). Acute Al toxicity occurs as a result of occupational exposure or chronic renal failure and is known to target the nervous system. Al is neurotoxic in animal models triggering the accumulation of neurofibrillary tangles and impairment of cognitive, behavioral, and motor functions. Al promotes Aβ aggregation, mitochondrial dysfunction, and triggers neuroinflammation (Fig. 10b, c) (24, 202, 309). However, conflicting results exist regarding the association of Al with any human disease, including AD (37, 359, 377).
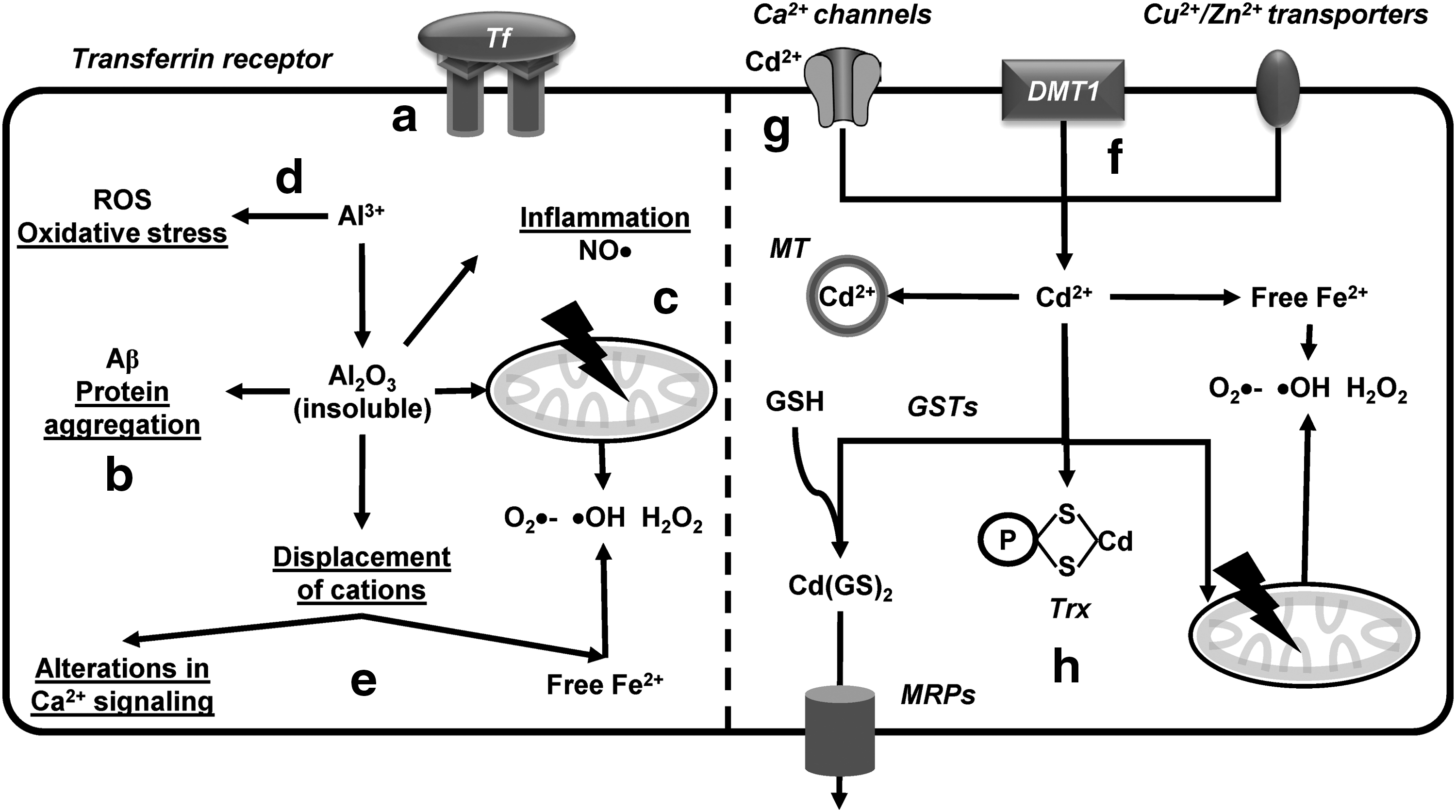
Al exists primarily in a trivalent state (Al3+). While Al has no redox capacity, Al toxicity is linked to oxidative damage. Al3+ has been proposed to react with H2O2 to produce Al superoxide radicals (AlO2 •−) that can deplete mitochondrial Fe and promote generation of ROS (Fig. 10d) (166). However, because of its high reactivity, Al is primarily found forming insoluble oxides whose toxicity seem to be related with the displacement of other biological cations (Ca2+, Fe2+, or Mg2+) (Fig. 10e) (377).
Cadmium
Cd is a transition metal whose use in industry has increased dramatically in the recent years. Cd is widely used in batteries, alloys, and pigments, and produced as a by-product from the extraction of other metals from ores. Food is the major source for Cd exposure as both animals and plants accumulate high levels of Cd. Inhalation is the prevalent route of Cd exposure due to industrial emissions and occupational activities (tobacco) (Supplementary Table S1). Cd neurotoxicity seems to occur only during development, before complete BBB formation, or in association with BBB dysfunction. Cd transport across membranes is thought to be mediated by molecular mimicry via several transporters and receptors for essential metals such as Cu/Zn transporters, DMT1, and Ca2+-channels (Fig. 10f) (117, 131, 174, 224, 339). Importantly, at high concentrations, Cd also has the ability to block Ca2+ currents (Fig. 10g).
Cd toxicity is linked to its ability to bind thiol containing molecules such as GSH, and protein-Cys (Trxs and MT) and as a consequence, displacement of redox-active metals and mitochondrial and metabolic dysfunction (Fig. 10h) (363). Cd detoxification of cells is facilitated by the activity of GST and the detoxification of GSH-Cd adducts via MRPs (Fig. 10h) (181, 339). Accordingly, resistance to Cd-toxicity is directly associated with the Nrf2-mediated antioxidant response (366).
Conclusions and Perspectives
Metals are important for brain function and human health. Thus, alterations in their content and/or distribution are expected to exert neurotoxicity. Both alterations in the homeostasis of essential metals and environmental exposure to xenobiotic metals can have silent chronic effects leading to neurodegeneration and neurological dysfunction (behavioral and cognitive alterations). The neurotoxic mechanisms by which metals impact neuronal or glial function are starting to become elucidated.
In this review, we have summarized how essential metals are trafficked in the brain. It is interesting to note that the mechanisms involved in metal transport and homeostasis are strongly linked to the chemical properties of each metal ion, while their coordination chemistry preferences also allow them to share some metal transport routes. For instance, Cu and Fe go through several redox cycles during their transport, while Mn and Zn remain in the same oxidation state. Metal trafficking in the cell is tightly regulated to control the high reactivity toward O2 of some metal ions, such as Fe2+ and Cu+, or to keep in solution otherwise insoluble species such as Cu+ and Fe3+. Moreover, while similar O-based ligand coordination preferences of Mn2+, Fe2+, and Fe3+ allow them to share some transport systems, the affinity of Zn2+ and Cu+ for Cys ligands makes MTs important players in their homeostasis. In fact, a close relationship between cellular redox environment and metal transport has been recently demonstrated for Cu and Fe (Figs. 1D and 4C).
Since metal trafficking machineries in neurons and astrocytes resemble those of other extensively studied mammalian cells, our understanding of intracellular metal homeostasis has advanced significantly. In contrast, metal trafficking at the synapse and the role in neuromodulation have just begun to be revealed, and point to a close inter-relationship among Zn, Cu, and Fe. For example, the synaptic release of Zn and Glu ultimately leads to the activation of NMDAR and the activation of signaling pathways that lead to postsynaptic Cu release and Fe uptake. Clearly, the close interplay between these metals at the synapse must play an important role in neuromodulation, while disturbed metal trafficking in neurodegenerative diseases would impact these processes.
We have also illustrated the differential alteration of metal homeostasis that occurs in neurodegenerative disorders as well as the key metal-protein interactions that might be involved in protein aggregation and metal-mediated oxidative damage. Understanding these interactions at the molecular level will shed light into the role of essential metals in neurodegenerative diseases.
Metals are transported across the BBB and into brain cells by selective transport systems for essential metals, while xenobiotic metals hijack those transporters via molecular mimicry (Supplementary Fig. S1a). The ability of xenobiotic metals to be transported and/or react with cellular targets is strongly determined by their metabolism via reduction/oxidation reactions, methylation, or adduct formation (Mt0→Mt1→Mt2). Phase II enzyme systems, GSH/GST, and MTs are essential for the detoxification of metals (Supplementary Fig. S1b). Metals induce or enhance ROS and RNS formation leading to oxidative stress (Supplementary Fig. S1c). While oxidative damage is one of the causative mechanisms involved in cellular damage induced by metals, it is now clear that other redox processes participate as well. The intrinsic reactivity of xenobiotic metals with thiol and selenol groups (Supplementary Fig. S1d) and their capacity to displace essential metals (Supplementary Fig. S1e) are also central to their capacity to promote energy failure (mitochondrial dysfunction), protein damage/aggregation, and metabolic alterations that challenge neuro/glial function and survival and trigger excitotoxic and inflammatory processes (Supplementary Fig. S1f). Cells have the capacity to respond by activating redox- or metal-dependent transcriptional regulation of antioxidant and anti-inflammatory responses (Supplementary Fig. S1g).
The aim of this review was to provide an integrated overview of the recent advances regarding how dysfunctional metal ion homeostasis of essential metals and exposure to xenobiotic metals alter cellular function to promote chronic neurodegeneration and neurotoxicity. Although the mechanisms involved in these processes are still being elucidated, the studies highlighted here are a starting point toward a better understanding of the pathological consequences of alterations in metal ion homeostasis, justifying the need of further studies regarding their metabolism and its impact on cellular homeostasis, function, and survival.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health Grant P20RR17675, Centers of Biomedical Research Excellence (COBRE), the Interdisciplinary Grant from the Research Council, the Life Sciences Grant Program of the University of Nebraska-Lincoln, the Mexican Academy of Sciences (AMC) (R.F.), and the National Council for Science and Technology in Mexico (CONACYT) via grant 221134 (L.Q.). PhD fellowships to Y.P. (308512) and C.G.-L. (290116) were from CONACYT. This work was performed in partial fulfillment of the requirements for the PhD degree in the posgrado en Ciencias Biomédicas at the Universidad Nacional Autónoma de México.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.