
Abstract
Significance:
Adenosine deamination in transcriptome results in the formation of inosine, a process that is called A-to-I RNA editing. Adenosine deamination is one of the more than 140 described RNA modifications. A-to-I RNA editing is catalyzed by adenosine deaminase acting on RNA (ADAR) enzymes and is essential for life.
Recent Advances:
Accumulating evidence supports a critical role of RNA editing in all aspects of RNA metabolism, including mRNA stability, splicing, nuclear export, and localization, as well as in recoding of proteins. These advances have significantly enhanced the understanding of mechanisms involved in development and in homeostasis. Furthermore, recent studies have indicated that RNA editing may be critically involved in cancer, aging, neurological, autoimmune, or cardiovascular diseases.
Critical Issues:
This review summarizes recent and significant achievements in the field of A-to-I RNA editing and discusses the importance and translational value of this RNA modification for gene expression, cellular, and organ function, as well as for disease development.
Future Directions:
Elucidation of the exact RNA editing-dependent mechanisms in a single-nucleotide level may pave the path toward the development of novel therapeutic strategies focusing on modulation of ADAR function in the disease context. Antioxid. Redox Signal. 29, 846–863.
Introduction
A
Interestingly, RNA-binding proteins may regulate all aspects of RNA metabolism and together comprise 11% of the entire coding genome in bacteria, archaea, and eukaryotes (8) underlying the importance of RNA fate in the encoded protein repertoire. In particular, evolutionary studies have demonstrated that RNA modification enzymes are one of the most conserved classes among the three domains of life (8). RNA modifications expand the RNA alphabet from 4 basic nucleotides to a growing number of 140 nucleotides, which tag the RNA (coding and noncoding) and determine its fate by providing numerous regulatory switches.
Almost half a century has passed since RNA modifications were first described, but only recently, methodological advances on RNA biology enabled their investigation bringing them into the spotlight. Adenosine (A)-to-inosine (I) RNA editing, the main RNA editing type present in mammals (31), engaged the epitranscriptomic era (126) mainly due to its nature of recoding the genetic information by deaminating adenosine residues and turning them into inosines (substitutional modification). Inosines are recognized by cellular machineries as guanosines due to their similar chemical characteristics. Two of the three known members of adenosine deaminase acting on the RNA (ADAR1-3) enzyme family, namely ADAR1 and ADAR2, have been identified to catalyze this substitutional modification in common or distinct target RNAs and are widely conserved in nearly all metazoans (60) (Fig. 1).

The human genome consists of 1–2% coding genes; the remaining 98% are referred to as noncoding elements and often termed dark matter (23, 33). Interestingly, regulatory mechanisms have been mapped within the dark matter, which may account for complexity in higher primates (23) and be crucially involved in several human pathologies. Thus, intensive efforts have been focused on unraveling the molecular role of A-to-I RNA editing in dark matter derivatives, such as microRNAs (miRNAs), circular RNAs (circRNAs), long-noncoding RNAs (lncRNAs), and Alu repetitive elements. The molecular functions of ADAR1 and ADAR1-induced RNA editing on RNA metabolism have been recently elegantly reviewed in Ref. (92).
In this review, we will summarize the recent advances that place A-to-I RNA editing among the frontiers in the study of fundamental aspects of gene expression in homeostasis and disease, focusing on noninfectious diseases. We will primarily discuss the ADAR1/ADAR2-induced RNA editing effects with pathophysiological implications in mammals.
ADAR1-Mediated RNA Editing in Physiology
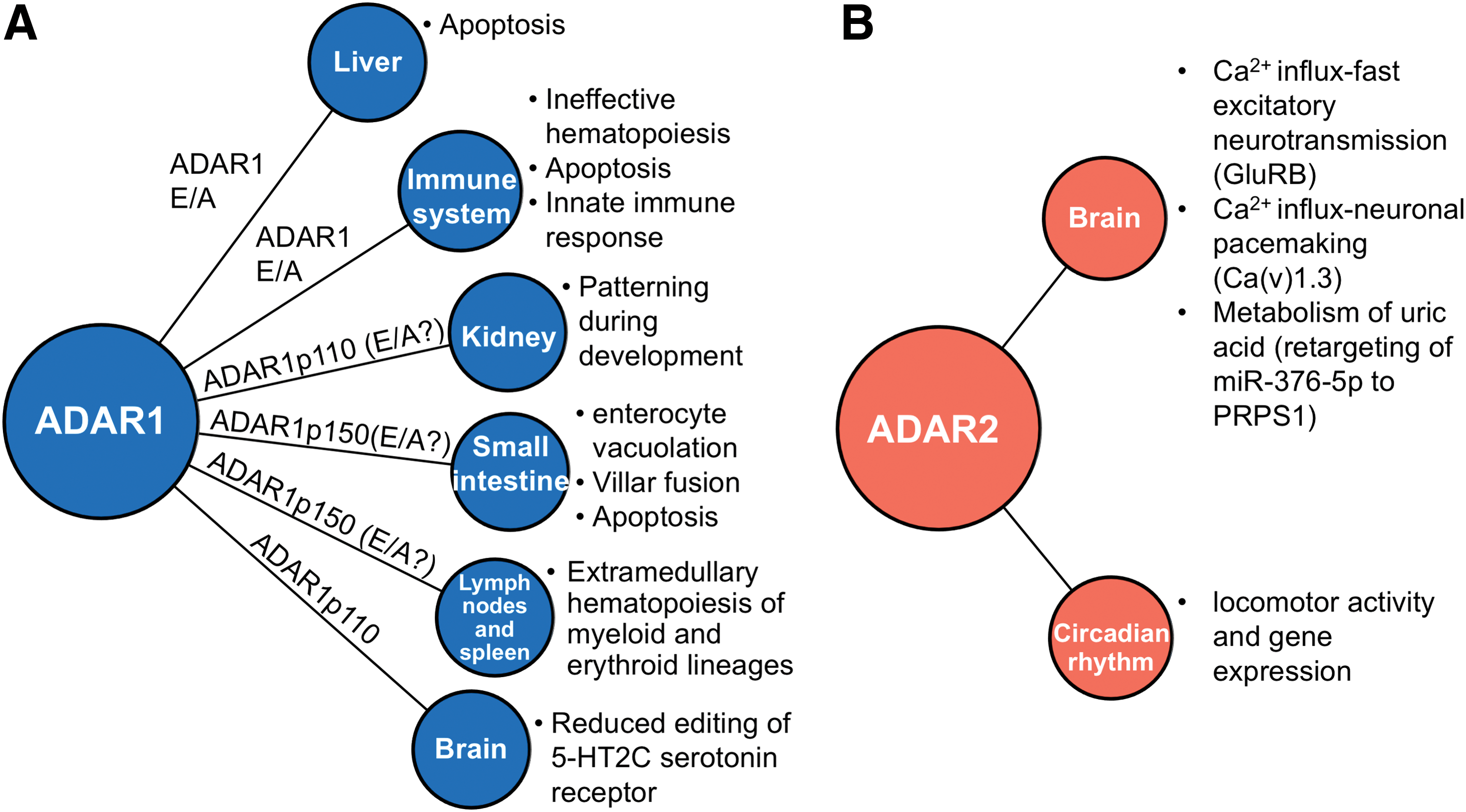
ADAR1 is indispensable for life since its genetic ablation results in in utero lethality of mouse embryos by embryonic day 12.5 (E12.5) associated with liver degeneration and anemia (143). Histological analysis of these embryos revealed widespread apoptosis, fetal liver disintegration, and ineffective hematopoiesis specifically with regard to erythroid and myeloid/granulomatous progenitors (45, 144). Taken together, these findings suggested an essential role of ADAR1 in hematopoiesis, organ homeostasis, and development.
However, given the binary nature of ADAR1, as an RNA-binding protein and an RNA editing enzyme, recent studies aimed to dissect the functional role of this enzyme in vivo. By applying an elegant experimental approach, Liddicoat et al. generated mice bearing a constitutive knockin of an RNA editing-deficient ADAR1 mutant (Adar1E861A/E861A ). Strikingly, these editing-deficient mice also died in utero around E13.5 displaying a similar phenotype (liver failure, defective hematopoiesis) and prominent activation of innate immune response (74), recapitulating, in principle, the initially observed phenotype of ADAR1 knockout (KO) mice (46). These findings underline that ADAR1-induced RNA editing is essential for homeostasis and life itself (Fig. 2).
Gene expression profiling of Adar1−/− embryos as well as Adar1−/− fetal liver showed the aberrant expression of interferon (IFN)-stimulated genes (ISGs) (45). Furthermore, in vitro studies on hematopoietic stem cells and hematopoietic progenitor cells revealed induction of an aberrant innate immune response in the absence of ADAR1, suggesting a new role for ADAR1 as a repressor of IFN signaling (46). In eukaryotic cells, pattern recognition receptors, such as retinoic acid inducible gene I (RIG-I) and melanoma differentiation-associated protein-5 (MDA5), trigger innate immune and inflammatory responses to counteract viral infections (6).
An initial in vitro observation that oligonucleotides containing inosine–uracil (I:U) base pairs (hallmarks of RNA editing events) suppress ISG expression by binding to MDA5 or RIG-I (139) suggested that ADAR1 and RNA editing may play a primary role in preventing sensing of endogenous double-strand RNAs (dsRNAs) as nonself by the innate immune system (Fig. 3). In contrast, other researchers propose that ADAR1-mediated suppression of innate immune response depends on its ability to bind dsRNA and physically contact RIG-I rather than on its catalytic activity (155).
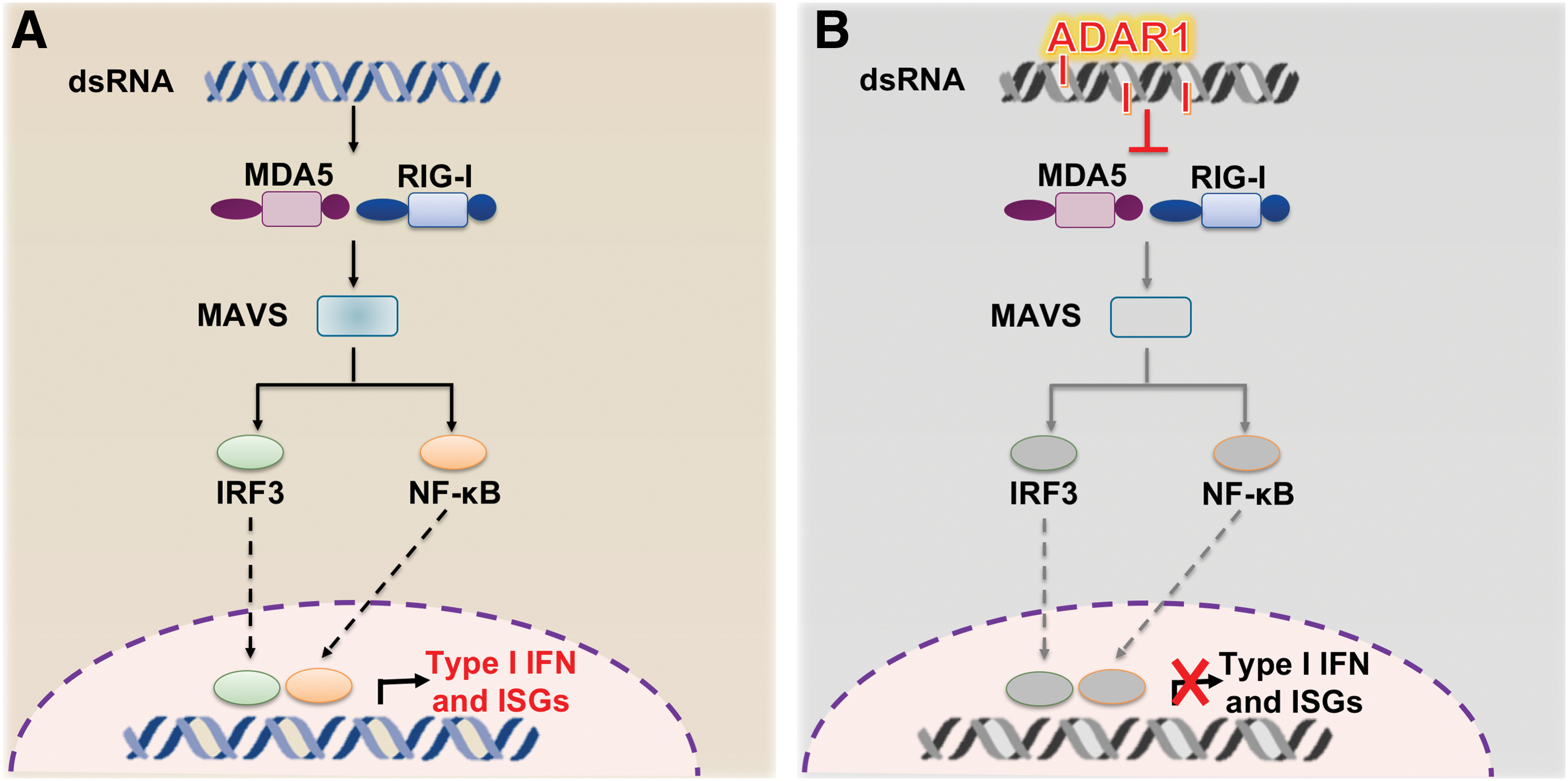
To rescue the embryonic Adar1−/− lethal phenotype and gain further insight on the underlying molecular mechanisms, a pioneering work by Mary O'Connell's group described the generation of a double mutant mouse for Adar1 and interferon alpha/beta receptor 1 (Ifnar1) (the common IFN-α and IFN-β receptor 1) or signal transducer and activator of transcription 1 (Stat1) [a key player of systemic IFN responses; reviewed in Ref. (125)] (81). In this way, the survival of Adar1 −/− /Ifnar1 −/− or Adar1 −/− /Stat1 −/− embryos was extended by 2–4 days compared with Adar1−/− embryos, but the embryos did not make it to birth, manifesting similar deficiencies to those observed in Adar1−/− (81). These results suggested that mouse phenotype rescue might be potentially achieved by blocking a more potent upstream mediator before innate immune response becomes prevalent.
Mitochondrial antiviral signaling protein (MAVS) is an adaptor protein that activates the transcription factors IRF3/IRF7 and NF-kB to induce the transcription of type I IFN and proinflammatory cytokines upon the recognition of viral dsRNA by cytosolic receptors RIG-I and MDA5 (42, 102, 149) (Fig. 3). Interestingly, Adar1 −/−/Mavs−/− double mutant embryos survived up to birth, but only to die a day (81) or maximum ten days later, with majority of the neonates dying within the first 2 days (101).
In the first study, histological analysis revealed apparently normal morphology of liver, heart, and other organs, but blood analysis indicated persistently elevated IFN and interleukin-1 levels (81). In the second study, severe defects in kidneys' architecture, disturbed intestinal homeostasis, lack of organization in lymphoid follicles in both lymph nodes and spleen, and defects in B cell maturation were present in double KO mutant mice (101). These findings suggested an MAVS-independent role of ADAR1 in hematopoiesis and an essential role of ADAR1 in multiple organ development and homeostasis (81, 101).
The overall observations supported the notion that knocking out an upstream dsRNA sensor, RIG-I or MDA5, might confer greater protection to aberrant innate immune response activation in the absence of ADAR1. Surprisingly, in the same study, Adar1−/−/Mda5−/− double KO mutants displayed a similar survival curve as the Adar1−/−/Mavs−/− mice and died postnatally within a week (101). However, Carl Walkley's group pursued this further by generating the Adar1E861A/E861A/Mda5−/− mutants that not only survived past weaning but also had no manifestations, apart from slightly smaller size compared with littermate controls (74). These breakthrough findings provided the basis for MDA5 to be considered as the primary sensor of endogenous unedited dsRNAs.
In parallel, a key question is which ADAR1 isoform contributes the most to the lethal phenotype (Table 1). As previously mentioned, two isoforms give rise to two ADAR1 proteins with distinct molecular weights, ADAR1p150 (long isoform, 150 kDa) and ADAR1p110 (short isoform, 110 kDa). The long isoform is induced by an IFN-responsive promoter utilizing a translational start site in exon 1A, while the short one is constitutively expressed using a promoter present in exon 1B or 1C and an alternative translational start site residing in exon 2 [(115), as reviewed in Ref. (11)].
ADAR, adenosine deaminase acting on RNA; DBD, DNA binding domain; dsRBD, double-stranded RNA binding domain; IFIH1, interferon-induced helicase C domain-containing protein 1; Ifnar1, interferon alpha/beta receptor 1; KO, knockout; MAVS, mitochondrial antiviral signaling protein.
In a first attempt to clarify this point, Ward et al. generated mice lacking the ADAR1p150 isoform by exclusively removing exon 1A, thereby preserving p110-specific exon 1B intact. Interestingly, these mice displayed the same phenotype as observed in the double ADAR1 KO mice (embryonic death at E11-12) (46), suggesting that the IFN-induced ADAR1 isoform may account for the lethal phenotype (146). Nevertheless, concerns with respect to the applied genetic strategy were raised, questioning whether ADAR1p110 protein levels are affected in the Adar1p150−/− mice participating in the phenotype (82).
A few years later, though, Pestal et al. using the same Adar1p150−/− strain demonstrated unaffected ADAR1p110 protein levels in isolated mouse embryonic fibroblasts consolidating the crucial role of ADAR1p150 in the lethal phenotype (101). Importantly, Adar1p150−/−/Mavs−/− mice survived to weaning, although smaller than their littermate controls, consistent also with the observation in Adar1E861A/E861A/Mda5−/− mice (74). Histological observations suggested that kidney morphological deficiencies present in Adar−/−/Mavs−/− were no longer manifested in Adar1p150−/−/Mavs−/− mice, while RNA editing analysis of an exemplary ADAR1-edited target (5-HT2C receptor) showed no decrease in RNA editing levels, indicating that the residual ADAR1p110 expression and editing activity (E/A) in these mice were sufficient for the recovery of kidney development (101). On the contrary, intestinal homeostasis and B cell development were still compromised in Adar1p150−/−/Mavs−/− mice, thereby attributing these specific roles to ADAR1p150 (101).
A series of studies addressing the complexed nature of the underlying cellular and molecular mechanisms are critically discussed in this excellent review (94) and more recently in Ref. (73, 92).
Despite these generous efforts, several questions are raised such as (i) What is the functional role of the nuclear ADAR1p110 isoform? An insightful recent study by Kazuko Nishikura's group suggests that stress-induced phosphorylation of ADAR1p110 leads to its translocation to the cytoplasm, where it orchestrates apoptosis in a Staufen1-dependent manner (111). (ii) What is the cell contribution to the lethal phenotype? Conditional inactivation-based strategies of ADAR1 in different cell types may provide further insights toward this direction. (iii) Is ADAR1 essential for all cell types and, if so, is the ADAR1p150/MDA5/IFN axis conserved in every cell type? For example, there are findings pointing out the importance of the ADAR1-RNA editing-independent function (97). (iv) Is MDA5 the primarily involved dsRNA sensor in every cell type or are there additional sensors of importance? Of note, Adar1−/−/RIG1−/− double mutant embryos were not recovered (101). (v) Which are the involved dsRNAs transmitting damage signals? Further studies are needed to address these questions in future.
ADAR2-Induced RNA Editing in Physiology
Editing of coding transcripts
Although ADAR2 is dispensable for embryonic development (49), it plays a central role in physiology (Fig. 2B) by regulating the function of a major target, the glutamate receptor, preserving thus primarily the neuronal homeostasis. Nuclear A-to-I RNA editing levels of neuronal transcripts in the brain have been shown to progressively increase after birth (34, 141). Recently, Marie Ohman's group ascribed this regulation to the accumulation of nuclear ADAR2 due to direct interaction of ADAR2 with importin-α4, which increases during neuronal maturation, and to the stoichiometrically increased interaction of ADAR2 with its stabilizing factor, the nuclear isomerase Pin1 (12). We may thus hypothesize that ADAR2 is a determining factor during brain development by marking neuronal transcripts and favoring their processing. Future in vivo studies may shed light on this notion.
As early as in 1991, Peter Seeburg's group described the first edited substrate of ADAR2, the glutamate receptor subunit B (GluRB) precursor messenger RNA (pre-mRNA) (Gria2) of the aminomethylphosphonic acid (AMPA) receptor family, with biological consequences for the ion flow in glutamate-gated channels (123). Two edited positions within the GluRB pre-mRNA have been attributed exclusively to ADAR2 and lead to recoding events in exon 11 (C

Accordingly, the physiological importance of this recoding event was confirmed by two independent in vivo studies that reported early onset epilepsy and premature death of mice heterozygous for an intron 11-modified GluRBΔECS allele with unedited Q/R site transcripts due to higher Ca2+ influx of AMPA receptors (14, 36). Subsequently, Adar2 −/− mice manifested seizures, already after P12, and similar dysfunctions, resulting eventually in postnatal death within 20 days from birth (49). Of note, a residual catalytically inactive, but with an intact RNA-binding domain, ADAR2 protein akin to exon skipping was present in these mice. Nevertheless, this protein may only account for less than 10% in Adar2+/+ mice and was not able to complement the mechanism (49).
The ultimate validation that GluRB is the major target of ADAR2 in homeostasis was provided by the rescue of Adar2
−/− mice lethality. Specifically, the same group inserted in Adar2
−/− mice genome a constitutively expressed pre-edited form of GluRB subunit at the Q/R site (49). Interestingly, the well-known channels for low-voltage Ca2+ influx and neuronal pacemaking, Ca(v)1.3, have been identified as another substrate of ADAR2 in vitro and in vivo (51). ADAR2 edits Ca(v)1.3 channels by inducing multiple substitutional editing events (IQDY→
Recently, Terajima et al. reported that Adar2 −/− mice exhibited disturbed rhythms in mRNAs due to the absence of RNA editing, as indicated by the unusual accumulation of CRY2, resulting in short-period rhythms in locomotor activity and gene expression (131). These findings support that ADAR2 participates actively in the coordination of the circadian clockwork.
Editing of dark matter
A revolutionary study, expanding the ADAR2 target repertoire to noncoding RNAs with functional consequences, revealed that ADAR2 alters the target selectivity of miR-376a-5p by editing its seed region (63) (Fig. 4). Specifically, the edited version of miR-376a-5p is redirected to target phosphoribosyl pyrophosphate synthetase 1 (PRPS1), an essential enzyme for purine metabolism and uric acid synthesis, in multiple positions within its 3′UTR, thereby repressing its expression and maintaining the normal uric acid levels in mouse brain cortex (63).
Accordingly, specifically in the brain cortex of wild-type mice, where the edited version of miR-376a-5p was detected with unaltered expression levels, PRPS1 showed up to twofold decreased expression levels compared with Adar2−/− mouse brain cortex (63). Interestingly, PRPS1 levels in the liver were not affected by Adar2 deletion, in congruence with the lack of the edited version of miR-376a-5p in this organ.
ADAR1-Induced RNA Editing in Autoimmunity and Inflammatory Diseases
A deregulated innate immune response is a key feature of autoimmune diseases. Specifically, type I IFN pathway activation has been implicated in the pathogenesis of various autoimmune diseases, including systemic lupus erythematosus (SLE), a prototypical autoimmune disease (10). Thus, monoclonal antibodies directed against IFN-α or IFNAR are currently in advanced phases of clinical trials for treatment of SLE (38, 64). The pivotal role of ADAR1 in innate immune response designates ADAR1-induced RNA editing as a factor worth exploring in the pathogenesis of autoimmune diseases. Taking into consideration that ADAR1 is induced by inflammatory cytokines such as tumor necrosis factor-α or IFN-γ (127, 154), which are known to be augmented during systemic autoimmune diseases and other inflammatory diseases, we hypothesize that RNA editing may play a pivotal role in inflammation.
Interferonopathies
Aicardi-Goutières syndrome (AGS) is a rare genetic type I interferonopathy, usually manifested as an early-onset encephalopathy, occurring due to different genetic causes, all of which lead to aberrant type I IFN expression (3, 71, 108). In accordance with the role of ADAR1 as a suppressor of type I IFN signaling, Yanick Crow's group first reported a census of mutations in Adar1 genetic locus, with the majority of them resulting in amino acid substitutions within the catalytic domain of the enzyme, in patients with AGS (109) [reviewed in Ref. (28, 29)]. A few years later, the same group elegantly showed that gain-of-function mutations in IFIH1 (MDA5) gene locus result in an induction of type I IFN signaling (107), in line with the suggested role of ADAR1/MDA5/IFN axis in innate immune response.
Harnessing the recently resolved crystal structures of human ADAR2 (83), which shares considerable similarity with ADAR1 deaminase domain (66, 84), AGS-causing mutations were mapped to the ADAR1 deaminase domain enabling the modeling of how exactly these mutations interfere with the interaction of ADAR1 with RNA substrates (37). These particular studies consolidate further the functional consequences of these mutations in AGS.
Bilateral striatal necrosis (BSN), another pediatric interferonopathy that may coexist with AGS (68), is clinically characterized by dystonic movement and has also been recently associated with Adar1 mutations (76). Strikingly, BSN patients with an Adar1 mutation exhibit a heightened type I IFN gene expression in contrast with those patients who had no mutation present in Adar1 locus (76). Interestingly, a case of unexplained spastic paraplegia was also attributed to the presence of Adar1 mutation (30).
Implications on SLE and other autoimmune diseases
Elevated levels of ADAR1 and particularly of ADAR1p150 isoform have been previously reported in peripheral blood mononuclear cells, T lymphocytes, and natural killer cells isolated from patients with SLE (27, 69, 70, 134). Taking into consideration the central role of activation of cytoplasmic RNA sensors in the pathogenesis of SLE, further studies exploring the potential contribution of ADAR1p150 in the pathogenesis of SLE are warranted. Whether the observed ADAR1 upregulation in an autoimmune environment (i.e. in AGS) is a counteractive mechanism to constrain the excess of the innate immune response or constitutes a pathogenic mechanism in autoimmune and chronic inflammatory rheumatic diseases remains to be elucidated in future studies.
ADAR1-Induced RNA Editing in Cancer
Hepatocellular carcinoma (HCC), a primary malignancy of the liver, is among the leading causes of cancer-related deaths worldwide, according to World Health Organization statistics found on Globocan (
Editing of coding transcripts
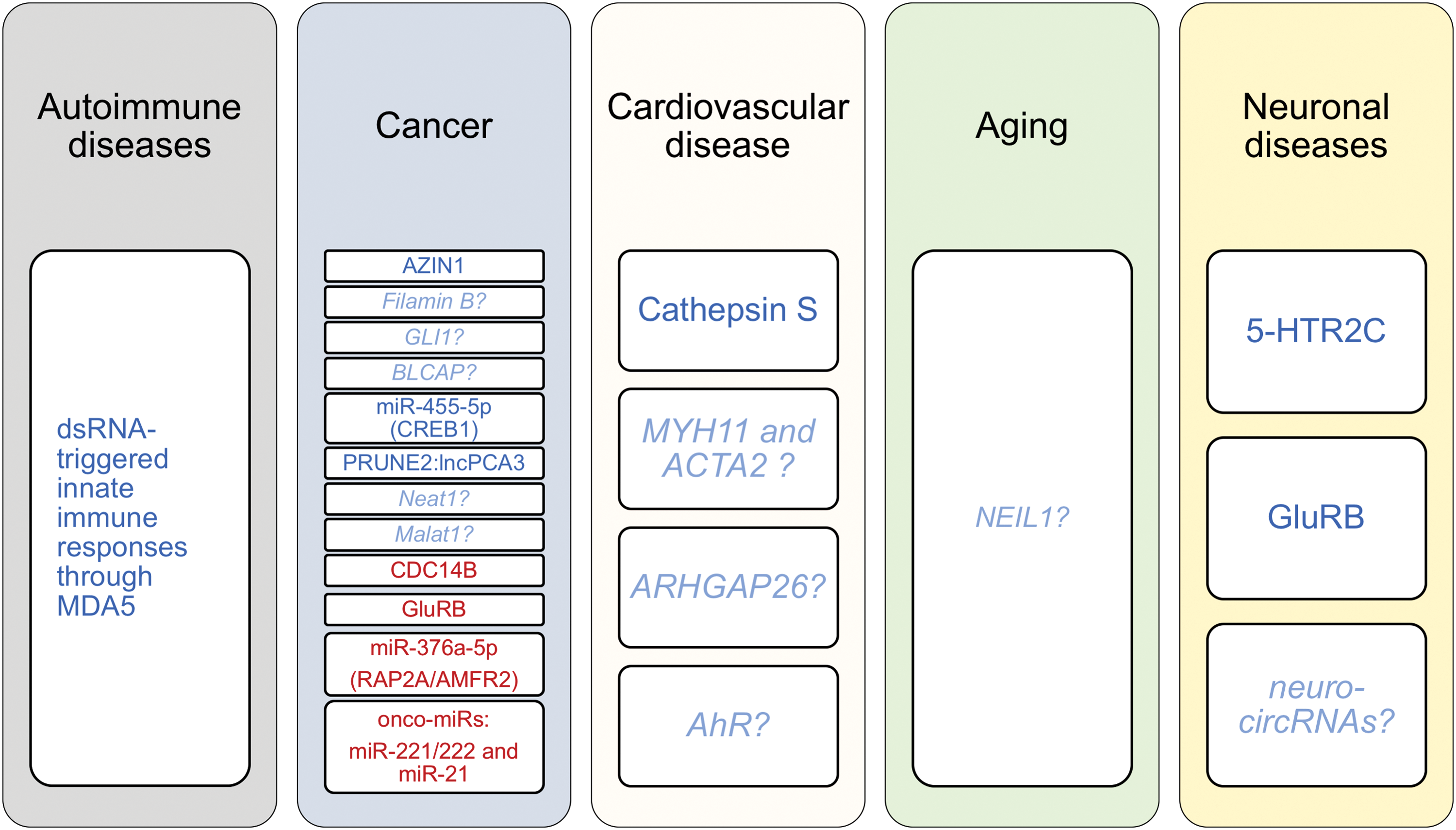
Interestingly, Chen et al. reported that the constitutively expressed nuclear ADAR1 isoform, ADAR1p110, edits antizyme inhibitor 1 (AZIN1) pre-mRNA shifting the cells toward a more tumorigenic phenotype, as studied in human HCC specimens and in a tumor animal model (19, 26). Specifically, the authors provide mechanistic insights supporting that a single recoding event (S367G) in the AZIN1 transcript induces a conformational change, which leads to enhanced AZIN1-antizyme direct interaction and thereby increases the levels of two key regulators of the cell cycle, ornithine decarboxylase and cyclin D1 (CCND1), augmenting (in this way) the tumor cell proliferation (19) (Fig. 4).
Compelling clinical evidence derived from 94 patients with HCC revealed increased RNA editing levels of AZIN1 mRNA (19). Another oncogenic target, namely filamin B (FLNB), is edited by both ADAR1 and ADAR2, promoting uncontrolled cancer cell proliferation in vitro (105). Future studies may elucidate the functional role of FLNB in tumor pathogenesis in vivo.
Glioma-associated oncogene 1 (GLI1) also holds a key role in cell proliferation during tumorigenesis and in embryonic patterning through Hedgehog signaling (26, 120). Of note, the nucleotide 2179 constitutes a highly edited site on GLI1 mRNA that results in an amino acid substitution (R→G) and changes the GLI transcription efficiency, thereby reducing GLI-dependent cellular proliferation (120). Additionally, it has been reported that ADAR1-mediated RNA editing regulates GLI1-dependent Dyrk1a-mediated phosphorylation, resulting in decreased transcription and reduced medulloblastoma cell growth (120).
Another study has shown that ADAR1p110-induced RNA editing of Gabra3 results in the suppression of breast cancer migration, invasion, and metastasis due to the lack of active AKT signaling (43). Moreover, bladder cancer-associated protein (BLCAP), which is highly conserved among species, is subjected to RNA editing by both ADAR1 and ADAR2 in the highly conserved amino terminus, leading to amino acid changes and thus resulting in alternative protein isoforms (40). On the other hand, the BLCAP RNA editing levels have been reported to be strongly decreased in astrocytomas and colorectal and bladder cancer (40) (Fig. 4).
Editing of dark matter
It has been previously proposed that loss of ADAR1 promotes tumor growth in metastatic melanoma (91). A few years later, Shoshan et al. documented that ADAR1 expression and function are impaired in melanoma by direct binding of the transcriptional factor CREB (121). More importantly, the authors showed that ADAR1 edits miR-455-5p with functional consequences in an experimental model of melanoma growth and metastasis in mice. Specifically, the study described two potential underlying mechanisms suggesting that ADAR1-dependent RNA editing of pri-miR-455 may result in (i) differential binding of Drosha and thus inhibit its maturation process and/or in (ii) altered miRNA targetome (retargeting) since edited miR-455-5p recognizes a different set of transcripts than the unedited miR-455-5p, which targets the cytoplasmic polyadenylation element-binding protein 1 (CPEB1) (121) (Fig. 4).
Taking together, these results suggest that ADAR1-mediated RNA editing promotes CPEB1 expression by impeding miR-455-5p targeting of CPEB1 transcript. This results in the suppression of experimental melanoma growth and metastasis in mice (121). Nevertheless, these intriguing findings raise several important questions that remain to be answered by future studies: (i) Which of the two suggested mechanisms accounts for the regulation in mice and in humans? (ii) Is this mechanism conserved between humans and mice? (iii) Could this mechanism bear clinical prognostic value that would put ADAR1-RNA editing in translational prospective?
Salameh et al. showed that ADAR1 edits a double-stranded RNA structure formed by the interaction of two RNA molecules, the intronic long noncoding RNA prostate cancer antigen 3 (lncPCA3), a highly specific and established biomarker for prostate cancer, and the pre-mRNA of prune homolog 2 (PRUNE2) (113). A-to-I RNA editing of the PCA3:PRUNE2 duplex results in decreased expression of both RNAs, lncPCA3 and PRUNE2 mRNA (113) (Fig. 4). In accordance with the previously documented PRUNE2 function as a tumor suppressor, the authors reported that this ADAR1-induced RNA editing-dependent mechanism augments malignant cell growth of prostate cancer cells (113).
Another long noncoding RNA, Neat1, which is essential for paraspeckle nuclear body formation (25, 114), has been documented to hold a key role in tumorigenesis and chemosensitivity mainly by directly interacting with p53 (2, 18, 52). Interestingly, it has been previously reported that NEAT1 directly interacts with p54nrb and actively participates in an ADAR1-RNA editing-dependent nuclear retention of the edited form of Lin28 mRNA, which is preferentially fished out by p54nrb (21), confirming the initial observations of another group (104). Xist (encoding X-inactive-specific transcript) and Malat1 (encoding metastasis-associated lung adenocarcinoma transcript 1) are two additional lncRNAs with a well-established link with tumorigenesis (9, 157). Nevertheless, whether direct RNA editing of these lncRNAs alters their molecular repertoire with functional consequences in tumor biology remains elusive.
Collectively, these findings underscore the necessity of balanced ADAR1-induced RNA editing during cancer progression (Fig. 5). More importantly, these studies preclude global ADAR1 targeting from a therapeutic perspective given its diverse effects on different substrates and cell types.
ADAR2-Induced RNA Editing in Cancer
Based on the essential function of ADAR2 during brain development (12, 49, 141), several studies have explored the role of ADAR2 in the pathogenesis of brain tumors (Fig. 5). Indeed, ADAR2-induced RNA E/A is severely hindered in patients with glioblastoma and this deficiency is associated with tumor progression (17, 79, 99, 133). Glioblastoma, a type of astrocytoma, is by far the most common and aggressive malignant brain tumor [reviewed in Ref. (96)].
Editing of coding transcripts
A series of in vitro and ex vivo studies from Angela Gallo's group showed that ADAR2 may inhibit glioblastoma cell proliferation and tumor growth by direct editing of phosphatase Cdc14b pre-mRNA and concomitant regulation of CDC14B/Skp2/p21/p27 axis in glioblastoma cell lines from adult and pediatric patients (41). These interesting findings encourage further studies to delineate the exact molecular mechanism and provide in vivo evidence highlighting the contribution of this pathway to the pathogenesis and progression of glioblastoma.
In line with the essential role of GluRB in neuronal physiology, reduced editing of GluRB transcripts at Q/R sites has been detected in patients with glioblastoma (79). Accordingly, Ca2+ influx caused by the presence of unedited GluRB subunit can augment glioblastoma invasiveness, revealing ADAR2 as a crucial parameter for glioma prognosis and a potential therapeutic target (55). Recently, Hundley and colleagues convincingly demonstrated that ADAR2 is antagonized by ADAR3 (the catalytically inactive member of ADAR family) for direct binding to GluRB pre-mRNA, thus contributing to the lower editing levels of GluRB in glioblastoma patients (95). Notably, this study provides, for the first time, a functional characterization of ADAR3 in relation to a disease context (95).
Editing of dark matter
Providing compelling in vitro and in vivo evidence, Choudhury et al. elegantly described that the previously reported downregulation of ADAR2 in glioblastoma tissues led to increased levels of the unedited version of miR-376a-5p and decreased edited miR-376a-5p with two severe biological consequences (24). The unedited version of miR-376a-5p targets RAP2A (a member of RAS family), lowering its expression levels that in turn lead to augmented glioma cell invasion (24). Simultaneously, the lower levels of the edited miR-376a-5p result in inefficient targeting of the autocrine motility factor receptor, thereby increasing its levels and further contributing to glioma cell invasion (24). ADAR2 has been suggested to be an miRNA surveillance officer since restored ADAR2 expression in glioblastoma cells attenuates RNA editing-dependent decrease of onco-miRNA levels, miR-221/222 and miR-21, in vivo (mouse brain) and in human glioblastoma cells (132).
Together, these studies suggest that reinforcement of ADAR2 RNA E/A may provide a promising therapeutic option in the treatment of glioblastoma. Encouraging results from newly developed approaches (44, 85, 86, 124, 140, 147) confirmed that site-directed ADAR2-induced editing of specific targets and nucleotides is not far from reality and preclinical applications. By equipping the ADAR2 deaminase domain with a scavenger platform (22-amino acid-long λ-N protein or SNAP-tag), which can also be light inducible (44), loaded with an antisense guide RNA to ensure the specificity of targeting, researchers corrected the editing levels of certain targets with satisfying efficiency both in cell culture (in vitro) and in Xenopus oocytes and annelid embryos (in vivo). Further studies exploring off-target and toxicity-related side effects, as well as seeking the application of these approaches in larger animals, are warranted.
ADAR1-Induced RNA Editing in Cardiovascular Disease
In the second decade of the 21st century, CVD remains the primary cause of mortality in the world with the Global Burden of Disease study reporting that one of three deaths worldwide is attributed to CVD, twice the number of cancer-related deaths (77). However, only recently, RNA editing has been associated with this important disease (127), which lacks efficient therapeutic strategies to date.
Specifically, we have recently described a previously unrecognized mechanism regarding ADAR1-induced clustered RNA editing within the 3 prime untranslated region (3′UTR) of cathepsin S (CTSS) mRNA, a proinflammatory gene with a well-established role among others in vascular function (116) and atherosclerosis (129), the main cause of CVD. The observed clusters of RNA editing lie within the 3′UTR of CTSS and specifically within two inverted Alu repeats (127), which form dsRNAs, a prerequisite for A-to-I RNA editing (22, 65, 106). RNA editing of the Alu dsRNAs within the CTSS 3′UTR induces bulges enabling human antigen R (HuR), a known single-stranded RNA-binding protein, to bind and confer stability on the bound mRNAs. This mechanism enhances CTSS expression in both mRNA and protein levels with functional consequences for vascular endothelial cells (Fig. 4).
Of note, hypoxia and inflammation were identified as inducers of ADAR1-mediated RNA editing of CTSS. Due to primate specificity of Alu sites precluding the use of an animal model, we investigated the clinical implications of this mechanism directly in human tissues from >300 subjects, reflecting various stages of CVD. We have recently revealed that ADAR1-induced RNA editing of CTSS is a novel regulatory mechanism driving CTSS mRNA expression in all stages of CVD, including subclinical atherosclerosis, coronary artery disease, aortic thoracic aneurysms, and advanced carotid atherosclerotic disease.
Based on additional observations that (i) HuR-binding motifs are enriched within the 3′UTRs of edited transcripts in close proximity to RNA editing sites and (ii) the expression of these transcripts is regulated by ADAR1, this mechanism may be extrapolated to a significant number of other affected targets that remain to be identified and studied with regard to their functional role in cardiovascular or other diseases (Fig. 5).
Interesting findings of a few other studies may also have important implications on the development of CVD. Smooth muscle cells (SMCs) show plasticity and switch from a contractile to a proliferative phenotype, promoting plaque formation in response to proatherogenic stimuli stemming from vascular endothelial or inflammatory cells. However, their presence within advanced plaques prevents the rupture of the fibrous cap dampening tremendous consequences [reviewed in Ref. (72, 130, 137)]. Interestingly, ADAR1-induced RNA editing may partially regulate phenotypic switch of SMCs (35), but not the contractile SMCs. Specifically, the authors suggested that during platelet-derived growth factor-BB-induced phenotypic switch, SMC marker pre-mRNAs, SMC myosin heavy chain (MYH11) and smooth muscle α-actin (ACTA2), undergo intronic RNA editing and are accumulated.
Simultaneously, they observed that the mature mRNA and protein levels of these markers are decreased, potentially due to alternative splicing, leading thus to maintenance of the SMC phenotypic switch (35) given that downregulation of these genes is a hallmark of this process [reviewed in Ref. (117)]. However, additional in vitro and in vivo studies clarifying the underlying mechanism (e.g. the presence of alternative spliced variants) are warranted to consolidate these interesting findings and define the role of ADAR1-induced RNA editing in SMC biology, which could be causatively involved in CVD.
RhoA and cell division control protein 42 homolog (Cdc42) are both members of the ubiquitously expressed Rho family (119). Rho GTPase-activating protein 26 (ARHGAP26) regulates the activity of RhoA by converting the active GTP-bound RhoA to the inactive GDP-bound form (142). Thus, it contributes to various Rho-related pathologies, including CVD (118). Of note, Wang et al. demonstrated that 3′ UTR ARHGAP26 is highly edited by ADAR1 disrupting the miR-30b-3p and miR-573 binding sites onto the transcript and thus enhancing the negative regulatory effect of ARHGAP26 on RhoA, as well as on its other target, Cdc42, a regulator of the cell cycle (128, 142). As a result, the absence of ADAR1-induced A-to-I RNA editing results in higher RhoA-GTP activity due to the loss of its negative regulatory mechanism (142).
Following similar mechanistic footprints, Nakano et al. reported that ADAR1 edits the 3′UTR of human aryl hydrocarbon receptor (AhR) mRNA (90) in a human cancer cell line. The edited form of AhR harbors a recognition motif for miR-378 leading to increased miR-378-binding to AhR mRNA, which results in repression of AhR mRNA and protein expression (90). Strikingly, AhR is known to be causatively involved in atherosclerosis (148). The findings of these studies encourage us to hypothesize that ADAR1-AhR and/or ADAR1-ARHGAP26 axis could represent additional mechanisms whereby RNA editing controls atherosclerosis. It is tempting to assess these regulatory mechanisms in the context of CVD in future studies.
Together, these studies underline the magnitude of RNA editing as an additional regulatory layer of CVD (Fig. 5). Identification of specific targets (e.g. CTSS, ARHGAP26, AhR) and editing sites along with recent technological advances (e.g. site-directed CRISPR/Cas9 genome editing) may provide a unique therapeutic strategy in the course of precision medicine.
ADAR1-Induced RNA Recoding of Deoxyribonucleic Acid Repair: Implications for Aging-Related Pathologies
Reactive oxygen species (ROS) comprise a highly reactive group of oxygen-based chemical species and unconstrained production of this group is associated with excessive oxidative stress, which is reflected among others by deoxyribonucleic acid (DNA) damage and eventually cellular dysfunction. Among the consequences of ROS production is the increased vascular tone, permeability, and in general, vascular endothelial dysfunction, which constitute therapeutic targets for CVD [reviewed in Ref. (89)], cancer (53, 135), and aging-related disorders (88).
Nei-like enzymes (NEIL)1-3 constitute part of the base excision repair mechanism of DNA and are particularly involved in the repair of oxidized base lesions, which are excessively generated in response to endogenous metabolic activities and oxidative stress (32). Interestingly, Peter Beal's group first reported that ADAR1 functionally regulates DNA repair by editing NEIL1 pre-mRNA (103, 156). Specifically, ADAR1-induced RNA editing causes an amino acid switch (K242R) harbored into the recognition loop of the enzyme. The authors further suggested that this recoding event renders the edited version of NEIL1 more efficient in removing a particular oxidized base lesion, namely guanidinohydantoin, while the unedited form preferentially removes thymine glycol oxidized base lesion. Nevertheless, it is still unclear how exactly RNA editing facilitates the altered properties of the enzyme and therefore further molecular and structural studies would deepen the understanding of this important mechanism.
Given that both forms of the enzyme, bearing distinct properties, are present in the cells (156), RNA editing can be considered as an endogenous on–off switch on NEIL1, which can be rapidly accessed in response to cell stimuli, thereby modulating DNA repair. Interestingly, Yeo et al. reported that NEIL1 RNA editing is significantly induced after IFN-α treatment of human glioblastoma cells due to the upregulation of the IFN-inducible isoform ADAR1 p150 (98). Along with a recent observation that ADAR1 levels are upregulated by H2O2 in neonatal cardiac myocytes (145), it would be tempting to hypothesize an imperative role for ADAR1 in oxidative stress cascade and aging-related disorders (Fig. 5) such as myocardial infarction. Nevertheless, future studies are called to consolidate this notion.
ADAR1-Induced RNA Editing in Neuronal Pathologies
To date, differential RNA editing levels have been associated with several neurological and neurodegenerative disorders (13, 50, 54, 78, 123, 138).
Editing of coding transcripts
One of the best studied edited transcripts, with a known role in neuronal biology, is 5-Hydroxytryptamine (serotonin) receptor 2C (HTR2C), a G-protein-coupled protein, which functions as a serotonin receptor (16). Interestingly, editing of two positions of HTR2C has been specifically attributed to ADAR1p110 (45, 101). As the edited form of this receptor exerts different biological properties, dysregulated A-to-I RNA editing levels of HTR2C have been linked to suicide (along with elevated ADAR1 levels in brain cortex) (93), mental disorders, Prader–Willi-like syndrome, and schizophrenia over the years (16, 57, 61, 87, 122) (Fig. 5). However, a complete functional characterization of the conserved ADAR1-dependent HTR2C edited sites is still pending.
Editing of dark matter
Due to their nature of forming inverted dsRNAs, Alu elements contribute to the formation of circRNAs in human subjects (136). Of note, ∼65,000 circRNAs are present in neuronal cell lines and in forebrain neurons of mice (110). Some of them are localized in the synapses, potentially contributing to synaptic plasticity and neuronal differentiation (110, 158). Alu region-based ADAR1-mediated RNA editing not only affects mRNA but also regulates circRNA biogenesis (56, 59, 110, 112, 136) (Fig. 4). Of note, Ivanov et al. reported that circRNAs are surrounded by Alu elements bearing multiple editing sites (56). Further corroborating this observation, another study showed that the number of circRNAs increases in the absence of ADAR1 (110). Thus, the two studies strengthened the hypothesis of the involvement of A-to-I RNA editing in circRNA biogenesis, which is currently under investigation [reviewed in Ref. (20)].
Considering that ADAR1 is well expressed in the forebrain, cerebral cortex, hippocampus, and diencephalon (58), ADAR1- RNA editing of Alu elements might suppress the biogenesis of circRNAs, thereby affecting the circRNA-dependent neuronal plasticity (158) (Fig. 5). Nonetheless, future investigations are required to elucidate how ADAR1-mediated A-to-I RNA editing orchestrates circRNA biogenesis and identify particularly influenced circRNAs, which are functionally involved in homeostasis and disease.
ADAR2-Induced RNA Editing in Neuronal Pathologies
The groundbreaking work of Seeburg and colleagues resulted in an outburst of studies pursuing the functional role of the neuro editor, ADAR2, in neuronal diseases (4, 5, 47, 48, 62, 100, 151 –153) (Fig. 5). Indeed, ADAR2 has been etiologically associated with amyotrophic lateral sclerosis (ALS) [recently reviewed in Ref. (150)], Alzheimer's disease (39), and epilepsia (67) by editing the Q/R site of GluRB subunit. Interestingly, Kwak's group achieved rescue of ALS manifested in motor neuron-restricted ADAR2 KO mice by targeted intravenous delivery of ADAR2 to motor neurons utilizing adeno-associated virus serotype 9 properties and synapsin I (SYNI) promoter for neuron-specific targeting (151). It would be tempting thus to envision that an application of the newly developed strategies, restoring the editing levels of GluRB in the central nervous system, would be a step forward toward personalized treatment of neurological diseases (44, 85, 86, 124, 140, 147).
Future Perspectives
The diverse functions of RNA editing observed in different tissue and disease contexts preclude the option of global ADAR manipulation strategies. Hence, a necessity of tissue/cell-specific targeting of ADARs/RNA editing arises. Such an approach will also shed light into which edited RNA species could be utilized as diagnostic, prognostic, or therapeutic targets in the near future, a question which still remains open. Toward this direction, the development of RNA-targeting CRISPR/Cas9 technologies (1) paves the way for RNA-specific therapeutic interventions.
In light of accumulating reports revealing the pivotal role of ADAR interactome in ADAR-dependent RNA metabolism (7, 97), future studies may be directed to provide a tissue/cell-specific ADAR interactome or potential cofactors, such as inositol hexakisphosphate—which Brenda Bass and colleagues previously reported for ADAR2 (80)—that will allow precise manipulation of RNA editing where required. Despite all the recent advances, there are major cellular processes, such as oxidative stress, whereby the contribution of RNA editing remains elusive.
Therefore, the dissection of the role of cell-specific RNA editing in RNA metabolism and cellular processes is an essential step toward a deeper understanding of the role of RNA editing in human physiology and disease. Taking into consideration that environmental stimuli, such as hypoxia and inflammation, regulate RNA editing, it is tempting to hypothesize that ROS may also control RNA editing or RNA editing may control the metabolism of transcripts that are critically involved in redox principles. Thus, future studies are needed to elucidate the role of adenosine-to-inosine RNA editing in redox biology.
Footnotes
Acknowledgments
The authors sincerely apologize to the authors whose work was not discussed in the present review due to space limitations. This work was funded by the ECCPS (Excellence Cluster Cardio-Pulmonary System) and the German Center of Cardiovascular Research (DZHK) to K.S.
Author Disclosure Statement
No competing financial interests exist.