
Abstract
Significance:
Various autoimmune syndromes are characterized by abnormalities found at the level of tissues and cells, as well as by microenvironmental influences, such as reactive oxygen species (ROS), that alter intracellular metabolism and protein expression. Moreover, the convergence of genetic, epigenetic, and even environmental influences can result in B and T lymphocyte autoimmunity and tissue pathology.
Recent Advances:
This review describes how oxidative stress to cells and tissues may alter post-translational protein modifications, both directly and indirectly, as well as potentially lead to aberrant gene expression. For example, it has been clearly observed in many systems how oxidative stress directly amplifies carbonyl protein modifications. However, ROS also lead to a number of nonenzymatic spontaneous modifications including deamidation and isoaspartate modification as well as to enzyme-mediated citrullination of self-proteins. ROS have direct effects on DNA methylation, leading to influences in gene expression, chromosome inactivation, and the silencing of genetic elements. Finally, ROS can alter many other cellular pathways, including the initiation of apoptosis and NETosis, triggering the release of modified intracellular autoantigens.
Critical Issues:
This review will detail specific post-translational protein modifications, the pathways that control autoimmunity to modified self-proteins, and how products of ROS may be important biomarkers of tissue pathogenesis.
Future Directions:
A clear understanding of the many pathways affected by ROS will lead to potential therapeutic manipulations to alter the onset and/or progression of autoimmune disease.
Introduction
A
Although not elaborated within this review, the sources of ROS at sites of tissue autoimmunity are many, including the invasion of activated immune phagocytic cells (neutrophils, macrophages, and dendritic cells) that are undeniably critical to the onset, progression, and tissue pathology of T1D as well as many other autoimmune syndromes. Though ROS-mediated cellular stress induces many protein post-translational modifications (PTMs) [reviewed in Ref. (121)], this work will focus on those specific pathways that are relevant in inflammatory autoimmune syndromes.
“Oxidative stress” can trigger direct modification of certain proteins, or protein motifs, or, alternatively, as secondary modifications due to indirect metabolic pathways affected by ROS. These secondary effects include the role of ROS in apoptosis, the generation of neutrophil extracellular traps (NETs; NETosis), and intracellular metabolic pathways, all of which may affect the outcome of autoimmune responses and/or inflammatory tissue pathology. Affected proteins may be altered in solubility, in their ability to be digested or cleared, or altered in immunogenicity. As illustrated in this review, oxidation can provoke a number of cellular changes at both the DNA and protein level, the latter of which includes both spontaneous and enzyme-mediated modifications to self proteins that are relevant biomarkers of tissue pathology and autoimmunity in T1D. Herein, we will describe elements of the initiation and progression of T1D immunity that are influenced by oxidative pathways, including various post-translational protein modifications, as well as the role of oxidation at the level of DNA transcription and translation.
Post-translational modifications of self-proteins in autoimmunity
One central role of the immune system is to differentiate responses of the “self” from the “non-self” proteome. A variety of regulatory pathways are in place to deplete the host of lymphocytes that respond vigorously to self-antigens that are present in secondary lymphoid organs, specifically the bone marrow and thymus. Although many self-proteins are found in the thymus (24), the PTMs of self-antigens (Tables 1 and 2) can create a novel autoantigenic proteome to which lymphocytes have never been exposed in either the thymus or peripheral lymphoid compartments. This concept, previously described as “autoantigenesis,” is a term described to proteins that “evolve” and acquire PTMs over the course of disease and provoke B and T cell autoimmunity leading to tissue pathology (31). This phenomenon is characteristic of many autoimmune diseases such as rheumatoid arthritis (RA), multiple sclerosis (MS), systemic lupus erythematosus (SLE), and T1D (Table 1) (30, 31). The PTM autoantigens illustrated in Tables 1 and 2 represent many of the notable modified self-proteins that trigger autoreactive B cell and T cell responses and, in many cases, are specific diagnostic biomarkers as well as a reflection of disease pathology. Other PTMs (Figs. 1 and 2) can be directly affected by tissue ROS and/or inflammatory microenvironments (carbonylation, methylation, isoaspartylation, deamidation), or be influenced by more indirect downstream pathways affected by ROS (acetylation, glycosylation, phosphorylation, citrullination).
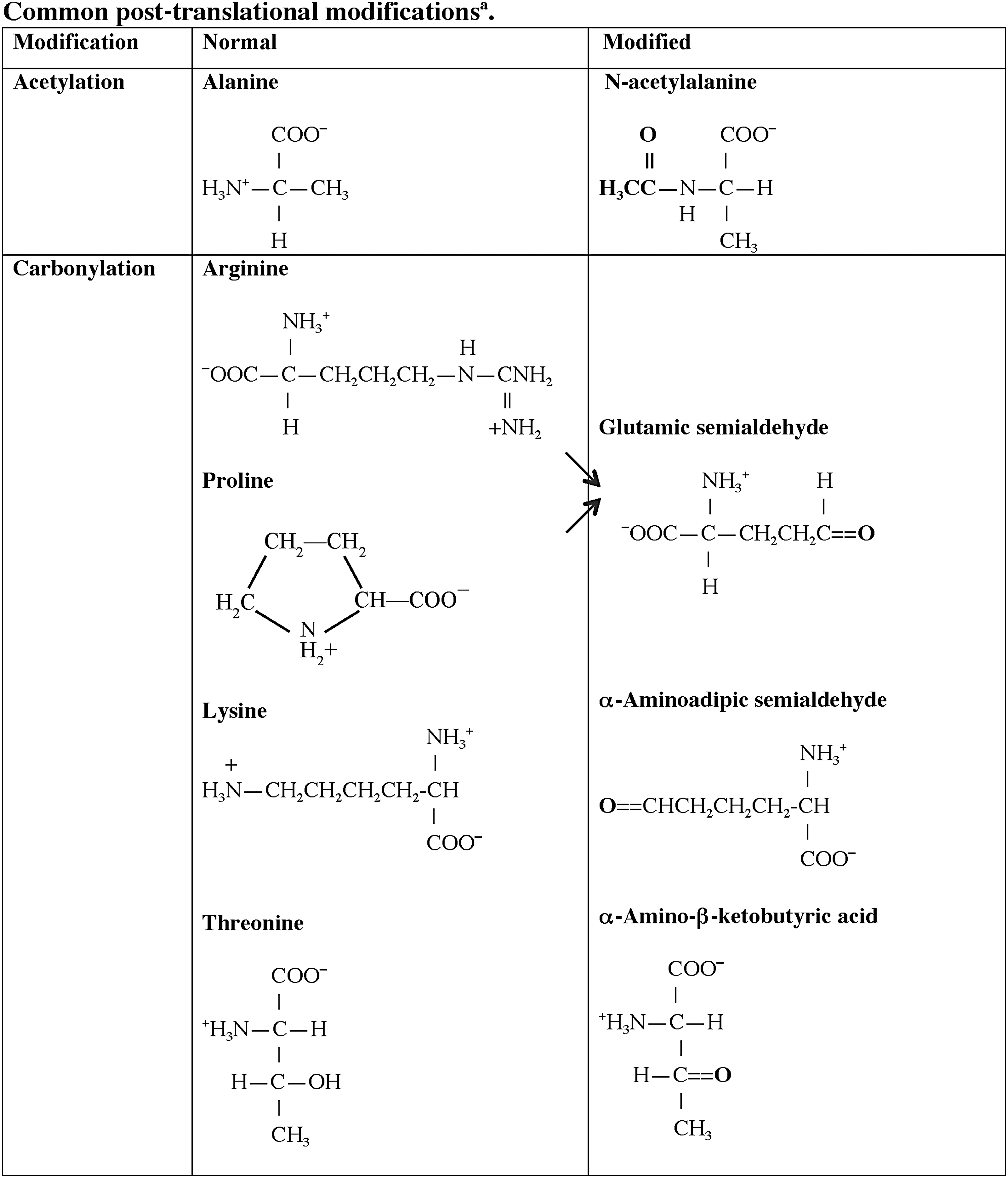
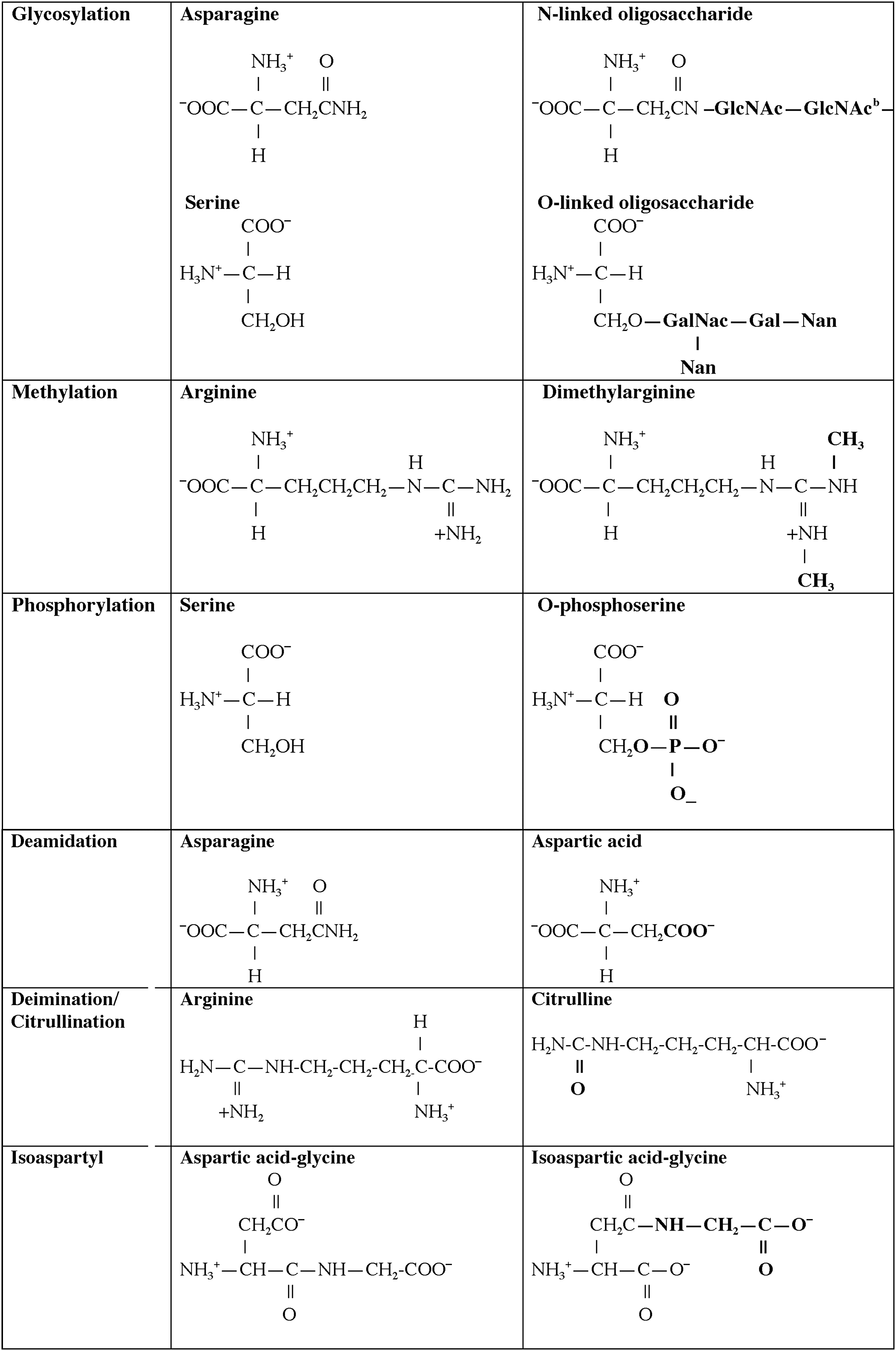
A1T1, alpha 1 anti-trypsin; CapZα1, F-actin capping protein alpha-1 subunit; CIA, collagen-induced arthritis; EAE, experimental autoimmune encephalomyelitis; GAD65, glutamic acid decarboxylase 65; GRP78, glucose-regulated protein 78; LDL, low-density lipoproteins; MBP, myelin basic protein; MS, multiple sclerosis; P4Hb, prolyl-4-hydroxylase beta; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus; T1D, type 1 diabetes.
IA-2, islet antigen-2; IAPP, islet amyloid polypeptide; ICA69, islet cell autoantigen 69; IGRP, islet-specific glucose-6-phosphatase catalytic subunit-related protein; SERCA2a, sarco/endoplasmic reticulum Ca2+ATPase; ZnT8, zinc transporter 8.
Citrullination
One well-studied illustration of autoimmunity to a post-translationally modified self-antigen are the autoantibodies arising to citrulline-modified proteins, notably in RA. Citrulline is a product of peptidylarginine deiminase (PAD) activity on arginine residues within peptides (8). More importantly, in the case of RA, PADs (PAD1, PAD2, PAD3, PAD4) are activated by Ca2+ ion deposition within an inflamed joint (78, 142) provoking synthesis of elevated levels of citrullinated self-proteins. Sera from patients with RA recognize many citrullinated autoantigens, including collagen, vimentin, fillagrin, and α-enolase (62, 141). The early onset period of RA is marked by anti-citrulline autoantibodies that reflect disease severity (62, 141). Anti-citrulline autoantibody titers are now routinely used for the diagnosis of RA (142). In particular, circulating citrulline, arising from arginine modification, is a byproduct in the synthesis of nitric oxide, itself, a product of reactive nitrogen species (41, 85, 150). As detailed herein (Fig. 1 and Tables 1 and 2) and in accompanying monographs of this issue, B and T cell responses directed at PTMs have become important diagnostic tools for many autoimmune diseases, including an emerging group of PTM biomarkers that are important in T1D (Table 2). A recent review from Nguyen and James (105) has carefully defined the biological implications of citrulline modifications and autoimmunity arising from pancreatic beta cell proteins in the development of T1D. In particular, both citrullinated glutamic acid decarboxylase 65 (GAD65) and glucose-regulated protein 78 (GRP78) elicit a vigorous B and T cell autoimmune response in human T1D and nonobese diabetic (NOD) mouse murine disease, respectively (92, 116). The latter studies were marked by a dramatic and aberrant upregulation of PADI2 in the islets of NOD mice, supported by genetic risk in the Idd25 locus of mouse chromosome 4. These citrulline modifications of T1D autoantigens were linked to cytokine and/or ROS stress effects on the endoplasmic reticulum, a recurrent theme in fostering many PTMs.
Carbonyl PTMs
Protein carbonylation is a major product of tissue proteins in response to oxidative stress. Increased carbonylation of self-proteins arises due to decreases in antioxidant defense pathways, increases in ROS production, or an inability to remove or repair oxidized self-proteins, as previously reviewed by Nystrom (106). The classical antioxidant (ROS) defense mechanisms, superoxide dismutase, catalases, and peroxidases, all protect against the induction of carbonyl modifications. Carbonylation is a metal-catalyzed (free iron) oxidative modification of the side chains of proline, arginine threonine, and lysine (Fig. 1). Carbonyl modifications are typically more difficult to induce relative to other oxidative modifications. This modification has many deleterious effects on various intracellular enzymatic mechanisms, and mitochondria are particularly vulnerable to ROS-induced carbonylation (106). Carbonylation is a marker of cellular senescence and is increased in aging cells and tissues. There is evidence to suggest that carbonylation is one protective mechanism of cells for directing damaged proteins into proteolytic degradation pathways, as these modified proteins are conformationally unstable. Carbonylation is an irreversible modification; thus, the biological functions of these modified proteins are unrepairable (32). All of these properties make carbonylated self-proteins recognized in an autoimmune response, particularly if they fail to find their way to normal cellular degradation pathways.
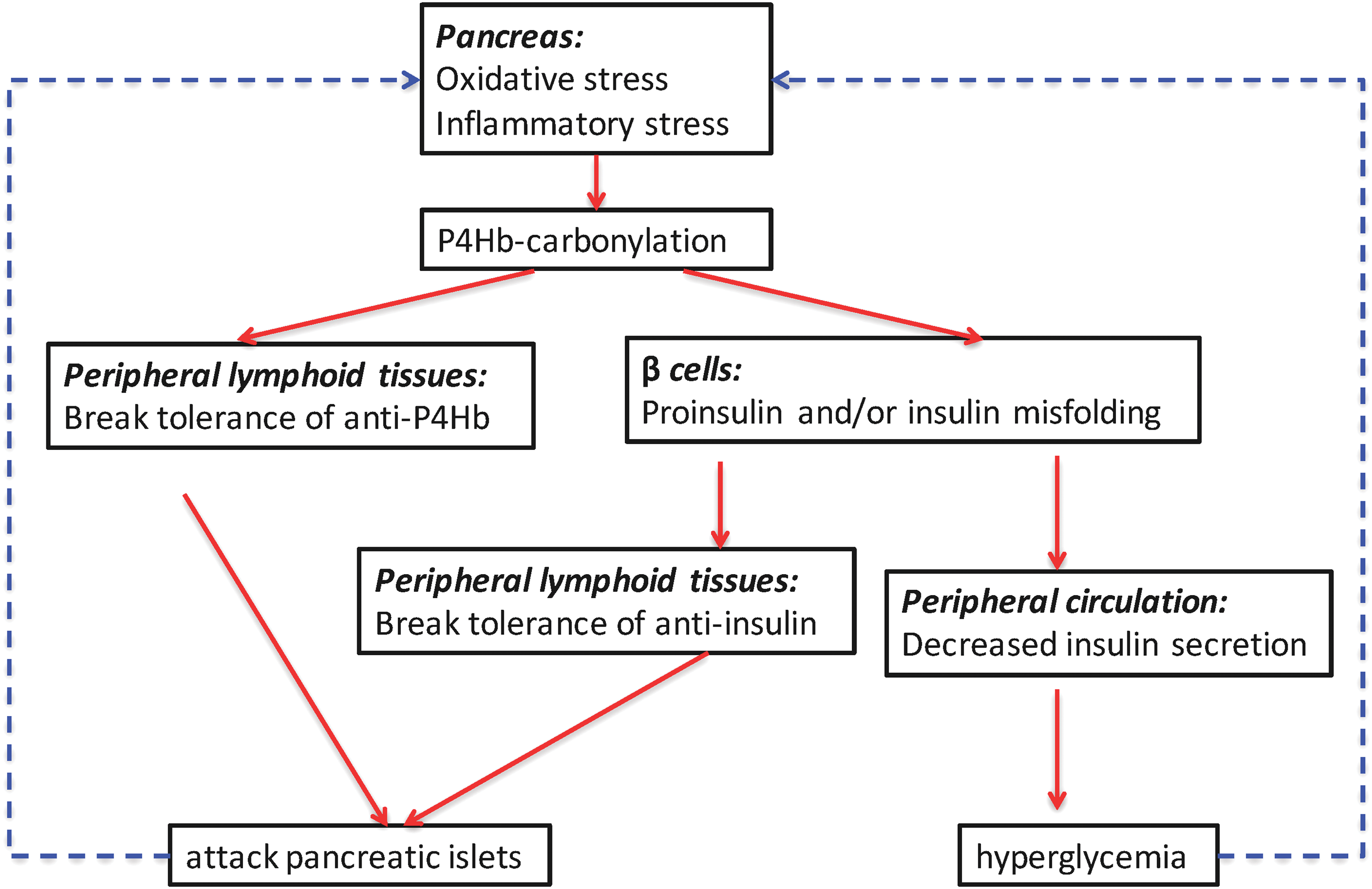
It has been observed that increases in ROS contribute to insulin resistance and metabolic dysfunctions in adipose tissue of both animal models and human type 2 diabetes (T2D) (36, 119, 124). Although publications have profiled carbonylated plasma proteins as potential biomarkers in T2D (35, 42), little is known about carbonylation in the progression of T1D. Our recently submitted studies from Yang et al. defined a group of pancreatic beta cell proteins with carbonyl PTMs, all bound by autoantibodies from human and NOD mice T1D antisera. Among this group were both novel and established biomarkers of T1D, including protein disulfide isomerase (PDI) isoforms, 14-3-3 protein isoforms, GRP78, chymotrypsinogen B, and malate dehydrogenase. Of interest, carbonylated prolyl-4-hydroxylase beta (P4Hb, also known as protein disulfide isomerase A1; PDIA1) was found to be an early autoantigen in both human and murine models of T1D. P4Hb is essential for the appropriate folding of insulin from pancreatic beta cells (112). Our data suggest a novel role of modified P4Hb, both as an early target of autoimmunity and as a pathway that provokes autoimmunity to insulin and/or proinsulin. In fact, autoimmunity to P4Hb always preceded autoimmunity to insulin in both NOD mice and human T1D, as defined by the pathway described in Figure 3. Carbonylation is amplified by either oxidative or cytokine stress to beta cells. Carbonylated P4Hb fails in its ability to accurately fold and process proinsulin to insulin. We hypothesize that misfolded proinsulin levels accumulate in the cell or serum and lead to linked autoimmune responses to insulin itself. Aberrant carbonylated P4Hb biological functions in the beta cell provide an explanation for recent observations of increased proinsulin to insulin ratios in the progression of T1D (115).
ROS and PTMs in the development of NETs
NETosis is a neutrophil destruction mechanism differing from the apoptotic pathway, initiated when neutrophils confront microorganisms, and generating chromatin webs from intracellular contents (22). The extruded DNA webs carry a number of bound bactericidal proteins (lactoferrin, elastase, proteinase 3, myeloperoxidase, cathepsin G, etc.) as well as histones and granule proteins. This pathway is marked by the release of mitochondrial DNA, a process dependent on cellular stress created by ROS, superoxide anion, hydrogen peroxide, myeloperoxidase, and NADPH oxidase. Patients who are deficient in myeloperoxidase cannot generate NETs. The mitochondrial electron transport chain is one source of intracellular ROS in neutrophils. With relevance to autoimmune responses, NETs are believed to be one source of immunogenic histone H2B. A study by Liu et al. (75) identified the PTMs originating from the histones of NETs. Autoantibodies were elicited to citrullinated forms of histone H3 and H4 and to forms of histone H2B that were either methylated or acetylated. The two pathways of cellular destruction, either apoptosis or NETosis, trigger unique PTMs, all of which may break immune tolerance to modified self-proteins. The role of NETs in T1D has yet to be fully resolved; one recent study illustrated a reduction in serum components of NETs (proteinase 3 and neutrophil elastase), consistent with a reduced overall neutrophil count in early onset T1D (111). Conflicting studies report increases in these same NET components in T1D (139).
A large number of PTM cytoplasmic proteins are the autoantigenic targets of autoantibodies in SLE (Table 1). Tissue pathology, inflammatory cytokines, and ROS create intracellular metabolic stresses that favor the generation of PTMs, including carbonyl modifications of self-proteins as detailed herein (31, 32). Pathways designed to clear inflammatory material (NETosis and apoptosis) may drive the PTM of self-antigens, being viewed as “foreign” to the immune system and are amplified by ROS. Although ROS are important in protecting cells from infectious agents (such as bacteria), ROS also induce aberrant PTMs within self-proteins that may be immunogenic in the host. The abnormal clearance of apoptotic or NETotic cellular debris is believed to be a principal source of autoantigens in lupus (46, 47, 65, 127). As noted earlier, NETs are enriched in various PTMs of self-proteins, including citrullination (58). In addition, specific lupus autoantigens, including nucleosome dsDNA, snRNPs, Ro/SSA, and La/SSB, are found within cell surface blebs in apoptosis (13), a reservoir of modified proteins. In addition, PTMs such as transglutamination, phosphorylation, proteolysis, and adenosine diphosphate-ribosylation are amplified in apoptosis (129). Notably, phosphorylated SR proteins (also known as pre-mRNA splicing factors) are targets of human SLE autoimmunity (104).
Isoaspartyl modification, the consequence of spontaneous, nonenzymatic isomerization of aspartic acid, is amplified in the presence of ROS as well as in cells that undergo necrosis and/or apoptosis (Fig. 1 and details below) (16, 19). Both lupus-prone MRL mice and human SLE patients have elevated titers of autoantibodies that react with isoAsp-modified histone H2B (27). Similarly, symmetric dimethyl arginines found in the C-terminus of the snRNP complex are amplified by ROS (12).
PTMs can trigger more extensive autoimmunity, such as epitope spreading
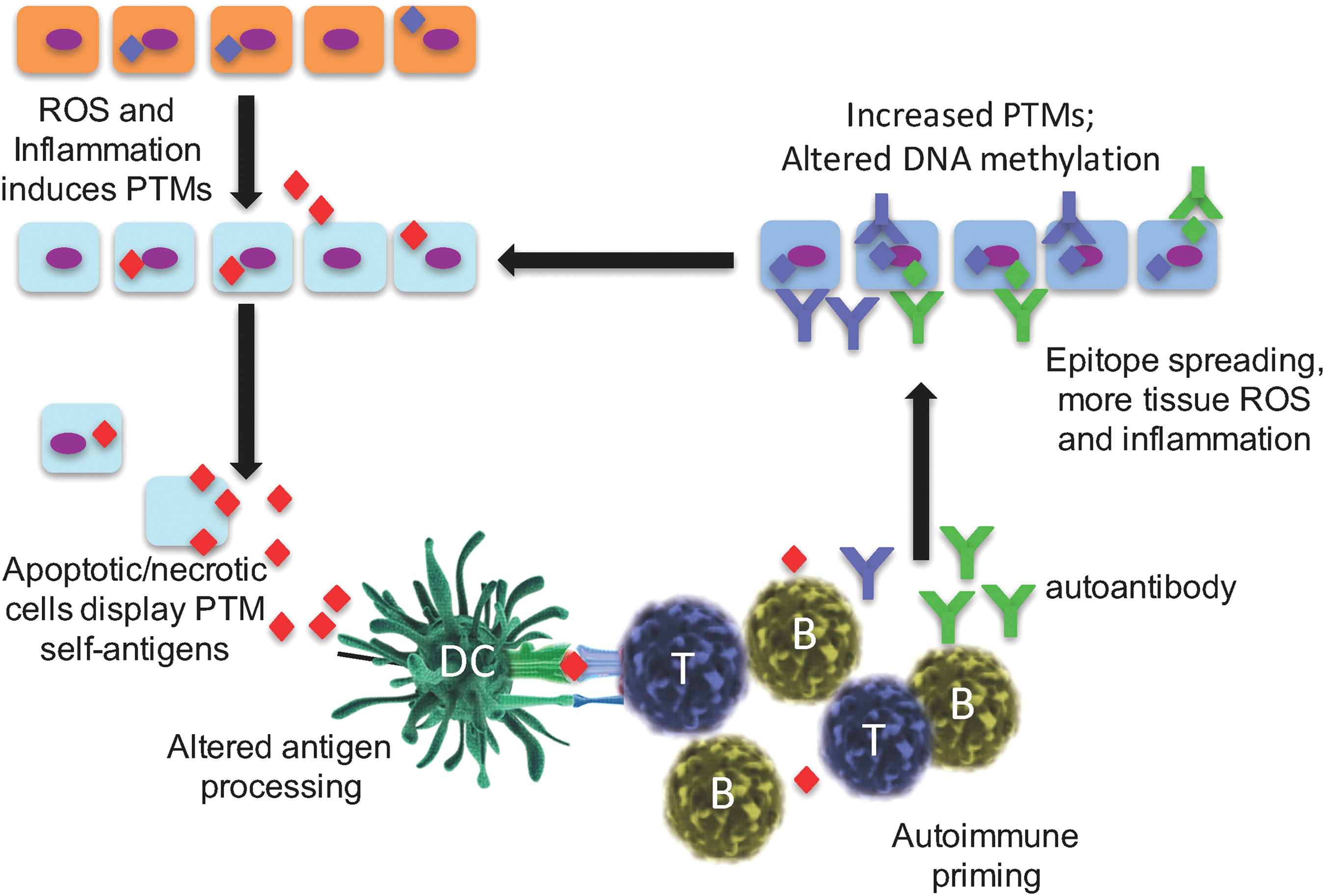
We and others have identified significant differences between the presence and specificity of T cell and B cell immunity that arise in response to ROS stress-induced PTM self-proteins (27 –31, 39, 45, 82, 87, 92). In particular, T cell responses to PTM determinants are most often specific for the modified peptide form only and do not bind the native (unmodified) protein or peptide. This concept is demonstrated in mice immunized with the isoAsp snRNP D where T lymphocytes bind only to the isoAsp-modified snRNP D peptide and are unresponsive to the native aspartic acid form of peptide (82). T cell autoimmunity to PTM self-proteins will be more fully addressed by other authors in this issue of Antioxidants and Redox Signaling. In contrast, B lymphocyte and autoantibody responses are typically promiscuous in their binding to both the modified and native self-protein. However, tolerance is often not only broken to the PTM-modified self-protein but also exhibits cross-binding to the native protein. This phenomenon may be due to the ability of antibodies to bind amino acid sequences adjacent to the PTM that is present in both modified and native protein sequences. As one example, human SLE and lupus-prone MRL/lpr mice have autoantibodies that are cross-reactive to both isoAsp and aspartic acid forms of histone H2B (27). It was found that autoimmunity is initiated with the PTM self-protein/peptide, and thereafter spreads in an intra- and extra-molecular manner to other determinants. Thus, the presence of a PTM in a self-protein amplifies “epitope spreading,” a mechanism in which the immunity amplifies to include determinants outside the site(s) that trigger the original immune response (Fig. 2) (70). As a consequence, there is both intra- and inter-molecular B and T cell epitope spreading to self-antigens in T1D, SLE, and MS (59, 82). Epitope spreading is associated with the development, progression, and severity of autoimmune disease (3). One favored mechanism is that modified self-antigens are recognized as foreign, thus triggering a restricted autoimmune response, even before the appearance of symptoms, followed by the subsequent accumulation of additional targeted determinants (Fig. 2). As noted earlier, necrotic and apoptotic cells are early sources of altered self-proteins and amplified by tissue stress, notably stress from ROS (38) or altered pH (39).
PTMs in antigen processing and presentation
Autoimmune responses develop to modified self-proteins due to defects in the negative selection of immune cells. It is clear that accurate antigen processing plays a major role in peptides that control negative selection (84). However, the presence of a PTM residue that is critical for cleavage changes the specificity of proteases and the types and sequences of self-peptides generated and, ultimately, presented to the immune system (Fig. 4). The presence of specific PTMs within a processed peptide also affects the binding to major histocompatibility complex (MHC). As one example, the absence of N-glycosylation of glutamate receptor subunit 3 in Rasmussen's encephalitis reveals sites that are susceptible to granzyme B cleavage, thereby creating a novel autoantigen (neoepitope) (37). Many proteases fail to recognize the β-peptide isoform between isoAsp residues and the adjacent carboxyl side amino acids (56). In addition, studies illustrated that proteins are resistant to asparagine endopeptidase cleavage as a result of the spontaneous deamidation of asparagine residues (100). Simply put, the proteolytic enzyme recognition of PTM self-proteins may generate novel repertoires of peptides during antigen presentation and the establishment of immune tolerance (Fig. 4). These observations were confirmed several years ago in studies of model proteins in immunity (79 –82), now confirmed by more recent work with disease-relevant PTM autoantigens. Cathepsin D, an enzyme in the antigen processing pathway, differentially cleaves substrate proteins based on the presence or absence of isoaspartyl protein modification (29). This phenomenon is also supported by the cleavage products of Granzyme B, again altered by the presence of post-translationally modified self-protein substrates, leading to neo-epitope presentation (14).

The biochemical processing within acidified lysosomal compartments of antigen-presenting cells (APCs) dictates the types of short peptides subsequently presented on both class I and class II MHC. Interesting studies by Ireland et al. (54) found that citrulline-modified peptides were generated, processed, and presented to CD4 T cells only in B cells that undergo autophagy. The presentation of unmodified peptides was unchanged. Thus, it was the autophagy pathway itself that presumably created the citrulline PTM that was subsequently presented in the priming of CD4 T cells. Our laboratory and others have demonstrated the unique ability of B lymphocytes as APCs in presenting specific self-peptides to T cells. For example, B cells transfer processed antigens to other APCs, including macrophages and dendritic cells (43, 44, 113). Thus, different APC subsets themselves may control the determinants generated and eventually presented by the immune system (20, 100).
Algorithms that predict motifs of how particular peptides will associate with MHC do not incorporate the presence of peptide PTMs. The “fit” of the PTM peptide for MHC differs significantly from native (unmodified) peptide. The MHC binding of some post-translationally modified T1D autoantigens has been studied and reviewed by McGinty et al. (91, 92). Also, different modifications of myelin basic protein (MBP) change the affinity of peptides for MHC compared with the corresponding wild-type peptide (21). Acetylated MBP peptide (Ac 1–11) stimulates pathogenic T cells in murine MS, though the unmodified peptide binds MHC with virtually identical kinetics. Similarly, isoaspartic acid residues in cytochrome c or snRNP D peptides bind MHC class II in a manner that is identical to the unmodified peptides (82), yet immune tolerance is maintained only to the native peptide. Alternatively, citrullinated-modified peptides of RA autoantigens, specifically vimentin, bind the high-risk allele, human leukocyte antigen (HLA)-DRB1*0401 greater than the native (unmodified) peptide (49). The overall message from these collective studies is that PTM self-peptides may or may not be processed and bound by MHC in a manner found with unmodified (native) peptide. Moreover, it cannot be predicted whether T cell receptors (TCRs) and B cell receptors will bind PTM self-peptides or proteins and cross-react with the corresponding unmodified antigen.
ROS alter the methylation of DNA and protein
Another emerging PTM of significance is intracellular methylation modification, a process now known to be essential in the normal functions of all immune cells. The enzymatic addition of methyl groups to substrates (including proteins, lipids, and DNA) occurs via the single methyl donor in cells, S-adenosyl-methionine (SAM). The family of methyltransferases includes both protein methyltransferases (PRMTs) and DNA methyltransferases (DNMTs). ROS alter all of these methylation pathways (10, 99, 145). For example, oxidation of DNA is required for DNA methylation pathways by ten-eleven translocations (TETs) (reviewed in Ref. 10).
The most well-studied transmethylation substrates in regulating epigenetics are histone proteins and, of course, DNA. However, other substrates of protein methylation are key regulators in a diverse array of cellular pathways, including lymphocyte signaling and differentiation pathways and the regulation of transcription. In fact, lymphocytes require accurate protein methylation during cellular activation compared with most other cell types (Table 3) (40). Protein methylation is required for efficient TCR signaling, and in the regulation of cytokine responses (including interferon [IFN]α/β-induced transcription via STAT1; Table 3). T cell growth and development relies on the accuracy of histone methylation and promotor region CpG dinucleotide sequences. Inhibition of protein arginine methyltransferase (PRMT) alters the ability of phosphorylated signal transducers and activators of transcription (STAT) in binding to DNA (103). Moreover, PRMT activity and Vav1 methylation is increased in T cells on CD28 costimulation (9). The recurring themes of this review are the effects of ROS in these various, and sometimes indirect, biological pathways. Recent studies have demonstrated that PRMT1, a major protein methyltransferase in humans, is downregulated by ROS in the microenvironment, causing the release of asymmetric dimethylarginine into the serum, a biomarker of tissue pathology (99).
CARM1, coactivator-associated arginine methyltransferase 1; NFAT, nuclear factor of activated T cells; PRMT, protein arginine methyltransferase.
Autoimmune disease is marked by abnormal methylation modifications, both protein and DNA. Defects in the methylation status of CpG dinucleotide elicit T cell autoimmunity, contributing to the pathogenesis of SLE (123). As mentioned earlier, symmetric dimethylated ribonucleoproteins, snRNP proteins D1 and D3, are autoantigens recognized by autoantibodies in SLE patients (12). Similarly, arginine-methylated MBP triggers the autoimmunity, in both the B and T cell compartments, in MS (110).
In T1D, it is attractive to hypothesize that DNA and/or protein methylation pathways are influenced by ROS as they are by inflammatory cytokines (IL-1β, IFNγ, or IL-6) in the pancreatic islet microenvironment (118). From the time that autoantibodies are first detected in individuals at genetic risk for T1D, the course of disease is highly variable (132). Many individuals do not progress to overt disease, and those who do may progress over different periods ranging from a few months to decades. This variability has largely focused on the immune effector and regulatory cells that are involved in β cell killing and maintenance of tolerance (11, 90). However, it is equally likely that β cells respond to the immune attack and environmental factors such as ROS in ways that may accelerate or retard disease progression (Fig. 5). In addition to the modification of these proteins, there may also be modifications of the enzymes responsible for the epigenetic changes themselves. TET2 was reported to be modified by acetylation during oxidative stress, and DNMT3a has been reported to be SUMOylated, which modulates its repression of transcription (74, 152). DNMT3a is known to control histone deacetylase (HDAC) gene expression and in the setting of inflammation, TET2 physically associates with certain HDACs (23). TET2 is required to resolve inflammation due to IL6 (72).

Rui et al. have studied individuals who were at very high risk for diabetes by virtue of finding two or more positive autoantibodies and dysglycemia (117, 118). Historically, ∼75% of these subjects progress to overt diabetes within 5 years. These studies measured the relative levels of unmethylated insulin INS DNA (released from dying β cells) in the serum compared with the amount of methylated INS DNA. It was found that the frequency of increased levels of unmethylated INS DNA measurements (taken about every 6 months) was low in the individuals at risk, and not significantly different in progressors and nonprogressors to diabetes. However, the frequency of elevated levels of unmethylated INS DNA was significantly higher in the very high-risk subjects, suggesting increased β cell killing in the prediagnosis period. These observations, and others from clinical studies of β cell function and pathology, have refined the kinetics of T1D progression, as originally reported by Eisenbarth in 1986.
In summary, studies of β cells during the progression of T1D in NOD mice and human β cells in vitro indicate that there is induction of DNMTs as well as PTMs that can remodel or affect survival of β cells (117, 118). An underlying theme to these studies is that the processes of epigenetic protein modifications are interconnected, possibly both in response to inflammatory mediators or more directly in which one process modifies the others. An overview of the converging pathways is illustrated in Figure 5.
Protein arginine methyltransferase
The first PRMT enzyme was identified, cloned, and characterized in 1996 (73). In the past 20 years, an additional 11 PRMT enzymes, with varying biological substrate activity, have been discovered, but all still utilizing SAM as the sole intracellular methyl donor. All of the PRMTs will methylate in a unique sequence pattern, based around glycine-arginine-rich domains. Four classes of PRMTs exist, depending on the unique methylation products that are catalyzed (Table 3). With relevance to autoimmunity, type I PRMTs catalyze asymmetric di-methylarginine modification and type II PRMTs will form symmetric di-methylarginine, both of which are known antigenic targets of lupus autoimmunity, notably the snRNP D protein (7). Other PRMT family proteins, described later, control many aspects of immune cell differentiation and function (Table 3).
The first well-studied methyltransferase, PRMT1, catalyzes more than 85% of arginine methylation within cells (7). PRMT1 regulates the production of T cell cytokines, both IL-2 and IL-4, via the methylation of Vav1, STAT1/6, and Akt/PKB in the T cell signaling pathway (9, 94, 103, 109) and by the regulation of transcription via nuclear factor of activated T cells (NFAT) (102). B lymphocyte differentiation is regulated by PI3K kinase activity that is optimized by PRMT1-mediated methylation. It is now clear that PRMT1-directed methylation of histones H4R2, H3, and H4 mediates DNA transcriptional activation (51, 71, 137). Yet other histone PTMs, including acetylation, in T lymphocytes are linked with disease flares in human SLE (50) whereas Th17 cell differentiation is regulated by PRMT2-mediated methylation (133). Reduced cellular methylation due to the loss of PRMT4 (coactivator-associated arginine methyltransferase 1 [CARM1]) alters normal thymocyte differentiation, chromatin remodeling, and possibly tolerance to self proteins (2). Finally, alterations in the SAM- and/or PRMT-mediated methylation pathways influence IL-2, IL-4, and IFNγ gene expression from T lymphocytes.
As detailed herein, ROS-mediated PTM within T cells alters the production of inflammatory cytokines that contributes to the onset of autoimmunity. Human SLE is marked by aberrant TCR signaling that is caused, in part, by an increase in phosphorylation of ERK, Syk, ZAP-70, and PI3K kinase activity (18, 101). As illustrated here, ROS may initiate a chain reaction of downstream effects of methylation and phosphorylation leading to aberrant T cell biology. The overall implications are that ROS-mediated alterations in PRMT methylation of histones contribute to increases in the serum levels of T lymphocyte products, including IFNγ, IL-4, and IL-17, inflammatory mediators in human SLE (1, 128) (Table 3).
Repair of PTMs induced by ROS
Isoaspartyl PTMs are created by a number of cellular stresses, including ROS. The efficient repair of cellular isoaspartyl PTMs is by the enzyme known as protein
In the murine model of human SLE, the MRL mouse experiences increased isoaspartyl PTM proteins in the kidney and brain, two sites of immune complex-mediated pathology (147). Increased isoaspartyl protein levels lead to T cell hyperproliferation, a phenotype of both human and murine SLE (28). We have recently identified the presence of isoAsp PTMs within several residues of the T cell signaling protein, ZAP70 (unpublished data), likely contributing to the aberrant T cell hyperproliferative phenotype in SLE.
Regarding T1D, the PIMT isoaspartyl repair enzyme is highly expressed in pancreatic beta cells and in the transformed insulinoma cell line, INS-1. Interestingly, the induction of PIMT expression delays the appearance of symptoms and reduces the severity of T1D in the BB rat model of T1D (135). The PIMT1 gene maps to a region, 6q24–25, linked to the IDDM5 site studied in genome analyses of human T1D (136). Four PIMT1 polymorphisms were found to be in linkage disequilibrium, with PCMT1 promotor activity increased in response to cytokine stimulation. Collectively, the work supports an interaction between PCMT1 and both HLA and SUM04 in the genetic risk for T1D. As yet, however, specific PIMT polymorphisms already defined to exist in humans (17, 25, 26) have not yet been clearly associated with any autoimmune syndrome. Moreover, the expression of PIMT protects from Bax-induced cellular apoptosis, perhaps yet another mechanism that evolved to prevent the release of isoaspartyl-modified self-proteins (52). We have recently observed significant increases in cellular isoaspartyl levels in the islets of NOD diabetogenic mice as well as in human pancreatic islets treated with physiologic levels of H2O2 or inflammatory cytokines (Yang and Mamula, unpublished data). The emerging picture is that ROS and/or cytokine-induced inflammation of tissues trigger various PTM pathways, followed by protection mechanisms initiated by the cell to both prevent its destruction and repair aberrant self-proteins. Unfortunately, the inflammatory storm found in autoimmune disease is often too vigorous to be impeded by these protective intracellular mechanisms. Further studies will be required to fully appreciate both the direct effects of ROS and more indirect pathways altered by ROS in T1D.
ROS, DNA methylation, and epigenetics in autoimmunity
Epigenetics refers to gene expression that is influenced and/or controlled by various modifications that arise during the transcription and translation of genetic loci (77). These epigenetic modifications collectively include DNA and histone methylation, histone acetylation/deacetylation, and nucleosome remodeling. As detailed earlier, post-translationally modified proteins, many of which are altered by ROS, are linked to alterations in DNA methylation. One study revealed that overall hypomethylation of DNA, in particular, methyl cytosine residues, is more frequent in SLE as compared with healthy individuals (5).
Relevant to human T1D, Rui et al. reported epigenetic modifications of β cells during progression of diabetes in NOD mice, the murine model of T1D (118). The study demonstrated specific methylation of exons in Ins1 and the promoter and exons of the Ins2 gene. More careful analyses revealed an inverse relationship between methylation states of Ins2 Exon1 and Ins2 mRNA levels in β cells. The study observed an induction of DNMT3a during disease progression, which was shown to account for the epigenetic changes by siRNA silencing. The progression of islet pathology was coincident with the induction of DNMT3a and the methylation of Ins2 DNA in response to inflammatory cytokines (IL-1β, IL-6, and IFNγ). DNMTs are critically involved in β cell differentiation, and subsequent studies suggested that there may be cellular changes resulting from the inflammation (cytokines or ROS) that may result from epigenetic modifications (117). A subpopulation of dedifferentiated β cells in the islets of NOD mice develop during progression of disease, noted by the loss of normal differentiation features of β cells and the acquisition of stem-like characteristics. The novel β cell subpopulation showed increased frequency of methylation of CpG sites in the Ins genes compared with normal β cells (unpublished). These β cells were resistant to immunologic killing, suggesting that the mechanism of epigenetic modification to inflammatory mediators may represent a cellular protective response.
In support of the studies in T1D described earlier, hypomethylation of DNA is a biological feature and profiling characteristic of a number of autoimmune maladies, including MS, Grave's disease, mixed connective tissue disease, ankylosing spondylitis Addison's disease, cancer, and Alzheimer's disease (48).
Concluding Remarks
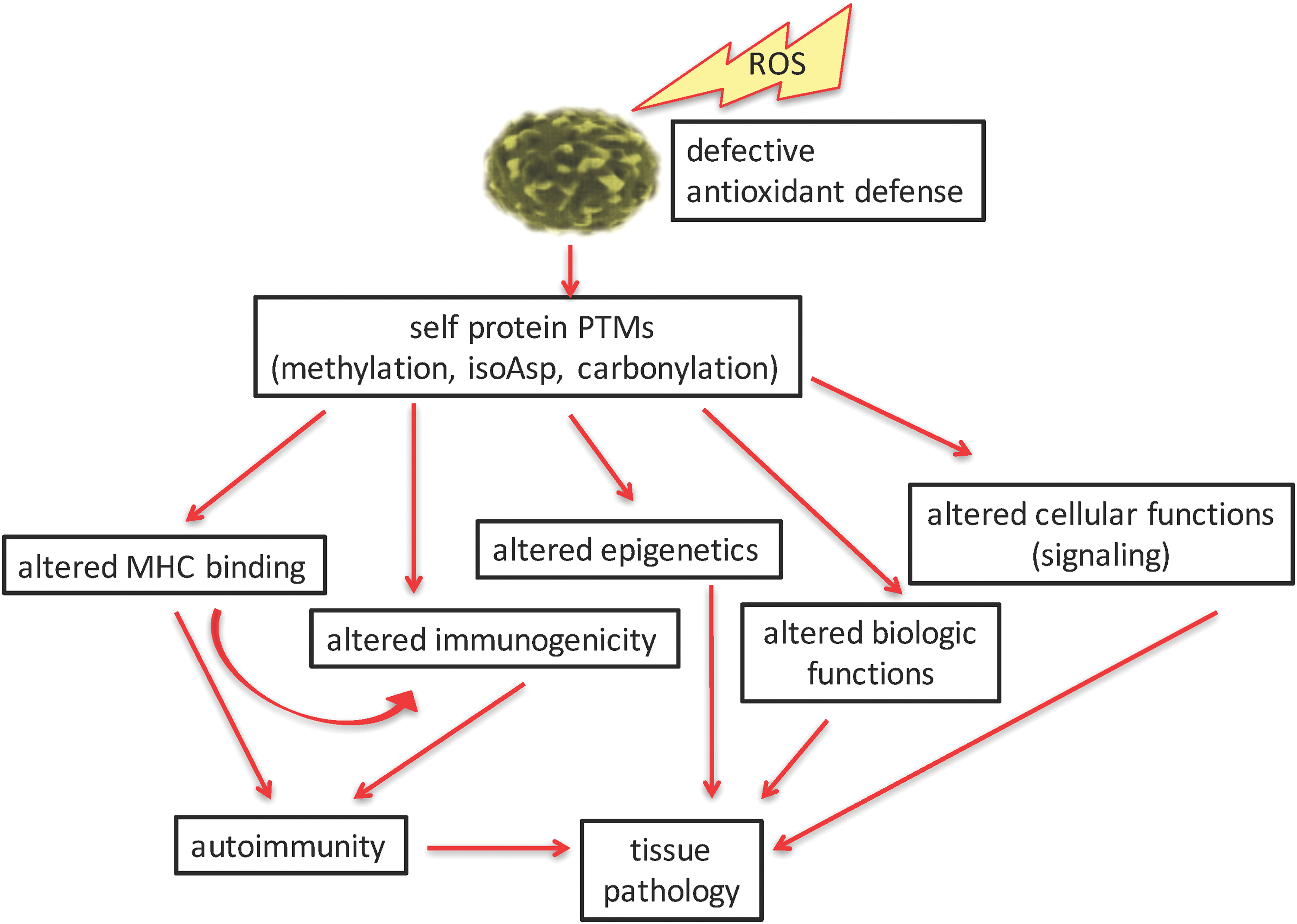
We have illustrated several specific ROS-influenced pathways that influence the onset and progression of autoimmunity (Fig. 6). Many of the PTMs and antigen processing pathways described herein are spontaneous in nature and beyond the prediction of genetics. It is now obvious that many PTMs and cellular pathways are affected by ROS. Indeed, it may appear that ROS are at the “center of the universe” of triggering autoimmunity. ROS trigger several PTMs of self-proteins in direct ways, such as with carbonyl or isoaspartyl modifications. Indirect pathways are affected by ROS, as with methylation of DNA or proteins and with amplifying enzyme-mediated PTMs (citrullination) and by triggering PTM repair mechanisms (PIMT). Although we have defined some specific PTMs in T1D, SLE, and many other autoimmune syndromes, this dynamic and quickly changing field does not allow us to enumerate and detail all published PTM autoantigens. Described herein, ROS-amplified PTMs lead to alterations in self-peptide binding to MHC, defects in immune tolerance induction, altered or defective biological functions of self proteins, changes in epigenetics, and even changes in cellular metabolic pathways (such as signaling). The end product of these pathways is the development of autoimmune-mediated tissue pathology (Fig. 6). The fine specificity of B and T cell autoimmunity, such as T cell subset analyses, and pathological responses to PTM self-proteins are detailed within other manuscripts of this volume of Antioxidants and Redox Signaling. Studies by Piganelli and colleagues (reviewed in this volume) have carefully defined the role of inflammatory and oxidated stress in pancreatic tissue and the endoplasmic reticulum in the course of T1D autoimmunity (86, 87). The emerging technologies in proteomics and tissue analyses will undoubtedly change the landscape of this field in the coming months and years. These analyses have already “modified” how the clinical community diagnoses and assesses the progression of disease, and tissue pathology. With the identification of specific biomarkers and an understanding of their origins, the field will now have potential therapeutic pathways as targets to modify these autoimmune diseases.
Footnotes
Acknowledgments
This work was supported by National Institutes of Health (AR-41032 and AI-48120 and), the Lupus Research Alliance, and the Juvenile Diabetes Research Foundation to M.J.M.