
Abstract
Significance:
It has been proposed that cancer cells are heavily dependent on their antioxidant defenses for survival and growth. Peroxiredoxins are a family of abundant thiol-dependent peroxidases that break down hydrogen peroxide, and they have a central role in the maintenance and response of cells to alterations in redox homeostasis. As such, they are potential targets for disrupting tumor growth.
Recent Advances:
Genetic disruption of peroxiredoxin expression in mice leads to an increased incidence of neoplastic disease, consistent with a role for peroxiredoxins in protecting genomic integrity. In contrast, many human tumors display increased levels of peroxiredoxin expression, suggesting that strengthened antioxidant defenses provide a survival advantage for tumor progression. Peroxiredoxin inhibitors are being developed and explored as therapeutic agents in different cancer models.
Critical Issues:
It is important to complement peroxiredoxin knockout and expression studies with an improved understanding of the biological function of the peroxiredoxins. Although current results can be interpreted within the context that peroxiredoxins scavenge hydroperoxides, some peroxiredoxin family members appear to have more complex roles in regulating the response of cells to oxidative stress through protein interactions with constituents of other signaling pathways.
Future Directions:
Further mechanistic information is required for understanding the role of oxidative stress in cancer, the function of peroxiredoxins in normal versus cancer cells, and for the design and testing of specific peroxiredoxin inhibitors that display selectivity to malignant cells. Antioxid. Redox Signal. 28, 591–608.
Introduction
O
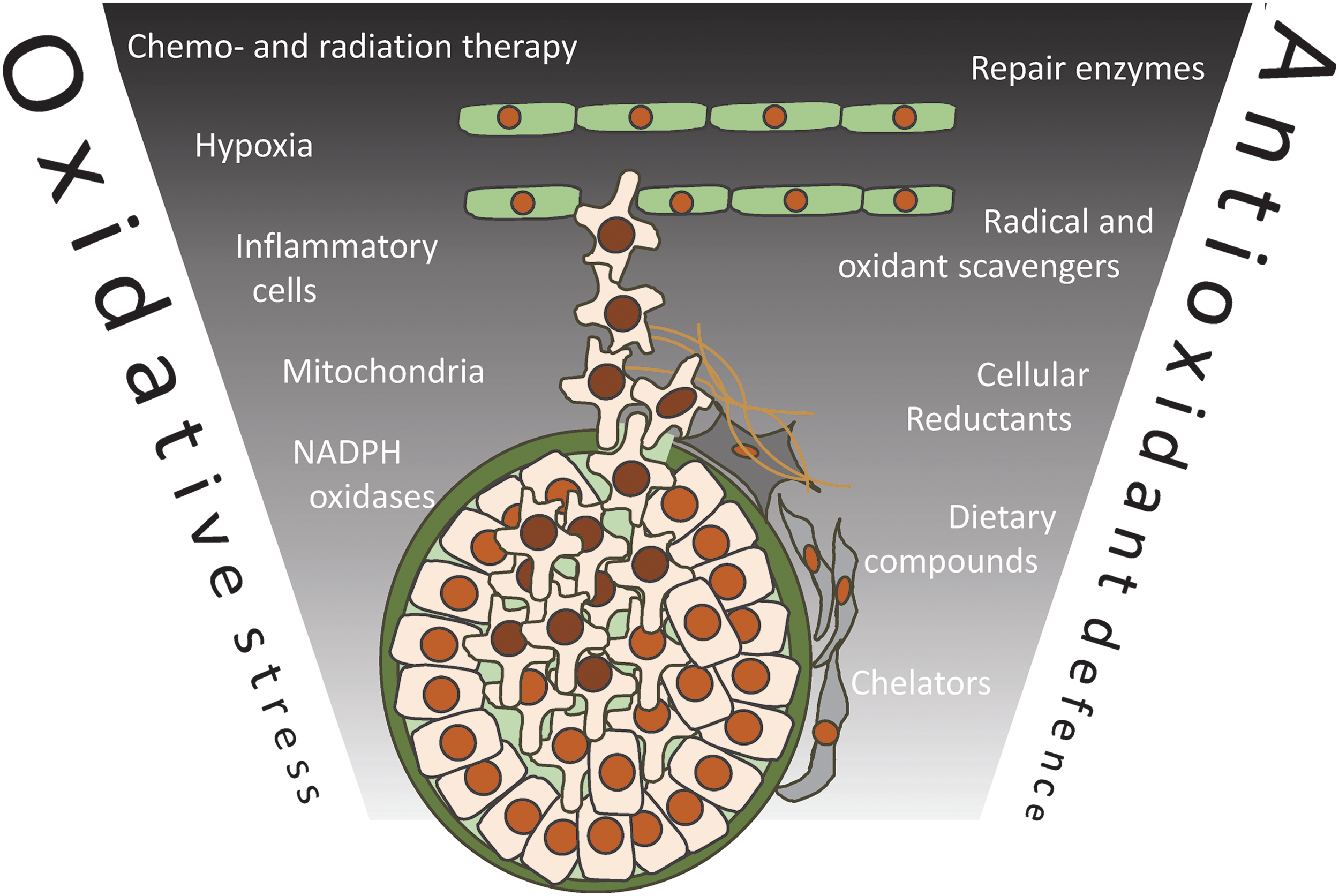
It is commonly believed that cancer cells are under more oxidative stress than normal cells. One early study reported high levels of hydrogen peroxide generation by human tumor cell lines (150), and subsequent studies have revealed that oncogenes such as Ras (171) and Bcr-Abl (106) increase hydrogen peroxide generation through either the constitutive activation of cellular NADPH oxidases, or indirectly as a consequence of the metabolic reprogramming that facilitates rapid tumor growth (48, 166). Early loss of tumor suppressor genes that influence antioxidant networks, for example p53, PTEN, FOXO, RB, or BRCA1, can also increase oxidative stress (79).
As tumors increase in size, an inadequate or unstable blood supply results in tumor hypoxia that increases oxidant production by mitochondria (104). The tumor microenvironment also includes growth factors, cytokines, and inflammatory cells that can combine to increase the levels of oxidative stress (143). This stress appears to increase during the first steps of invasion when cancer cells detach from the extracellular matrix, and it has been reported that some compounds with antioxidant properties promote the survival and migration of tumor cells (84). Cancer therapies, including radiation and some chemotherapeutic drugs, are also strong inducers of oxidative stress. In the ideal situation, this would be enough to kill all malignant cells; however, surviving cells may cope better with this stress, thereby contributing to the aggressive phenotypes of relapsed cancers.
High levels of oxidative stress lead to irreversible damage, whereas lower levels have more subtle effects on cell function. Indeed, hydrogen peroxide can promote cell proliferation by acting as a signaling molecule. Growth factors stimulate hydrogen peroxide production from membrane-associated NADPH oxidases, and redox-sensitive cysteine residues in many different proteins, including phosphatases, kinases, and transcription factors, are regulated by reversible oxidation (174) (Fig. 2). Increased exposure of tumor cells to hydrogen peroxide could, therefore, act as a proliferative signal.

Redox homeostasis is dependent on oxidant generation and the combined activity of the cellular antioxidants. These antioxidant networks are tightly controlled, with nucleated cells that are able to respond to oxidative stress by upregulating their defenses. One key player in this response is the transcription factor Nrf2, which induces the expression of various elements of the thioredoxin (Trx) and glutathione reduction systems, in addition to promoting iron sequestration and xenobiotic detoxification (128).
Nrf2 activation occurs on the redox-dependent dissociation from Keap1, which constrains Nrf2 to the cytoplasm (109). It has been reported that cancer cells express higher levels of different antioxidants, including Trx (17, 133) and glutathione-based defenses (21, 124), and superoxide dismutase (20), consistent with an adaptive response associated with increased oxidative stress. This is a controversial area, however, with other studies reporting either limited alterations or lower antioxidant expression (39, 113). Interestingly, some cancers have Keap1 mutations that result in constitutively higher Nrf2 activity without requiring oxidative stress (43, 76, 145). Overall, Nrf2 appears to be both a tumor suppressor and an oncogene (98). In addition to Nrf2, cellular antioxidants can be influenced by other transcription factors, including HIF1 (183), FOXO (164), and p53 (8).
This review focuses on the peroxiredoxin family of antioxidant proteins and their involvement in cancer initiation and progression. Peroxiredoxins are abundant thiol-dependent peroxidases that are highly efficient at reducing hydrogen peroxide, peroxynitrite, and other hydroperoxides (11, 25, 174, 181). Mammalian cells express six different peroxiredoxins. Peroxiredoxin 1, 2, and 6 are localized in the cytosol and nucleus; peroxiredoxin 3 is exclusively expressed in the mitochondria; peroxiredoxin 4 in the endoplasmic reticulum (ER); and peroxiredoxin 5 in the cytosol, mitochondria, and peroxisomes (180). Each peroxiredoxin has a conserved cysteine (peroxidatic) at the active site that reacts with peroxides to form a sulfenic acid (Fig. 3). Neighboring residues in the peroxiredoxin active site dramatically increase the reactivity of the peroxidatic cysteine, resulting in rate constants with hydrogen peroxide of 106–108 M −1 s−1, which are up to 1 million times higher than for most other thiol proteins (125).

The fate of the initial sulfenic acid differs depending on the peroxiredoxin. The typical 2-Cys enzymes (peroxiredoxins 1–4) exist as homodimers, and the sulfenic acid reacts with a second cysteine (resolving) on the adjacent monomer to form an interchain disulfide. Peroxiredoxin 5 is an atypical 2-Cys protein in which the resolving cysteine is present on the same subunit, resulting in an intramolecular disulfide after a reaction with hydrogen peroxide. The peroxidase cycle of the 2-Cys peroxiredoxins is typically completed through reduction by the Trx/thioredoxin reductase (TrxR)/NADPH or glutaredoxin systems (126). In contrast, peroxiredoxin 6 is a 1-Cys protein in which the sulfenic acid formed on a reaction with hydrogen peroxide is directly reduced by low-molecular-mass thiols or ascorbate. In addition to antioxidant activity, the peroxiredoxins also appear to influence signaling pathways that have a redox-dependent component (28).
Studies with peroxiredoxin 1 knockout mice have also revealed a dramatic increase in tumor formation, consistent with peroxiredoxin 1 having a role in protecting cells from carcinogenic oxidants (107). However, peroxiredoxin expression is increased in many tumors, potentially providing malignant cells with a survival advantage and making them therapeutic targets.
Peroxiredoxin Knockout Animals and Tumor Formation
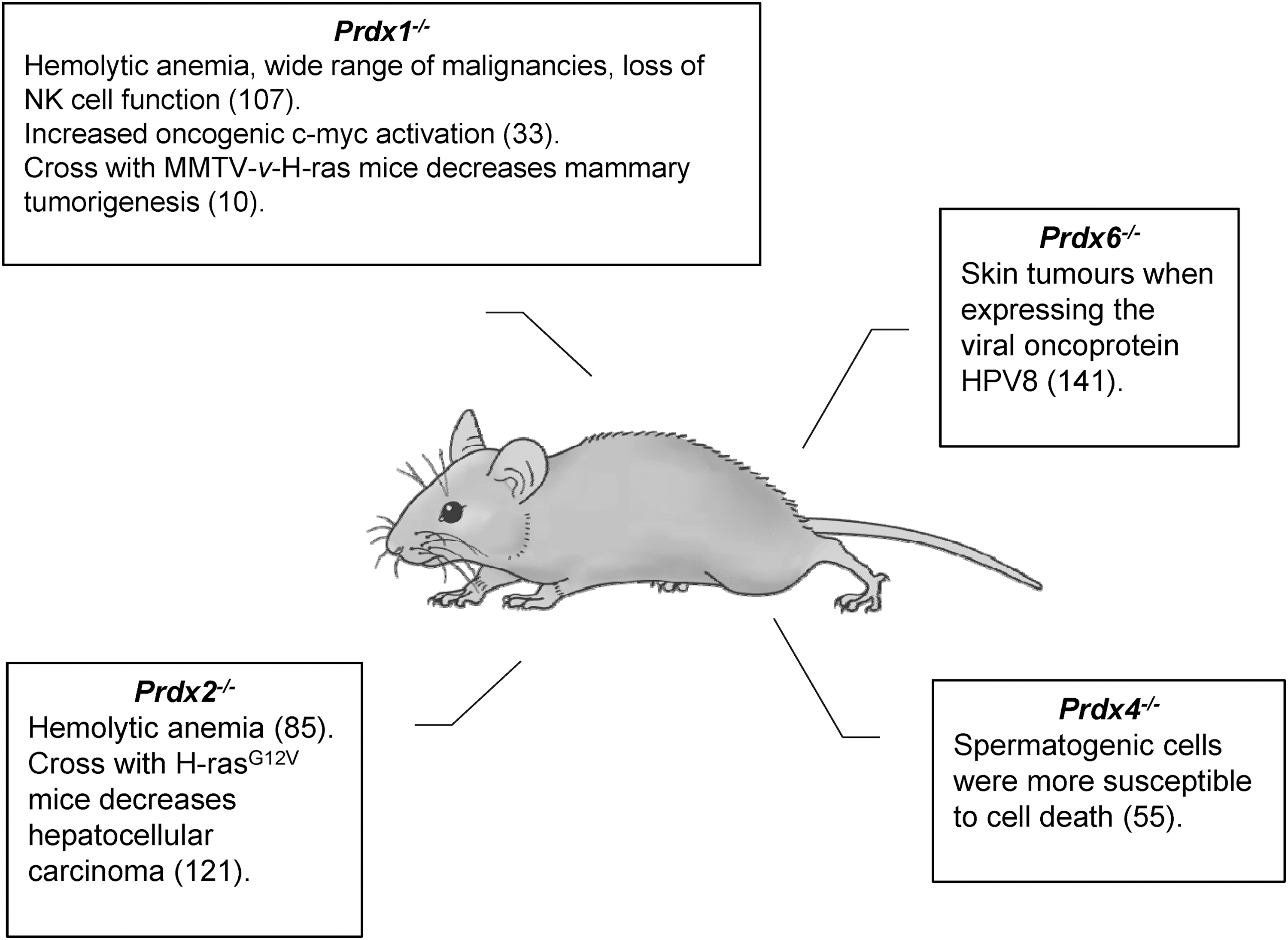
Knockout mice have provided valuable insight into the role of peroxiredoxins in cancer. Mice harboring single deletions of Prdx1–4 or 6 are viable and display disparate cancer phenotypes (33, 55, 85, 101), whereas Prdx5−/− mice have yet to be studied (Fig. 4). The most dramatic phenotype relates to peroxiredoxin 1, with Prdx1+/− and Prdx1−/− mice displaying a wide range of malignancies including lymphoma, hepatocellular carcinoma, fibrosarcoma, osteosarcoma, islet cell adenomas, and adenocarcinomas of lung and breast in addition to hemolytic anemia (107). Loss of peroxiredoxin 1 expression was observed in many Prdx1+/− mouse tumors, but it was not the result of gross chromosomal rearrangements or deletion of the wild-type peroxiredoxin 1 allele.
Examination of isolated mouse embryonic fibroblasts (MEFs) found that 8-oxo-2′-deoxyguanosine levels were heightened in Prdx1−/− when compared with wildtype, and became further elevated when cultured Prdx1−/− cells were incubated with exogenous hydrogen peroxide (107). Prdx1−/− mice generated independently by another group also showed heightened levels of dichlorofluorescein oxidation in knockout MEFs and erythrocytes (33), which is indicative of disrupted redox homeostasis (173). Increased oxidative modifications of DNA nucleosides were found in the brain, liver, and spleen, which support a protective role for peroxiredoxin 1. Surprisingly, fluorescence microscopy suggested nuclear localization of oxidized dichlorofluorescein in Prdx1−/− MEFs (33). One explanation is that loss of peroxiredoxin 1 leads to increased hydrogen peroxide levels in the nucleus, but more in-depth investigation is required. Peroxiredoxin 1-deficiency has also been associated with features of loss of heterozygosity mutation frequency that is tissue specific and contributes to tumorigenesis (107, 135).
The inherent susceptibility of Prdx1−/− mice to tumorigenesis in the absence of exogenous cancer drivers contrasts with the disruption of Prdx6 in mice. Prdx6−/− mice did not develop spontaneous cancers, but if they harbored expression of the viral oncoprotein HPV8 they exhibited an increased number of skin tumors (141). Transgenic expression of oncoproteins in Prdx1−/− mice also promote increased tumor burden as MMTV-v-H-Ras mice with Prdx1 deleted displayed increased tumor incidence after 36 weeks compared with wild-type mice (107). Binding of peroxiredoxin 1 to the lipid phosphatase PTEN has been shown to inhibit oxidative inactivation, thereby regulating AKT activity (10). Mammary epithelial cells harvested from MMTV-v-H-Ras mice before tumor emergence displayed elevated active AKT and oxidized PTEN when peroxiredoxin 1 was deleted. Further experiments with shRNA against Prdx1 in Pten−/− MEFs supported repression of H-RAS and ERBB-2-induced transformation through PTEN in a peroxiredoxin 1-dependent manner. Prdx1−/− MEFs have also been shown to be more susceptible to RAS transformation, which has been hypothesized to be due to deregulation of MYC (10, 103).
In contrast to peroxiredoxin 1, deletion of Prdx2 in mice did not cause spontaneous neoplasia or decreased natural killer cell-induced cell lysis, but it was associated with hemolytic anemia (85). Further evidence of the divergence between peroxiredoxin 1 and 2 in the development of cancer in vivo was demonstrated when Prdx2−/− mice were crossed with H-rasG12V mice. These mice had reduced hepatic tumor incidence and developed smaller sized tumors when compared with Prdx2+/+ H-rasG12V mice (121).
Studies performed in Saccharomyces cerevisiae have observed increases in oxidative stress accompanied by a range of genomic perturbations on deletion of the peroxiredoxin 1 ortholog, TSA1 (176, 177). Loss of TSA1 leads to an enhancement of DNA mutations, genomic instability, and gross chromosomal rearrangements (50, 51, 134). Further exploration of this phenotype found cells deficient of TSA1 constitutively activate the DNA damage checkpoint and generate an overabundance of dNTPs (152). The combination of deletions to TSA1 and double-strand break repair genes was found to induce synthetic lethality, which could be rescued when TSA1 and RAD51 double mutant cells were grown under anaerobic conditions (134). The limitation of available oxygen (27) suggests that TSA1-deficient survival could be due to reduced intracellular oxidant production in the mitochondria, ER, and peroxisome. These studies also suggest that TSA1-dependent reduction of intracellular hydrogen peroxide is an important protective mechanism to maintain genomic integrity.
Further studies in yeast shed light on the non-redundant roles of human peroxiredoxin 1 and 2 to maintain genomic stabilization. The genomic rearrangements and synthetic lethality phenotypes of TSA1 mutant cells combined with RAD6 or RAD51 deletion could be rescued by the insertion of Prdx1, but not Prdx2, into the TSA1 promoter in S. cerevisiae (54). This observation suggests that the role of peroxiredoxin 1 in genomic integrity is unique and independent of transactivation elements, despite the fact that peroxiredoxin 1 and peroxiredoxin 2 are highly similar proteins, with 78% sequence similarity and apparently similar peroxidase activity.
Peroxiredoxins in Human Tumors
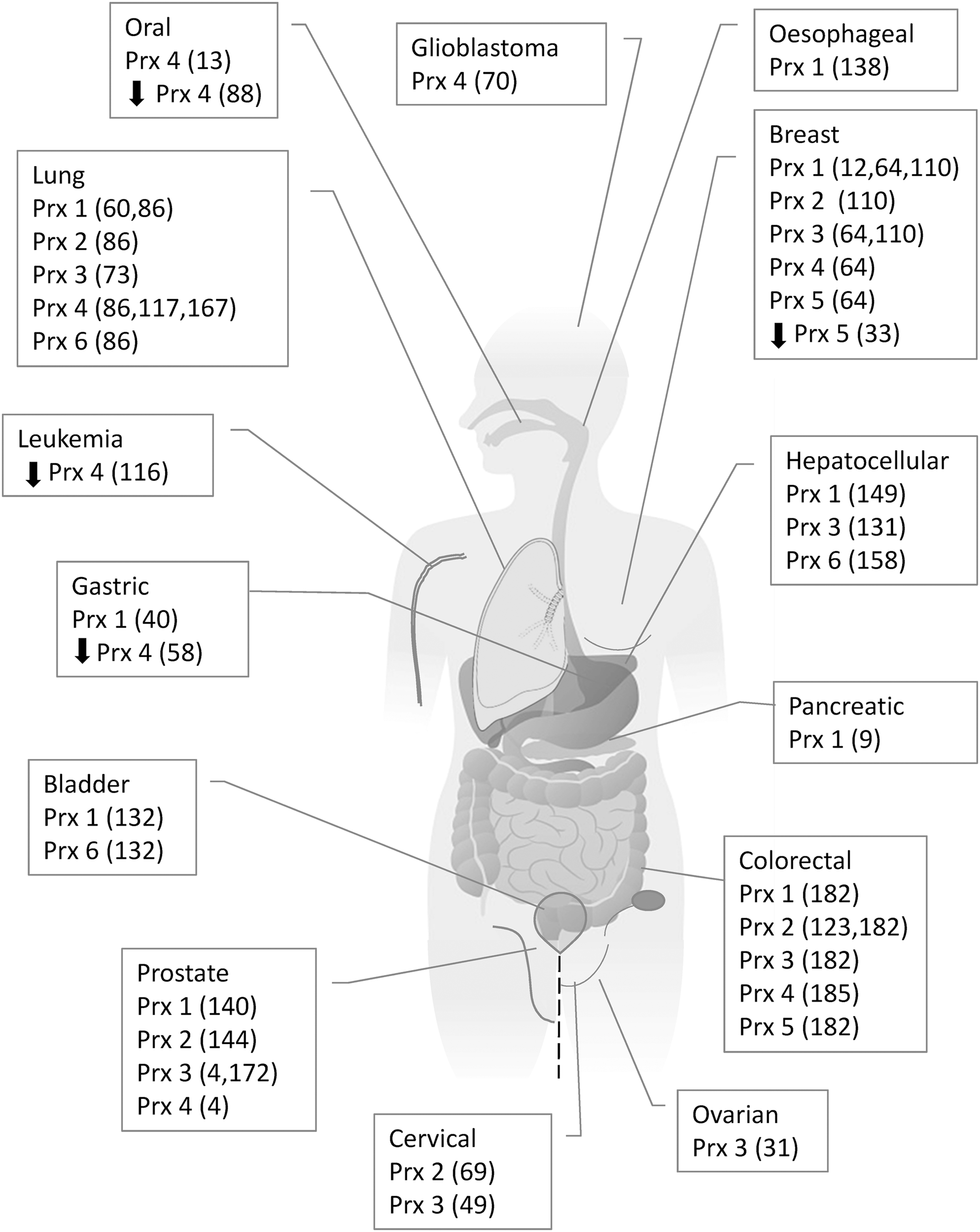
The emergence of neoplastic phenotypes in knockout mice suggests that peroxiredoxins act as tumor suppressors (107). This is consistent with a reduction of peroxidase activity increasing oxidative damage and tumorigenesis. However, analyses of peroxiredoxin expression in human tumors and cell lines derived from tumors are variable but typically show increased peroxiredoxin expression (Fig. 5). As with the knockout mice, however, there seem to be differences between the peroxiredoxin family members.
Peroxiredoxin 1
Expression and survival studies
Increased expression of peroxiredoxin 1 is observed in several human cancers types, including breast (12, 64, 110), colorectal (182), hepatocellular (149), gastric (40), lung (60, 86), malignant mesothelioma (74), esophagus (138), pancreatic (9), and prostate (140). Higher-grade malignancies are associated with high expression levels of peroxiredoxin 1 in breast cancer (12). Peroxiredoxin 1 is associated with numerous aggressive features of hepatocellular cancer, including larger and increased number of tumors, increased microvascular invasion, advanced Edmondson grade, higher serum α-fetoprotein, and advanced tumor node metastasis staging (149). The elevated expression of peroxiredoxin 1 reported in bladder cancer tissue compared with bladder mucosa of normal controls or normal surrounding tissue has been correlated with development, progression, and recurrence of the cancer (132).
Peroxiredoxin 1 expression in gastric cancer is positively correlated with the epithelial-mesenchymal transition (EMT) markers, which is an important step in invasion and metastasis (40). Vascular endothelial growth factor (VEGF), a pro-angiogenic factor that partly mediates angiogenesis through its effects on endothelial cell proliferation, migration, and differentiation, has also been positively correlated with peroxiredoxin 1 expression in both pancreatic (9) and hepatocellular cancer (149). High expression of peroxiredoxin 1 has also been significantly correlated with poor survival in non-small cell lung cancer (68), and associated with tumor progression in pancreatic cancer (140).
In addition to the published literature, we have used a Kaplan–Meier plotter tool (
The association between peroxiredoxin mRNA level and the overall survival of patients with breast, gastric, lung, or ovarian cancer was analyzed on public data of Km plotter (
HR, hazard ratio; N.S., not significant.
Mechanistic studies
EMT involves several related signaling pathways, including transforming growth factor beta (TGF-β) 1 (15), nuclear factor kappa-B (NFκB) (52), Notch (153), and Wnt/β-catenin pathway (47, 82). TGF-β1 stimulates tumor invasion and metastasis via induction of EMT, and silencing peroxiredoxin 1 expression in lung adenocarcinoma cells inhibited TGF-β1-induced EMT and cell migration, whereas peroxiredoxin 1 overexpression enhanced both processes (41).
Knockdown of peroxiredoxin 1 expression by shRNA reduced angiogenesis and the growth of tumors derived from human prostate in cancer cells, and it was associated with lowering levels of VEGF, tumor necrosis factor (TNF)-α, interleukin, TGF-α, and TGF-β (140). Further, the addition of recombinant peroxiredoxin 1 to noncancerous endothelial cells stimulated cell proliferation, migration, and differentiation. This effect was ablated by treatment with an inhibitor of Toll-like receptor (TLR) 4, known to stimulate the expression of VEGF through a TLR4-Akt signaling pathway (140). More evidence of peroxiredoxin 1-dependent cell signaling modulation in prostate cancer comes from a study investigating aggressive prostate cancer cell lines that showed that elevated levels of peroxiredoxin 1 caused greater androgen receptor (AR) activation in response to hypoxia/reoxygenation. These studies also reported that hypoxia increases AR activity in prostate cancer cells, and that peroxiredoxin 1, which is upregulated in response to hypoxia, interacts with AR to enhance the expression of androgen-regulated genes (119, 120).
A strong link seems to exist between peroxiredoxin 1 and NFκB, a transcription factor commonly deregulated in cancer. Depletion of peroxiredoxin 1 in estrogen-deficient breast cancer cells decreased COX-2 expression by reducing NFκB occupancy at its upstream promotor region (162). Silencing of peroxiredoxin 1 expression in T24 bladder cancer cells not only significantly suppressed growth and promoted apoptosis but also reduced phospho-NFκB, p50, and p65 protein expression (61). This suggests that peroxiredoxin 1 promotes NFκB signaling in cancer cells. In support of this, tumor hypoxia upregulated peroxiredoxin 1 expression, NFκB translocation, and DNA binding affinity in oral squamous carcinoma cells; whereas peroxiredoxin 1 knockdown decreased NFκB translocation and DNA binding activity (188).
Other studies have also linked peroxiredoxin 1 expression changes with hypoxic conditions in both cell culture and tumor models. For example, hypoxia has been shown to increase nuclear localization of Nrf2, and binding to regions of the peroxiredoxin 1 promoter (71) and to increase expression peroxiredoxin 1 (and also peroxiredoxin 2) in lung cancer cells (66). Peroxiredoxin 1 expression, NFκB translocation, and DNA binding affinity were upregulated and heme oxygenase (HO-1) was downregulated in response to hypoxia in oral squamous carcinoma cells. When peroxiredoxin 1 was knocked down in these cells, the expression level of HO-1 was increased, whereas NFκB translocation and DNA binding activity were decreased after hypoxia or hypoxia/reoxygenation (188). In a range of xenograft tumors derived from oral squamous carcinoma cells, peroxiredoxin 1 expression was highest in the larger, more hypoxic tumors. In addition, HO-1 and NFκB DNA binding activities were enhanced compared with those with a smaller diameter (2 mm) (188).
Activation of the AP-1 signaling pathway is well documented in promoting the growth, proliferation, invasion, and metastasis of respiratory epithelial cancers (63). Knockdown of peroxiredoxin 1 reduced the activation of c-Jun phosphorylation and repressed the AP-1 mediated promoter activity in a lung cancer cell line in culture (60). Knockdown also significantly reduced subcutaneous tumor growth and blocked metastasis in mouse xenograft models derived from a lung cancer cell line, and the overexpression of peroxiredoxin 1 enhanced the malignancy of these lung cells in both culture and mouse xenografts models (60).
Resistance to cancer therapeutics
Peroxiredoxin 1 has been associated with resistance to anticancer treatments in thyroid and lung cancers. Proteasome inhibitors are a novel class of antitumor agents with pre-clinical and clinical evidence of activity against malignancies (99). High levels of peroxiredoxin 1 expression were found in thyroid cancer cells treated with proteasome inhibitors, and peroxiredoxin 1 knockdown resulted in accelerated proteasome inhibitor-induced cell death (30). Similarly, depletion of peroxiredoxin 1 was able to dose dependently increase the cytotoxicity of docetaxel through suppression of FOXO1, which significantly reduced the growth rate and the inhibitory effects of docetaxel in lung cancer cells (53).
Peroxiredoxin 2
Expression and survival studies
Peroxiredoxin 2 expression is elevated in breast (110), lung (86), malignant mesothelioma (74), colorectal (123, 182), and cervical (69) cancers compared with normal tissue. High peroxiredoxin 2 expression in colorectal tumors is significantly associated with advanced tumor nodes metastasis staging, poor tumor differentiation, and increased lymph node metastasis (123). Elevated peroxiredoxin 2 was reported in high-grade cervical intraepithelial neoplasia and in cervical cancer tissue, compared with low-grade and normal cervical tissue, suggesting that peroxiredoxin 2 could be a marker for cervical cancer progression (69). Increased expression of peroxiredoxin 2 was observed in head and neck tumor tissues isolated from the patients who did not respond to radiation therapy, and peroxiredoxin 2 expression was weak in tissues from patients with regressed tumors (118). Similar to peroxiredoxin 1, increased peroxiredoxin 2 mRNA levels are associated with decreased survival in breast and lung cancer, and improved survival in gastric cancer, but do not relate to survival in ovarian cancer (Table 1).
Mechanistic studies
Similar to peroxiredoxin 1, peroxiredoxin 2 has been reported to regulate AR activity in the nucleus and the cytoplasm, and to be involved in the proliferation of AR-expressing prostate cancer cells (140). Overexpression of peroxiredoxin 2 in castration-resistant prostate cancer cells increased the levels of AR transactivation in the cytoplasm, whereas silencing peroxiredoxin 2 reduced the expression of androgen regulated genes and suppressed growth by inducing cell-cycle arrest at the G1 phase (144). Dysregulation of the Wnt/β-catenin pathway has been implicated in many cancers, and the stabilization of β-catenin results in a transcriptional response that is believed to be critical in tumorigenesis (77). In colorectal cancer cells, peroxiredoxin 2 knockdown suppressed Wnt/β-catenin signaling by elevating GSK-3b activity and disturbing the nuclear translocation of β-catenin, resulting in the downregulation of the target genes c-myc and survivin (92).
Hypoxia-inducible factors (HIFs) control the transcription of genes that are crucial for the pathogenesis of cancer and other human diseases (142), and the transcriptional activity of HIFs increases rapidly on exposure to hypoxia. Peroxiredoxin 2 has been reported to interact with HIF-1α and HIF-2α in hypoxic cervical cancer cells and inhibit their transcriptional activity (93). Prolonged hypoxia increases the nuclear translocation of peroxiredoxin 2 and impairs HIF-1 and HIF-2 binding to the hypoxia response elements of a subset of HIF target genes (93). Peroxiredoxin 2 is a direct HIF target gene and its expression is induced by prolonged hypoxia (93). Collectively, these results suggest that peroxiredoxin 2 is involved in a feedback inhibition mechanism during hypoxia.
Resistance to cancer therapeutics
High levels of peroxiredoxin 2 expression are associated with resistance to chemotherapy, and they can be reversed by peroxiredoxin 2 knockdown. For example, elevated peroxiredoxin 2 levels were observed in gefitinib-resistant (GR) A549 lung cancer cells compared with non-resistant cells. GR/A549 cells showed lower dichlorofluorescein fluorescence, and increased JNK phosphorylation and cell cycle progression, which was reversed by depletion of peroxiredoxin 2. In addition, mice transfected with GR/A549 cells in which peroxiredoxin 2 had been knocked down showed a decrease in tumor growth, JNK phosphorylation, and Bcl-2 expression, as well as increased caspase-3 cleavage compared with control cells (81). Higher peroxiredoxin 2 expression was measured in an MCF-7-derived breast cancer cell line that is radiation resistant (MCF + FIR3) compared with a radiation-sensitive control cell line (MCF + FIS4) (161), but there was no change in peroxiredoxin 1, 3, 5, and 6 expression. As expected, peroxiredoxin 2 depletion in MCF + FIR3 cells reversed radiation resistance, whereas overexpression of peroxiredoxin 2 increased resistance in the MCF + FIS4 cell line (161). In a follow-up study, peroxiredoxin 2 knockdown by siRNA increased sensitivity to ionizing radiation by altering cellular thiol status, inhibiting Ca2+ efflux from the cells, and perturbing the intracellular Ca2+ homeostasis (29). In support of this, peroxiredoxin 2 knockdown decreased resistance to radiation treatment in a head and neck cancer cell line (118), enhanced cisplatin-induced cell death in gastric cancer cells (186), and increased the susceptibility of colorectal cancer cells to hydrogen peroxide-mediated apoptosis (92).
Peroxiredoxin 3
Expression and survival studies
Mitochondrial peroxiredoxin 3 is elevated in breast (64, 110), cervical (49), colorectal (182), prostate (4, 172), hepatocellular (131), ovarian (31), and lung (73) cancer tissue where Nrf2-dependent overexpression has been identified (73). In cervical cancer, peroxiredoxin 3 expression correlates positively with the proliferation marker Ki67 (49). Similarly, in breast tissue, peroxiredoxin 3 expression was positively correlated with proliferating cell nuclear antigen (PCNA) (19). Hepatocellular cancer cell lines and metastatic lesions also exhibited higher expression of peroxiredoxin 3 in hepatocellular tissue that was poorly differentiated when compared with normal liver tissue (131). In prostate tissue, peroxiredoxin 3 expression was associated with increased tumor stage (4) and in ovarian cancer, metastatic cancer samples were shown to have the highest levels of peroxiredoxin 3 expression (31). However, elevated mRNA expression does not indicate shorter survival in breast cancer or lung cancer (Table 1).
Mechanistic studies and response to cancer therapeutics
Mitochondrial peroxiredoxin 3 is a direct target of c-myc and is required for mitochondrial homeostasis during c-myc-induced proliferation, apoptosis, and neoplastic transformation (178). Further, peroxiredoxin 3 is required for maintaining mitochondrial mass and membrane potential (178). Various studies suggest that knockdown of peroxiredoxin 3 compromised cell function and sensitized cells to cytotoxic agents. For example, depletion of Prx3 in HeLa cells resulted in increased sensitivity to staurosporin and TNF-α (14). Silencing of peroxiredoxin 3 inhibited cell proliferation and cell-cycle progression in breast cancer cells (19). In liver cancer cells, knockdown of peroxiredoxin 3 significantly enhanced the ability of hydrogen peroxide to inhibit proliferation and promote apoptosis (165). In mouse thymoma cells, overexpression of peroxiredoxin 3 (111) protected against apoptosis caused by hypoxia, hydrogen peroxide, t-butylhydroperoxide, and the anticancer drug imexon (111). The proliferation rate of ovarian cancer cells after peroxiredoxin 3 knockdown was reduced significantly both under normal cell growth conditions and when the cells were treated with cisplatin (31). This knockdown was also associated with enhanced induction of apoptosis by cisplatin through inhibition of NFκB signaling pathways (31).
HIF-1α is speculated to decrease peroxiredoxin 3 expression in some colorectal cancers by acting as a transcriptional repressor (183). Knockdown of HIF-1α increased peroxiredoxin 3 expression and lowered the proliferation rates of colorectal cancer cells, with knockdown of peroxiredoxin 3 restoring proliferation (183). Surprisingly, given the surge of interest in metabolic reprogramming during cancer, there is very little information on the impact of altered peroxiredoxin 3 expression on mitochondrial metabolic pathways.
Peroxiredoxin 4
Expression and survival
Endoplasmic reticular peroxiredoxin 4 has been reported to be elevated in a number of lung (86, 117, 167), breast (64), glioblastoma (70), oral cavity squamous cell (13), prostate (4), and colorectal (185) tumors. Expression level in colorectal tumors correlates with depth of invasion and lymph node metastasis, and it is associated with short survival time (185). In prostate tissue, peroxiredoxin 4 expression was significantly associated with increased tumor stage and a higher Gleason sum score (4). Significantly higher levels of peroxiredoxin 4 were also observed in squamous cells from oral cavity cancers compared with adjacent non-tumor epithelia (13). Compared with 31 corresponding primary tumors, peroxiredoxin 4 expression was elevated in metastatic lymph node samples (13).
In addition, both univariate and multivariate analyses showed an association of peroxiredoxin 4 overexpression with poorer prognosis, suggesting peroxiredoxin 4 to be a possible prognostic marker for oral cavity squamous cell carcinoma (13). In contrast, however, another study showed that peroxiredoxin 4 levels were found to be decreased in oral squamous cell carcinoma compared with normal tissues (88). Decreased peroxiredoxin 4 levels were also reported for human stomach adenocarcinoma tissue (58), and acute promyelocytic leukemia (116) compared with their normal counterparts. Our survival analysis indicates no relationship of peroxiredoxin 4 expression with survival associated with lung or ovarian cancer, yet increased peroxiredoxin 4 expression is associated with shortened survival in breast cancer and prolonged survival in gastric cancer (Table 1).
Mechanistic studies and resistance to cancer therapeutics
Sulfiredoxin reactivates hyperoxidized peroxiredoxins and is reported to have the highest affinity toward peroxiredoxin 4 (167). Interestingly, knockdown of peroxiredoxin 4 or sulfiredoxin reduced anchorage-independent colony formation, cell migration, and invasion of human lung cancer cells (167). Mechanistic studies revealed that the sulfiredoxin–peroxiredoxin 4 axis is required for sufficient activation and/or amplification of certain phosphokinase signaling cascades, including the AP-1/MMP9 axis, CREB, and MAPK pathways (167). Disruption or enhancement of the sulfiredoxin–peroxiredoxin 4 axis in a lung cancer cell line led to reduction or acceleration of tumor growth and metastasis in a mouse xenograft (167). Similarly, cell migration and invasiveness were attenuated in an oral cavity squamous cell carcinoma cell line on knockdown of peroxiredoxin 4 (13), and reducing peroxiredoxin 4 expression in glioblastoma decreased cell growth and radio resistance and increased levels of dichlorofluorescein, DNA damage, and apoptosis (70). In a syngeneic orthotopic transplantation model, peroxiredoxin 4 depletion decreased glioblastoma infiltration and significantly prolonged mouse survival (70).
Peroxiredoxin 4 has been shown to interact with HIF-1α and HIF-2α in hypoxic cervical cancer cells. Prolonged hypoxia increased the nuclear translocation of peroxiredoxin 4 and as a result, peroxiredoxin 4 impaired HIF-1 and HIF-2 binding to the hypoxia response of elements of a subset of HIF target genes, thus inhibiting gene transcription in cells exposed to prolonged hypoxia (93).
Peroxiredoxin 5
To date, there are only a limited number of studies investigating a role for peroxiredoxin 5 in cancer, and these provide conflicting data. For example, one study found peroxiredoxin 5 overexpression in breast cancer to be associated with larger tumor size, positive lymph node status, and shorter survival (64); whereas another report showed that peroxiredoxin 5 was the only member of the peroxiredoxin family that was significantly downregulated in breast tumors (34). Peroxiredoxin 5 elevation was found in colorectal (182) and malignant mesothelioma (74) tumors compared with non-malignant tissue. In addition, peroxiredoxin 5 was significantly elevated in colorectal cancer patients with stage 3 or lymph node metastasis-positive cases (182). Increased peroxiredoxin 5 mRNA was associated with lower survival in breast cancer, but with improved survival in gastric and lung cancer (Table 1). The role of this peroxiredoxin in response to cancer therapies has not been investigated.
Peroxiredoxin 6
Expression and survival
Peroxiredoxin 6 is overexpressed in hepatocellular (158), lung (86), malignant mesothelioma (74), and bladder (132) cancer. Increased peroxiredoxin 6 mRNA is associated with lower survival in breast and ovarian cancer, but with improved survival in gastric and lung cancer (Table 1).
Mechanistic studies and resistance to cancer therapeutics
Expression of PCNA, VEGF, cyclin-dependent kinases, and the activity of mitogen-activated kinases were increased in a xenograft mouse model injected with A549 lung cancer cells overexpressing peroxiredoxin 6, and decreased in cells overexpressing a catalytic variant (44). This suggests that peroxiredoxin 6 peroxidase activity is driving lung cancer progression. Peroxiredoxin 6 involvement was reported in the resistance of ovarian, gastric, and cervical cancer cells to cytotoxic agents (16, 115, 158). For example, peroxiredoxin 6 overexpression inhibited cisplatin-induced apoptosis in ovarian cancer cells (115), and peroxiredoxin 6 knockdown promoted peroxide-induced cytotoxicity in liver cells (158).
Metastatic gastric cancer cells can overexpress peroxiredoxin 6, which renders them more resistant to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) than primary cancer cells. One study shed more light on how peroxiredoxin 6 inhibits apoptosis by showing that it binds to caspase-10, thus reducing TRAIL-induced death-induced signaling complex formation and subsequent caspase activation. Further, knockdown of peroxiredoxin 6 sensitizes cervical cancer cells to TRAIL-induced cell death (16).
Summary of peroxiredoxin expression data
The majority of published studies have focused on peroxiredoxins 1–4, with the first three of these typically overexpressed in a range of human cancers, and peroxiredoxin 4 displaying variability depending on the cancer type. Caution should be taken in interpreting the expression data for peroxiredoxins 3 and 4 as the studies do not account for differences in mitochondrial or ER volume between samples. If, for example, the tumors had increased mitochondrial numbers, there would be a corresponding increase in peroxiredoxin 3 levels without a change in concentration of the enzyme. The higher expression of the cytoplasmic peroxiredoxins can more confidently be translated to high cytoplasmic concentrations. In some studies, higher expression was positively correlated with more aggressive tumors and decreased patient survival, and knockdown of the peroxiredoxins often diminished the aggressive phenotypes of different tumors and sensitized them to cytotoxic agents. This implicates the peroxiredoxins as potential therapeutic targets.
The mechanism of action by which the peroxiredoxins enhance tumor survival is not clear. These proteins have antioxidant activity through catalyzing the reduction of hydroperoxides, but their biological properties seem considerably more complex than this (see below), and it is difficult to directly ascribe any effects of altered expression to antioxidant properties. Indeed, several of the studies described above talk more about protein interactions of the peroxiredoxins and their effects on gene expression than peroxidase activity. Also, since peroxiredoxin 1 and 2 have very similar peroxidase activities, other biological properties may explain the diverse phenotypes associated with altered expression.
Peroxiredoxins As Signaling Proteins
Although peroxiredoxins were initially recognized as antioxidant proteins, it has become apparent that they can influence various cell signaling pathways, including those that regulate proliferation and cell death (42). Although the mechanisms of redox signaling are not fully understood, signaling itself involves generation of hydrogen peroxide, or a related oxidant, in response to a stimulus, and resultant reversible activation or inactivation of critical thiol proteins in the signaling pathway. Due to their high reactivity and abundance, the peroxiredoxins will be major targets of intracellular hydrogen peroxide (25, 174), and therefore important regulators of peroxide-dependent signaling pathways.
One of the first hints for a regulatory function of peroxiredoxins came from studies that showed the sulfenic acid formed at the peroxidatic cysteine can also react with a second molecule of hydrogen peroxide to form a sulfinic acid in a process termed hyperoxidation or overoxidation (Fig. 3). This inactivates peroxiredoxin peroxidase activity until the enzyme is slowly reduced by sulfiredoxin. Intriguingly, structural motifs that appear to promote hyperoxidation have been selected for during eukaryote evolution (180). Increased sensitivity to hyperoxidation is achieved by slowing disulfide formation between the peroxidatic and resolving cysteines, thereby increasing the lifetime of the sulfenic acid (130). Since hyperoxidation compromises the antioxidant function of peroxiredoxins, it is proposed that increased sensitivity to hyperoxidation is associated with a gain of signaling function (57, 180).
Various possible roles have been proposed for peroxiredoxins in redox signaling (25, 146, 155, 157, 175, 180). One is that peroxiredoxins regulate steady-state levels of hydrogen peroxide in specific regions of the cell, thereby competing with redox-sensitive signaling proteins and dampening the signal. If the generation rate is sufficiently high for the peroxiredoxins to become hyperoxidized, this can lead to an increase in hydrogen peroxide and the potential to oxidize less reactive thiol proteins (Fig. 6A). However, modeling predicts that the diffusion of hydrogen peroxide will still not allow its levels to reach that necessary for direct oxidation of these less reactive thiols (155).
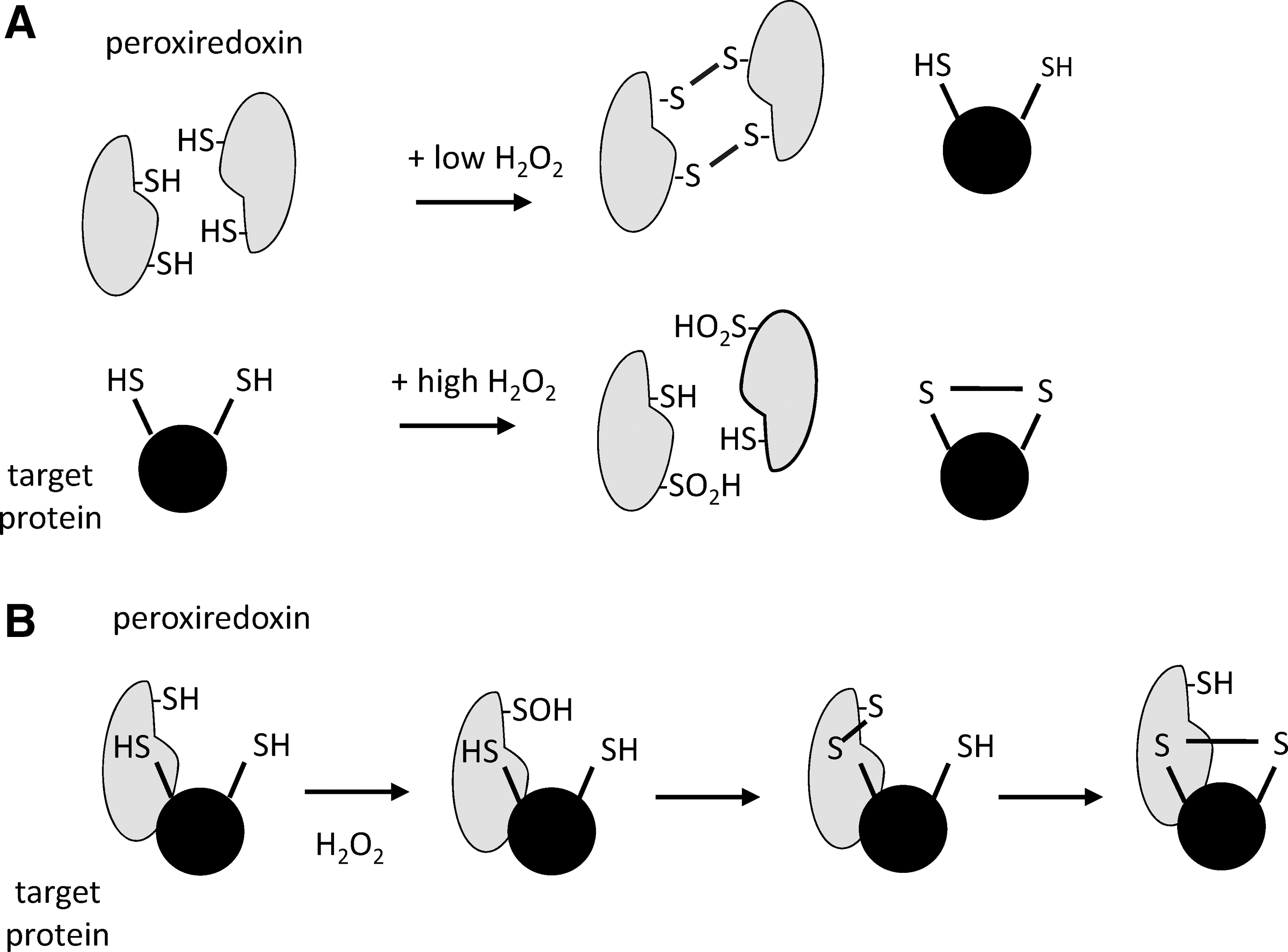
An alternate model, in which the peroxiredoxins facilitate the oxidation of target proteins through a relay mechanism (Fig. 6B), is receiving increased attention. Many of the phosphatases, kinases, and transcription factors that can be oxidized in cells only react slowly with hydrogen peroxide, and it is proposed that the sulfenic acid formed at the active site of peroxiredoxins forms a disulfide with a cysteine on an interacting protein, leading to oxidation. Selectivity is provided by peroxiredoxin protein interactions rather than reactivity of the target protein. This mechanism could explain the evolution of 2-Cys peroxiredoxins that are more susceptible to hyperoxidation, that is, more stable sulfenic acid intermediates produced during the catalytic cycle having greater opportunity to oxidize other proteins. The first example of this mechanism was discovered to explain oxidation of the Yap1 transcription factor in S. cerevisiae (28), and more recently, two examples have been reported in mammalian cells: peroxiredoxin 1-dependent oxidation of the kinase ASK1 (59), and peroxiredoxin 2-dependent oxidation of the STAT3 transcription factor (146). More research is required to determine the prevalence and significance of this mechanism.
Peroxidase activity involves peroxiredoxin homodimers arranged in a head-to-tail fashion, whereas the peroxiredoxin dimers further associate to yield decameric (pentamer of dimers) or dodecameric (hexamer of dimers) structures (179). These units can also form lateral columns consisting of multiple stacked rings (97, 179). The dimer-to-oligomer equilibrium is influenced by the concentration and redox state of the peroxiredoxins, including hyperoxidation, and also by phosphorylation (56), pH (78), and ionic strength (75). In general, reducing conditions promote decamer formation, oxidation favors the dimer, and hyperoxidation stabilizes the higher-order oligomeric structures (89, 122). These complex oligomeric structures have been proposed to influence signaling in a manner independent of oxidation of associated proteins. The higher-order structures of some peroxiredoxins have been shown to act as chaperones, protecting proteins from denaturation during oxidative stress (1, 57).
A number of peroxiredoxin binding partners have been identified (139), especially for peroxiredoxin 1, and many of these are cancer related. Peroxiredoxin 1 binds to c-Abl and inhibits c-Abl-dependent phosphorylation (170). Peroxiredoxin 1 also binds to PTEN, inhibits its oxidation, and protects against PTEN/Akt-mediated tumorigenesis (10). Besides PTEN, peroxiredoxin 1 has been shown to protect other protein tyrosine phosphatases from oxidation, including MKP5, the phosphatase that acts on MAPK kinase p38 and JNK, thereby inhibiting senescence in breast cancer cells (156). Interestingly, although stress signaling induces peroxiredoxin 1 expression, the protein directly regulates stress signaling by interacting with proteins of that signaling cascade. For example, association of peroxiredoxin 1 with the tumor suppressor, Ste20-like kinase 1 (MST1) has been observed in oxidant-treated cells (102, 137). Others observed that although peroxiredoxin 1 was not required for MST1 activation, it became inactivated due to MST-1-dependent phosphorylation (137).
Other interactions of peroxiredoxin 1 include binding to p66shc, thereby reducing the ability of the p66CH2CB tetramer to induce mitochondrial rupture (36). The interaction of peroxiredoxin 1 with cMyc is believed to inhibit specific oncogenic transcriptional activities of c-Myc (103). Peroxiredoxin 1 also interacts with the AR in prostate cancer cells, thus promoting transactivation of AR (120), whereas peroxiredoxin 2 is associated with the PDGF receptor (18). Peroxiredoxin 1 participates in the glutathione-S-transferase pi/JNK complex formation and is believed to suppress JNK signaling in ionizing radiation-induced cell death (72). Peroxiredoxin 1 also interacts with macrophage migration inhibitory factor, with inhibition of tautomerase activity only occurring under oxidizing conditions (62). Peroxiredoxin 2 binds to the protein disulfide isomerase, ERp46, but only when the peroxiredoxin 2 is hyperoxidized (114). Also, recently, the interaction of Prx1 with FOXO3 has been described to require Prx1 catalytic and resolving cysteines to control FOXO3 function (45).
Development of Peroxiredoxin Inhibitors
Reports of increased oxidative stress in tumors have led to exploration of therapeutics that target the antioxidant systems of cancer cells. One approach is to use compounds that disrupt redox homeostasis, such as sulfasalazine (38), 6-nicotinamide (5), benzophenanthridines (83, 160), buthionine sulfoximine (112), and arsenic trioxide (91). An alternate approach is to target members of the peroxiredoxin family, and several groups are currently investigating novel peroxiredoxin inhibitors.
One of the first reported peroxiredoxin inhibitors was adenanthin, a naturally occurring diterpenoid that induces differentiation in acute promyelocytic leukemia cells and shows some efficacy in leukemia and hepatocellular carcinoma models (46, 90). Tagged adenanthin was shown to bind the resolving cysteine of peroxiredoxin 1 and 2 in cells, and to inhibit the peroxidase activity of these proteins in vitro (90). A subsequent study, however, has shown that adenanthin is not a potent peroxiredoxin inhibitor, but it preferentially promotes peroxiredoxin oxidation through effects on the Trx and glutathione-dependent reduction systems (147). Another natural compound, the thiazole antibiotic thiostrepton, was shown to bind and inhibit the activity of mitochondrial peroxiredoxin 3 (108). Thiostrepton displays biological activity in malignant mesothelioma models (26, 108). An intriguing mechanism of inhibition has been proposed, in which thiostrepton crosslinks the peroxidatic and resolving cysteine of one active site in peroxiredoxin 3, but only if the other active site in the homodimer is already oxidized (26). Targeting of a partially oxidized peroxiredoxin would provide selectivity toward cells under increased oxidative stress.
Screening approaches have unearthed two small-molecule inhibitors of peroxiredoxin 1: AMRI-59 (184) and H7 (168). AMRI-59 displayed IC50 values of 12–33 μM against recombinant peroxiredoxins 1–3, whereas H7 inhibited peroxiredoxin 1 with an IC50 of 8 μM. H7 appeared to be selective for peroxiredoxin 1 over other peroxiredoxins, and it induced differentiation of leukemia cells to a greater extent than observed with normal cord blood or bone marrow mononuclear cells (168). In vivo studies further supported its ability to differentiate leukemia cells at doses of 10 and 20 mg/kg. AMRI-59 inhibited reduction of the disulfide form of the peroxiredoxins without affecting Trx or TrxR activity. It was able to slow proliferation and kill A549 lung adenocarcinoma cells, with peroxiredoxin 1 overexpression protecting these cells. Xenograft experiments with A549 cells showed reductions in tumor volume and weight after treatment with 25 and 50 mg/kg AMRI-59.
An alternate approach is the use of compounds that indirectly impact peroxiredoxins by inhibiting Trx, TrxR, and sulfiredoxin that are responsible for maintaining peroxiredoxins in their reduced form (105). Considerable effort has gone into the development and use of TrxR inhibitors (91, 163), and several approved chemotherapeutics, including arsenic trioxide (91), cyclophosphamide (163), and cisplatin (2), have been identified to inhibit TrxR. Other TrxR inhibitors that have been identified to have antitumor activity in vivo include auranofin (96) and motexafin gadolinium (95). The gold complex auranofin is a particularly effective TrxR inhibitor (35). Though initially approved for use in rheumatoid arthritis, it is currently being explored as an anticancer agent (67, 94, 96, 100). The Trx1 inhibitor 1-methylpropyl 2-imidazolyl disulfide (PX-12) has been found to inhibit tumor growth in a range of cancers, including breast (169), colorectal (159), liver (87), and lung (187) cancer, as well as acute myeloid leukemia (151) and multiple myeloma (136), but further development of this compound has been questioned (3). Since Trx plays an important role in many biological processes, it will be difficult to ascribe any effects directly to impairment of peroxiredoxin function. In contrast, the only known function of sulfiredoxin is to reduce hyperoxidized peroxiredoxins. The sulfiredoxin inhibitor J14 can cause accumulation of hyperoxidized peroxiredoxins and activate apoptosis with doses of 20 μM (65). In vivo xenografts of lung cancer cells in mice showed reduced tumor growth when administered 50 mg/kg J14.
Concluding Remarks and Future Challenges
It is clear that peroxiredoxins have a significant influence on the development and progression of cancer. Genomic disruption in mice often results in increased carcinogenesis, whereas elevated peroxiredoxin expression is commonly observed in human tumors. Knockout mice have germ line mutations and, therefore, do not reflect what happens during the initiation of human cancers, but even so, these two observations appear to be contradictory. They can be explained by a two-phase model in which oxidative stress is carcinogenic and therefore removal of an antioxidant will promote tumorigenesis, whereas established tumors and metastatic cells are under increased oxidative stress and require increased antioxidant expression to survive. This latter scenario may also explain how high-dose antioxidant supplementation can increase metastasis in animal models (84).
The role of peroxiredoxins in cancer is likely to be more complex than this. Peroxiredoxin expression is not consistently increased in tumors, and in some cases decreased peroxiredoxin expression is observed, or lower expression correlates with decreased survival. The peroxiredoxins are part of a network of enzymes that function together to maintain redox homeostasis, and the expression of individual family members has to be considered in the context of Trx and TrxR expression, NADPH levels, and other peroxide detoxification mechanisms. Also, the cellular actions of the peroxiredoxins are only just beginning to be revealed. Their oligomeric structures are more complicated than is required for reduction of hydroperoxides, and there is growing evidence that peroxiredoxins regulate signaling pathways through their physical interactions with other proteins, and are themselves regulated by post-translational modifications, including oxidation and phosphorylation. Clearly, more studies investigating peroxiredoxin biochemistry in conjunction with biological functions are needed. This is especially important if novel therapeutic avenues building on peroxiredoxin inhibition are pursued.
Knockout studies indicate that there is little redundancy between the peroxiredoxins, despite the different family members having similar catalytic activity. Both peroxiredoxin 1 and peroxiredoxin 2 are abundant cytoplasmic proteins, but peroxiredoxin 2 is unable to compensate for the loss of peroxiredoxin 1, and vice versa. This may be because the two proteins have different sets of interaction partners and different signaling roles. An increased understanding of peroxiredoxins as antioxidants and signaling proteins will therefore be important for interpreting the mechanisms underlying the knockout and expression data.
It is commonly stated that tumor cells either generate or are exposed to higher levels of oxidants (150, 154). Determining whether this translates to increased oxidative damage usually relies on measurement of endpoint markers such as DNA oxidation or increased oxidation of exogenous probes. The typical 2-Cys peroxiredoxins (peroxiredoxins 1–4) provide alternative biomarkers of redox homeostasis (24, 129). Since oxidation results in an intermolecular disulfide, Western blotting of non-reducing gels enables detection and quantification of peroxiredoxins that are in their reduced and oxidized forms. The peroxiredoxins are mostly reduced in cells, but the addition of small amounts of hydrogen peroxide (22) or redox-active compounds (6, 24), ischemia-reperfusion injury (80), or initiation of cell death signaling (6, 7, 23) can cause transient or sustained oxidation depending on the cell type and stimulus. A challenge in this regard is a proper experimental redox analysis of peroxiredoxins. The peroxiredoxins have to be alkylated before cell lysis to prevent artefactual oxidation, thus samples have to be collected fresh (24, 32). To date, the methodology has not been applied to the assessment of tumor samples, but this information would complement expression studies.
Similarities of peroxiredoxins can be found with the tumor suppressor p53. Both are stress responders; their loss in vitro and in vivo promotes malignant transformation, and a loss of heterozygosity accelerates oncogenesis. In contrast to p53, which is found to be mutated in more than 50% of human tumors, peroxiredoxin 1 is rarely found to be mutated in cancer (Table 2) (37), though its deletion rate is low and quite similar to p53 (not shown). Also, research of the past 15 years indicates that many p53 mutants act as oncogenic proteins (148). Thus, it is possible that instead of acquiring oncogenic mutations, peroxiredoxins may undergo post-translational modifications such as phosphorylation or oxidation, thereby supporting oncogenesis. Future studies are needed to address this possibility.
The Cancer Genome Atlas data sets that are publically available in the cBIOPortal (
Finally, we need to examine whether the peroxiredoxins qualify as drug targets for the treatment of cancer. The proposal is that if cancer cells are more reliant on their antioxidant defenses for survival, then inhibition of these defense systems would have a more significant impact on malignant rather than normal cells. The evidence for this proposal is still lacking but warrants more in-depth investigation. It will be important to know that secondary malignancies and anemic phenotypes do not develop as a consequence of peroxiredoxin inhibition, or whether peroxiredoxins perhaps play a role in cancer cell dormancy. Some of the current generation of inhibitors lack selectivity for the peroxiredoxins, and for others it is unclear as to how exactly they are functioning. Thus, further studies should compare the role of individual peroxiredoxins in tumor initiation versus tumor progression. The development of inhibitors with sub-micromolar potency against specific peroxiredoxin family members will be important for exploring the feasibility of this strategy.
Footnotes
Acknowledgments
The authors would like to thank Prof. Christine Winterbourn for her critical reading of the article; and the University of Otago and the Health Research Council of New Zealand for funding.