
Abstract
Significance:
Head and neck squamous cell cancer (HNSCC) is a complex disease characterized by high genetic and metabolic heterogeneity. Radiation therapy (RT) alone or combined with systemic chemotherapy is widely used for treatment of HNSCC as definitive treatment or as adjuvant treatment after surgery. Antibodies against epidermal growth factor receptor are used in definitive or palliative treatment.
Recent Advances:
Emerging targeted therapies against other proteins of interest as well as programmed cell death protein 1 and programmed death-ligand 1 immunotherapies are being explored in clinical trials.
Critical Issues:
The disease heterogeneity, invasiveness, and resistance to standard of care RT or chemoradiation therapy continue to constitute significant roadblocks for treatment and patients' quality of life (QOL) despite improvements in treatment modality and the emergence of new therapies over the past two decades.
Future Directions:
As reviewed here, alterations in redox metabolism occur at all stages of HNSCC management, providing opportunities for improved prevention, early detection, response to therapies, and QOL. Bioinformatics and computational systems biology approaches are key to integrate redox effects with multiomics data from cells and clinical specimens and to identify redox modifiers or modifiable target proteins to achieve improved clinical outcomes. Antioxid. Redox Signal.
Introduction
A
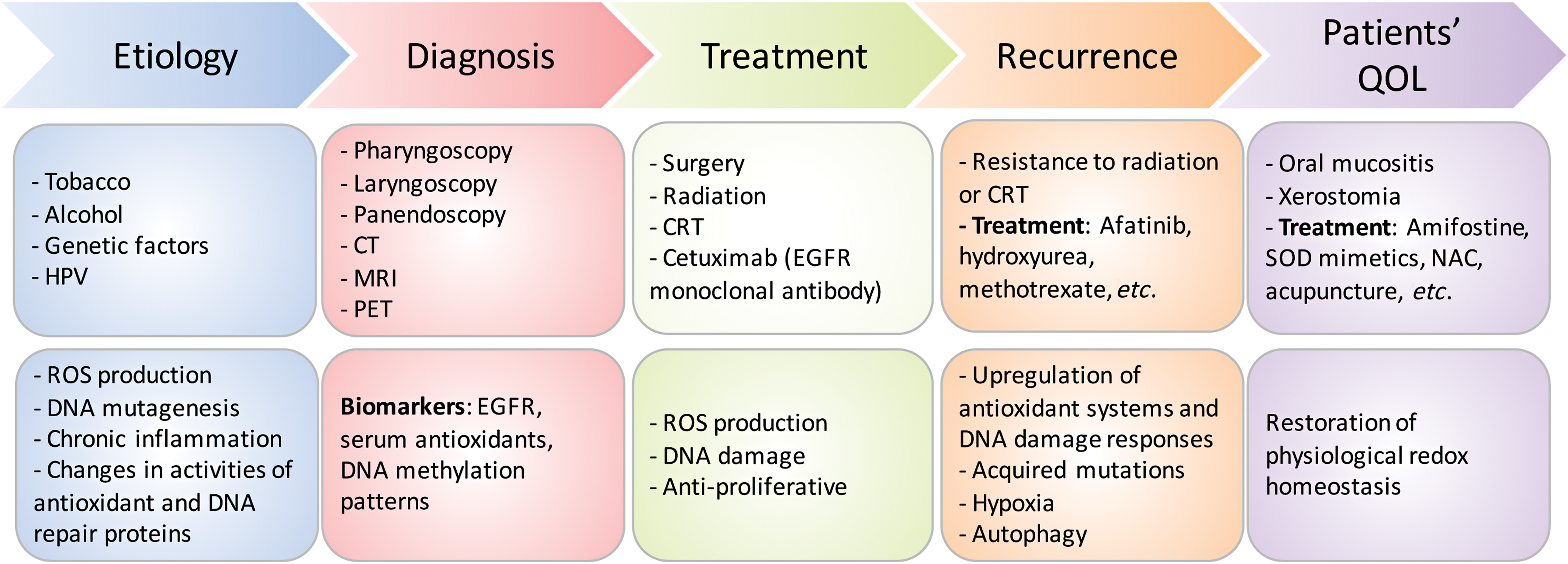
Prevention: Redox Effects in HNSCC Etiology
Multifactorial interactions between environment and genetic mutations play a critical role in the development and complexities of staging HNSCC. Key etiologic factors associated with HNSCC are tobacco smoking, alcohol consumption, and genetics. More recently, human papillomavirus (HPV) emerged as an important etiologic factor, in particular for oropharyngeal HNSCC (91, 242, 306). These environmental stressors induce acute or chronic shifts in redox homeostasis promoting inflammation and neoplastic transformation.
Tobacco smoke
It is estimated that 42% of the HNSCC deaths worldwide can be attributed to smoking alone (153). Studies show that each puff of smoke contains 5000 carcinogenic compounds that produce 1015 free radical molecules in the gas phase (121). Once inhaled, these compounds are metabolized either by the cytochrome P450 (CYP) super family of proteins (CYP1A1, CYP1A2, CYP2E1, and CYP2A6) leading to DNA adduct formation or by glutathione S-transferases (GSTs; GSTM1, GSTT1, and GSTP1) leading to secretion (85). The activities of both CYP and GST families of enzymes depend on the availability of reducing equivalents controlled by the nicotinamide adenine dinucleotide phosphate [NAD(P)H]/NAD(P)+ or reduced/oxidized glutathione (GSH/GSSG) redox couples, respectively. Exposure to cigarette smoke increases oxidative damage rate by 30–50% (183), inducing either cell death by activation of ceramide-mediated apoptosis or cell proliferation by activation of epidermal growth factor receptor (EGFR) (120). Prolonged exposure to cigarette smoke also leads to increased GSSG, which has two consequences: (i) limits the availability of reduced GSH needed for the detoxification activity of GSTs and (ii) activates inflammatory pathways, including nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) and nuclear factor-erythroid 2-related factor 2 (NRF2) (85, 111). These result in increased gene expression, release of cytokines (121), and onset of chronic inflammation. The shift to a more oxidative environment could potentially further enhance the activity of EGFR through oxidation at Cys797 in the active site of this enzyme as reported in recent studies (229) and further detailed hereunder.
Alcohol consumption
Alcohol consumption in conjunction with tobacco smoking increases the risk for HNSCC (91, 242, 306). Excessive alcohol consumption increases mucosa permeability to toxins and reduces epithelial thickness, leading to damage of vital structures and tumor formation (193). Ethanol, for example, can be metabolized to carcinogenic acetaldehyde by alcohol dehydrogenase in the mitochondria or NADPH-dependent CYP2E1 in the microsomal ethanol oxidizing system (187). Catalase (CAT), an antioxidant enzyme with the main function of catalyzing H2O2 dismutation into H2O and O2, can also oxidize ethanol to acetaldehyde in peroxisomes in the presence of H2O2. Similar to ROS species, acetaldehyde interacts with proteins and nucleic acids to form adducts, which interfere with critical cellular functions such as DNA synthesis and repair (43).
Genetic factors
The relationship between genetic factors and ROS metabolism is not inherently obvious, but a strong case can be made by the data summarized here. Variant alleles of genes involved in carcinogen metabolism, alcohol and folate metabolism, and DNA repair and cell cycle control are thought to play a role in development of HNSCCs. Meta-analyses have focused on carcinogen metabolism enzymes such as CYP super family of enzymes and the antioxidant enzymes GSTs, introduced previously (2, 169). For example, several studies demonstrated an increased risk of HNSCCs in individuals with GSTP1 I105V mutation associated with increased DNA oxidative products (156, 198, 225). Presence of the null GSTMμ1 genotype versus the positive genotype also increases the risk for HNSCC (225). Genetic variants of cell cycle control gene, TP53, are also common in HNSCCs and increase HNSCC risk by 75% (36, 185). The G to C polymorphism in codon 72 of exon 4 results in an arginine to proline substitution in TP53. Although both variants are wild-type (WT), the proline/proline genotype was shown to be less effective in suppressing cellular transformation and showed a higher risk for HNSCC than individuals with the arginine/arginine genotype (36, 185). Striking ethnic differences have been observed in the frequencies of these variants, with the proline allele showing a latitude gradient from 0.17 in a Swedish population to 0.63 in a Nigerian population (18, 274). The role of 1-C/folate metabolism in maintaining the reduced NADPH pool has been reported in several publications (102, 289). Relationships between polymorphisms in folate metabolism enzymes (e.g., methylenetetrahydrofolate reductase [MTHFR], serine hydroxymethyltransferase [SHMT]), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and immune response factors (e.g., interleukin [IL]-8, toll-like receptor 10 [TLR10]), and HNSCC risks have been investigated (115, 218, 246, 304). However, the number of such studies is limited, and it is difficult to draw definite conclusions.
Human papillomavirus
The incidence of HNSCCs caused by tobacco and excessive alcohol consumption is decreasing, whereas there is an increase in incidence of HNSCCs in younger white individuals and nonsmokers due to infection with “high-risk” HPVs (e.g., HPV subtype 16, which accounts for 87% of all HPV+ tumors) (51, 80). Of note, HPV− tumors do not necessarly imply virus-free HNSCCs and the tumors may be positive for other viruses such as EBV, HCV, HSV, or HIV. The HPV viral genes are divided into “early” genes (E1–7) and “late” genes (L1, L2). Two “early” genes (E6, E7) are considered tumorigenic. Mechanisms involve suppression of p53 and retinoblastoma protein (pRb) function and induction of oxidative stress (180, 196, 316) by activation of NADPH oxidase 2 (NOX2) (196) and by suppression of antioxidant proteins superoxide dismutase 2 (SOD2) and glutathione peroxidase 1/2 (GPX1/2) (316). Consequently, ROS accumulation in HPV+ cells contributes to DNA damage and genomic instability needed for HPV-induced tumorigenesis and synergizes with IR by forming clustered lesions that are highly lethal (196), in keeping with the clinical observation that patients with HPV+ head and neck tumors have a more favorable outcome after chemoradiation therapy (CRT) than patients with HPV− tumors (6, 101). In turn, a more oxidative environment promotes several stages of HPV infection, including viral entry, DNA replication, and integration into the host genome (83, 317). In the case of HNSCCs, it has been shown that HPV16 utilizes a tissue-spanning redox gradient to facilitate viral maturation, and a complete viral life cycle can occur in oral (tonsil) tissue (151). HPV vaccines such as Gardasil® and Cervarix®, which are currently being evaluated for prevention of HPV-related HNSCCs, (135) are expected to decrease the incidence of HPV+ HNSCCs though their efficacy under increased oxidative stress environments remains to be tested. Common diagnostic methods for HPV positivity in HNSCCs include detection of viral DNA with in situ hybridization or polymerase chain reaction (PCR), and detection of p16 (a surrogate marker for HPV infection) protein expression with immunohistochemistry [reviewed in Chai et al. (51), Mirghani et al. (206), and Venuti and Paolini (303)]. With each of these methods providing different information and having their own specific limitations, there is currently no consensus on the optimal way to identify HPV-related HNSCC.
Detection and Diagnosis: Redox Biomarkers in HNSCC Detection
HNSCC patients can present with various precancerous conditions and lesions depending on the tumor location within specific areas of the head and neck. The most common symptoms presented by HNSCC patients include chronic sore throat, difficulty swallowing, a change or hoarseness in the voice, and a lump or sore that does not heal. There are currently no biomarkers for early HNSCC detection. Although preclinical studies to identify possible markers for early cancer detection have been reported (e.g., EGFR, serum antioxidant CAT, and GSH levels), these remain exploratory (126). DNA methylation patterns appear gradually due to environmental influences (i.e., tobacco and alcohol use), and are thus considered as potential source of biomarkers. Epigenetic modifications are essential for regulation of cell cycle control (i.e., p16 and p14), DNA repair, and apoptosis. Longitudinal studies reported higher hypermethylation of p16 in precancerous lesions leading to increased tumor progression compared with hypomethylation of p16 in precancerous lesions leading to tumor regression (46, 131). Clearly, the GSH levels and epigenetic DNA methylation are mechanistically connected as S-adenosylmethionine (SAM), the substrate for DNA methyltransferases and other methyltransferase enzymes, is synthesized from methionine, which is also part of the GSH biosynthesis through the redox-regulated transsulfuration pathway (136). Given the redox shifts associated with the etiology of HNSCC supported by the value of utilizing CAT and GSH antioxidant biomarkers for early detection, a need for development of redox positron emission tomography (PET) imaging methods for HNSCC early diagnosis is emerging. This has not yet been explored in preclinical or clinical studies in relation to HNSCC. Such PET imaging probes may include [18F]fluorothymidine probe for H2O2 or other yet unexplored biomarker indicators of redox shifts (48).
Treatment: Redox Modulators of Standard of Care and Emerging Therapies for HNSCC
Standard of care
The treatment plan for patients with HNSCC is determined from three parameters: (i) location of tumor, (ii) stage of cancer, and (iii) person's age and overall performance status regardless of HPV status (89, 92). Surgery followed by fractionated radiotherapy is the standard of care for resectable primary and secondary malignancy with the goal of obtaining tumor-free surgical margins (132). However, negative surgical margins often result in removal of normal tissue causing impairment of critical functions, such as chewing and swallowing, and an adverse QOL (132). In many cases, due to presence of high risk of relapse factors such as positive margins and/or the presence of extracapsular invasion of the positive lymph nodes by cancer cells, surgery is followed up with aggressive CRT to kill remaining tumor cells. Patients generally undergo fractionated doses of 2 Gy each in 5 weekly sessions for 6–6½ weeks for a total dose of 60–66 Gy (27). Patients with unresectable tumors or on whom an organ sparing approach is possible receive radiation therapy (RT) or most often CRT with an even higher dose of RT of 70–72 Gy for 7 weeks (116).
Based on large randomized clinical trials and meta-analysis, cisplatin is considered the standard radiosensitizing agent for definitive or adjuvant RT. When used in combination with radiotherapy, cisplatin is given at 100 mg/m2 every 3 weeks during the course of RT (15). However, in recurrent tumors or for palliative care, other chemotherapeutics such as taxanes, hydroxyurea, and the antifolates methotrexate or pemetrexed have been utilized as well as radiosensitizers (129, 258). Antifolates, such as methotrexate and the newer drug pemetrexed, were reported to sensitize tumors to RT in both preclinical and clinical studies (149, 199, 257). These drugs inhibit the enzyme dihydrofolate reductase, which is essential for DNA synthesis and connects the 1-C/folate metabolism to NAD(P)H/NAD(P)+ balance, GSH biosynthesis, ROS and epigenetics through biosynthesis of SAM, a substrate for DNA methyltransferases, already described. Consistent with this notion, methotrexate also exhibits potent immunosuppressant activity through ROS accumulation and decreased GSH biosynthesis (234). Normal tissue toxicities associated with methotrexate or pemetrexed therapy are typically reduced in the clinic by coadministration of folate/B9 and vitamin B12 supplements, without compromising the efficacy of treatment (41, 69, 219). What all these chemotherapeutics have in common is the connection to the central redox metabolism network, which sets the balance between the intracellular ROS (E0 = approximately −0.3 to 2.5 V; e.g., H2O2 + 1.78 V) and the NAD(P)H/NAD(P)+ (E0 = −0.32 V) reserve (245). Depletion of the NAD(P)H pool and the ensuing increase in ROS kill cancer cells by DNA damage and induction of cell death pathways.
Therapies targeting EGFR
EGFR is overexpressed in 90% of HNSCC patients (19, 222), making EGFR a possible target to address locally advanced or metastatic HNSCCs (116). EGFR is a transmembrane glycoprotein with tyrosine kinase activity (91) serving as a critical signaling hub for tumor cell growth, angiogenesis, and invasion (Fig. 2). Currently, cetuximab (Erbitux®) is the only targeted therapy approved by the Food and Drug Administration (FDA) to be used in combination with RT for locally advanced HNSCC (38). Cetuximab is a recombinant immunoglobulin G1 (IgG1) monoclonal antibody directed at the extracellular domain of EGFR to block ligand-mediated activation of the EGFR pathway (288) (Fig. 2). It has been reported that cetuximab combined with RT improves progression-free survival (PFS) from 14.9 to 24.4 months and median OS rates from 20.3 to 54.0 months, but does not decrease rates of distant metastases in patients with locally advanced HNSCCs compared with RT alone (7, 15). Cetuximab is also FDA approved for use as a single agent or in combination with chemotherapy in metastatic HNSCCs. Cetuximab combined with platinum and 5-fluorouracil (5-FU) was tested in the EXTREME clinical trial. The study showed improved outcome in all end points including (i) increased PFS from 2.7 to 4.2 months, (ii) increased OS from 8.0 to 9.2 months, (iii) 1 -year survival rate from 31.7% to 38.6%, and (iv) locoregional control from 60% to 80% with no compromise in QOL (266). Other monoclonal antibodies, similar to cetuximab such as nimotuzumab (128, 186), zalutumumab, duligotuzumab (104), and panitumumab, have proved promising in preclinical research. One significant take away from the duligotuzumab study is that HPV− patients had higher response rates to both cetuximab and duligotuzumab than HPV+ patients (104). In general, newly developed EGFR monoclonal antibodies combined with radiotherapy or CRT have not improved oncologic outcomes or demonstrated superiority over CRT but promising attempts at enhancement continue.

Emerging therapies for HNSCCs
Despite decades of research, the best treatment modality yielding significant efficacy with reduced side effects, improved organ function, and overall QOL has yet to be determined. Most research has focused on identifying radiation-sensitizing therapies with decreased side effects. There are many new emerging therapies on the horizon, including anti-EGFR monoclonal antibodies already described, tyrosine kinase inhibitors (TKIs), and other targeted drugs, as well as immunotherapies.
Tyrosine kinase inhibitors
EGFR TKIs target the receptor catalytic domain (Fig. 2). Overall, lack of survival benefit for EGFR TKIs alone or in combination with radiotherapy or CRT in clinical trials has hindered their FDA approval for HNSCCs. For example, erlotinib (Tarceva®) and gefitinib (Iressa®) clinical trials have shown modest single agent response rate of only 4% and 10%, in recurrent or metastatic HNSCCs. However, in combination with cisplatin, the response improved to 21% but the combination of cisplatin, radiation, and erlotinib has not been well tolerated in HNSCC clinical trials (273, 281). Second generation EGFR TKIs, such as afatinib (Giotrif®), have shown increased cellular potency against EGFR (98, 204). Afatinib is an irreversible pan-EGFR TKI targeting Cys797 in the active site of EGFR. Studies have shown, however, that this cysteine site is also redox sensitive. Its oxidation to sulfenic acid state increases the activity of EGFR (229), and under chronic oxidative stress conditions induces resistance to afatinib (296). The impact of EGFR redox state on the response to afatinib and other targeted agents needs to be further investigated in the clinical context as RT or CRT may also induce oxidation of EGFR. There are at minimum three major redox mechanisms by which a drug could be rendered ineffective or result in unwanted toxicity when used in clinic (Fig. 3): (i) cancer drugs can be modified by tumor ROS (intrinsic or generated by cotherapies such as radiation or chemotherapeutics; e.g., erlotinib), (ii) molecular targeted drugs or the protein target itself may be modified by tumor ROS (e.g., EGFR), and (iii) drug-induced changes in tumor redox state can drive changes in the cellular phenotype (epithelial/mesenchymal, stem-cell like, etc.), which, in turn, would impact tumor progression and response to RT or CRT (14). Consideration of these effects along with others, which have been outlined recently (70), is expected to significantly improve the success rate of translating lead bench compounds into successful cancer therapeutics in clinic. Other TKIs targeting vascular endothelial growth factor (VEGF) and mechanistic target of rapamycin (mTOR) are being studied but have not provided any evidence of significant therapeutic efficacy in HNSCC patients. Tools to better predict patient responses to EGFR inhibitors will provide new opportunities for increased efficacy and newly identified mutations in the EGFR catalytic domain that confer sensitivity to EGFR TKIs promise to open new doors to use EGFR TKIs in the future.

Exploiting NAD(P)H quinone oxidoreductase 1 bioactivatable drugs for radiosensitization of HNSCCs
The powerful potential of quinone oxidoreductase 1 (NQO1) bioactivatable drugs to sensitize NQO1 overexpressing human head and neck cancer cells in culture and xenografts in athymic nude mice to radiation was recently reported (177), consistent with findings in other NQO1 overexpressing human cancer cells (39, 40, 93, 177, 226). Importantly, synergy noted between radiation and NQO1 bioactivatable drugs avoids the only normal tissue toxicity noted with these drugs, methemoglobinemia (a condition characterized by elevated levels of hemoglobin that contains the ferric [Fe3+] form of iron). As these series of drugs are relatively new and are tightly connected to both ROS metabolism and NAD(P)H reserve, we present this class of compounds in more detail hereunder.
NQO1 expression specificity
In normal tissue, NQO1 plays a role as a Phase 2 detoxification enzyme, performing two-electron oxidoreduction of most quinones. The enzyme converts quinones to their hydroquinone forms, which are then conjugated within the cell and exported. However, there are a few specific quinones that undergo a rapid futile cycle response, whereby NQO1 creates an unstable hydroquinone that spontaneously and rapidly converts back to its original compound using two oxygenation steps and creating two molecules of superoxides (Fig. 4) (235). For example, mitomycin C and streptonigrin can be converted to active drugs by NQO1, but these agents are not solely dependent on NQO1 and they are converted to active DNA-modifying agents through this reaction (267). There are currently three classes of futile cycle-bioactive quinones: (i) deoxynyboquinones (DNQ) (147), (ii) KP372 agents (328), and (iii) β-lapachone (235) with β-lapachone being the prototype drug (235). The term “NQO1 bioactivatable compounds” has been applied to these specific quinones (52 –54, 147, 148, 209). In the NQO1 catalyzed reaction, β-lapachone treatment uses 60 moles NAD(P)H and creates 120 moles of superoxide within a 2 min period (Fig. 4). DNQ and its related derivatives cause five- to eightfold more superoxide and NAD(P)+ formation in the same timeframe (147). Measurements of superoxide formation and NAD(P)+ formation for KP372 have not been determined (328).
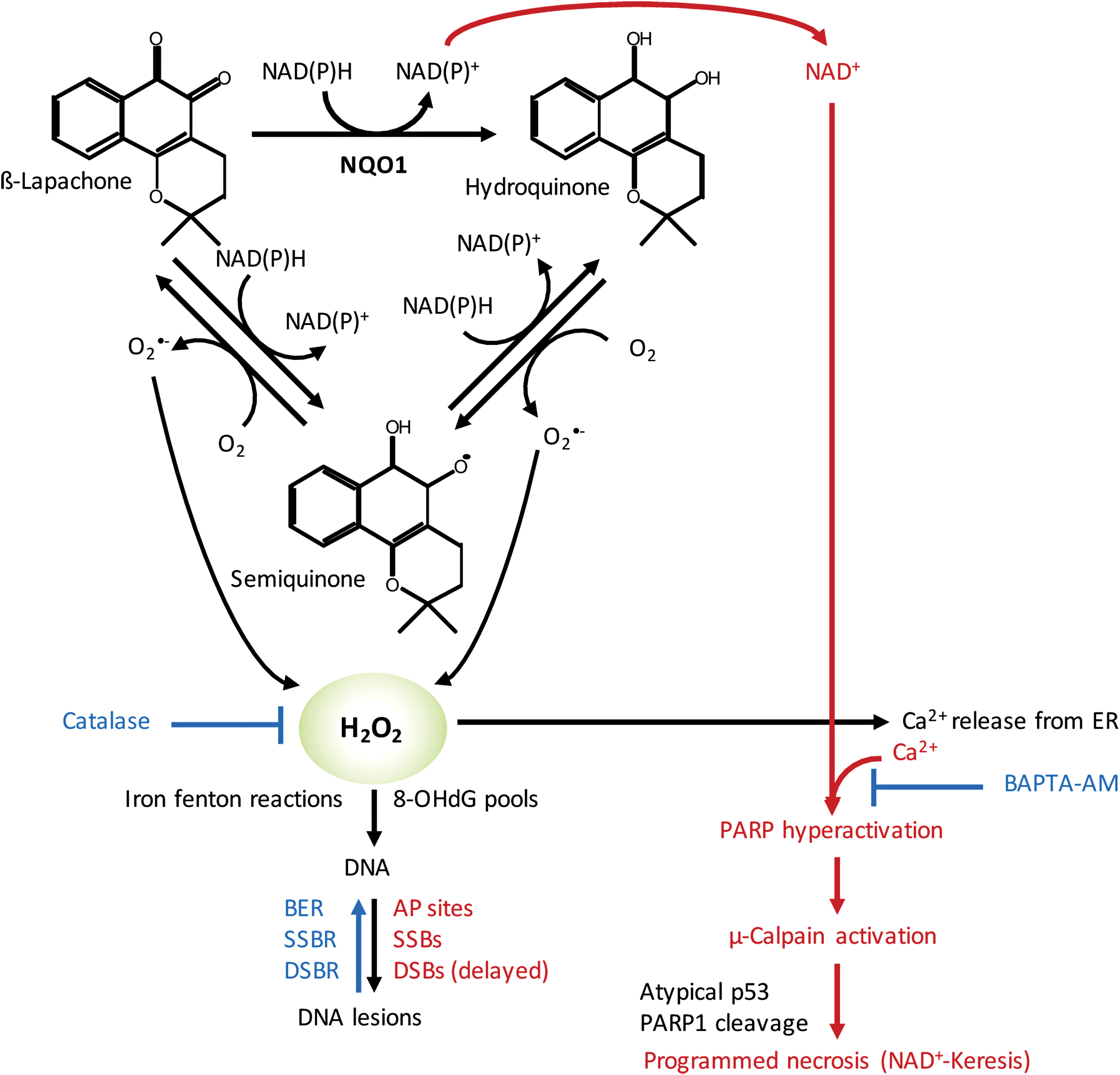
NQO1-dependent calcium release
A key feature of the cell death response caused by NQO1 bioactivatable drugs is that calcium release from otherwise inert central endoplasmic reticulum (ER) core stores is required for the specific programmed necrotic pathway referred to as NAD+-Keresis (177, 284, 285). Cell death does not require extensive formation or accumulation of poly ADP ribose (PAR) moieties generated by PAR glycohydrolase (PARG), since coadministration of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors and loss of NAD+ levels increased programmed necrosis induced by β-lapachone or isobutyl-deoxynyboquinone (IB-DNQ) (147, 177, 209). Instead, pretreatment with a specific calcium chelator, such as BAPTA-AM, caused the specific inhibition of NQO1-dependent cell death by these bioactivatable quinones (Fig. 4) (147, 177, 284, 285).
CAT detoxification
In HNSCCs, as well as in pancreatic, lung, prostate, and breast cancers, CAT levels were found to be lower in tumor than in associated normal tissue within human patients (52 –54, 148, 177). This generates an extensive therapeutic index for these cancers that can be exploited using NQO1 bioactivatable drugs (52 –54, 148, 177). Forced overexpression of CAT (>1000 U of cell membrane permeable polyethylene glycol [PEG]-CAT) can suppress NQO1-dependent DNQ or β-lapachone lethality in these cancers (Fig. 4); however, these levels are well beyond CAT expression typically seen in these cancers under endogenous conditions. This strongly suggests that NQO1 can generate more than “saturable” levels of H2O2 within human cancers that overwhelm the enzyme's capacity for degrading H2O2. The accumulated H2O2 can pass through membranes and easily damage neighboring NQO1-deficient cancer cells (29). In contrast, normal cells plated along side NQO1 overexpressing cancer cells are protected due to their elevated CAT levels. These findings supported by in vivo data suggest a potent NQO1-dependent bystander effect capable of suppressing the emergence of NQO1 cells growing from mixed cultures (47). Such bystander effects are likely to play a role in the radiosensitization responses observed in HNSCC xenografts (177).
DNA damage created by NQO1 futile recycling
DNA damage analyses of NQO1+ overexpressing cancer cells exposed to β-lapachone or IB-DNQ (or DNQ) revealed extensive DNA lesion formation by alkaline comet analyses, whereas little to no DNA lesions were noted after the same treated cells were examined using neutral comet assays (23, 24, 54, 93, 147). These data showed that the initial DNA damage created by NQO1 bioactivatable drugs is DNA single-strand breaks (SSBs) and apurinic/apyrimidinic (AP) site generation from incorporation of 8-oxo-deoxyguanine (8-OHdG), a result of H2O2-induced 8-oxoguanine-dGTP pools (54, 147, 177), as well as other altered bases. H2O2-induced Fenton reactions are also likely to ultimately induce DNA SSBs and double-strand breaks (DSBs). However, these lesions appear to be protected by hyperactivation of poly ADP ribose polymerase (PARP), which occurs immediately after exposure of cells to β-lapachone or IB-DNQ. PARP hyperactivation in this context is prevented by BAPTA-AM or dicoumarol (NQO1 inhibitor) and is not apparent in NQO1-deficient cells (e.g., resulting from NQO1 polymorphisms, *2 [C609T] and *3 [C465T]) (148, 177). Importantly, PARP1−/− cells lack these responses, but are not immune to overall cell death after exposure to NQO1 bioactivatable drugs. Instead, NQO1+/PARP1−/− cells treated with NQO1 bioactivatable drugs, or NQO1+ cancer cells exposed to PARP inhibitors, undergo a synergistic apoptotic cell death responses caused by DNA SSBs converted to DSBs due to inaccurate DNA replication, without a major loss in NAD+ or adenosine triphosphate (ATP) (148).
PARP hyperactivation and subsequent μ-calpain-dependent programmed necrosis
The massive level of DNA lesions, in concert with Ca2+ release from the ER, induces hyperactivation of PARP in NQO1+ cancer cells exposed to the NQO1 bioactivatable drugs. Increased NAD+ pools in these cells resulting from the oxidation of NADH in the futile cycle (Fig. 4) are rapidly degraded by the hyperactivated PARP. Such responses are not present in NQO1-deficient cells or in exposed NQO1+ cancer cells treated with dicoumarol, a fairly specific NQO1 inhibitor. NAD+ pools, aside from the sequestered mitochondrial NAD+ level, are degraded within 20–35 min in most NQO1+ cancer cells, leading to concomitant ATP losses within 30–40 min, μ-calpain activation with 8–24 h, terminal deoxynucleotidyl transferase dUTP nick end labeling positive staining within 24–48 h, and loss of attachment and programmed necrosis within 72 h of exposure to NQO1 bioactivatable drugs (Fig. 4) (23, 24, 28, 47, 52 –54, 93, 176, 235, 236, 284, 285).
Combined radiation and NQO1 bioactivatable drugs
IR creates a spectrum of DNA lesions, including DNA DSBs, SSBs, AP sites, and DNA-protein crosslinks. DSBs are potentially the most lethal lesions, with only one DSB able to cause lethality (22, 50). The formation of DSBs among a range of SSBs and AP sites (also known as a multidamaged site) is enough to synergistically hyperactivate PARP when treated immediately with NQO1 bioactivatable drugs after exposure to IR. The formation of SSBs and AP sites after radiation, to which PARP binds with high affinity (239), synergistically interacts with the DNA lesions created by NQO1 bioactivatable drug exposure (exclusively SSBs and AP sites) to hyperactivate PARP activity (93, 148). Indeed, efficient synergy responses were noted with five doses of 1 Gy radiation and 5 mg/kg β-lapachone intravenous treatments (∼10-fold below the maximum tolerated dose of β-lapachone alone). As already mentioned, hyperactivation of PARP leads to massive NAD+ and ATP losses, which then prevent the repair of DSBs and greatly suppress NAD+, ATP, and, in theory, carbon metabolism (which has not been monitored to date). As a result, NQO1+ cancer cells, including from HNSCCs, can be efficiently killed by radiation and NQO1 bioactivatable drug combination therapy. The cell death pathway occurs specifically in NQO1+ cancer cells, independent of any oncogenic drivers, and causes a unique programmed necrotic death, NAD+-Keresis (93, 148, 177, 209). Similar to the treatment with the NQO1 bioactivatable drug alone, responses are only noted in NQO1 overexpressing cancer cells (177) with no responses in NQO1-deficient cells or normal tissue (93, 177). The therapeutic responses were also independent of the HPV or p53 status (177).
Future studies
The exact metabolic changes occurring in NQO1+ cancer cells after exposure to radiation and NQO1 bioactivatable drug treatments are not known. The role of released and circulating calcium in PARP hyperactivation, cell metabolism, and DNA repair has not been elucidated and is extremely important for radiation responses, since pretreatment with BAPTA-AM can suppress the synergistic response of combined radiation and NQO1 bioactivatable drug therapies (93, 177). Understanding the roles of genes that can suppress the responses is also unexplored, and genes involved in base excision repair (BER), DSB repair, ROS scavenging, and suppressing calcium ER release signaling are the most likely to strongly affect the response in NQO1+ cells. Currently, there are four clinical trials using a β-lapachone prodrug (as ARQ761) ongoing or planned. The monotherapy trial with ARQ761 is just about completed and responses to the agent were only noted in NQO1-expressing cancers, with no or little responses noted in NQO1-deficient cancers, most notably small cell lung cancers. Ongoing is a clinical trial treating NQO1+ pancreatic cancer patients in combination with abraxane and gemcitabine, wherein synergy was previously noted (162). Soon to open clinical trials are with sensitizing agents, including methoxyamine (54) and PARP inhibitors (148). Finally, another more potent NQO1 bioactivatable drug IB-DNQ should enter clinical trials within 1–2 years.
Immunotherapy
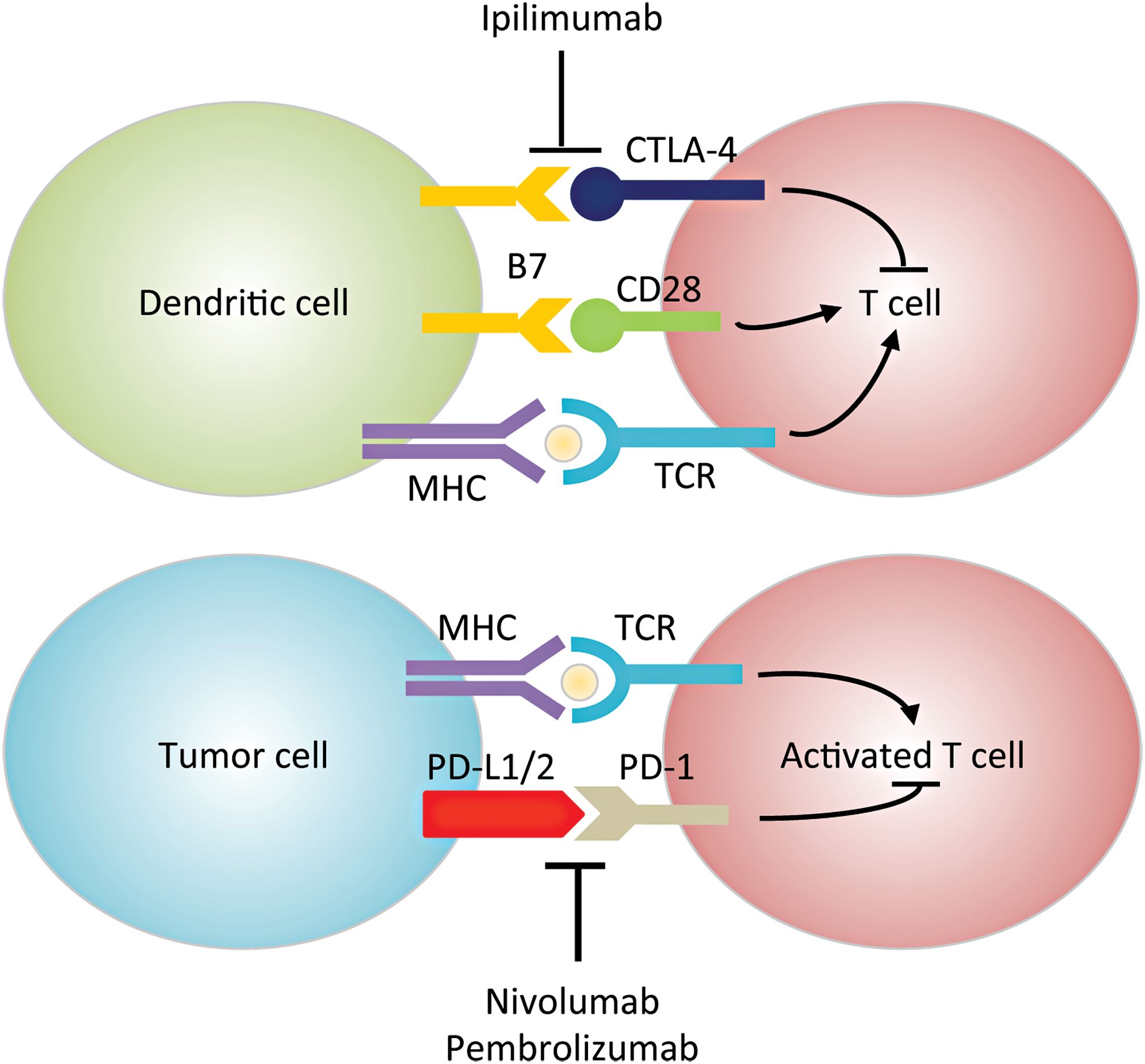
HNSCC is considered an immunosuppressive disease because of its ability to (i) dysregulate the cytokine profile (e.g., chemokine and VEGF overexpression), (ii) impair immune effector cell functions (e.g., upregulating IL-6), and (iii) cause abnormalities in tumor-associated antigen presentation (107). The interplay between the tumor and the host's immune system has become an increased area of research for HNSCC therapeutics. Tumor progression or relapse is believed to be associated with the inability of the immune system to eliminate cancer (255, 301). Immune checkpoints are critical in regulating T cell response, and coinhibitory molecules are new exciting targets of inhibition for HNSCC. The two most common targets include programmed cell death protein 1 (PD-1) or cluster of differentiation 279 (CD279), and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4 or CD152) (Fig. 5). Since ROS affects many functions of T cells such as activation, differentiation, and apoptosis [reviewed in Belikov et al. (20), Chen et al. (59), and Nathan and Cunningham-Bussel (215)], modulation of ROS levels could potentially enhance or impede immunotherapy efficacy. For example, it has been shown that increasing ROS synergizes with the tumoricidal activity of PD-1 blockade by strongly activating mitochondrial function of tumor reactive cytotoxic T lymphocytes (56). This activation signals through mTOR, AMP-activated protein kinase (AMPK), and peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α). Small-molecule activators of AMPK and mTOR, or PGC-1α, also synergize with the PD-1 blockade therapy, whereas none of these activators alone has any effects on tumor growth. These findings provide novel combinatorial treatment strategies for patients who are less responsive to PD-1 immunotherapy alone. In contrast, as shown using multiple graft-versus-host-disease models, PD-1 signaling promotes apoptosis in alloreactive T cells by increasing ROS in a fatty acid oxidation-dependent manner, whereas PD-1 blockade, which decreases ROS, renders cells less susceptible to subsequent metabolic inhibition (292). Based on these findings, adjuvant therapies relying on increased ROS levels (e.g., glucocorticoids, methotrexate, and cyclophosphamide) will likely be undermined in T cells after PD-1 blockade. There are currently two promising PD-1 inhibitors for HNSCC, pembrolizumab (Keytruda®) and nivolumab (Opdivo®), which have shown efficacy irrespective of tumor programmed death-ligand 1 (PD-L1) expression (>1%) or HPV status (106). As of now, no CTLA-4 monoclonal antibodies have FDA approval for HNSCC treatment (95). However, there are several open-label dose escalation clinical trials with durvalumab (anti-PD-L1 monoclonal antibody) and ipilimumab or tremelimumab against CTLA-4. Preclinical data also suggest that there is increased efficacy when PD-1 and CTLA-4 immunotherapies are combined because they regulate T cell induction and maturation at different phases (95).
Resistance to Standard of Care Therapy: Redox Controlled Mechanisms of Resistance to HNSCC Therapies
Suppression of ROS and upregulation of DNA-damage response have emerged as major mechanisms of resistance to CRT across cancers. Thus, it is not surprising that a high number of emerging drugs directly target these pathways (e.g., PARP, DNA-dependent protein kinase [DNA-PK], and BER). PARP1, in particular, impacts both ROS and DNA-damage response by consuming intracellular NAD+ to modify critical nuclear proteins by PARylation. Inhibition of PARP increases intracellular NAD+ reserve while decreasing the NADH/NAD+ balance in cancer cells and contributing to the synergy of interaction with CRT. In addition, the effectiveness of PARP inhibitors in tumors defective in DNA-damage repair is well documented (240, 272). EGFR is overexpressed in HNSCCs and thus constitutes an important therapeutic target both because of its function in cell proliferation and because of its recently discovered role in DNA DSB repair with PARP (213). Surprisingly, the redox state of EGFR in the tumor environment is not currently being considered as a prognostic indicator of response to EGFR-targeted therapies when used alone or in combination with CRT. Studies have shown a significant role of ROS, driven by NAD(P)H/NAD(P)+ ratio and cholesterol/lipid rafts content, in the response to both radiation treatment and EGFR inhibitor erlotinib in vitro and clinical tumor specimens of HNSCCs (14, 205). These and other redox-dependent mechanisms of resistance to RT and chemotherapies are presented hereunder.
Resistance to radiation in HNSCC
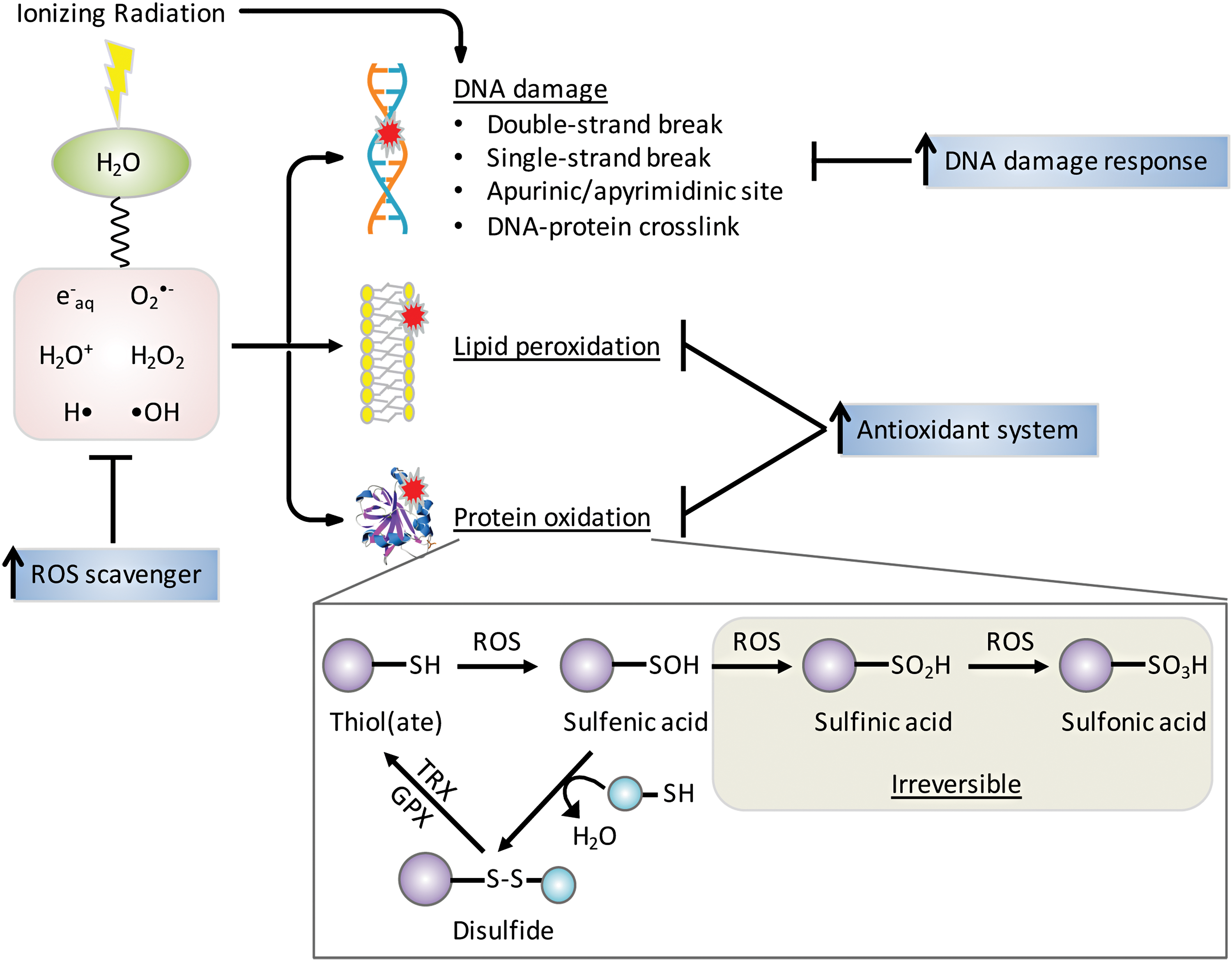
The mechanisms of damage by IR were reviewed recently (243) and are only briefly summarized here. The major types of IR are alpha and beta particles, X-rays, and gamma rays, and all are capable of causing cellular damage and are thus used for therapeutic purposes (87). The adult human body is composed of 60% of H2O that absorbs energetic radiations leading to ROS production (Fig. 6). Gamma irradiation of cellular H2O rapidly generates ROS, including hydroxyl radical (•OH) and ionized water (H2O+) as well as hydrogen radical (H•) and hydrated electrons (e− aq). Within one picosecond, O2 •− and H2O2 are formed as secondary ROS products and this chemical cascade generates cell-damaging molecules (271). These ROS and their progenies attack macromolecules (DNA, RNA, lipids, and proteins), induce mitochondrial ROS production, and upregulate NOX expression (11, 71, 94). Most IR-induced protein modifications are irreparable and critically affect protein stability. Reversible oxidation of cysteine amino acids in critical signaling molecules, such as kinases and phosphatases, can lead to amplification or dampening of signaling pathways and in some cases may synergize or antagonize the efficacy of targeted therapies when used in combination with RT. Under moderate oxidative stress, cysteine residues can be oxidized to relatively unstable sulfenic acid (-SOH), which can be further oxidized to -SO2H and -SO3H, or form mixed disulfides (S-thiolation) with other protein thiol groups (-SH) or low molecular mass thiols such as GSH (68). Disulfides are readily reduced to thiols by disulfide reductases in coupled reactions such as thioredoxin (TRX)/thioredoxin reductase (TR) or GPX/GSH/glutathione reductase (GR) with NADPH as ultimate provider of reducing equivalents (140) (Fig. 6). Loss or gain of function by disulfide formation is a regulatory mechanism by which proteins mediate the cellular response to oxidative stress. For example, phosphatase and tensin homologue (PTEN), which is mutated in HNSCC, has a catalytic Cys124 residue that when oxidized leads to disulfide bond formation with a neighboring Cys79 residue. The disulfide bond inactivates the phosphatase and protects the cysteine from hyperoxidation, enabling reactivation of PTEN by TRX (174). Redox-regulated proteins with signaling, metabolic, or epigenetic functions have been reviewed recently (86, 181, 224).
Radiation resistance is a major clinical problem for HNSCC patients compounded by origin, location, and tumor grade that limits tumor control and ultimately affects patient QOL (232). A tumor is considered radioresistant if there is either lack of tumor response or partial response resulting in recurrence a few weeks after an initial complete response. Tumor size, location, grading, and stem-like cell population play a critical role in driving radiation resistance. For example, larger, more differentiated tumors have a worse overall prognosis due to hypoxic cores, and differentiated tumors have the ability to accelerate repopulation capacity during radiotherapy (268). Rapid repopulation depends on the stem-like cells ability to activate checkpoints and DNA repair to enhance self-renewal and results in differentiation into heterogeneous cells (100). There are several well-studied mechanisms of resistance to radiation involving redox processes associated with hypoxia, autophagy, intracellular pathway alteration, and metabolic reprogramming, as described hereunder.
Hypoxia
The cytotoxic effects of radiotherapy depend heavily on the presence of molecular O2 to react with free radicals to produce ROS and irreparable DNA damage. However, under hypoxic conditions, O2 availability is low due to either reduced perfusion or starvation of necessary O2 and nutrients (144). It is estimated that HNSCCs contain three zones where O2 tensions fluctuate between anoxic (0% O2), hypoxic (1%/7 mm Hg O2), and normoxic (8%/60 mm Hg O2) (302). A study measuring oxygen partial pressure (pO2) in 28 HNSCC patients reported an 80% postradiotherapy PFS correlation with a median oxygen tension of pO2 > 10 mm Hg (42). Transcription factors, such as hypoxia-inducible factors (HIFs), respond to changes in O2 availability in the cellular environment, making them attractive targets in cancer therapeutics. HIF-1α is considered most active during initial intense short period of hypoxia (i.e., <0.1% O2 and <24 h), whereas HIF-2α is active under longer more chronic periods of hypoxia (i.e., <5% O2 and >24 h) (259). Elevated expression of HIF-1α and HIF-2α is associated with radiation resistance in HNSCCs (166). Unfortunately, there are limitations to accurately detecting hypoxia as most exogenous markers can only detect chronic hypoxia, and endogenous markers may be upregulated due to stress factors that are not hypoxia related (17). However, PET studies using (18F)–fluoroazomycin arabinoside (FAZA) to measure hypoxia and predict PFS showed that hypoxic tumors had a lower PFS of 60% than nonhypoxic tumors PFS of 93% (261). The clinical trial attempts at increasing tumor oxygenation before and during radiotherapy have included red blood cell transfusion, erythropoietin administration, and hyperbaric oxygen treatment, but all have ended in conflicting or inconclusive results (223). Inhibitors against HIFs such as acriflavine, YC-1, endostatin, and TNP-470 have poor toxicity profiles and lack increased efficacy (173, 188).
Autophagy
Autophagy is the cellular recycling mechanism responsible for degrading dysfunctional cellular organelles in living cells to provide building blocks for metabolites and synthesis of macromolecules as a source of energy. Cell survival or death depends on the severity and length of autophagy: although cells utilize autophagy as a prosurvival mechanism for maintaining cellular homeostasis under starvation or other stress stimuli, extensive or prolonged autophagy can activate apoptosis or apoptosis-independent autophagic cell death (81, 191). Many lines of evidence support a role of ROS in both protective autophagy and autophagic cell death. The most direct demonstration of a redox-regulated mechanism in autophagy involves oxidation and inactivation of autophagy-related protein 4 (ATG4). Specifically, starvation-induced mitochondrial H2O2 oxidizes ATG4 at Cys81, which inactivates its delipidating activity on ATG8, thus allowing for autophagosome formation (251). Other possible mechanisms of redox regulation of autophagy include (i) S-glutathionylation of AMPK (332), which inhibits mTORC1 and activates unc-51-like autophagy activating kinase 1 (ULK1) to promote autophagy (96, 130, 163) and (ii) upregulation of proautophagic protein Beclin-1 (60, 90). ROS is involved in autophagic cell death, wherein it can be upstream of autophagy (60), or is accumulated as a result of autophagy-mediated specific degradation of CAT (325). In addition, autophagy can regulate ROS through NRF2/Kelch-like ECH-associated protein 1 (KEAP1)/p62 interactions, where degradation of KEAP1 by selective autophagy facilitated by autophagic adaptor p62 releases transcription factor NRF2 to enter the nucleus, leading to subsequent expression of antioxidant proteins (165, 171, 286). Given the dual role of autophagy in cell survival and death and the complex interplay between autophagy and ROS, it is not surprising that autophagy has been regarded as both a tumor promoter and a tumor suppressor, and its role in cancer cell response to therapies can be both protective and cytotoxic (10, 61, 119, 175, 283, 290, 315, 323). Nevetheless, one popular hypothesis suggests that during early tumor development, autophagy is anticarcinogenic through removal of damaged organelles and protein aggregates (195, 208). For example, short-term exposure to arecoline, similar to nicotine, increases ROS and inflammatory cytokines, leading to autophagy activation and removal of DNA damage or protein aggregates to prevent tumor formation. After tumor formation or radiation-induced oxidative stress, autophagy is upregulated and used by the tumor as a survival mechanism (143). However, the exact mechanism by which autophagy contributes to radiation resistance is unclear. Some studies suggest autophagy is regulated by HIFs to clear ROS-producing damaged organelles and protein aggregates too large for proteasomal removal (327). Overall, autophagy seems to have a dual role in cancer development and progression, making it a potential therapeutic target and biomarker to determine tumor stage. It is worth noting that mitophagy, which refers to the selective degradation of damaged or surplus mitochondria, also crosstalks with ROS and plays a role in cancer progression (62, 167, 184). On the one hand, ROS, produced by damaged mitochondria, RT, or chemotherapy, may trigger mitophagy to protect cells from oxidative stress and cell death (34, 112, 310). Suppression of mitophagy leads to overproduction of ROS and further damage to the mitochondria, which then produces more ROS in a vicious cycle (168). In this regard, mitophagy serves as a tumor suppressor to maintain the integrity of the mitochondrial pool and cellular homeostasis, supported by the observations that loss or downregulation of several mitophagy proteins promotes tumor growth (26, 63, 64, 105, 114, 238). On the other hand, mitophagy provides a survival mechanism for tumor cells, which already have higher ROS than normal cells and are likely more dependent on mitophagy to manage ROS and maintain functional mitochondria (62), suggesting that inhibition of mitophagy represents a promising antitumor strategy in combination with conventional cancer treatment (i.e., mitophagy inhibitors as radio- or chemosensitizers). However, studies showed conflicting results as to whether it is inhibition or induction of mitophagy that kills cancer cells (167). Just as autophagy, mitophagy is a double-edged sword in cancer biology and further research is needed to correctly target mitophagy for cancer intervention.
Reprogramming of cellular signaling and metabolism
Radiation-induced cell death is accompanied by alterations in intracellular pathways primarily involved in DNA repair and cell cycle, proliferation, apoptosis, and metabolic reprogramming. These insults can lead to radiation resistance over time. For example, p53 regulates cell cycle arrest through cyclin-dependent kinase inhibitor 1 (CDKN1A) stimulation and programmed cell death in response to environmental stimuli. In the presence of cellular stressors, p53 may undergo post-translational modifications, including phosphorylation, acetylation, and PARylation that lead to its activation. During cell cycle arrest in G1 phase, cells are able to repair DNA before its replication to avoid propagation of nucleic acid alterations (307). However, p53 may not be able to rescue a cell after repetitive insults, leading to damage and apoptosis induction through upregulation of Bcl-2-associated X protein (BAX) and p53-upregulated modulator of apoptosis (PUMA) (324). It is estimated that 50% of HNSCC patients have small mutations in the gene encoding TP53 (i.e., nonsense, missense, insertions, or deletions of nucleotides), leading to inactivation or absence of protein (278). These alterations impair the cells ability to arrest cell cycle and activate apoptosis, leading to more genetic mutations, genetic instability, and clonal selection of radiation-resistant cells. The origin and location of HNSCCs in combination with the presence of TP53 mutations further increase tumor heterogeneity, resulting in decreased survival and increased locoregional failure. As previously described, EGFR overexpression is correlated with poor prognosis in HNSCCs. Radiation can induce autophosphorylation of EGFR, causing protection from apoptosis (252), increased cell proliferation, and tumor repopulation after radiotherapy (230). EGFR and p53 expression and activity in HNSCCs are extremely important to therapeutic response. For example, HPV+ tumors are generally characterized by WT p53 and lower EGFR expression than HPV− tumors (49, 170), resulting in high chemoradiosensitivity. The metabolic reprogramming of cancer cells through the Warburg effect is also mediated by HIF-1α. Activation of glycolytic enzymes, pyruvate dehydrogenase kinase 1 (PDK1) (164) and pentose phosphate pathway (PPP) glucose-6-phosphate dehydrogenase (G6PD), by HIF-1α causes downregulation of aerobic respiration and upregulation of the PPP, leading to increased production of NADPH and ribonucleotides (231, 311) to protect cells against apoptosis (327) during radiation-induced hypoxia and oxidative stress. HIF-2α also regulates the expression of cytochrome c oxidase (COX) and SOD2 to increase mitochondrial electron transport chain efficiency and suppressing ROS generation (122, 123). Thus, tumor cell signaling and metabolism are connected by key enzymes and transcription factors to enhance resistance to RT and chemotherapy by regulating energy production, proliferation, and apoptosis.
Upregulation of antioxidant system
Human body has developed complex antioxidant defense mechanisms against endogenous- and exogenous-induced oxidative stress (Fig. 7). When ROS increases, cellular transcription factors, NRF2 and NF-κB, are activated to help increase production of antioxidants (12). Enzymatic (e.g., SOD, peroxiredoxin [PRX], and CAT) and nonenzymatic [e.g., GSH and NAD(P)H] antioxidants are some of the most well-studied intracellular defenses against oxidative damage. In addition to their antioxidant properties, several of these molecules are increasingly recognized as signaling molecules (262).
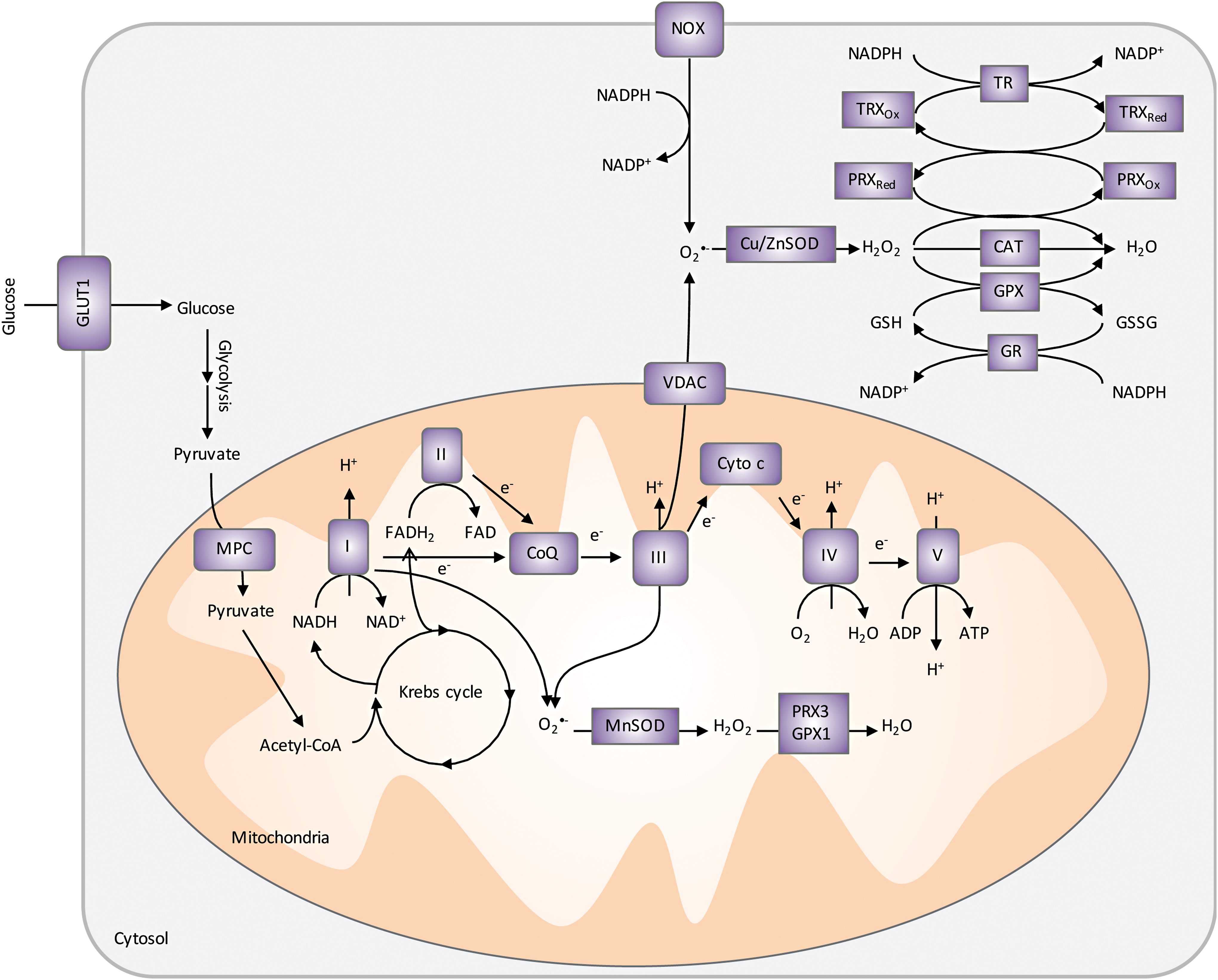
SODs are the major superoxide scavengers in the cells, catalyzing the dismutation of superoxide radicals to H2O2 and molecular oxygen. There are three forms of SODs, each encoded by a separate gene: CuZnSOD (SOD1) located mainly in the cytosol, MnSOD (SOD2) located in the mitochondrial matrix, and EcSOD (SOD3) located in the extracellular region of the cell. The role of SOD2 in cancer progression is controversial [reviewed in Holley et al. (137 –139) and Miriyala et al. (207)]. Although many studies show a reduction in SOD2 expression in tumors compared with normal tissues suggesting a tumor suppressor activity (65, 78, 146, 221), there is also abundant evidence supporting that SOD2 promotes cancer progression to a more aggressive stage (e.g., increased SOD2 activity was detected in advanced HNSCCs) (66, 145, 152, 192, 249, 294). This dichotomy is reflected in a study using a chemically induced skin cancer mouse model, in which SOD2 expression, mediated by transcription factors Sp1 and p53, was decreased in the early stage but increased at late stages of skin cancer (88). To reconcile the opposing roles of SOD2 in cancer progression, one has to take into consideration that by catalyzing the dismutation reaction, SOD2 not only controls levels of superoxides but also contributes to H2O2 accumulation in cells (44). In other words, SOD2 is both an antioxidant and a pro-oxidant, whose role in cancer progression is likely a result from concerted efforts with other antioxidant enzymes. In fact, based on this dual role of SOD2, strategies aiming at increasing SOD2 expression and/or activity have proven to be important adjuvants to radiotherapy, sensitizing tumor cells to radiation while protecting normal tissues, as discussed in Survival and QOL: Improving HNSCC Patients' QOL with Redox Modifiers section.
PRXs are a family of thiol-dependent peroxidases that catalyze the reduction of H2O2, alkyl hydroperoxides, and peroxynitrite (ONOO−). PRXs also function as regulators in growth factor signaling and cell cycle progression by virtue of their inactivation, which enables localized build-up of H2O2 and protein oxidation. In this case, the transient inactivation of PRXs occurs by either hyperoxidation (PRX-SO2/3) (319, 320) or phosphorylation at key tyrosine or threonine residues (318). For example, cells stimulated by EGFR can cause PRX1 association with lipid rafts and subsequent inactivation through selective phosphorylation at tyrosine 194 by Src kinase (318). The inactivation allows H2O2 to accumulate near lipid rafts and mediate cell signaling. Therefore, PRXs play a critical role as a sensor and transducer of H2O2 signaling in cells. It has been shown that oxidized PRX2 forms a redox relay with transcription factor signal transducer and activator of transcription factor 3 (STAT3), resulting in STAT3 oxidation and attenuation of transcriptional activity (275). In HNSCC cells, PRX2 expression increases in cells after treatment with radiation and its overexpression blocks cells from radiation-induced cell death (227). Importantly, STAT3 is also expressed at high levels in HNSCCs and current therapeutics such as cetuximab lacks the ability to abrogate STAT3 activity. Combined inhibition of EGFR and control of redox metabolism could provide a path to improve cetuximab and other EGFR-targeted therapies.
GSH is synthesized in the cytosol by sequential reactions catalyzed by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS). Cysteine availability is the rate-limiting factor in GSH biosynthesis and it can be either synthesized in cells from methionine through the activities of cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE or GCL) or imported by the cystine/glutamate transporter system (xCT). GSH donates reducing equivalents (H++e−) to molecules such as cysteine disulfides in oxidized proteins to shift their redox state to reduced forms. As a result, GSH becomes oxidized to GSSG, which can be either excreted from the cell or reduced by GR in an NADPH-dependent manner. In addition, glutathione's protective roles against oxidative stress include (i) acting as a cofactor of detoxifying enzymes, (ii) participating in amino acid transport, (iii) scavenging hydroxyl radical and singlet oxygen, and (iv) regenerating other antioxidants back to their active forms (197). The ratio of reduced GSH versus oxidized GSSG is an indicative measure of oxidative stress in the cell (154).
TRX is a disulfide-containing redox protein with two redox active cysteines that can be oxidized to a disulfide and present in all subcellular organelles (159). TRX is reduced to its active state TRX-(SH)2 by TR in an NADPH-dependent manner (312). In addition to its function in the PRX catalytic cycle, TRX plays critical roles in controlling cell growth through redox regulation of DNA synthesis, cell signaling, and apoptosis by regulating transcription factors and kinase cascades. For example, TRX acts as a reducing cofactor for ribonucleotide reductase, the first unique step in DNA synthesis and methionine sulfoxide reductase, which reduces methionine sulfoxide to methionine (13). TRX also controls redox-sensitive points during apoptosis, including activating caspases by reducing cysteines (297) and by binding to apoptosis signal-regulated kinase 1 (ASK1) to create an inactive complex that when oxidized is activated and released from TRX (293). Overexpression of TRX has been shown to protect cancer cells from oxidative stress-induced apoptosis and provide a survival and growth advantage to tumors (298). Owing to the widespread redundancy between the TRX and GSH systems, strategies often target both of them to achieve powerful and synergistic antitumor effects (21, 133, 160, 194). In addition, dual inhibition of the TRX and GSH systems sensitizes cancer cells to chemotherapeutics (103, 250, 270). Remarkably, additional inhibition of NRF2 on top of GSH and TRX results in even more HNSCC cell death, particularly in cisplatin-resistant cells (247).
NAD(P)H is the ultimate donor of reducing equivalents in cells and a key substrate for TR and other antioxidant enzymes. In addition to its function in the maintenance of the redox homeostasis in cells, it is also necessary for synthesis of nucleic acids and lipids (300). NADPH is generated in cells via glucose and glutamine metabolism through the PPP enzymes G6PD and 6-phosphogluconate dehydrogenase (6PGD), conversion of pyruvate to malate by malic enzymes, conversion of isocitrate to α-ketoglutarate by isocitrate dehydrogenases 1 and 2 (IDH1 and 2), and through 1-C/folate metabolism. Transcription factors NRF2 and p53 modulate NADPH production by increasing transcription of NADPH-generating enzymes and upregulating TP53-induced glycolysis and apoptosis regulator (TIGAR), which inhibits glycolysis and promotes PPP (124). As previously described, NADPH is necessary to maintain reduced GSH and TRX during oxidative stress. Therefore, attenuation of the PPP or downregulation of NADPH production in tumors would reduce or inhibit the cells' ability to handle ROS and inhibit critical biosynthetic pathways.
Resistance to chemotherapy in HNSCC
Chemotherapy induces cell death primarily via DNA damage and apoptosis. Defective apoptotic initiation may cause drug resistance (190). Cisplatin is the most commonly used chemotherapy for HNSCC but has significant patient variability related to outcome, efficacy, and toxicity. To overcome disease resistance, cisplatin is used in combination with other chemotherapeutics, RT, or drug-targeted therapy. However, as the disease-free period shortens, the response to platinum-based therapy decreases, resulting in a platinum-resistant or a refractory disease state. The primary mechanism of resistance to cisplatin at low levels (i.e., 10- to 15-fold above baseline) is due to altered DNA repair. At intermediate and high levels of cisplatin resistance, the resistance is due to reduced cellular accumulation and cytosolic inactivation of cisplatin (3) (Fig. 8).
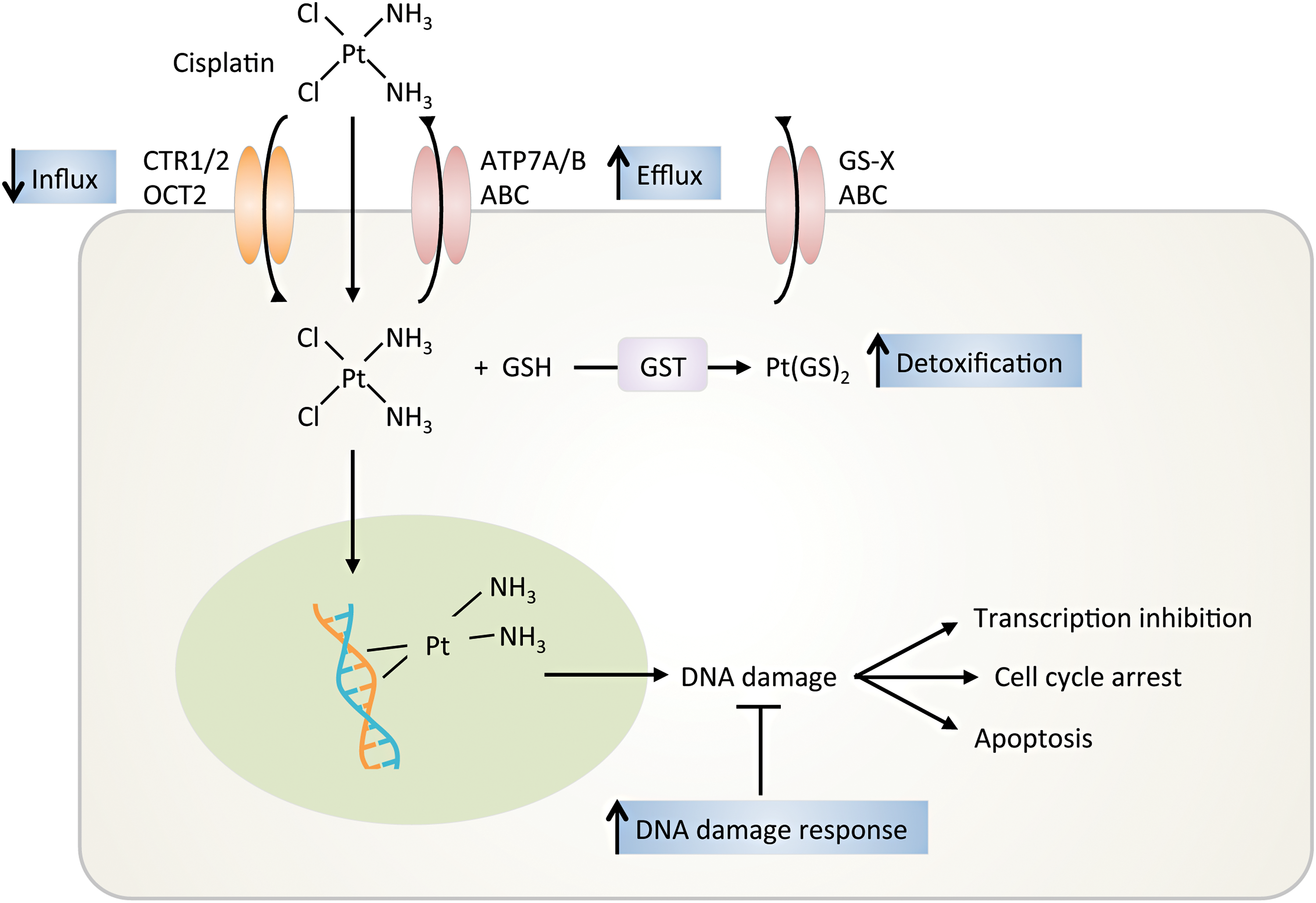
DNA damage alteration
The balance of DNA damage versus DNA repair determines cancer cell death versus survival after cisplatin therapy (202). DNA excision repair protein, excision repair cross-complementation group 1 (ERCC1), complexed with xeroderma pigmentosum complementation group F (XPF), catalyzes the incision of the damaged DNA strand, the rate limiting step in the nucleotide excision repair pathway. High ERCC1 and XPF expression is indicative of increased removal of DNA platinum adducts and has a linear relationship with resistance to cisplatin in many cancers, including HNSCCs (322). Polymorphisms have been identified in ERCC1 and XPF but they have not correlated with increased sensitivity or resistance.
Platinum influx and efflux
Moderate platinum resistance is mediated by decreased uptake or increased export, thereby reducing platinum concentration (161). Cisplatin can passively diffuse the cell membrane or is transported by copper transporter proteins 1/2 (CTR1/2) and the organic cation transporter 2 (OCT2). Cisplatin-resistant cells show decreased levels of CTR1 messenger RNA (mRNA) and reduced levels of copper, but increased levels of CTR2, suggesting that CTR1 to CTR2 ratio may be a useful biomarker for determining cisplatin response (155). Cisplatin is exported out of the cell by P-type ATPase transporters (e.g., ATP7A and ATP7B) or by ATP-binding cassette (ABC) transporters. These transporters are trafficked to the cell membrane to remove excessive copper (3). As expected, increased ATP7A and ATP7B levels are associated with poor response to cisplatin (178).
Platinum conjugation
Cisplatin resistance can also be due to conjugation with protein thiols (e.g., metallothionein and GSTs such as GST-pi and GST-Mμ), leading to increased solubility, increased cellular export, and less DNA damage. In HNSCCs, there is a correlation between high expression of GST-pi and cisplatin resistance (217).
Other mechanisms of chemoresistance
HIFs have been implicated in increased resistance to chemotherapeutics by increasing drug efflux and reducing DNA damage. HIF-2's ability to inhibit apoptotic pathways and activate antiapoptotic signaling pathways results in enhanced cyclin D2 expression, causing improved growth and resistance to DNA damage (122, 123). Multidrug resistance 1 (ABCB1) is an ABC (ATP-binding cassette) transporter protein, which can traffic substances across cellular membranes and cause efflux of many xenobiotics (214). It was also shown in colon and gastric cancer that ABC transporter, ABCG2, mediates resistance to chemotherapies under hypoxic conditions (79). Acid ceramidase (AC) plays a role in ceramide metabolism by converting ceramide into sphingosine to prevent ceramide-induced apoptosis. AC is overexpressed in 70% of HNSCC malignant tissue and causes resistance to chemotherapeutic agents. An in vitro study demonstrated that AC silencing regulates expression of WT or mutant p53 by post-transcriptional processing and caspase-dependent mitochondrial apoptosis, increasing response to chemotherapy (210).
Resistance to EGFR-targeted therapies
Monoclonal antibodies and TKIs target EGFR at the ligand binding or the catalytic domain, respectively, causing their efficacy and cytotoxicity profiles to be different. Cetuximab resistance is associated with dysregulation of the internalization and degradation processing of EGFR. Forty-two percent of HNSCCs express an in-frame deletion in the ligand-binding portion of the receptor, resulting in a truncated EGFR-variant 3 (EGFRvIII) to be expressed (276). EGFRvIII is constitutively phosphorylated, independent of ligand binding causing resistance to monoclonal antibodies, which target the ligand binding domain (276) (Fig. 2). STAT3 is also expressed at higher levels in HNSCCs with EGFRvIII. Cetuximab lacks the efficacy to abrogate EGFRvIII constitutive activation and STAT3 activity. In contrast, resistance to TKIs is mostly attributed to (i) activation of redundant kinase signaling pathways such as tyrosine-protein kinase Met (c-MET) and other EGFR family members and (ii) EGFR-independent activation of downstream signaling pathways such as phosphatidylinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway (74, 182, 237). For example, recent studies have suggested that overexpression of other members of the EGFR family, such as human epidermal growth factor receptor 3 (HER3), is involved in resistance to EGFR TKIs (25). HER3 lacks tyrosine kinase activity but can be phosphorylated by c-MET or other receptor tyrosine kinases (75). c-MET is commonly overexpressed, mutated, or has increased number of gene copies in 60% of HNSCCs (182). Mesenchymal-epithelial transition (MET) amplification drives HER3-dependent activation of PI3K/Akt signaling bypassing the EGFR inhibition by TKI (263, 277). Similarly, inhibitory PTEN mutations seen in many cancers including HNSCC (263, 277) or persistent oxidation result in activation of Akt pathway and downstream effectors (237).
Survival and QOL: Improving HNSCC Patients' QOL with Redox Modifiers
Owing to the location of the tumor, HNSCC patients often suffer from impairments in swallowing, breathing, and speaking as side effects of RT or CRT, greatly affecting their QOL. Preventing and controlling these complications can not only improve the QOL of HNSCC patients but also allow them to continue or receive higher doses of treatment, achieving overall better cancer management. There are many aspects of preventing and treating oral complications associated with HNSCC treatment, including pain control, nutritional support, and oral hygiene, just to name a few. Here we highlight the use of ROS modifiers in managing oral mucositis and xerostomia.
Oral mucositis is a common side effect observed in HNSCC patients receiving RT or CRT. The pathogenesis of oral mucositis was defined to have five biological stages: (i) initiation, (ii) primary damage response, (iii) signal amplification, (iv) ulceration, and (v) healing (279). ROS, as a result of the chemotherapy and/or RT, is involved in the first two stages by causing direct mucosal tissue damage and activating NF-κB and ceramide pathways that further result in apoptosis and tissue injury. Management of oral mucositis has been largely palliative, although targeted therapeutic interventions exploiting the dependency of oral mucositis on ROS generation are emerging. Amifostine, originally developed by U.S. military to protect soldiers from radiation exposure, is thought to act as a thiol-based free radical scavenger to protect HNSCC patients against radiation- and chemoradiation-induced toxicity. However, due to conflicting results on its ability to reduce oral mucositis, insufficient evidence of benefit, and strong toxic side effects, a guideline regarding the use of this agent in oral mucositis in chemotherapy or RT patients could not be established (216, 241). Currently, FDA only approves amifostine for prevention of xerostomia in HNSCC radiotherapy and for prevention of nephrotoxicity in cisplatin-based chemotherapy in ovarian cancer.
Another group of ROS inhibitors with potential therapeutic benefit in treating oral mucositis involves SOD activity. Small-molecule SOD mimetic M40403 has shown efficacy in attenuating radiation-induced oral mucositis in a hamster model (212). Its enantiomer, GC4419, has completed a Phase 1b/2a clinical trial with positive results of reducing the incidence, severity, and duration of oral mucositis in HNSCC patients receiving CRT (5), and is currently being evaluated in a randomized Phase 2 clinical trial to assess its effect on radiation-induced oral mucositis in patients with HNSCC (NCT02508389). In addition, a novel manganese porphyrin SOD mimetic (MnBuOE-2-PyP5+, BMX-001) has shown radioprotective properties on normal tissue by reducing radiation-induced mucositis, xerostomia, and fibrosis, while acting as a radiosensitizer in a mouse model of HNSCCs (8, 35). Given its differential effect on normal tissue versus tumor (16, 313, 314), BMX-001 is currently under a Phase 1a/b clinical trial to test its ability to reduce radiation-induced mucositis and xerostomia in HNSCC patients receiving RT (NCT02990468). Along the same lines, overexpression of human SOD2 in the oral cavity mucosa by plasmid/liposome delivery prevented radiation-induced mucositis in a mouse HNSCC model (127). For details regarding different physical properties, distribution, and chemistry of these SOD mimetics, readers are referred to Batinic-Haberle et al.'s review (this Forum).
Finally, RK-0202, a proprietary matrix for topical application in the oral cavity, consists of an antioxidant N-acetylcysteine (NAC). In a Phase 2 clinical trial, RK-0202 significantly reduced the incidence of severe oral mucositis in HNSCC patients treated with radiotherapy (55). NAC also showed efficacy in reducing the incidence and total duration of oral mucositis in leukemia patients undergoing allogeneic hematopoietic stem cell transplant and high-dose chemotherapy (211). Other ROS inhibitors with promising antioral mucositis effects include benzydamine (99, 157, 264), vitamin E (97, 308), and allopurinol (321).
Xerostomia or dry mouth is a common side effect associated with RT or CRT. It is a consequence of the hypofunctioning salivary gland, which is damaged by these therapies. Standard treatment options for xerostomia include saliva substitutes and stimulating agents, such as pilocarpine hydrochloride, a cholinergic agonist. Recent treatment has included novel antioxidants, such as lecithinized SOD and Tempol, and has shown positive results in mouse models (76, 287). Interestingly, acupuncture has also been reported to alleviate xerostomia (117, 269). The underlying mechanism is unknown but believed to be mediated by decreasing ROS levels (265).
Bioinformatics and Computational Systems Biology Approaches to Integrate Redox Effects in HNSCC Management
The diverse tissue sources (e.g., laryngeal, oropharyngeal, and salivary gland) implicated in HNSCCs create challenges in the development of systems biology approaches for studying etiology of HNSCCs and prediction of effective treatment strategies. Compounding the tissue diversity is the distribution of genetic drivers in HNSCCs (45, 282, 309). Compared with other cancers, for example, genomic analysis of allelic mutations in HNSCCs indicates lesions across numerous genes rather than a small set of dysregulated drivers of carcinogenesis (282). Targeted deep sequencing of 51 oncogenes highlighted the diversity of mutations in HNSCCs that are related to the underlying patient population (tobacco users and HPV+) (118). Although the occurrence of shared gene mutations across samples analyzed by whole-exome sequencing was low, recurrent mutations of TP53, PI3KCA, PTEN, CASP8, and HRAS were enriched across the sample set (282). Strikingly, these proteins have been implicated in redox-regulated processes either as direct protein thiol targets or as accessories to redox enzyme regulation in other cell types and biological contexts (4, 73, 134, 256). The motivation for using computational models for precision medicine approaches for targeting of redox-dependent signaling pathways is further supported by the two examples given hereunder.
EGFR and ROS formation
Systems biology modeling of the EGFR receptor network has resulted in refined understanding of regulation and optimal drug targets in cancers in general and specific to breast cancer and nonsmall cell lung cancer (NSCLC) (37, 58, 113, 253, 254); however, these types of models have not been applied extensively to HNSCCs. Regulatory features gleaned from these models can potentially be generalized to HNSCCs. For example, a large-scale kinetic model of EGFR signaling identified HER3 (ErbB/EGFR3) as the optimal inhibitory target for monoclonal antibody modulation of PI3K signaling (254). In a computational and experimental analysis of HER2 overexpression in breast cancers, extracellular signal-regulated kinase (ERK) activity was more robust against variability in parameter values than Akt, which may reflect drug targeting of Akt to be more advantageous in heterogeneous tumors or across a broad patient population. Zhou et al. built upon these findings to include features of EGFR-induced NOX activation to examine regulatory features of ROS in oncogene addiction in NSCLC cells (329). This model predicted that crosstalk between Akt and ROS-activated ASK1 suppresses proapoptotic mechanisms during sustained EGFR signaling. Disruption of this crosstalk by the EGFR TKI gefitinib resulted in enhanced apoptosis that could be modulated by vitamin C (ascorbate); however, the matching of simulations to experiments was limited due to the lack of detail of additional redox-regulated mechanisms that would be impacted by the NOX activation. Interestingly, a large prospective study found an inverse correlation between the intake of vitamin C and the incidence of HNSCCs (84). A body of experimental evidence implicates EGFR-induced NOX activation in the thiol oxidation of multiple proteins within this receptor tyrosine kinase network, including EGFR itself, PTEN, protein-tyrosine phosphatase 1B (PTP1B), and tyrosine-protein phosphatase nonreceptor type 11 (SHP2) (203, 229, 296). Furthermore, proinflammatory cytokines such as IL-6 are reported to be modulated in a NOX4-dependent manner in HNSCC cells, and incorporation of tumor cell migration and invasion as additional phenotypic endpoints beyond cytoxicity in network simulations would provide insight into the role of ROS generated by NOX during different therapies (110). Despite these limitations, the authors propose from their systems-level analysis that oncogene-addicted cancers may require combinatorial treatment to target survival signals and enhance proapoptotic mechanisms, such as elevation of ROS. The applicability of these systems level features of cancer regulation to HNSCCs is unknown. For instance, isolation of the ERK or Akt module was reported to exhibit switch-like behavior that was very different from the behavior of the intact system, suggesting that parameter sensitivity and ligand thresholds are very context-specific (58). Thus, the nuances in expression levels of this complex network and the multiple nodes of cross-regulation via ROS may render cetuximab or erlotinib dose responses to regulate EGF signaling very differently from all of the other cancer modeling studies to date.
Transcriptomic analysis of 60 HNSCC patient samples identified 4 distinct subtypes of patients that were loosely categorized as EGFR-related, mesenchymal-enriched, normal epithelium-like, and high antioxidant defense (67). The latter subset of patients exhibited a gene signature of upregulated xenobiotic, pentose phosphate, and redox enzymes such as GPX2 and TR1. This profile corresponds to other transcriptional analyses of epithelium from cigarette smokers and clustered the furthest from the EGFR-related subset. Targeting EGF signaling may be suboptimal for HNSCC patients based upon history of tobacco use.
STAT3 as a redox sensor
A recent stratification of HNSCC patient outcomes using big-data approaches leveraged somatic mutation information with prior knowledge derived from protein–protein and immune signaling databases (179). This methodology resulted in three classes of patients that mapped into different Cox survival profiles, yet distributed smoking status, HPV status, and other clinical metrics similarly. Instead, the algorithm identified different protein subnetworks associated with mutations within the patient groups, with STAT3 being a key “hidden” linker between the network that associated with best prognosis. STAT3 activation and upregulation are associated with loss of growth control in early-stage HNSCC (125), and blockade of STAT3 signaling has demonstrated antitumor efficacy in preclinical models of HNSCCs (1, 30, 260). Notably, STAT3 has been identified as the first mammalian signaling protein capable of serving as a redox relay with PRX2, resulting in an attenuation of Janus kinase/STAT signaling due to either endogenous or exogenous H2O2 levels (275). Thus, transcriptional activity of STAT3 is dependent on the PRX/TRX system and indirectly coupled to NADPH for reducing equivalents. To date, no computational modeling has been done to explore the role of STAT3 in translating elevated tumor levels of ROS into transcriptional changes; implementing these effects in pre-existing three-dimensional computational models of tumor growth and hypoxia (189) would be insightful in the spatial contributions to chemotherapeutic resistance.
Models of redox-based drug mechanisms of action for HNSCC therapies
To date, very little computational modeling has been performed that incorporates redox-based mechanisms of action for the common chemotherapeutics used clinically for HNSCCs (e.g., cisplatin, 5-FU, and methotrexate). One study was able to determine a 10-gene signature to predict response of HNSCC patients to 5-FU induction chemotherapy by analyzing HNSCC biopsies with whole-genome microarrays and quantitative reverse transcriptase PCR (295). Some of the candidate genes identified play a role in response to redox stress such as heat shock protein 40 and TRX domain containing 9 (295). Another notable example is a computational network model, consisting of 44 ordinary differential equations with mass action kinetics, which incorporated cisplatin effects on apoptotic signaling pathways, DNA damage, and oxidative stress (142). Time and concentration-dependent effects of cisplatin are accounted for through explicit description of OCT2-mediated cellular uptake. The model provided insight into the three-way crosstalk between death receptor, mitochondrial, and ER stress contributions to apoptosis in a generic cell. The lack of literature-derived parameter values for a mechanistic model such as this precludes the use of this approach in HNSCCs unless concerted effort to quantify rates of caspase activation, Bcl-2 homologous antagonist/killer (BAK)/BAX binding, etc. is undertaken in a consistent experimental setting. Other examples of ROS production induced by chemotherapeutics, such as doxorubicin (108, 109), highlight the use of in vitro measurements for establishing cancer-specific parameters.
Models of HPV infection and progression
A spatial model of HPV infection and progression within the stratified squamous epithelium was developed to consider the role of stochastic division rates of stem-like progenitor cells in viral persistence/clearance within infected tissue (248). Although this model primarily focuses on the balance between immune responses and cellular dynamics for viral clearance, the framework provides an opportunity to couple progression from HPV infection to sustained neoplastic growth and account for viral-induced cellular oxidation (57, 200), which is spatially defined within a tumor mass. Model-based simulations that account for recent immunotherapeutic advances (280, 299) and susceptibility to redox-modulating therapeutics in HPV+ versus HPV− cancers could also feasibly incorporate T cell–HNSCC interactions, as recently developed in a computational analysis for HIV–HPV coinfection (305).
Bioinformatics analysis of the Cancer Genome Atlas and the Cancer Proteome Atlas HNSCC data
A growing number of studies are using data archived through NIH programs such as the Cancer Genome Atlas (TCGA) and the Cancer Proteome Atlas (TCPA). For example, a novel cascade propagation (CasP) subtyping approach, using TCGA data, was developed to investigate codependent immune signaling pathways in HNSCCs by using dynamic network modeling followed by stratifying somatic mutations. Using CasP subtyping, HNSCC patients were stratified into two distinct groups, including (i) patients with mutations in TLR signaling who have better OS and (ii) patients with mutations in T and B cell receptor signaling who have poorer survival (179). Also, another study identified genomic and/or epigenomic changes that lead to STAT3 activation in HNSCCs using data generated by the TCGA and the TCPA programs (233). The authors identified that STAT3 expression is associated with disease stage, nodal status, tumor size, PFS, and OS in early stage oral HNSCCs. These types of modeling techniques offer opportunities to better understand molecular signaling and identification of biomarkers as well as opportunities for therapeutic escalation/deescalation.
Concluding Remarks
The research reviewed here provides clear evidence of the critical role played by altered redox homeostasis at all stages of HNSCC from development and early detection to treatment and improvement of patients' QOL. There is an established connection between etiologic factors, ROS, and inflammation as driving mechanisms of HNSCCs. There remain, however, certain aspects of HNSCC etiology that are just starting to be investigated and are critical for disease prevention and early detection. These include (i) synergy of interaction between currently acknowledged drivers of HNSCCs (e.g., HPV and smoking), (ii) the possibility of other coinfections acting as cofactors with HPV [e.g., Chlamydia, another sexually transmitted infection with cofactor function in HPV+ cervical cancer (330) and with potential for upregulating host cell EGFR and ROS (228)], and (iii) the role of oral microbiota in HNSCC development. Composition of oral microbiota and associated biomarkers was shown relevant for HNSCC development and early detection (172) and for predicting severity of oral mucositis induced by RT (331). Detailed mechanisms of how bacterial metabolism interacts with current etiologic factors and standard of care HNSCC therapies require further investigation. Redox mechanisms remain relevant in this context as oral microbiota plays a key function in nitrate metabolism (e.g., high in cruciferous vegetables like broccoli), which ultimately contributes to blood nitric oxide levels and controls cancer-relevant processes such as neovascularization (150).
Accumulation of ROS as a mechanism to kill cancer was applied successfully over decades through the use of RT. Toxicities associated with treatment and damage to normal tissue have been, however, concerns from the very beginning. Earlier methods at the turn of the 20th century employed glass tubes placed in vicinity of the tumors to deliver radiation in a method known as interstitial brachytherapy (72). Current technologies such as hybrid imaging and image-guided treatment devices allow for better targeting of radiation to the tumor, largely avoiding damage of normal healthy tissue. Nevertheless, metabolic and signaling changes such as those enabling suppression of radiation-induced ROS and DNA damage impair efficacy of RT and CRT. As NADH and NADPH species are the ultimate donors of reducing equivalents fueling ROS degradation and energy metabolism, it is critical to learn how to manipulate these species with spatial and temporal resolution within tumors to increase sensitivity to treatment (Fig. 9). Although some tools to selectively detect and manipulate NADH, NADPH (31 –33, 291, 328), and specific ROS species are currently available (in some cases with subcellular resolution) [e.g., Kelso et al. (158) and Robb et al. (244)], more are needed to enable basic and translational studies on this subject. NQO1 bioactivatable drugs are examples of how NAD(P)H-consuming and O2 •−/H2O2-producing reactions can be manipulated to achieve tumor kill alone or in combination with RT. In particular, NQO1 substrates capable of futile cycling, like the β-lapachone described here, are exciting new leads for further development into therapeutics.
Targeted therapies, such as EGFR kinase inhibitors and others, continue to remain of interest for treatment of HNSCCs. It is unfortunate to date that the impact of tumor redox microenvironment on the efficacy of response to small-molecule inhibitors is not being routinely considered. Similarly, more consideration needs to be given to physiologic redox modifiers such as aging, diet, and exercise on cancer development and response to treatment. Although a significant body of literature exists on the role of these factors in cancer development and incidence (9), less is known with regard to their impact on regulation of response to RT, CRT, targeted drugs or immunotherapies (220, 326), and QOL post-treatment (201).
There are currently two SOD mimetics under clinical trials as radioprotective agents in HNSCC treatment (GC4419 and BMX-001). This is the first time metal complexes are in clinical trials and target cellular redox environment. Although these drugs are indeed redox active, it is also likely that they act through functions other than SOD mimicking, such as inhibiting NF-κB and activating NRF2, which has been demonstrated for BMX-001 (16). For such functions and mitochondrial localization of BMX-001, readers are referred to Carroll and St Clair's review in this Forum.
In summary, there is strong evidence connecting redox metabolism with all stages of HNSCC management. The location of HNSCCs brings new opportunities for exploiting critical redox modifiers generated through activation of biologically active dietary molecules by oral microbiota to prevent or decrease HNSCC progression, improve response to therapies, and patients' QOL.
Footnotes
Acknowledgments
The authors would like to acknowledge the following funding support: NIH/NHLBI Training Grant T32 HL091797 (J.M.), NIH/NCI R33 CA1777461 (C.M.F.), and U01 CA215848 (M.L.K., C.M.F., and D.A.B.).