
Abstract
Aims:
Peroxiredoxin 4 (PRDX4) is a member of the peroxiredoxin family of antioxidant enzymes. Previously, we reported that PRDX4 can restrain the initiation and progression of nonalcoholic steatohepatitis by reducing local and systemic reactive oxygen species (ROS) levels. Oxidative stress is recognized as a key factor in hepatocarcinogenesis, and a high ROS level has also been found in hepatocellular carcinoma (HCC). Here, our aim is to investigate roles of PRDX4 in the initiation and progression of HCC.
Results:
In this study, for hepatocarcinogenesis, wild-type (WT), PRDX4 knockout (PRDX4−/y ), and human PRDX4 transgenic (hPRDX4+/+ ) mice were given a weekly intraperitoneal injection of diethylnitrosamine for 25 weeks. The HCC incidence was higher in PRDX4−/y mice than in WT or hPRDX4+/+ mice. Intrahepatic and circulating oxidative stress and inflammatory cell infiltration in the liver were obviously decreased in hPRDX4+/+ mice, compared with WT mice. Furthermore, in our cohort study, human HCC specimens with low expression of PRDX4 had higher ROS levels and a highly malignant phenotype, which was associated with a reduced overall survival, compared with those with high PRDX4 expression. However, in human HCC cell lines, PRDX4 knockdown led to a rapidly increased intracellular ROS level and suppressed cell proliferation, inducing cell death.
Innovation and Conclusion:
Our results clearly indicate that PRDX4 has an inhibitory effect in the initiation of HCC, but a dual (inhibitory or promoting) role in the progression of HCC, suggesting the potential utility of PRDX4 activators or inhibitors as therapy for different stages and phenotypes of HCC.
Introduction
Peroxiredoxin 4 (PRDX4),
Targeting antioxidants for cancer therapy remains a very controversial issue. The relationship between peroxiredoxins and cancer has garnered widespread attention for specific antioxidant properties. However, the role of peroxiredoxin 4 (PRDX4) has rarely been investigated in cancer. Here, we investigated the role of PRDX4 in hepatocellular carcinoma (HCC), suggesting that PRDX4 has an inhibitory effect in the initiation of HCC but a dual (inhibitory or promoting) role in the progression of HCC, and highlight the importance of antioxidant enzymes in the pathogenesis of cancer. These findings provide novel insights into the role of PRDX4 in cancer and evidence to support the application of antioxidase activators or inhibitors in cancer therapy.
In our previous series studies, elevated levels of PRDX4 were observed in the serum and certain tissues of human and mice with chronic inflammatory diseases, and the overexpression of human PRDX4 (hPRDX4) in mice markedly reduced the local and systemic levels of ROS and suppressed the development and progression of these diseases (14, 17, 32, 34). In particular, we found that PRDX4 can prevent the initiation and progression of nonalcoholic steatohepatitis by reducing the expression of oxidative stressors and inflammation, suggesting a beneficial role of PRDX4 in the liver (32, 34).
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide and the third-most common cause of cancer death, with a rising incidence (52). The initiation of HCC mainly arises from a context of inflammation caused by chronic liver disease (4). Chronic hepatic inflammation will lead to increased oxidative stress. The latter is becoming recognized as a key factor in the progression of chronic liver disease and hepatocarcinogenesis. Recruited inflammatory cells and activated Kupffer cells can produce a large amount of ROS that can conversely affect these cells, causing the further release of chemical mediators, enhancing oxidative stress and increasing the likelihood of hepatocarcinogenesis (30, 53). Thus, the interaction between oxidative stress and persistent inflammatory stimuli plays an important role in the initiation of HCC (11, 43).
Diethylnitrosamine (DEN) is often used as a hepatic carcinogen in the murine model, since DEN-induced HCC has similar histologic and genetic features to human HCC (25). There is evidence that DEN causes the initiation of HCC, depending on the mechanism, with oxidative stress and the inflammation response playing important roles (23, 29). Therefore, a potential role of PRDX4 in the pathogenesis of DEN-induced HCC is of particular interest.
ROS overproduction has been also demonstrated in HCC. Cancer cells have an increased rate of ROS production with an altered redox environment, resulting in higher basal ROS levels in cancer cells than that in their normal counterparts. However, the Warburg effect enables cancer cells to evade excess ROS generated by glucose oxidation from mitochondrial respiration, leading to moderate oxidative stress, which protects cancer cells against ROS-mediated anoikis and allows them to gain a survival advantage over normal cells (28). The higher-than-normal ROS levels that do not damage cancer cells are suggested to be related to tumor growth, angiogenesis, and metastasis (16, 27, 41). However, in an environment of increased ROS production, cancer cells still experience a high level of oxidative stress (31). The excess production of ROS caused by various factors produces severe oxidative stress, which is still toxic to cancer cells and can induce cell death through cell prodeath pathways (5). Thus, the role of PRDX4 in HCC therapy is attracting increasing interest.
In this study, through DEN-induced hepatocarcinogenesis in PRDX4 genovariation mice, immunohistochemistry (IHC) staining of PRDX4 in human HCC tissues, and PRDX4 small interfering RNA (siRNA) transfection to human HCC cell lines, we investigated the role of PRDX4 in the initiation and progression of HCC.
Results
Deficiency of PRDX4 enhances hepatocarcinogenesis in mice
There were no significant differences in the appearance or body weight of wild-type (WT), PRDX4 knockout (PRDX4−/y ), and human PRDX4 transgenic (hPRDX4+/+ ) mice. The mRNA and protein expression levels of hepatic hPRDX4 and the serum level of hPRDX4 were high in hPRDX4+/+ mice (Supplementary Fig. S1). To determine the role of PRDX4 in hepatocarcinogenesis, a weekly intraperitoneal (i.p.) injection of DEN (35 mg/kg) or saline (control) was administered to 3- to 4-week-old WT, PRDX4−/y , and hPRDX4+/+ male mice, and based on our pilot studies (Supplementary Fig. S2), animals were sacrificed after 25 injections of DEN or saline. None of the mice that received a weekly i.p. injection of saline developed HCC (Supplementary Fig. S3A). A significant increase in the HCC incidence rate was observed in PRDX4−/y mice, compared with WT or hPRDX4+/+ mice, while no marked difference in HCC incidence was found between WT and hPRDX4+/+ mice (Table 1 and Supplementary Fig. S3A, B). Accordingly, the serum level of alpha-fetoprotein (AFP) was increased in DEN-treated PRDX4−/y mice. Chronic liver disease may induce the production of AFP; thus, an increased AFP level was also observed in the serum of WT mice after DEN treatment (Supplementary Fig. S4). To further determine the role of PRDX4 for hepatocarcinogenesis, a long-term observation of 24 months was carried out in WT, PRDX4−/y , and hPRDX4+/+ male mice under natural conditions. Consistent with the DEN-induced HCC model, a dramatic increase in tumor incidence was observed in 24-month-old PRDX4−/y mice, while no tumor incidence occurred in age-matched WT and hPRDX4+/+ mice (Table 1 and Supplementary Fig. S5).
Rate of Hepatocellular Carcinoma Development in Wild-Type, PRDX4−/y , and hPRDX4+/+ Mice Twenty-Five Weeks After Diethylnitrosamine Treatment or Twenty-Four Months Under Natural Condition
Bold indicates p < 0.05.
DEN, diethylnitrosamine; hPRDX4, human peroxiredoxin 4; hPRDX4+/+ , human PRDX4 trangenic; PRDX4, peroxiredoxin 4; PRDX4−/y , PRDX4 knockout; WT, wild-type.
Multiple tumor nodules were observed in the liver of PRDX4−/y mice (Fig. 1A), and histologically, DEN-induced tumors were hepatocyte tumors with the typical features of HCC, including enlarged round hyperchromatic nuclei, high nuclear/cytoplasmic ratios, and moderate micro- or macrovesicular fat globules in the cytoplasm (Fig. 1B). Azan and silver staining showed the carcinoma cells are not surrounded by reticulin fibers, but adjacent liver cells were clearly surrounded by reticulin fibers (Fig. 1C).

Overexpression of PRDX4 protects liver from DEN-induced injury in mice
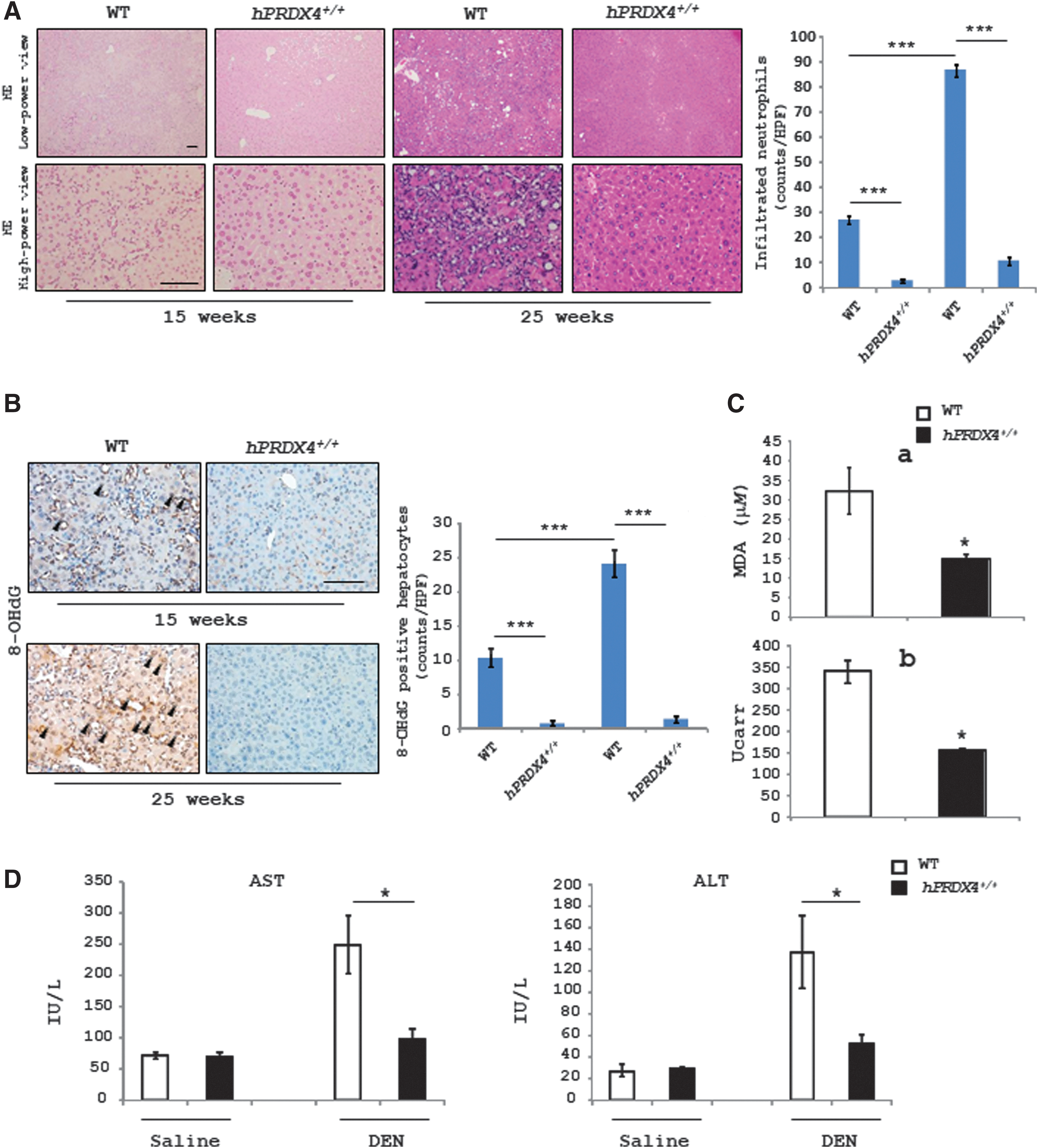
Although there were no significant differences in the HCC incidence between WT and hPRDX4+/+ mice, the number of infiltrated neutrophils was less in the liver of hPRDX4+/+ mice than in WT mice exposed to DEN for 15 or 25 weeks (Fig. 2A). Furthermore, the number of 8-hydroxy-2′-deoxyguanosine (8-OHdG)-positive hepatocytes in the liver was significantly lower in hPRDX4+/+ mice at 15 or 25 weeks after DEN treatment than in WT mice (Fig. 2B), and the level of thiobarbituric acid reactive substances (TBARS) (Fig. 2C[a]) and H2O2 (Fig. 2C[b]) in the serum was also lower in hPRDX4+/+ mice than in WT mice at 25 weeks after DEN treatment (Fig. 2C), indicating that the overexpression of hPRDX4 had reduced the local and circulating oxidative stress status in mice treated with DEN. The level of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in the serum was also lower in hPRDX4+/+ mice than in WT mice at 25 weeks after DEN treatment (Fig. 2D). These results suggest that PRDX4 can protect hepatocytes from DEN-induced injury that is closely associated with the initiation of HCC.
Low expression of PRDX4 with high ROS levels was correlated with disease progression and a reduced overall survival in human HCC
To evaluate the clinical significance of PRDX4 in human HCC, we investigated the PRDX4 expression by IHC staining of 86 human HCC tissues. Based on the IHC staining scores evaluated by three professional pathologists, cases were divided into two groups—low- and high-PRDX4 groups—using a receiver operating characteristic curve (Table 2; Fig. 3A). The clinicopathological characteristics were compared between the two groups (Table 2). The low-PRDX4 group exhibited larger tumor size and more portal and hepatic vein invasion, than the high-PRDX4 group, suggesting that tumors with low PRDX4 expression had more aggressive characteristics than those with high PRDX4 expression. Accordingly, a Kaplan–Meier's survival analysis (Fig. 3B) indicated that the low-PRDX4 group had a significantly reduced overall survival compared with the high-PRDX4 group. Furthermore, using IHC staining, we evaluated the expression of 8-OHdG as a marker for oxidative stress to determine the status of oxidative stress in tumor tissues (Fig. 3C) and found that the 8-OHdG level was higher in the low-PRDX4 group than in the high-PRDX4 group (Fig. 3D). Thus, PRDX4 may suppress tumor growth and invasion by downregulating the intracellular ROS level of cancer cells, which is associated with the disease prognosis.

Peroxiredoxin 4 Expression and Clinicopathologic Factors in Patients with Hepatocellular Carcinoma
Bold indicates p < 0.05.
HBV, hepatitis B virus; HCV, hepatitis C virus; NA, not available; SD, standard deviation.
Downregulation of PRDX4 enhances the ROS level but represses cell proliferation in HCC cell lines
To further determine the role of PRDX4 in HCC cells, we knocked down its expression in PLC/PRF/5 and HepG2 cells using three kinds of PRDX4-specific siRNAs. Real-time polymerase chain reaction (PCR) showed that siRNAs (HSS116214) were the most efficient at knockdown of PRDX4 (Fig. 4A). A significant reduction in the PRDX4 mRNA and protein expression was observed at days 1 and 3, respectively, after transfection with PRDX4 siRNAs (HSS116214) (Fig. 4B). Viability assays using the Cell Counting Kit-8 showed that the proliferation of PLC/PRF/5 and HepG2 cells was significantly suppressed after PRDX4 expression was downregulated (Fig. 4C). Fluorescence microscopy revealed that the number of dihydroethidium (DHE)-positive (Fig. 4D) and 2′,7′-di-chlorofluorescein (DCF)-positive (Fig. 4E) cells was much greater among HCC cells transfected with PRDX4 siRNAs than among those cells transfected with negative siRNAs. These results support the notion that PRDX4 has very important roles in redox homeostasis of HCC cells, which may affect cancer cell proliferation.

PRDX4 knockdown affects the HCC cell survival
A flow cytometry assay showed no significant difference in the number of apoptotic cells 3 days after transfection with PRDX4 or negative siRNAs. However, the ratio of sub-G1 was higher at 5 and 7 days after transfection with PRDX4 siRNAs than after that with negative siRNAs (Fig. 5A). In addition, the number of terminal deoxynucleotidyl transferase end-labeling (TUNEL)-positive cells was significantly greater among HCC cells transfected with PRDX4 siRNAs than among those transfected with negative siRNAs (Fig. 5B). In line with the increased apoptosis, a large number of PRDX4-knockdown cells were found in the medium at 7 days after transfection, while cells with normal PRDX4 expression remained firmly attached to the culture dish (Fig. 5C). Supporting these observations, Western blotting showed that the protein expression of cleaved caspase-3 was significantly higher in PRDX4 siRNA-transfected HCC cells than in negative siRNA-transfected HCC cells (Fig. 5D).

The intracellular ROS level is also closely associated with another cell prodeath pathway: autophagy. A significant increase in the number of CYTO-ID-positive cells was observed in those cells transfected with PRDX4 siRNAs compared with negative siRNAs (Fig. 6A, B). An ultrastructural analysis also showed that a large number of autophagosomes and/or autolysosomes were observed in PRDX4-knockdown HCC cells (Fig. 6C and Supplementary Fig. S6). The analysis of protein expression of some autophagy-related genes showed that the expression of microtubule-associated protein light chain (LC) 3-II was increased, while that of autophagy-related gene (Atg) 4 was decreased at 5 days after HCC cells were transfected with PRDX4 siRNAs (Fig. 6D). These data indicate that increased oxidative stress caused by downregulating PRDX4 expression triggered cell death pathways through not only apoptosis but also autophagy.

Discussion
In this study, our results showed that the deficiency of PRDX4 promoted DEN-induced hepatocarcinogenesis in mice, low PRDX4 expression enhanced tumor growth and invasion with a reduced overall survival in human HCC, but downregulation of PRDX4 suppressed cell proliferation and induced cell death in human HCC cell lines. We suggest that PRDX4 may play important roles in the initiation and progression of HCC.
All the time, it is widely accepted that PRDXs act as tumor “preventers” (35). Indeed, loss of PRDXs could promote carcinogenesis in mice. For example, PRDX1-knockout mice exhibit a shortened life span owing to be predisposed to the development of hemolytic anemia and several malignant cancers (36). Deficiency of PRDX6 in mice enhanced the susceptibility to 7,12-dimethylbenz[a]anthracene/12-O-tetradecanoylphorbol 13-acetate- or human papilloma virus 8-induced skin tumorigenesis (44). Similar to these data, in the present study, a markedly higher HCC incidence was observed in PRDX4−/y mice 25 weeks after i.p. injection of DEN weekly, compared with WT or hPRDX4+/+ mice. Whereas overexpression of PRDX4 reduced intrahepatic and circulating ROS level and suppressed leukocyte infiltration in the liver of mice exposed to DEN. A large amount of ROS generated by the P450-dependent enzymatic system during the process of DEN biotransformation can affect hepatocyte replication, proliferation, and apoptosis through multiple signal pathways (3, 38, 46), leading to hepatocarcinogenesis. In addition, oxidative stress can accelerate the infiltration and activation of inflammatory cells and the excessive release of inflammatory factors, which can enhance DEN-induced hepatocarcinogenesis (33). Our previous results have indicated that PRDX4 can prevent endothelial dysfunction and decrease expression of adhesion molecules and inflammatory cell migration (14, 17, 32, 34). Therefore, combining these results suggests that PRDX4 may have an efficient effect in inhibiting ROS/inflammation-related hepatocarcinogenesis.
A large number of clinical evidences have indicated that PRDXs also play important roles in malignant progression of many cancers and have potential clinical implications in cancer therapy, where PRDXs function as a tumor suppressor or promoter (7, 13, 37, 42, 47, 48). The overexpression of PRDX4 was identified in oral and prostate cancer tissues and was found to be associated with a positive pN status and tumor cell proliferation (9, 51), while the expression of the antioxidant protein is downregulated in acute promyelocytic leukemia (39). Interestingly, a report showed that the high expression of PRDX4 in tumor tissues was significantly correlated with higher recurrence rates and shorter disease-free survival in patients with lung squamous cell carcinoma, but not in patients with lung adenocarcinoma (18). Our clinicopathological analysis displayed that low PRDX4 expression promoted tumor growth and invasion with a higher ROS level, which is closely related to a reduced overall survival, implying PRDX4 may be a tumor suppressor in HCC. Whereas our in vitro data showed that downregulation of PRDX4 expression inhibited cell proliferation and induced death in HCC cell lines, suggesting PRDX4 may also be a promoter of HCC. These results may seem contradictory, but they should be plausible because of the “two-faced” property of ROS. A great number of studies confirm that moderate oxidative stress can lead to cancer progression, whereas severe oxidative stress will cause cancer cell death (1, 10). In current in vitro experiments, a sudden decrease of PRDX4 expression led to a mass rapid accumulation of intracellular ROS, probably causing severe oxidative stress, directly resulting in cell growth inhibition and death. However, in human HCC tumors with low PRDX4 expression, a possible explanation is that an adaptable redox homeostasis may exist and enable cancer cells to tolerate a higher level of ROS that contribute for malignant progression (49), suggesting PRDX4 may have a key role in the process of new redox homeostasis formation caused by high metabolism of cancer cells. Our current understanding of the function of PRDX4 is still lacking in cancer, and further research must be undertaken to better understand these findings.
Inducing cancer cell apoptosis is a widely acceptable strategy for cancer therapy (6), since cancer cells may be more sensitive to the toxic generated by the accumulation of excess ROS by inducing big amounts of ROS or damaging the ROS scavenging capacity of cells, potentially inducing cell death. Indeed, our results showed that downregulation of PRDX4 expression enhanced intracellular ROS level and induced cell apoptosis in HCC cell lines, which is also supported by other research (21). Interestingly, in this study, besides apoptosis, we also found PRDX4 is also closely associated with another cell prodeath pathway, autophagy (12). Many anticancer treatments have been shown to activate autophagy, and therapeutic induction of autophagy-related cell death is obtaining widespread attention in cancer therapy strategies (8, 55). Our in vitro results confirm that enhanced ROS levels caused by PRDX4 knockdown led to the inactivation of Atg4, which promoted lipidation of Atg8 (LC3), a key step during autophagosome formation (45), thereby increasing autophagy activity in HCC cells. Similar results were also observed in breast cancer cells treated with carnosol that induced ROS-related autophagy and apoptosis leading to cell death (2). However, the consideration is probably oversimplified, as the complexity of cellular effects has been observed in various anticancer treatments, where ROS-induced autophagy causes either cell death or drug resistance or both, showing autophagy can also protect against cellular stress (40). In addition, emerging evidence suggests the multiple layers of crosstalk between autophagy and apoptosis, depending on interactions among the crucial proteins involved in them (24, 56). At present, we cannot delineate that an increase in autophagy accelerated cell death or was only a protect response to oxidative stress. Further studies are necessary to confirm the role of autophagy and its relationship with apoptosis in HCC.
There are still some research limitations in the present study. (i) Although we suggested an inhibitory effect of PRDX4 in hepatocarcinogenesis, DEN treatment used in this study did not produce a significant difference in HCC incident between WT and hPRDX4+/+ mice. A lower dose and a longer induction of DEN may be needed to identify the difference in hepatocarcinogenesis between these mice. (ii) Our IHC results showed obvious differences in the expression levels of PRDX4 between adjacent noncancerous liver tissues. Thus, it may be interesting to analyze PRDX4 expression in a larger number of normal liver and HCC samples and to monitor these patients with regard to HCC initiation and progression. (iii) PLC/PRF/5 and hepG2 cells were also transfected with PRDX4-plasmid DNAs to upregulate expression, but overexpression of PRDX4 did not affect cell survival, probably due to these cells originally having a high PRDX4 level. As far as we know, high PRDX4 expression is observed in almost all of the HCC cell lines (Gene Expression Omnibus database), and thus, primary culture from HCC tumors with low PRDX4 expression may be a good way to identify the effect of additional PRDX4 in HCC progression and clinical outcome.
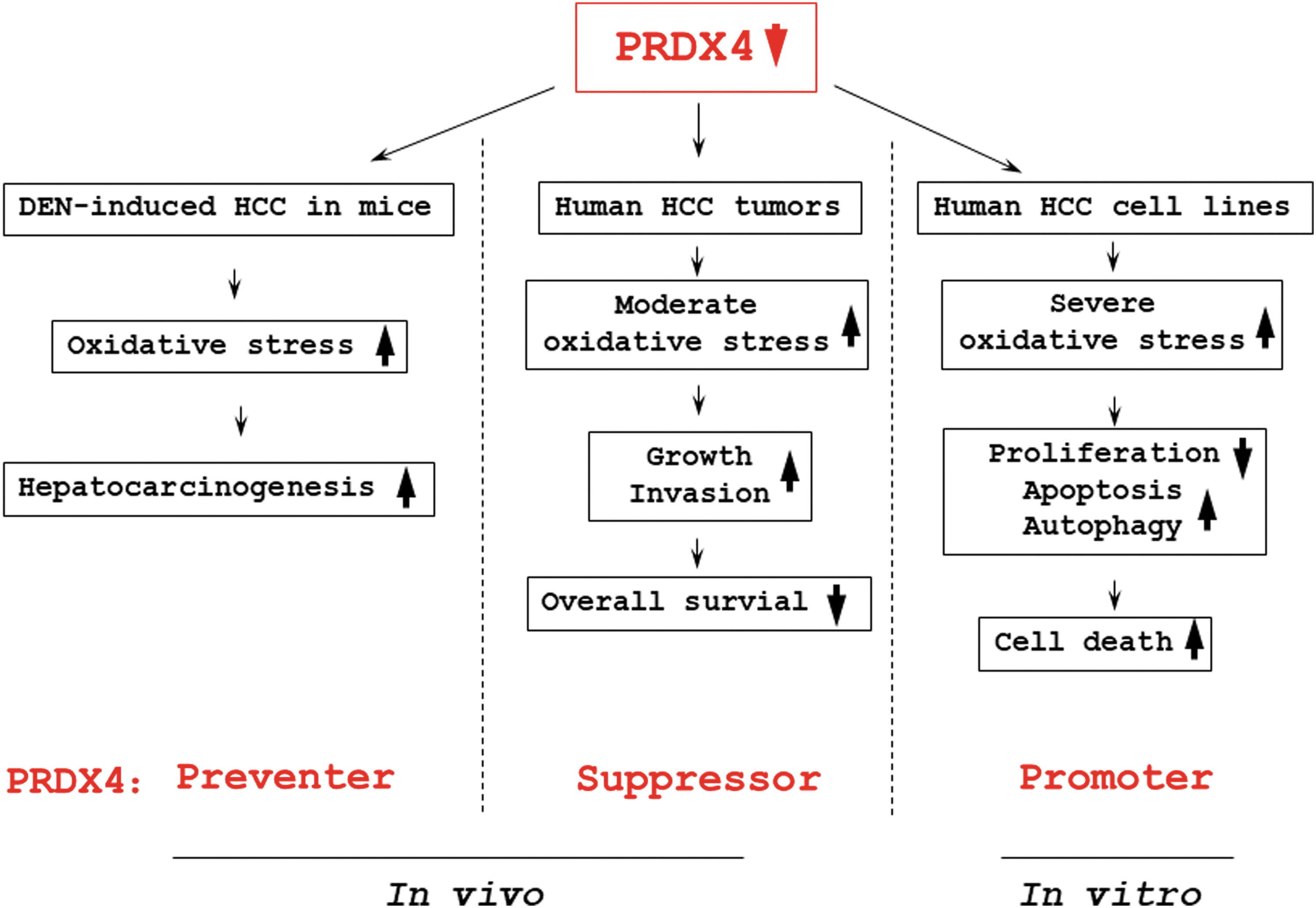
In summary, our present data indicate that PRDX4 can restrain DEN-induced hepatocarcinogenesis in mice by reducing intrahepatic and circulating oxidative stress, as well as the inflammation response in the liver. However, due to the contradictory property of ROS, PRDX4 plays a dual role in the progression of HCC, promoting the survival of cancer cells but inhibiting the rapid growth and invasion of tumor. We summarized the roles of PRDX4 in HCC in Figure 7. These results may provide a novel treatment strategy for HCC and highlight the importance of antioxidant enzymes in the pathogenesis of cancer.
Materials and Methods
Animals and treatment
PRDX4 knockout (PRDX4−/y ) and hPRDX4 transgenic (hPRDX4+/+ ) mice were generated and genotyped as previously described (14, 17, 19, 32, 34). Briefly, for the generation of PRDX4−/y mice, PRDX4 genomic DNA was cloned from a b129/SVJ mouse genomic library (Stratagene, CA). Blastocysts containing embryonic stem cells in which homologous recombination had occurred were implanted into the uteri of pseudopregnant C57BL/6 female mice. For the construction of hPRDX4+/+ mice, based on a published sequence (GenBank accession no. NM_006406), hPRDX4 cDNA was amplified and cloned into the pGEM-T easy vector system (Invitrogen, Life Technologies Japan Ltd., Tokyo, Japan). The entire nucleic acid sequence was microinjected into the male pronuclei of one-cell C57BL/6 female mouse embryos. C57BL/6 inbred mice were used as control animals. These mice were maintained in a temperature- and light-controlled facility under a standard 12-h light/12-h dark cycle, fed standard rodent chow, and given water ad libitum. For hepatocarcinogenesis, experiments were performed using 3- to 4-week-old male WT, PRDX4−/ , and hPRDX4+/+ mice weighing 10–14 g, who were given a weekly i.p. injection of DEN (35 mg/kg) or saline (negative control) for 25 weeks. In addition, the HCC spontaneous rate was observed in these mice under natural conditions for 24 months.
All protocols were approved by the Ethics Committee of Animal Care and Experimentation, Kagoshima University, Japan, and were performed according to the Institutional Guidelines for Animal Experiments and the Law (no. 105) and Notification (no. 6) of the Japanese Government. The study also conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Patients and HCC specimens
Eighty-six clinical tissue samples of HCC with clinicopathological information were collected from HCC patients who underwent hepatic resection at Kagoshima University Hospital between 2008 and 2012 in this study. All formalin-fixed, paraffin-embedded specimens were used for the IHC study (22). A 5-year follow-up was conducted after operation. The Ethics Committee of Kagoshima University approved the experimental and research protocols.
Cell culture
The human HCC cell lines PLC/PRF/5 and HepG2 were purchased from Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). These cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal calf serum and maintained in a humidified atmosphere at 37°C and 95% air/5% CO2.
siRNA transfection
Three kinds of siRNAs specific for PRDX4 (Stealth siRNAs HSS116212, HSS116214, and HSS173720) were purchased from Invitrogen (Carlsbad, CA). The cells were plated in six-well plates and cultured in the growth media at ∼60% confluence, incubated for 24 h, and transfected for 72 h with 20 nM siRNA duplexes in Lipofectamine® RNAiMAX and Opti-MEM medium (Life Technologies, Carlsbad, CA). Stealth RNAi™ siRNA Negative Control was used as a negative control (Stealth RNAi; Life Technologies).
Histology and IHC
For histocytological analyses of human HCC specimens and mouse liver, images of staining with hematoxylin and eosin, silver or azan, or IHC sections were captured and quantified using the NanoZoomer Digital Pathology Virtual Slide Viewer software program (Hamamatsu Photonics Corp., Hamamatsu, Japan) (20). To detect hPRDX4 expression in human HCC tissues, we used a rabbit anti-human polyclonal antibody (1:100; Affinity BioReagents, Golden, CO), incubated at 4°C overnight, and a goat polyalkaline phosphatase secondary antibody (Vulcan Fast Red; Biocare Medical, Concord, CA). To analyze the ROS/oxidative stress levels in cancer cells of mouse liver or human HCC tumor tissues, we used an anti-8-OHdG monoclonal antibody (1:200; Japan Institute for the Control of Aging, Fukuroi, Japan), and staining scores were evaluated by three professional pathologists. The endogenous peroxidase activity was not blocked for the analysis of oxidative stress markers. Silver staining and azan staining were used for the observation of reticulin fibers around the tumor cells in PRDX4−/y mice.
Blood sample measurements
TBARS are a well-established method for evaluating lipid peroxidation, and were used as an index of lipid peroxidation and oxidative stress. To assess a second and more quantitative marker of oxidative stress, we measured the TBARS levels using a TBARS Assay Kit (Cayman Chemical Company, Ann Arbor, MI) in serum collected from DEN- or saline-injection mice. Results are expressed as -μM malondialdehyde. The serum levels of hPRDX4 (hPRDX4 ELISA Kit; Abnova, Taipei, Taiwan) and AFP (Mouse AFP ELISA kit; Cloud-Clone Corp., Houston) in DEN-treated mice were measured using ELISA kits according to the manufacturers' instructions. The serum levels of ALT and AST were measured using an automatic blood analyzer JCA-BM6070 (BioMajesty™; Japan). For the assessment of the hydroperoxide levels, a diacron reactive oxygen metabolite test was performed using an FRAS4 System (H&D, Parma, Italy), as described previously (50). The units of this measurement are expressed as UCarr (Carratelli unit) values, where one unit corresponds to 0.8 mg/LH2O2.
Real-time reverse transcription-PCR
Total RNA from HCC cells was extracted using the Total RNA Extraction Miniprep System (Viogene BioTek, New Taipei City, Taiwan) and stored at −80°C until analysis. Total RNA was converted into cDNA using a High Capacity RNA-to-cDNA Kit (Life Technologies). The cDNA was analyzed by a LightCycler® 96 (Roche Diagnostics, Tokyo, Japan) and subjected to 40 cycles of amplification using TaqMan gene expression assays (Life Technologies). Each sample was analyzed in triplicate in separate wells for PRDX4 and ribosomal 18S genes. The average of three threshold cycle values for the target and 18S genes was calculated, and then analyzed using the comparative Ct method. Custom-made primers and TaqMan probe for PRDX4 gene amplification were purchased from Life Technologies (Assay ID: Hs01056076_m1).
Western blotting
Proteins (20 mg) isolated from HCC cells were separated by sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred to Immun-Blot polyvinylidene difluoride membranes (Bio-Rad Laboratories, K.K., Tokyo, Japan) using a semidry blotter (26). After transfer onto polyvinylidene difluoride membranes, the membranes were blocked with 5% nonfat milk in TBST (20 mmol/L Tris-HCl [pH 7.6], 150 mmol/L sodium chloride, and 0.1% Tween 20) for 1 h at room temperature (RT) and then incubated overnight at 4°C with primary antibody diluted in Can Get Signal solution 1 (Toyobo, Osaka, Japan) and with a horseradish peroxidase-conjugated goat anti-rabbit antibody (MP Biomedicals, Santa Ana, CA) for 1 h at RT. The following primary antibodies and dilutions were used: rabbit polyclonal antibody to hPRDX4 (1:1000; Santa Cruz Biotechnology), rabbit monoclonal antibodies to Atg4 (D62C10), LC3 (D3U4C), caspase-3 (8G10), cleaved caspase-3 (5A1E), and β-actin (13E5) (Cell Signaling Technology, Danvers, MA) (Supplementary Figs. S7–S10). Protein expression was detected with SuperSignal West Pico chemiluminescent substrate or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific). Densitometry analysis was performed with NIH ImageJ software version 1.43.
Cell proliferation assay
The viability of cells was measured using the CCK-8 method according to the manufacturer's instructions. After transfection with siRNAs, 3 × 103 PLC/PRF/5 or HepG2 cells were seeded in one well of 96-well plates, respectively, allowed to attach for 16 h, and observed for viability for 72 h. The absorbance at 450 nm was measured using a microplate reader. Six replicate wells were used for each group.
Detection of intracellular ROS
The intracellular ROS level was assessed with two oxidation-sensitive fluorescent dyes, DHE (Molecular Probes, Eugene, OR) and CM-H2DCF-DA (chloromethyl 2′, 7′-dichlorodihydrofluorescein diacetate), (Molecular Probes) for HCC cell lines. PLC/PRF/5 cells were transfected with PRDX4 or negative siRNA for 72 h, and then, cells were rinsed quickly with Krebs–Ringer solution and incubated with freshly prepared 5 μM DHE or CM-H2DCF-DA for 5 min, respectively. After staining, we quantified the number of positive cells (those with increased DHE- or DCF-associated fluorescence in 10 randomly selected fields per section; original magnification: × 400), as described previously.
Flow cytometric analysis of cell cycle and apoptosis
The HCC cells were transfected with PRDX4 or negative siRNA for 3, 5, and 7 days, then collected, and washed twice with cold phosphate-buffered saline (PBS). Cells were fixed in 70% ethanol at −20°C and then washed with PBS. The cells were then incubated with phosphate/citrate buffer (pH 7.8) at RT for 30 min and resuspended in 1 mL of propidium iodide solution (50 μg/mL) containing 50 μL of RNase A solution (10 mg/mL). After suspensions were incubated for 30 min on ice, DNA content was analyzed using a CyAn-ADP flow cytometer (Beckman Coulter, Brea, CA).
Detection of autophagy
The autophagic vacuoles in HCC cells transfected with siRNAs were determined using a Cyto-ID autophagy detection kit 2.0 (Enzo Life Sciences, Farmingdale, NY) according to the manufacturer's instructions.
TUNEL staining
To determine the number of apoptotic cells in PRDX4- or negative siRNA-transfected PLC/PRF/5 cells, we conducted TUNEL assays using the in situ Cell Death Detection Kit, POD (Roche Applied Science, Lewes, United Kingdom). After staining, the number of positive cells was quantified as described above.
Ultrastructural analysis
PLC/PRF/5 cells transfected with PRDX4 or negative siRNAs were fixed with 2.5% glutaraldehyde in 0.1 M Na cacodylate, pH 7.4 for 2 h, postfixed with 2% aqueous osmium tetroxide for 2.5 h, and subsequently stained en bloc in 2.5% uranyl acetate (in water) overnight before dehydration and embedding in Eponate 12 resin (Ted Pella, Inc., Redding, CA). Thick (1 μm) sections of the embedded cells were examined at the light microscopic level. The cell blocks were further trimmed to obtain thin sections (80 nm), stained with a saturated solution of uranyl acetate (15 min) followed by Reynold's lead citrate (8 min), and viewed on a transmission electron microscope (JEM-1200EX; JEOL Ltd., Tokyo, Japan).
Statistical analyses
All statistical analyses were performed with EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan), the graphical user interface for R (The R Foundation for Statistical Computing, version 2.13.0, Vienna, Austria). Categorical variables were compared using χ2 tests or Fisher's exact tests. A Kaplan–Meier's analysis was used to analyze the overall survival, and survival curves were compared using log-rank tests. Statistical analyses were performed for continuous variables using Student's or Welch's t-test. All statistical tests were two sided, and differences and correlations values of p < 0.05 were considered statistically significant.
Footnotes
Acknowledgments
We thank Orie Iwaya, Mai Tokudome, and Yoshie Jidouhou for their expert technical assistance; we thank the patients for taking part in this study. This study was supported by grants from the National Natural Science Foundation of China (No. 81402490) (to X.G.), Natural Science Foundation of Hebei Province (No. H2016206170) (to X.G.), High-level talent support project of Hebei Province (No. CG2015003011) (to X.G.), the Kodama Memorial Fund for Medical Research, Kagoshima, Japan (to S.Y. and A.T.), and by Grants-in-Aid for Scientific Research (16K08750) from the Ministry of Education, Culture, Sports, Science and Technology, Tokyo, Japan (to S.Y.).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.