
Abstract
Background:
Cardiovascular diseases have been associated with stress in the endoplasmic reticulum (ER) and accumulation of unfolded proteins leading to the unfolded protein response (UPR). Reactive oxygen species (ROS) such as superoxide and H2O2 derived from NADPH oxidases have been implicated in the pathogenesis of cardiovascular diseases. ROS have also been associated with ER stress. The role NADPH oxidases in the UPR is, however, not completely resolved yet.
Aim:
In this study, we investigated the role of p22phox, an essential component of most NADPH oxidases, in the UPR of endothelial cells.
Results:
Induction of ER stress increased p22phox expression at the transcriptional level. p22phox was identified as novel target of the UPR transcription factor ATF4 (activator of transcription factor 4) under ER stress conditions by promoter analyses and ChIP. Depletion of ATF4 and p22phox diminished the levels of superoxide and H2O2 under ER stress conditions. On the contrary, p22phox was instrumental in increasing eIF2α phosphorylation and subsequent ATF4 expression on induction of ER stress by chemicals, oxysterols, or severe hypoxia in vitro and in vivo, leading to increased expression of CHOP and activation of effector caspases.
Innovation:
p22phox is a novel target of ATF4 in response to ER stress, which can promote the PERK-ATF4 branch of the UPR in vitro and in vivo.
Conclusion:
p22phox-dependent NADPH oxidases are important mediators of ER stress driving the UPR.
Reactive oxygen species (ROS) have been associated with endoplasmic reticulum (ER) stress and the unfolded protein response (UPR). In this study, we show that in endothelial cells, p22phox-dependent NADPH oxidases contribute to superoxide/H2O2 production under ER stress conditions. p22phox is a novel target of the UPR gene activator of transcription factor 4 (ATF4), which mediates its upregulation under ER stress conditions. p22phox on the contrary contributes to the UPR under ER stress conditions, including 7-Keto-cholesterol loading and severe hypoxia, by upregulating ATF4 and CHOP and activating caspases. Thus, p22phox initiates a positive feedback loop under ER stress conditions, promoting the UPR and limiting cellular survival.
Introduction
V
ATF6 is an ER-transmembrane-activating transcription factor. On ER stress, ATF6 translocates to the Golgi compartment, where the active cytosolic part is released and translocated to the nucleus activating the transcription of UPR genes including X-box binding protein 1 (XBP1) (23). The ER transmembrane glycoprotein IRE1 contains both kinase and RNase activities in the cytoplasmic domain. ER stress leads to autophosphorylation and activation of IRE1 RNase activity, which initiates then the splicing of XBP1 mRNA generating a mature XBP1 mRNA (23, 26). Also, XBP1 induces UPR genes, including PDI, which was shown to have a regulatory function on NADPH oxidases in smooth muscle cells (18).
Activation of the ER transmembrane protein kinase PERK phosphorylates the α-subunit of the translation initiation factor 2α (eukaryotic initiation factor 2 alpha subunit [eIF2α]) thus diminishing the formation of translation initiation complexes, subsequently leading to the attenuation of general translation. Paradoxically, translation of mRNAs with a lower requirement for eIF2α and the translational initiation complex are enhanced. Among them, the activating transcription factor ATF4, which regulates UPR genes, is preferentially translated. On cessation of ER stress, the protein phosphatase 1 (PP1) dephosphorylates eIF2α, thus restoring protein translation (23). On prolonged ER stress, IRE1 initiates a phosphorylation cascade leading to activation of c-Jun N-terminal kinases (JNK), mitochondria/Apaf1-dependent caspases, and subsequently apoptosis (30).
Reactive oxygen species (ROS), such as superoxide, H2O2 or other reactive species, have been implicated at various stages in the ER stress response in particular as toxic molecules inducing ER stress and the UPR (44). On the contrary, there are some hints that increased ROS levels may also be a consequence of ER stress although the sources of ROS and mechanisms involved are not well understood.
One major source for ROS such as superoxide and H2O2 in the endothelium and other vascular cells is the family of NADPH oxidases (13, 14, 17). The first member of this protein family—gp91phox or NOX2—was discovered in neutrophils where it forms together with p22phox the cytochrome b558 as the catalytic core of the NADPH oxidase (1, 11). Recently, additional homologues of NOX2 have been identified in nonphagocytic cells (2, 8, 12, 32), through which NOX1, NOX3 and NOX4 form, like NOX2, a membrane-bound complex with p22phox, thus making this small protein an essential component of most NADPH oxidases. Interestingly, NOX2, NOX4, and p22phox have been found not only at the plasma membrane but also in the ER in endothelial cells and other cells (2, 18, 30, 32, 41).
Although NADPH oxidases have been suggested to be linked to ER stress, their specific role in such a condition is not well elucidated. While controversial data exist regarding the role of NOX4 in the ER stress response (37), the role of p22phox, which is essential for most NADPH oxidases, has not been clarified. In this study, we show that chemical induction of ER stress upregulated p22phox expression and increased the levels of superoxide and H2O2 dependent on p-eIF2α and ATF4. Induction of p22phox and generation of superoxide and H2O2 were instrumental in promoting the UPR in chemically induced ER stress conditions, and also in response to oxysterols and severe hypoxia in vitro and in vivo eventually mediating a proapoptotic process.
Results
p22phox regulates ER stress-induced ROS generation
ER stress was induced in human microvascular endothelial cells (HMEC-1) using the sarco/endoplasmic reticulum calcium ATPase inhibitor thapsigargin, which resulted in splicing of XBP1 (Fig. 1A), as well as in a transient increase in the levels of phosphorylated eIF2α (p-eIF2α), ATF4, and cleaved ATF6 (Fig. 1B), important mediators of the ER stress response. Concomitantly, thapsigargin increased dihydroethidium (DHE) fluorescence within 30 min to 4 h of treatment (Fig. 1C). Treatment with cell-permeable superoxide dismutase (PEG-SOD) or its mimetic TEMPOL completely diminished this response, indicating the generation of superoxide anion radicals (Fig 1D). Similarly, the N-glycosylation inhibitor tunicamycin not only transiently elevated p-eIF2α, ATF4, and cleaved ATF6 protein levels (Fig. 1E) but also increased superoxide generation in a time-dependent manner (Fig. 1C).
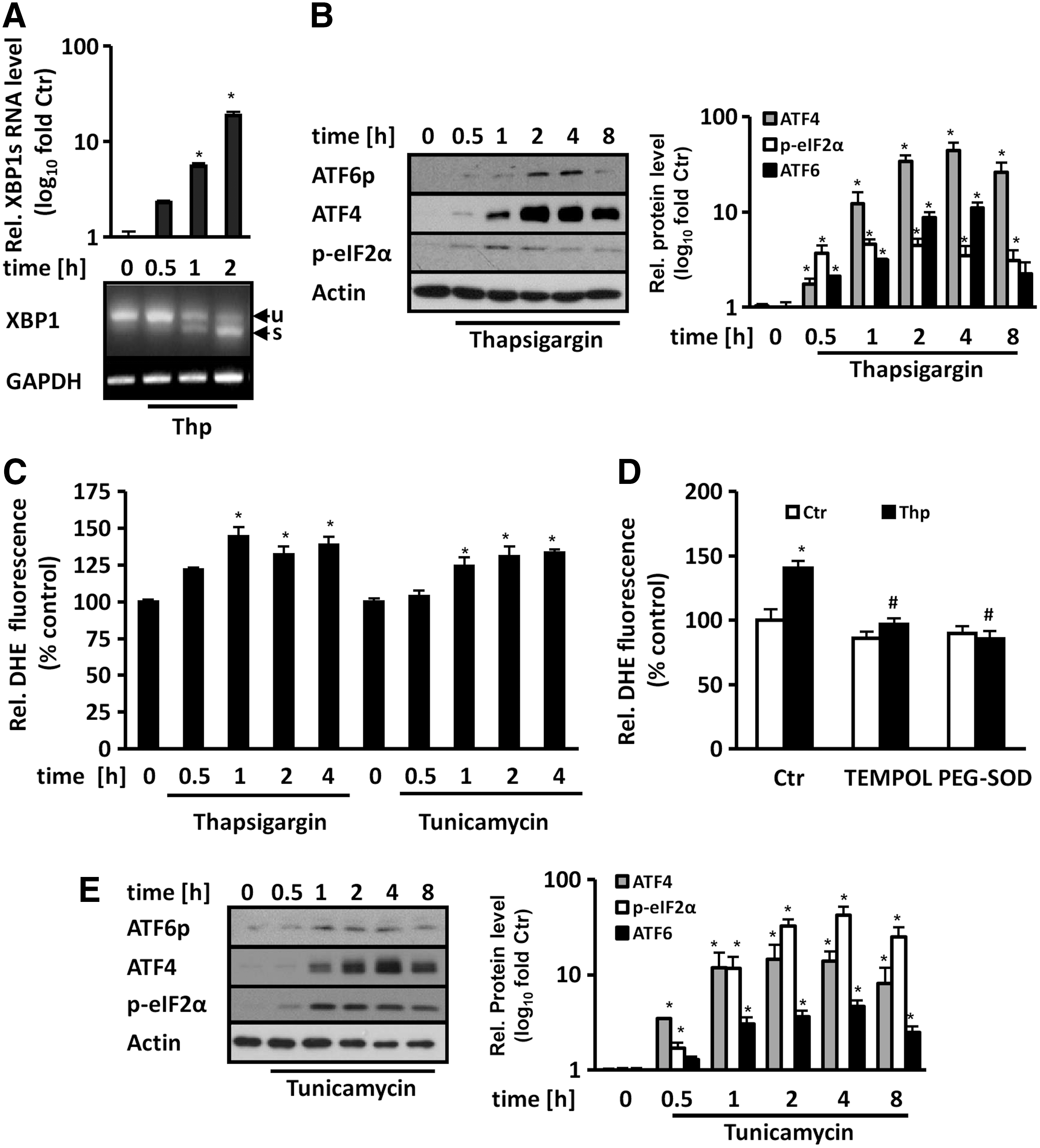
In addition, in human umbilical vein endothelial cells (HUVEC), thapsigargin and tunicamycin time dependently increased superoxide levels as determined by DHE fluorescence (Supplementary Fig. S1; Supplementary Data are available online at
As we and others previously could show that p22phox, which is important for most NADPH oxidases, is present in the ER of endothelial cells (32, 41), we thus tested whether p22phox is involved in ER stress-induced superoxide generation. To this end, we depleted endothelial cells from p22phox by RNA interference, resulting in reduced superoxide generation under ER stress conditions induced by thapsigargin or tunicamycin treatment (Fig. 2A, B).

Furthermore, treatment with thapsigargin not only enhanced the mRNA levels of GADD34 (growth arrest and DNA damage-inducible protein 34), a known ER stress responsive gene (26), but also those of p22phox (Fig. 2C).In addition, thapsigargin and tunicamycin increased p22phox protein levels (Fig. 2D, E). Similarly, treatment with the oxysterol 7-Keto-cholesterol, which also has been shown to induce the UPR (30), not only increased, as expected, p-eIF2α, ATF4, and cleaved ATF6 levels as signs of the UPR but also upregulated p22phox protein levels and p22phox-dependent superoxide generation (Fig. 2F and Supplementary Fig. S3). These data show that ER stress induces p22phox expression and p22phox-dependent superoxide generation in endothelial cells.
ER stress induction increases p22phox expression via ATF4
Next, we tested whether upregulation of p22phox by ER stress involved de novo transcription. Pretreatment with actinomycin D—an inhibitor of transcription—prevented the increase in p22phox protein levels in response to thapsigargin stimulation (Fig. 3A). As the transcription factors ATF4, XBP1, and ATF6 are the main mediators of the ER stress response, we tested their involvement in ER stress-induced p22phox expression. Depletion of ATF4 by siRNA prevented p22phox upregulation in response to ER stress (Fig. 3B). Silencing of XBP1 or ATF6, however, did not affect p22phox levels (Fig. 3B, C). Moreover, overexpression of ATF4 enhanced not only p22phox protein levels (Fig. 3D) but also catalase-inhibitable DCF fluorescence indicative of H2O2 generation (Fig. 3E). On the contrary, silencing of ATF4 reduced DCF fluorescence under ER stress (Fig. 3F), indicating that ATF4 is the physiological regulator of p22phox on ER stress.

ATF4 activates the p22phox promoter by binding to a proximal ATF site
To further decipher the role of ATF4 in regulating p22phox expression, we performed bioinformatic analyses of the 6 kb upstream region of the human CYBA (cytochrome b alpha peptide) gene coding for p22phox. Several potential ATF binding sites were identified, but only one was located in a proximal region at position −568 bp upstream of the translational start (Supplementary Fig. S4).
To test whether this ATF site is active, a region of the p22phox gene containing 1174 bp upstream of the translational start was cloned into a luciferase reporter plasmid. Overexpression of ATF4 increased luciferase activity, while mutation of the putative ATF binding site in the p22phox promoter abolished this response (Fig. 4A). Similarly, thapsigargin induced luciferase activity of the wild-type construct, but not of the mutated construct (Fig. 4B). In addition, silencing of ATF4 prevented p22phox promoter activation in response to ER stress (Fig. 4C). Importantly, direct binding of ATF4 to the p22phox promoter was demonstrated by chromatin immunoprecipitation (ChIP) analysis, and thapsigargin treatment enhanced binding of ATF4 to the p22phox promoter (Fig. 4D).

ATF4 pathway of the ER stress response enhances p22phox expression by direct activation of the p22phox promoter
To dissect whether activation of eIF2α is involved in p22phox regulation, dephosphorylation of eIF2α was blocked by treatment with salubrinal, an inhibitor of phosphatases that act on eIF2α (4). As expected, salubrinal enhanced phosphorylation of eIF2α and ATF4 protein levels, and also increased the levels of p22phox in a time-dependent manner (Fig. 5A). In line, salubrinal promoted superoxide generation, which was reduced by siRNA-mediated depletion of p22phox (Fig. 5B).

In addition, salubrinal enhanced luciferase activity of the wild-type p22phox promoter construct, but not of the construct mutated at the ATF4 binding site (Fig. 5C). Similarly, silencing of ATF4 prevented the salubrinal-mediated activation of the p22phox promoter (Fig. 5D). These data further demonstrate that blocking of general translation by preventing activation of eIF2α induces p22phox expression and p22phox-dependent superoxide generation via ATF4.
p22phox is important for induction of the UPR
To further verify the importance of ROS such as superoxide and H2O2 and p22phox-dependent NADPH oxidases in the regulation of the UPR, cells were treated with 50 μM H2O2. This treatment increased eIF2α phosphorylation and upregulated ATF4 and p22phox protein levels (Fig. 6A). Treatment with the cell-permeable SOD mimetic TEMPOL prevented thapsigargin-induced eIF2α phosphorylation and reduced significantly the upregulation of ATF4 indicating that superoxide anion radicals contribute to this response (Fig. 6B). In line, silencing of p22phox in HMEC-1 (knockdown efficiency 68.7% ± 8.4%) and HUVEC (knockdown efficiency 58.1% ± 16.4%) prevented the induction of eIF2α phosphorylation and upregulation of ATF4 by thapsigargin (Fig. 6C and Supplementary Fig. S5).
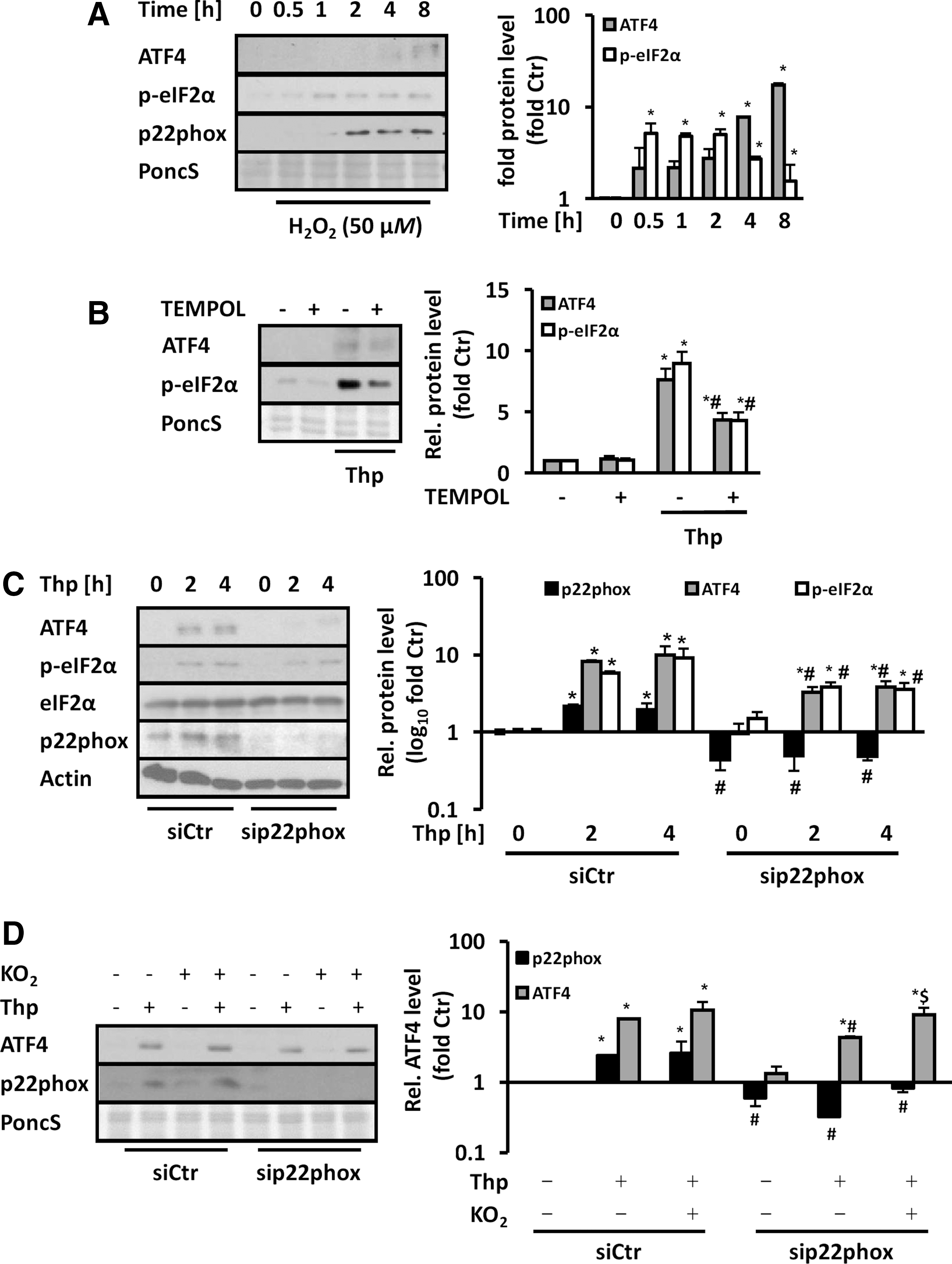
Similarly, 7-Keto-cholesterol-induced eIF2α phosphorylation and ATF4 protein levels were diminished by p22phox depletion (Supplementary Fig. S6). In contrast, overexpression of p22phox increased phosphorylation of eIF2α and ATF4 protein levels, while addition of TEMPOL prevented this response indicating that p22phox-dependent superoxide generation promoted this response (Supplementary Fig. S7). In line, application of the superoxide donor KO2 to thapsigargin-stimulated endothelial cells resulted in recovery of ATF4 protein levels in p22phox-depleted cells (Fig. 6D). These findings suggest that ROS such as superoxide or H2O2, respectively, derived from p22phox-dependent NADPH oxidases, are able to promote the UPR.
NOX4 contributes to thapsigargin induction of the UPR
As endothelial NADPH oxidases primarily involve NOX2 and NOX4 as partners of p22phox, we tested the effects of ER stress on the regulation of these proteins. Similar to p22phox, NOX2 and NOX4 protein levels were increased by thapsigargin treatment (Fig. 7A). Depletion of p22phox, however, reduced NOX2 levels in thapsigargin-stimulated endothelial cells, while NOX4 levels were not altered (Supplementary Fig. S8). Depletion of NOX2 (66.9% ± 11.5% knockdown efficiency) or NOX4 (70.6% ± 5.9% knockdown efficiency) decreased superoxide levels in response to thapsigargin (Fig. 7B).
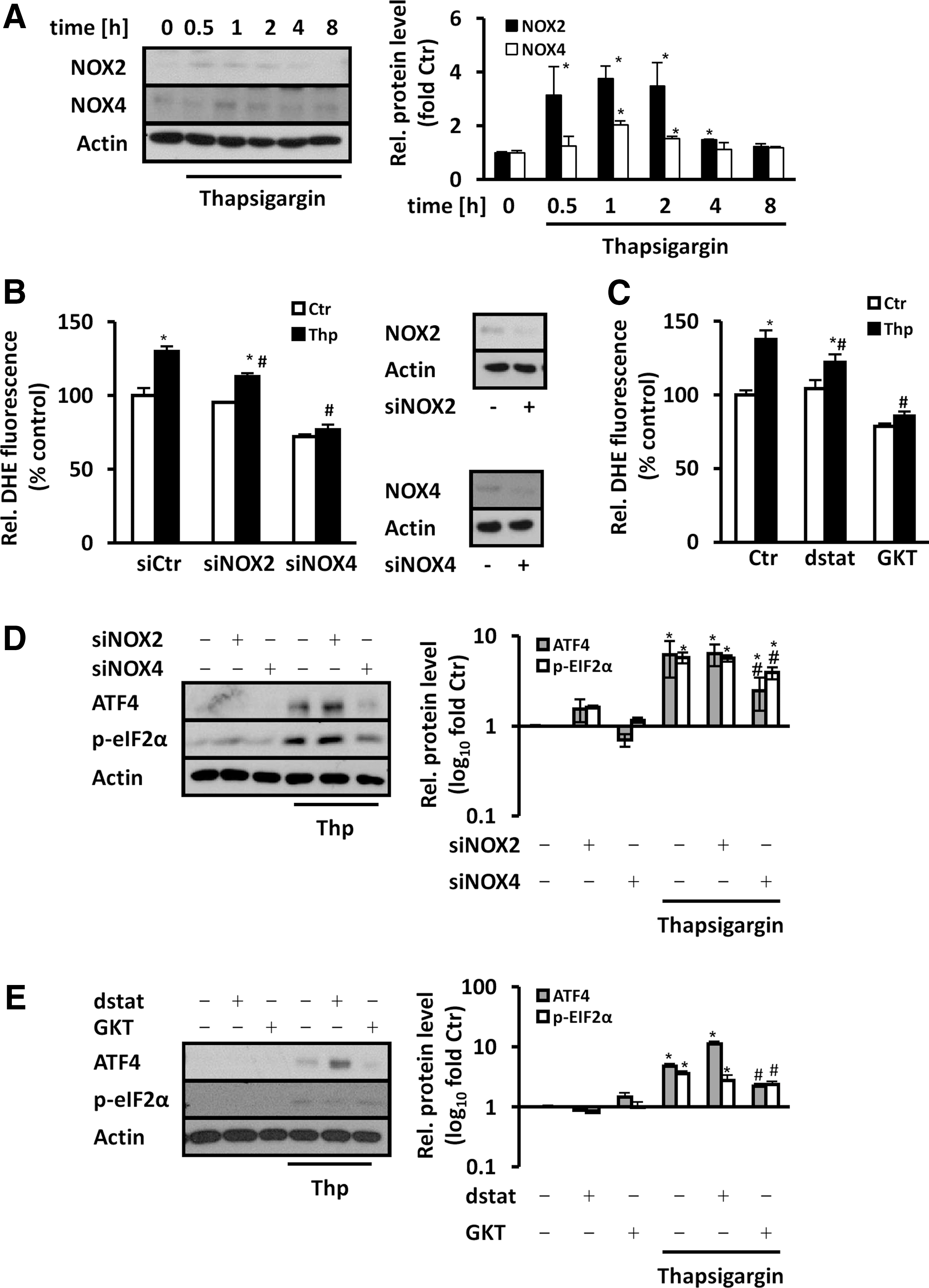
Similarly, treatment with gp91ds-tat, which inhibits NOX2-dependent NADPH oxidase, or GKT137831, which inhibits NOX4 (and NOX1)-dependent NADPH oxidases, reduced thapsigargin-induced superoxide generation (Fig. 7C). In line, GKT137831 treatment reduced thapsigargin-induced p-eIF2α and ATF4 levels, while application of gp91ds-tat was not effective (Fig. 7D). Moreover, upregulation of p-eIF2α and ATF4 in response to thapsigargin was reduced in NOX4-depleted cells, but remained largely unaffected in NOX2-depleted cells (Fig. 7E).
These findings indicate that NOX2 and NOX4 could act as partners with p22phox to generate superoxide/H2O2 under ER stress conditions, while NOX4-dependent NADPH oxidase would be more prominent in the regulation of p-eIF2α and ATF4 levels.
p22phox regulates UPR-induced apoptosis
In the next step, we tested the role of p22phox in downstream effects of the UPR. Exposure to thapsigargin decreased Alamar Blue fluorescence compared to control cells, indicating reduced metabolic activity in endothelial cells under ER stress conditions (Fig. 8A). Depletion of p22phox partially recovered the loss in metabolic activity under ER stress conditions. Next, we tested whether p22phox is involved in the regulation of the ATF4 target gene CHOP (CCAAT/-enhancer-binding protein homologous protein) under ER stress conditions. CHOP levels were indeed enhanced under ER stress conditions, but reduced in p22phox-depleted cells (Fig. 8B).
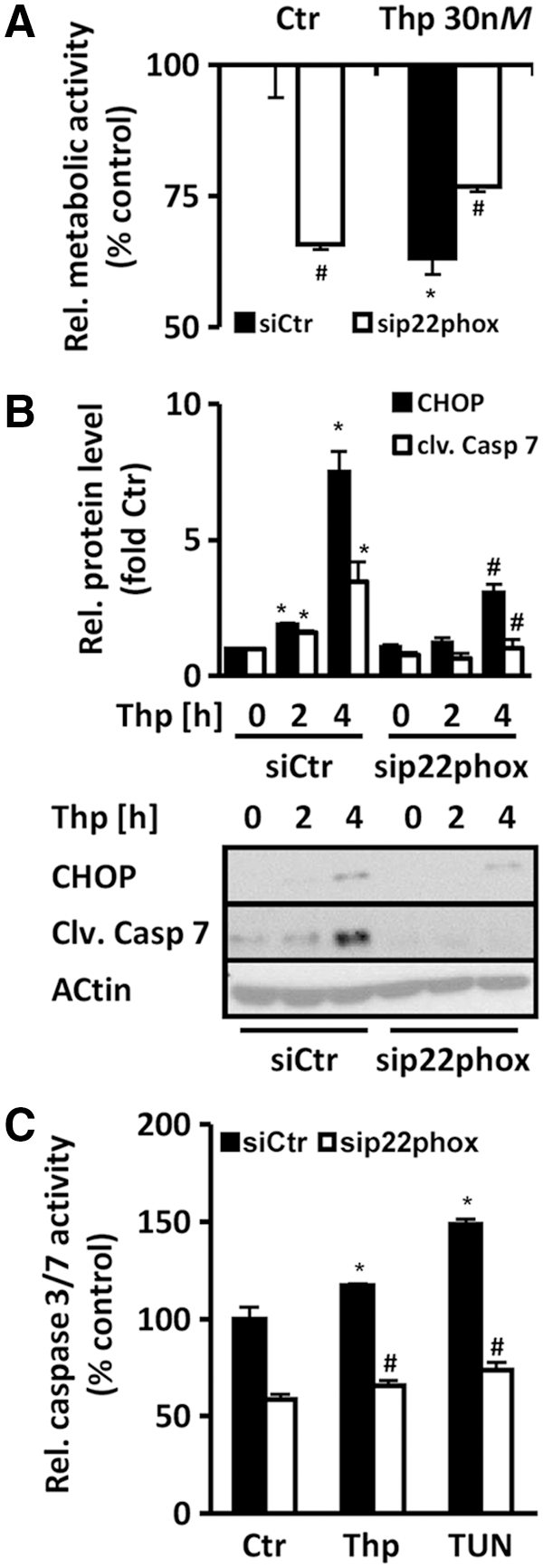
As CHOP is a proapoptotic factor, we tested whether p22phox is involved in caspase activity in response to ER stress conditions. Cleaved caspase 7 levels were increased after ER stress induction, but reduced in p22phox-depleted cells (Fig. 8B). In line, ER stress also induced caspase 3/7 activity in endothelial cells (Fig. 8C), which was reduced in p22phox-silenced cells. These findings indicate that p22phox promotes ER stress-induced UPR and apoptosis.
Hypoxia and UPR
In addition to 7-Keto-cholesterol, we investigated whether p22phox also plays a role in the response to severe hypoxia, a pathophysiological condition that we and others previously showed to induce ER stress and the UPR (19, 35). As expected, severe hypoxia (0.1% O2) increased eIF2α phosphorylation and ATF4 induction as well as p22phox levels (Fig. 9A). These responses were abrogated in p22phox-deficient HMEC-1 (Fig. 9B). In addition, cleaved caspase 7 levels were also reduced in p22phox-deficient cells.
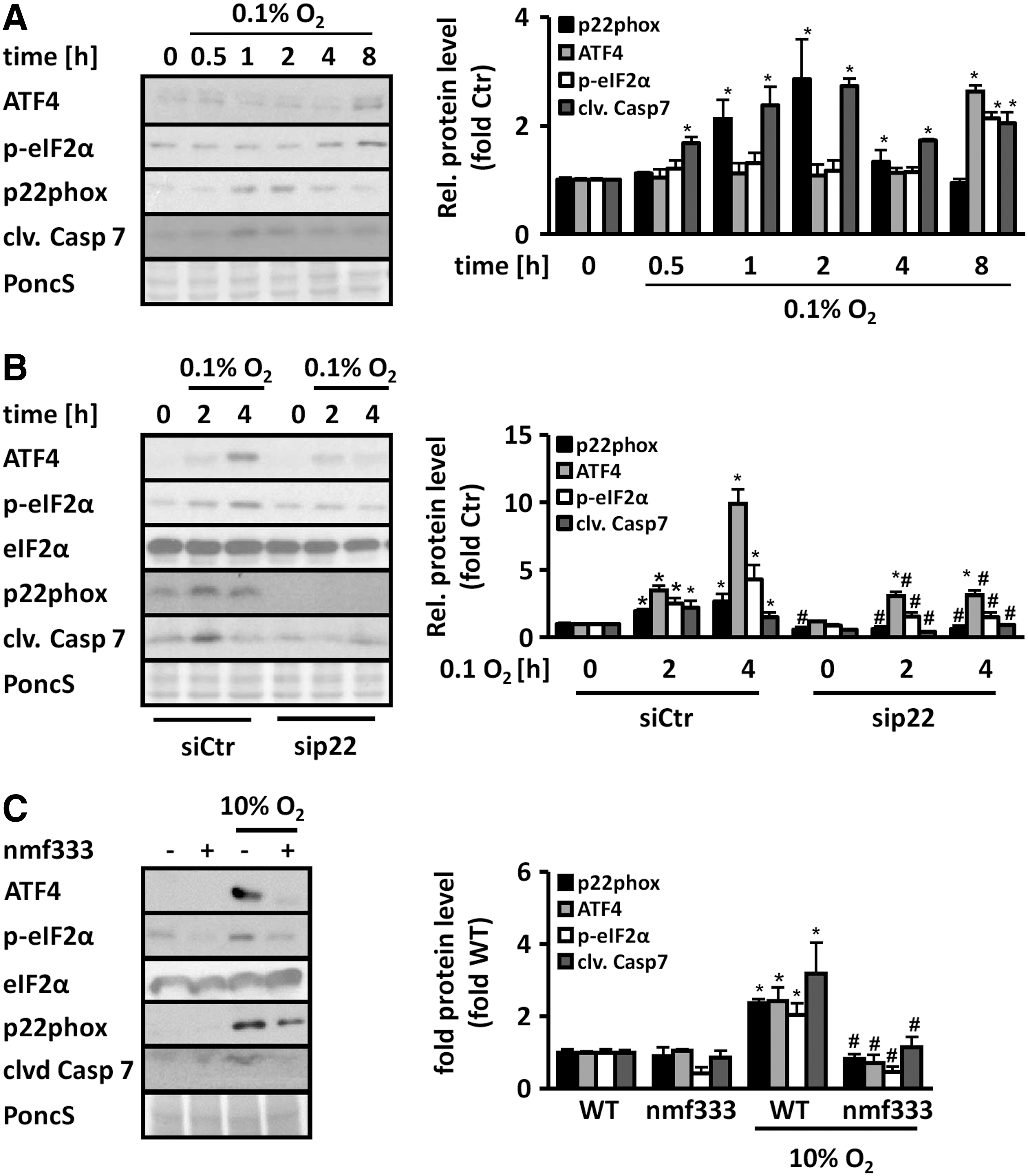
Similarly, in lungs from mice exposed to chronic hypoxia, eIF2α phosphorylation and ATF4 induction as well as p22phox expression were enhanced (Fig. 9C). However, in p22phox-deficient mice, these responses were blunted. Moreover, increased levels of cleaved caspase 7 were observed in hypoxic wild-type mice, but not in p22phox-deficient mice (Fig. 9C). These findings indicate that p22phox plays a role in the induction of the UPR also in the in vivo situation of ER stress.
Discussion
The UPR is an important mechanism to maintain cellular integrity in various adverse conditions leading to ER stress. In this study, we could show that in endothelial cells, p22phox-dependent NADPH oxidases are important signaling hubs in the UPR, which can promote the UPR, but can also be targeted and induced by the UPR.
This is based on our findings that (i) ER stress upregulated p22phox mRNA and protein levels; (ii) p22phox was identified as an ATF4 target gene that required activation of the p-eIF2α-ATF4 pathway under ER stress conditions for expression; (iii) ER stress induced the levels of ROS such as superoxide and H2O2 dependent on the NADPH oxidase subunit p22phox; (iv) p22phox and ROS such as superoxide and H2O2 were essential for UPR activation in response to ER stress induced by thapsigargin, tunicamycin, 7-Keto-cholesterol, or severe hypoxia in vitro and in vivo; and (v) p22phox promoted ER stress-mediated caspase activation in vitro and in vivo.
The UPR has been previously associated with different ROS such as superoxide or H2O2 and oxidative stress (3, 25) since the ER requires an oxidizing environment to promote protein folding, which has been mainly attributed to the ER-oxidoreductase 1 (ERO1) (16, 39). In this study, we show that the NADPH oxidase subunit p22phox contributes to enhanced levels of superoxide/H2O2 in response to chemical ER stress inducers and to 7-Keto-cholesterol in endothelial cells, and that NOX2 and NOX4 can be relevant partners in this response. In line, NOX4 and NOX2 have been previously shown to contribute to H2O2 or superoxide generation in response to ER stress induced by tunicamycin in cardiomyocytes and endothelial cells, and by cholesterol loading and 7-Keto-cholesterol in macrophages and human aortic smooth muscle cells (10, 21, 30, 37), further confirming that p22phox-dependent NADPH oxidases contribute to increased levels of superoxide and H2O2 in response to ER stress.
Our data further indicate that ER stress and the p-eIF2α/ATF4 arm of the UPR were associated with an upregulation of p22phox, as silencing of ATF4, but not of ATF6 or XBP1, was able to diminish p22phox expression under ER stress conditions. Using promoter analyses and ChIP, we identified an ATF4 binding site in the proximal p22phox promoter, which was sensitive to ER stress, thus adding p22phox to the array of ATF4 target genes in the UPR. In line, depletion of ATF4 decreased ER stress-induced generation of superoxide/H2O2, further emphasizing our findings that ATF4 is a major physiological regulator of p22phox in response to ER stress.
Furthermore, ATF4 depletion prevented ER stress-induced generation of superoxide/H2O2 similar to p22phox depletion, while overexpression of ATF4 not only increased p22phox levels but also induced superoxide/H2O2 generation. Previous studies showed that ATF4 depletion also reduced DCF fluorescence—indicating the generation of H2O2 in thapsigargin- or tunicamycin-treated murine embryonic fibroblasts (15) and in cortical neurons (20) after prolonged UPR. This delayed response was accompanied by reduced levels of antioxidative enzymes such as SOD. These findings suggest that short-term induction of ROS such as superoxide/H2O2 within the first hours of UPR is mainly an NADPH oxidase-mediated process, whereas on prolonged ER stress, ROS levels increase due to downregulation of antioxidant enzymes and upregulation of other enzymes such as ERO1α able to increase ROS (38).
Previously, NOX2 and NOX4 have been shown to be upregulated in response to ER stress induced by chemicals or high cholesterol load in vascular cells, cardiomyocytes, and macrophages (10, 21, 30, 37). Induction of ER stress by high cholesterol increased NOX2 mRNA levels in macrophages involving Ca2+/calmodulin-dependent protein kinase II, ERO1α, inositol 1,4,5-triphosphate receptor type 1, and JNK (21), and in smooth muscle cells involving IRE1 and JNK (30). In line, in our study, thapsigargin treatment increased NOX2 and NOX4 protein levels, while depletion of both subunits decreased thapsigargin-induced generation of superoxide in endothelial cells. Similar to p22phox in our study, NOX4 induction in response to chemical ER stress was dependent on ATF4 in cardiomyocytes (37). These studies further emphasize the importance of p22phox-dependent NADPH oxidases for ER stress-induced generation of ROS such as superoxide and H2O2.
Our findings further show that p22phox-dependent NADPH oxidases can activate the PERK/p-EIF2α/ATF4 pathway of the UPR by promoting phosphorylation of eIF2α and induction of ATF4 under ER stress conditions. While TEMPOL prevented p22phox-dependet upregulation of p-eIF2α and ATF4, application of a superoxide donor/H2O2 generator reversed the decrease in ATF4 in p22phox-depleted cells, indicating that superoxide/H2O2 generated by a p22phox-dependent NADPH oxidase promoted the UPR. Thus, our results suggest that p22phox is part of a positive feedback loop, which maintains the phosphorylation of eIF2α and induction of ATF4 to prevent unsuccessful protein translation in the earlier phase of the ER stress response.
It has been reported that induction of ATF4, but not of XBP1, in response to high cholesterol-induced ER stress was suppressed in NOX2-deficient macrophages. However, this response did not affect the PERK-p-eIF2α pathway, but rather PKR activation (21). In contrast, in our study, silencing or blocking of NOX2 did not prevent ATF4 induction by thapsigargin in endothelial cells. Whether this discrepancy is related to the cell type and/or different time frames remains to be further elucidated. However, in our study, silencing or blocking of NOX4 inhibited the ER stress-induced UPR similar to the situation with p22phox. A similar role of NOX4 was shown in response to chemical ER stress in cardiomyocytes. This response involved inhibition of PP1 by NOX4 (37). Whether p22phox is also involved in such a mechanism remains to be further elucidated.
In addition to chemical ER stressors and 7-Keto-cholesterol, we could show that severe hypoxia (0.1% oxygen) induces eIF2α phosphorylation and ATF4 in endothelial cells. Our findings further indicate that p22phox is involved in hypoxia-induced ER stress in endothelial cells, as depletion of p22phox diminished eIF2α phosphorylation and ATF4 induction, Importantly, increased phosphorylation of eIF2α and upregulation of ATF4 were also observed in lungs from mice exposed to chronic hypoxia, and these responses were blunted in p22phox-deficient mice.
While severe hypoxia has been previously shown to promote the UPR in tumors and endothelial cells (35, 43), and ER stress has been described in animal models of chronic hypoxia (40), our results are the first to link p22phox-dependent NADPH oxidases to ER stress and UPR regulation under hypoxia.
Our findings further indicate that induction of p22phox promotes apoptosis in response to chemical ER stress and hypoxia, since p22phox deficiency prevented activation of the effector caspases 3 and 7 under these conditions. Moreover, the ER stress-induced reduction in metabolic activity was improved in p22phox-deficient endothelial cells.
The PERK-ATF4 branch of the UPR has been shown to promote programmed cell death via different mechanisms (36), but mainly via induction of the transcription factor CHOP (45). CHOP can inhibit the expression of the antiapoptotic gene Bcl2 (27) and enhances the expression of proapoptotic genes, including death receptor 5 (DR5), which was reported to activate caspase 8- and caspase 3-dependent apoptosis on ER stress (24, 42).
In line, p22phox increased not only ATF4 but also CHOP levels in response to ER stress in our study, suggesting that p22phox limits the prosurvival response of the UPR under ER stress conditions. Although our data did not provide sufficient evidence that NOX2 is involved in the regulation of p-eIF2α and ATF4 in endothelial cells, others could show that NOX2 mediated apoptosis via activation of CHOP in macrophages exposed to high cholesterol load, and NOX2-deficient mice were protected against the development of apoptosis in the kidney in response to tunicamycin injection (21).
Similar to our study, demonstrating that NOX4 in addition to p22phox is involved in the regulation of p-eIF2α and ATF4 under ER stress conditions, depletion of NOX4 reduced the UPR, decreased CHOP expression, and protected against cell death in cholesterol-stimulated smooth muscle cells (30). In contrast, in tunicamycin-treated NOX4 knockout mice, kidney injury and apoptosis were increased despite decreased levels of ATF4 (37). While the reasons for these conflicting data regarding the role of NOX4 in the survival response to ER stress are not clear to date, our data in p22phox-deficient cells and mouse models suggest that targeting p22phox-dependent NADPH oxidases might enhance the prosurvival response of the UPR under ER stress conditions.
Collectively, our data show that p22phox is upstream and downstream of the p-eIF2α-ATF4 branch of the UPR (Fig. 10), suggesting that p22phox-dependent NADPH oxidases promote the UPR under ER stress conditions possibly counteracting the prosurvival function under hypoxic and other stress conditions. Thus, selective therapeutics targeting p22phox-dependent NADPH oxidases might be helpful in preventing UPR-induced disease states related to hypoxia and other ER stress conditions.

Materials and Methods
Chemicals
Thapsigargin and tunicamycin were from Merck Millipore (Darmstadt, Germany). Salubrinal was from Santa Cruz (Heidelberg, Germany), 7-Keto-cholesterol and TEMPOL were from Biomol (Hamburg, Germany), NOX2 inhibitor gp91ds-tat and nonfunctional scrambled gp91ds-tat were from Eurogentec (Liege, Belgium), and NOX1/NOX4 inhibitor GKT137831 was from Ambinter (Orleans, France). All other chemicals were from Sigma (Taufkirchen, Germany).
Cell culture
The HMEC-1 (ATCC CRL-3243) were grown in MCDB131 medium (PAN Biotech, Aidenbach, Germany) supplemented with 2 mM
Plasmids and transfection
A 1174 bp fragment of the CYBA gene 5′ flanking region directly upstream from the translational start site was amplified by PCR and subcloned into pGL3-BASIC (Promega, Mannheim, Germany) to create pGL3-p22phox-1.2K (p22-1.2K) using the following primers: Forward: 5′-TCTCTGAGAAAGGAGCTCAG-3′. Reverse: 5′-AAACTCATGACGACACGA ACCCGGCTG-3′. A putative ATF binding site at −568 bp (acctggtGAcgggaggccccc) was mutated to acctggtACcgggaggccccc using the QuikChange Mutagenesis Kit (Promega) revealing pGL3-p22phox-1.2Kmut (p22-1.2Kmut). For knockdown experiments, the following siRNAs were used: p22phox id: SI03078523, NOX2 id: SI03028424, NOX4 id: SI02642500 (all three Qiagen), XBP1 id: s14913, ATF4 id: 122372, ATF6 id: 115889 (all three Ambion, Darmstadt, Germany). As negative control, the AllStars Negative Control siRNA (Qiagen) was used. In addition, plasmids coding for a short hairpin RNA against ATF4 (target sequence: 5′-GGTGGCCAAGCACTTCAAA-3′), p22phox, and control (2) were used. For transfection, Lipofectamine 3000 (Life Technologies GmbH, Darmstadt, Germany) was used as described (35).
Luciferase assay
HMEC-1 were transfected using Lipofectamine 3000 (Life Technologies) after seeding in 24-well plates (25,000 cells/well). Twenty-four hours after transfection, cells were treated with thapsigargin (30 nM) or salubrinal (50 μM) for 8 h. Luciferase assay was carried out as described (31).
ROS measurements
HMEC-1 were pretreated or not with inhibitors for 30 min followed by treatment with thapsigargin, tunicamycin, or salubrinal. Generation of ROS was then detected using the fluoroprobe dihydroethidium (DHE; Life Technologies) or 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein di-acetate acetyl ester (CM-H2DCFDA; Life Technologies) as described (31). After stimulation, cells were washed with HBSS (PAN), loaded with DHE (50 μM) or CM-H2DCFDA (8.5 μM) for 10 min, and fluorescence was analyzed using a microplate reader (Tecan, Crailsheim, Germany). The fluorescence signal was normalized to viable cells using Alamar Blue assay (Life Technologies).
PCR/quantitative PCR analysis
After ER stress, RNA was extracted and reverse-transcribed using random primers as described (35). For detecting spliced and unspliced XBP1 mRNA, splicing area spanning primers were used (forward: 5′-CCT TGT AGT TGA GAA CCA GG-3′, reverse: 5′-GGG GCT TGG TAT ATA TGT GG-3′). GAPDH primers were already described (6). PCR products were separated on 1.5% agarose gels and stained with ethidium bromide. For visualization, a GelDoc 2000 system was used (Bio-Rad, Munich, Germany).
Quantitative PCR was performed using primers for p22phox (forward: 5′-CACAGCTGGGCGCTTCA-3′, reverse: 5′-TCCAGCAGGCACACAAACAC-3′), GADD34 (forward: 5′-GCCCAGAAACCCCTACTCAT-3′, reverse: 5′-GCCAGGAAATGGACAGTGAC-3′), and β-actin (7). Quantitative PCR analysis was performed as described (7) using the Perfecta SYBR Green FastMix (VWR, Darmstadt, Germany) in a Rotor-Gene 6000 (Corbett Life Science, Australia). Quantification was performed using ΔCT calculation.
Western blot
Western blot was carried out using 50 μg cell lysate as described (31) using antibodies against p22phox, NOX2 (both Active Bioscience GmbH, Hamburg, Germany), p22phox, β-actin, ATF4 (all Santa Cruz), XBP1, CHOP, eIF2α, p-eIF2α, cleaved caspase 7 (all New England Biolabs, Frankfurt, Germany), and ATF6 and NOX4 (both Abcam, Cambridge, United Kingdom). All unprocessed Western blots are presented in Supplementary Figure S9.
Bioinformatic and ChIP analysis
Bioinformatic analysis was performed using MatInspector Tool (Genomatix, Munich, Germany) (5). Chromatin immunoprecipitation analysis was performed as previously described (35). Briefly, HeLa cells were fixed at room temperature with formaldehyde. After stopping with 0.125 M glycine, cells were pelleted, washed with phosphate-buffered saline, lysed, and sonicated. Chromatin was precipitated with an anti-ATF4-protein A agarose bead complex for 12 h at 4°C. Immunocomplexes were washed, eluted in buffer (50 mM Tris, pH 8, 10 mM EDTA, and 1% SDS) followed by RNase A and proteinase K treatment. Precipitated DNA was purified with phenol/chloroform and ethanol and amplified by quantitative PCR using primers flanking the putative ATF4 site of the p22phox promoter (forward: 5′-TGC AGA GGT GTT CCC GGG CT-3′, reverse: 5′-GTC CCG TGT CAA CCA GGC GT-3′).
Metabolic activity and caspase assays
Cellular metabolic activity was tested by Alamar Blue (Life Technologies) in accordance with the manufacturer's protocol. Equal numbers of cells were transfected and seeded. Before treatment, cells were loaded with Alamar Blue and allowed to be reduced by intracellular reduction equivalents. Reduced Alamar Blue fluorescence was measured at 555 nm excitation and 585 nm emission in a microplate reader (Tecan).
Caspase 3/7 activity was measured using the Rh110 Caspase-3/7 Assay Kit (Anaspec, San Jose, CA) in accordance with the manufacturer's manual. Briefly, equal numbers of cells were seeded in 96-well plates and transfected. Twenty-four hours after transfection, assay buffer was added containing (Asp-Glu-Val-Asp)2–rhodamine (Rh) 110, which is cleaved by caspases 3 and 7 liberating Rh110 to generate a fluorescence signal. Fluorescence intensity is proportional to caspase 3/7 activity. Fluorescence was measured at 490 nm excitation and 520 nm emission wavelength in a 96-well plate reader (Tecan).
Animals
Mice containing a point mutation in the Cyba gene (nmf333 mice) resulting in a loss of p22phox protein were obtained from Jackson Laboratories (Bar Harbor, MN) (A.B6 Tyr +-Cybanmf333 /J) (28). All mice were of genetic C57BL/6 background. In all experiments, littermates from the same breeding pair were used as controls. For genotyping, genomic DNA was isolated from the ear. Genotyping was performed by PCR as described (28) using forward (5′-dCAGATGCCCACTGACTGCTA-3′) and reverse (5′-dCGAGCCACAGTACAGCTTCA-3′) PCR primers followed by digestion with BslI (New England Biolabs). The wild-type allele produces digestion products of 202 and 89 bp. The nmf333 allele produces digestion products of 162, 89, and 40 bp. All animal experiments were approved by the Regierung von Oberbayern.
Statistical analyses
Values presented are mean ± standard error if not otherwise stated. For qPCR analysis, REST analysis was used (33). For other statistical analyses, GraphPad Prism software (GraphPad Software, La Jolla, CA) was used. Results were compared by one-way ANOVA followed by Tukey's multiple comparison test or by Student t-test. p < 0.05 was considered statistically significant.
Footnotes
Acknowledgments
The authors thank Tobias Ziegler for help with the in vitro experiments, Daniela Koska for technical assistance, Michael Weitnauer for providing constructs, Maria Isabel Castellanos for providing cells, and Alexandra Sipol for providing chemicals. This work was supported by DFG GO709/4-5 and DZHK (German Centre for Cardiovascular Research, 81 × 2600510) and BMBF (Acidox, Epiros). Benjamin Trautz was a recipient of a stipend from the Medical Faculty, Technical University Munich (Translational Medicine).
Author Disclosure Statement
No competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.