
Abstract
Significance:
Properly controlled intracellular Ca2+ dynamics is crucial for regulation of neuronal function and survival in the central nervous system. The endoplasmic reticulum (ER), a major intracellular Ca2+ store, plays a critical role as a source and sink for neuronal Ca2+.
Recent Advances:
Accumulating evidence indicates that disrupted ER Ca2+ signaling is involved in neuronal cell death under various pathological conditions, providing novel insight into neurodegenerative disease mechanisms.
Critical Issues:
We summarize current knowledge concerning the relationship between abnormal ER Ca2+ dynamics and neuronal cell death. We also introduce recent technical advances for probing ER intraluminal Ca2+ dynamics with unprecedented spatiotemporal resolution.
Future Directions:
Further studies on ER Ca2+ signaling are expected to provide progress for unmet medical needs in neurodegenerative disease. Antioxid. Redox Signal. 29, 1147–1157.
Introduction
I
In the present review, we summarize current knowledge concerning the relationship between ER Ca2+ dynamics and neuronal cell death. We then discuss the use of recently developed genetically encoded ER Ca2+ indicators (114) to further understand the role of ER Ca2+ dynamics in neuronal cell death.
Neuronal ER Ca2+ Signaling
The ER plays a key role in not only protein synthesis and transport but also as a Ca2+ source and sink for influencing intracellular Ca2+ signals (6, 124). High concentrations of Ca2+ and Ca2+-binding proteins within the ER lumen render the organelle a large-capacity Ca2+ reservoir (96). Intraluminal free Ca2+ concentration within the ER ([Ca2+]ER) reaches the millimolar range, which is far higher than nano- to micromolar ranges of cytosolic Ca2+ concentration ([Ca2+]cyt) (115). This large concentration gradient across the ER membrane is a driving force for Ca2+ release from the ER to the cytosol. In neurons, fine tubular ER networks extend throughout cellular compartments, including dendritic spines (40, 72, 107), allowing involvement of ER Ca2+ signaling in multifarious functions, including synaptic plasticity and synaptic maintenance (4, 33, 61, 91). Proper regulation of Ca2+ release channels and pumps in the ER membrane is crucial for normal ER Ca2+ signaling.
Two types of Ca2+ release channels (inositol 1,4,5-trisphosphate [IP3] receptors [IP3Rs] and ryanodine receptors [RyRs]) mediate Ca2+ mobilization from the ER. A wide variety of neurotransmitter or neurotrophin receptors mediate IP3 generation through Gq-coupled activation of phospholipase Cβ or tyrosine kinase-mediated activation of phospholipase Cγ (8) (Fig. 1). IP3 is not the sole ligand of IP3R, but cooperative activation with Ca2+ is required for channel opening (11, 47) (Fig. 1). Among three mammalian subtypes of IP3R (IP3R1, 2, and 3), IP3R1 is the major subtype expressed in CNS neurons (27, 32).
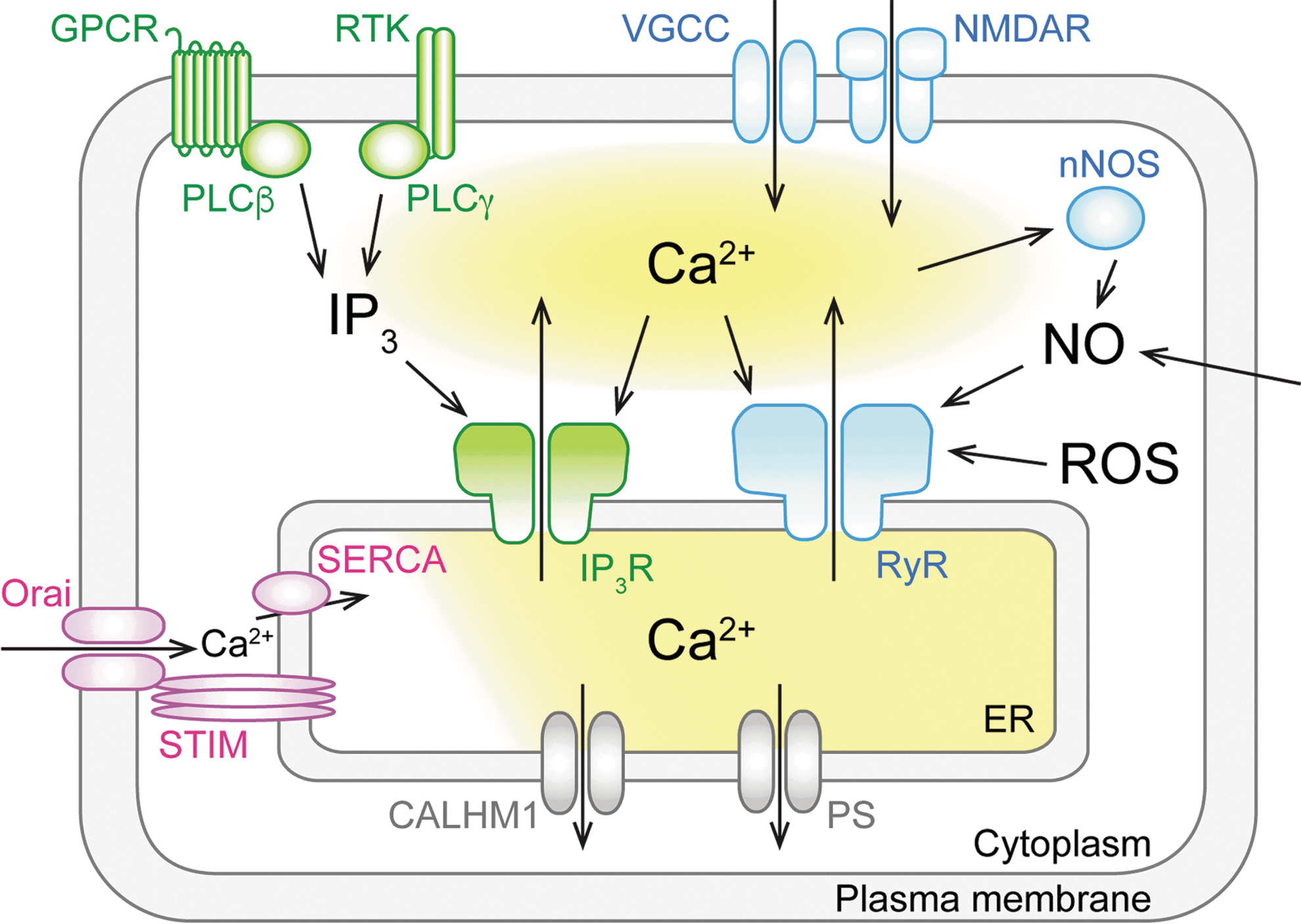
RyR is a Ca2+ release channel activated by Ca2+, and which mediates Ca2+-induced Ca2+ release (CICR). Thus, Ca2+ influx via VGCC or ligand-gated channels, including the N-methyl-D-aspartate receptor, triggers neuronal CICR and subsequent Ca2+-dependent events (25, 26, 54) (Fig. 1). Three mammalian subtypes of RyR (RyR1, 2, and 3) are expressed throughout the brain with relatively minor differences in expression pattern (81). It has been postulated that there are intrinsic modulators of RyR opening, such as cyclic ADP-ribose (77), although their significance is debated (31, 105). Reactive oxygen species are also known as a critical modulator of RyR (2, 71, 75, 121) (Fig. 1). Furthermore, nitric oxide (NO)-induced activation of RyR1 is involved in neuronal Ca2+ signaling and cell death (51, 78) (Fig. 1).
High levels of [Ca2+]ER are maintained by sarco-ER Ca2+ ATPase (SERCA) pumps. SERCA transports Ca2+ from the cytosol to the ER lumen against a large concentration gradient by hydrolyzing ATP (92) (Fig. 1). SERCA is crucial for Ca2+ refilling into the ER after Ca2+ release via IP3R or RyR. Treatment with an SERCA inhibitor such as thapsigargin and cyclopiazonic acid effectively depletes Ca2+ from the ER, suggesting that basal SERCA activity is required to counteract basal Ca2+ leakage from the ER. SERCA activity is regulated by ER-resident transmembrane proteins, including phospholamban, sarcolipin, myoregulin, endoregulin, and another regulin (1, 60, 69). The SERCA pump includes three gene products, SERCA1, SERCA2, and SERCA3. Two isoforms of SERCA2 (SERCA2a and 2b) as well as SERCA3 are expressed in the CNS (3, 124).
The ER is also involved in an active Ca2+ refilling mechanism that is evoked in response to ER Ca2+ depletion. Decreased [Ca2+]ER induces Ca2+ influx across the plasma membrane. This event, called store-operated Ca2+ entry (SOCE), enhances ER replenishment via SERCA-dependent Ca2+ uptake (Fig. 1). SOCE is mainly mediated by the ER-plasma membrane interaction via stromal interaction molecule (STIM) proteins in the ER membrane and Orai channels in the plasma membrane (67, 97, 126) (Fig. 1). Two subtypes of STIM (STIM1 and 2) localized within the ER membrane sense [Ca2+]ER via their intraluminal domain and multimerize on decreased [Ca2+]ER to activate Orai channels in the plasma membrane via an interaction between the cytosolic domains of STIM and Orai molecules (106). Neurons show active Ca2+ influx via VGCC and ligand-gated channels, in turn enhancing ER Ca2+ filling (14, 28, 29, 35, 41, 44, 95, 108). However, STIM-dependent ER Ca2+ filling is also crucial to maintain ER Ca2+ content in neurons (41, 113).
Disrupted Ca2+ Loading in the ER and Neuronal Cell Death
Under physiological states, [Ca2+]ER is maintained within a certain range by the balance between Ca2+ uptake and release. Disruption in ER Ca2+ loading state (i.e., depletion or overloading of Ca2+) may lead to pathological reactions (Fig. 2). It has been established that [Ca2+]ER depletion inhibits the protein folding mechanism and induces the unfolded protein response, which is followed by apoptotic cell death (22). Depletion of Ca2+ within the ER and the resulting ER stress response are implicated in ischemia/reperfusion-induced neuronal death (22, 23, 58, 76, 90). Although the precise mechanism remains elusive, NO-dependent processes are, at least in part, involved in ischemia/reperfusion-induced ER Ca2+ depletion (16, 23, 51, 58) (Figs. 2 and 3).


Sustained disruption of intracellular Ca2+ homeostasis involving imbalanced ER Ca2+ loading states (namely, the Ca2+ hypothesis) has been postulated as a critical factor in the etiology of Alzheimer's disease (AD) (7, 10, 17, 62). Presenilins (PS) are the catalytic component of γ-secretase complexes, which cleave amyloid precursor protein to produce amyloid-β. Mutations in PS are linked to some forms of familial AD (FAD), and thus, PS are recognized as critical players in the amyloid hypothesis (46, 101). In contrast, a crucial role for PS in the Ca2+ hypothesis has been documented. Neuronal attenuation of SOCE is one of the hallmark phenotypes of FAD-linked PS mutations (65, 127). Attenuation of SOCE, involving downregulation of STIM1 and STIM2, induces destabilization of dendritic spine morphology in hippocampal neurons (113, 122, 130), which may lead to disrupted synaptic functions underlying memory and cognitive deficits in AD. Prolonged disruption of synaptic functions is likely to provoke neurodegeneration (30).
Excessive ER Ca2+ loading is proposed as a cause of attenuated SOCE in FAD-linked PS mutations (65). In line with this hypothesis, PS can form putative Ca2+ leak channels on the ER membrane, and FAD-linked mutations in PS cause disruption of Ca2+ leakage from the ER, resulting in excessive ER Ca2+ loading (123) [Fig. 2, but see also Shilling et al. (103)]. It has also been reported that SERCA activity is enhanced by PS1, and that an FAD-linked PS1 mutant has a greater SERCA enhancing effect than wild-type PS1, which may lead to excessive ER Ca2+ loading (37). While excessive ER Ca2+ loading is associated with FAD-linked PS mutations in the above studies, it should be noted that unchanged or even decreased ER Ca2+ loading by PS mutants has also been reported (15, 20, 74, 128).
The calcium homeostasis modulator 1 (CALHM1) is another protein of interest in the Ca2+ hypothesis of AD. CALHM1 is localized in both the plasma membrane and ER membrane, and has been shown to form a Ca2+ leak channel in the plasma membrane (24). CALHM1 may also function as a Ca2+ leak channel in the ER, as overexpression of CALHM1 reduces ER Ca2+ content (34). Similarly, a polymorphism in the CALHM1 gene reduces Ca2+ permeability and also increases amyloid-β levels and is thereby linked to AD risk (24, 59, 80, 99). Therefore, it is possible that CALHM1 mutation disrupts ER Ca2+ leakage and results in excessive ER Ca2+ loading in the same manner as PS mutations (Fig. 2).
Disrupted Ca2+ Release and Neurodegeneration
The relationship between disrupted Ca2+ release function and neurodegenerative disease has been reported. Excessive or attenuated Ca2+ release via IP3R and RyR is implicated in several neurodegenerative diseases (Table 1 and Fig. 2).
FAD, familial Alzheimer's disease; HD, Huntington's disease; IP3R, inositol 1,4,5-trisphosphate receptor; RyR, ryanodine receptor; SCA, spinocerebellar ataxia.
Increased activity and/or expression of IP3R (20, 21, 104) and RyR (18, 100, 110) have been observed in cells expressing PS with FAD-linked mutations. Exaggerated Ca2+ release is expected to exacerbate Ca2+-dependent mechanisms involved in AD pathogenesis (82). Enhanced IP3R and RyR function in cooperation with excessive ER Ca2+ loading (associated with FAD-linked PS mutations) may further exaggerate Ca2+ release from the ER.
Huntington's disease (HD) is an autosomal dominant genetic disorder that is caused by CAG triplet (CAG repeat) expansion in the huntingtin (Htt) gene (38). Insertion of polyglutamines coded by CAG repeats changes the properties of Htt protein allowing toxic interactions with various target molecules (52), for example, mutant Htt protein directly binds to IP3R1 (120) (Fig. 2). This interaction increases the affinity of IP3R1 for IP3 and enhances Ca2+ release from the ER (120), which has been suggested to induce Ca2+ overloading in mitochondria and trigger apoptosis of striatal neurons (119, 129). Not only the enhancement but the impairment of IP3R1 function is also implicated in HD. The impairment of IP3R1-mediated Ca2+ release due to the reduced interaction between IP3R1 and GRP78, an ER chaperone, is reported in striatal neurons of HD model mice (43). The involvement of RyR is also reported. The enhanced spontaneous Ca2+ release (Ca2+ leak) via RyR is implicated in neuronal cell death induced by mutant Htt (116).
Spinocerebellar ataxias (SCAs) are genetic disorders caused by either a recessive or dominant gene. SCA2 is caused by CAG repeat expansion in the ataxin-2 gene, and results in cerebellar Purkinje cell (PC) degeneration (38). Mutant ataxin-2 protein binds to IP3R1, increasing the affinity of IP3R1 for IP3 and enhancing IP3-induced Ca2+ release (55, 68) (Fig. 2). The effect is similar to that of mutant Htt protein described above. IP3-induced Ca2+ release can be blocked by overexpressing IP3 5-phosphatase (5ppase), an IP3 hydrolyzing enzyme (33, 53, 61, 63, 73). This 5ppase-dependent blockade of IP3 signaling in PCs of SCA2 transgenic mice alleviates the SCA2 phenotype (55). These results support the notion that enhanced IP3-induced Ca2+ signaling underlies SCA2 pathogenesis.
Another form of neurodegenerative disease, SCA3, is caused by CAG repeat expansion in the ataxin-3 gene, which results in neuronal degeneration in the substantia nigra and pontine nuclei (38). Here, the mutant form of ataxin-3 specifically binds and enhances IP3R1, similar to mutant Htt and ataxin-2 proteins (19) (Fig. 2). Furthermore, RyR inhibition by dantrolene prevents neuronal loss and alleviates motor coordination deficits in mutant ataxin-3-expressing mice (19). Therefore, overactivation of IP3R1- and RyR-mediated Ca2+ release from the ER may be involved in SCA3 pathogenesis. Furthermore, genetic studies of human SCA15 have revealed total or partial deletion as well as a missense mutation of the IP3R1 gene (39, 57, 64).
Neuronal Cell Death Induced by NO-Induced Activation of RyR
Excessive neuronal activation by the excitatory neurotransmitter, glutamate, often leads to neuronal cell death. This pathological process is referred to as excitotoxicity and is encountered in various disease states, including brain ischemia and epileptic seizures. Brain NO concentration is increased owing to activation of NO synthase (NOS) both during ischemia and after reperfusion (70). NO generated by neuronal-type NOS (nNOS or NOS1) is implicated in excitotoxic neuronal death, because excitotoxic neuronal death is ameliorated in NOS1-knockout (KO) mice in disease models such as the middle cerebral artery occlusion (MCAO) model of stroke (45, 51) (Fig. 3) and kainic acid (KA) model of temporal lobe epilepsy (89).
How does an increase in NO concentration lead to neuronal death? When NO is simultaneously generated along with superoxide, peroxynitrite is also generated, which is a highly cytotoxic oxidant implicated in neuronal death (12, 36, 87, 117). On the contrary, it has been shown that NO may modulate the function of various proteins through S-nitrosylating cysteine residues on target proteins (42, 49, 83) (Fig. 4A). Recent studies indicate that S-nitrosylation-mediated modulation of RyR1 function is involved in excitotoxic neuronal death (51, 78).
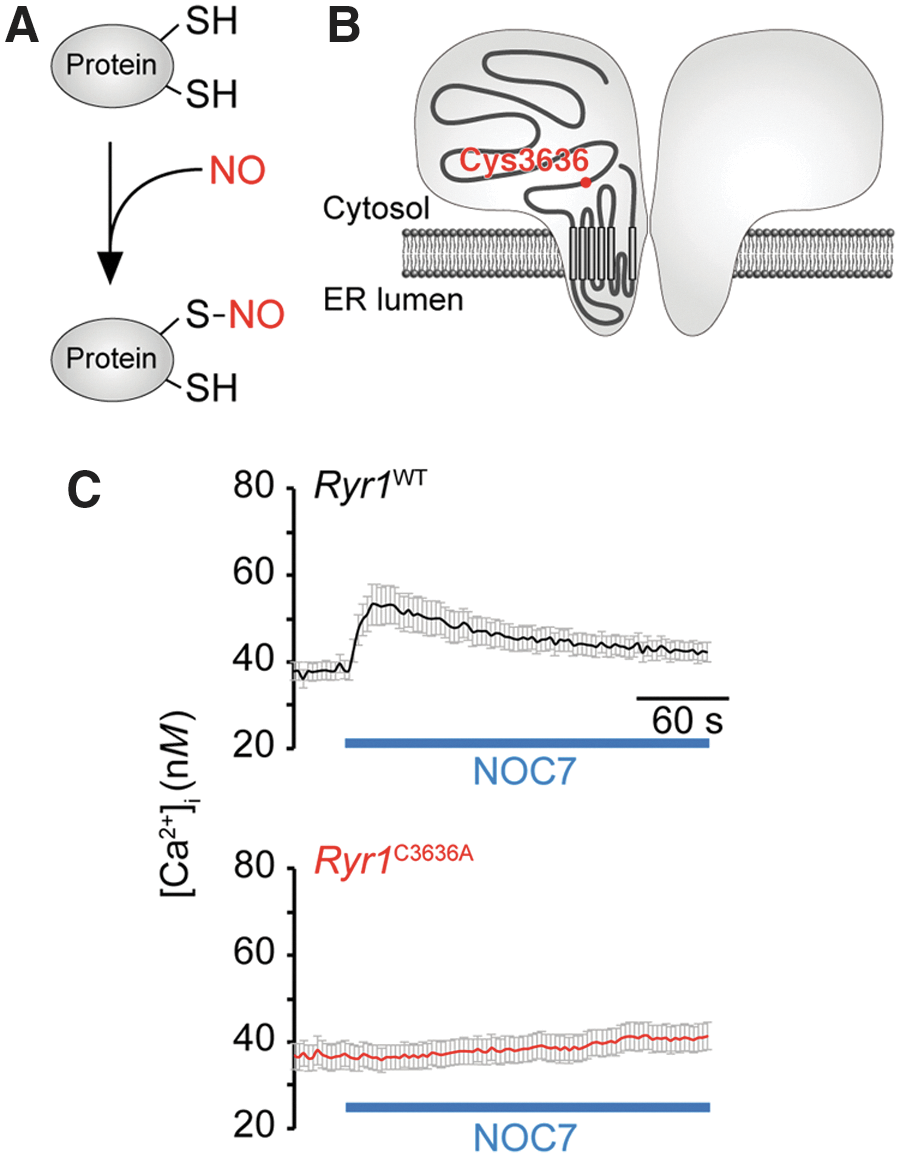
The effect of NO on RyR1 function was first studied in skeletal muscle cells, in which abundantly expressed RyR1 is involved in excitation/contraction coupling. Accordingly, S-nitrosylation of one of the cysteine residues (Cys-3635 in rabbit/human RyR1) was shown to enhance channel opening (94, 111, 112). On increased NO concentration, increased [Ca2+]i owing to Ca2+ release via RyR1 has been observed in central neurons, and is hereafter referred to as NO-induced Ca2+ release (NICR) (51). The effect of NO has been studied in other RyR subtypes, with NO found to activate both RyR1 and RyR3, but not RyR2 (51).
Because RyR1 expression overlaps with NOS1 in various brain regions, and because NOS1 is a key molecule in excitotoxicity, involvement of NICR in excitotoxicity has been investigated. Indeed, there was a hint that RyR1 is associated with excitotoxicity, because dantrolene (an NICR inhibitor) ameliorates both brain infarction after MCAO (66) and seizure-induced neurodegeneration (5, 85, 93). When the effect of dantrolene was compared between wild-type and NOS1-KO mice, the ameliorating effect of dantrolene was absent in NOS1-KO mice (51) (Fig. 3). These results are consistent with the notion that NICR is involved in excitotoxicity.
To further clarify the role of NICR in excitotoxicity, NICR was genetically silenced in mice in which Cys-3636 (corresponding to Cys-3635 in rabbit/human) of RyR1 was replaced with alanine (Ryr1 C3636A mice) (78) (Fig. 4B). Appropriately, NICR was lost in cultured neurons prepared from Ryr1 C3636A mice (Fig. 4C). These NICR-deficient mice bleed and grow normally. When they are subjected to KA-induced epilepsy, their seizure score is the same as wild-type mice, indicating that their susceptibility to seizures is unchanged (Fig. 5A). Neuronal damage was observed in the CA3 region of the hippocampus in wild-type mice after KA-induced epilepsy (Fig. 5B) but was significantly ameliorated in Ryr1 C3636A mice (Fig. 5B). Furthermore, application of dantrolene also ameliorated KA-induced neuronal damage (Fig. 5B). These results indicate that NICR via RyR1 is involved in excitotoxicity and that NICR inhibitors may have therapeutic value in prevention of excitotoxicity. It remains to be clarified how NICR in neurons leads to neuronal death. Prolonged depletion of ER Ca2+ due to NICR may play a role.

Probing Ca2+ Dynamics Within the ER
As reviewed above, ER Ca2+ loading state, which determines the driving force for Ca2+ mobilization and regulates function of ER-resident proteins, is crucially involved in neuronal cell death. Therefore, direct monitoring of neuronal ER Ca2+ dynamics using genetically encoded ER Ca2+ indicators (13, 48, 79, 88, 98, 118, 125) may be instrumental in advancing our understanding of associated pathologies. In a pioneering work, one of those indicators was used to study ER Ca2+ dynamics in cultured fibroblasts expressing FAD-linked PS mutants (74), although further improvements of spatiotemporal resolution of ER Ca2+ imaging are needed for studies on in situ or in vivo neurons.
Recently developed ER Ca2+ indicators, for example, CEPIAs (calcium-measuring organelle-entrapped protein indicators) provide high-resolution visualization of ER Ca2+ dynamics in various types of cells, including neurons (115) (Fig. 6A). G-CEPIA1er (86) was used to visualize synaptically evoked ER Ca2+ dynamics in cerebellar PCs, which possess a well-developed ER network with a high density of IP3R (72, 102) (Fig. 6B). In response to parallel fiber (PF)-dependent IP3 production, transient and focal decreases in [Ca2+]ER in dendrites and spines were observed in PCs (Fig. 6B). Refilling of ER Ca2+ after PF-induced local depletion mainly occurred through lateral Ca2+ diffusion within the ER network, together with a minor contribution of SERCA-dependent Ca2+ reuptake (Fig. 6B). Repetitive climbing fiber (CF) stimulation, which induces dendrite-wide Ca2+ spikes via VGCC (56), results in an accumulating increase in [Ca2+]ER throughout the dendrite (Fig. 6C). CF-induced responses reach a plateau after 10–20 stimulation pulses, likely because [Ca2+]ER reaches a new equilibrium during repetitive Ca2+ spikes (Fig. 6C). Elevated [Ca2+]ER returned to basal levels within a few minutes after cessation of CF inputs (Fig. 6C), which is consistent with previous observations showing that the potentiating effect of preceding depolarization lasts a few minutes (14, 35, 44, 108). Thus, G-CEPIA1er enables visualization of synaptically evoked ER Ca2+ dynamics at the individual spine level.
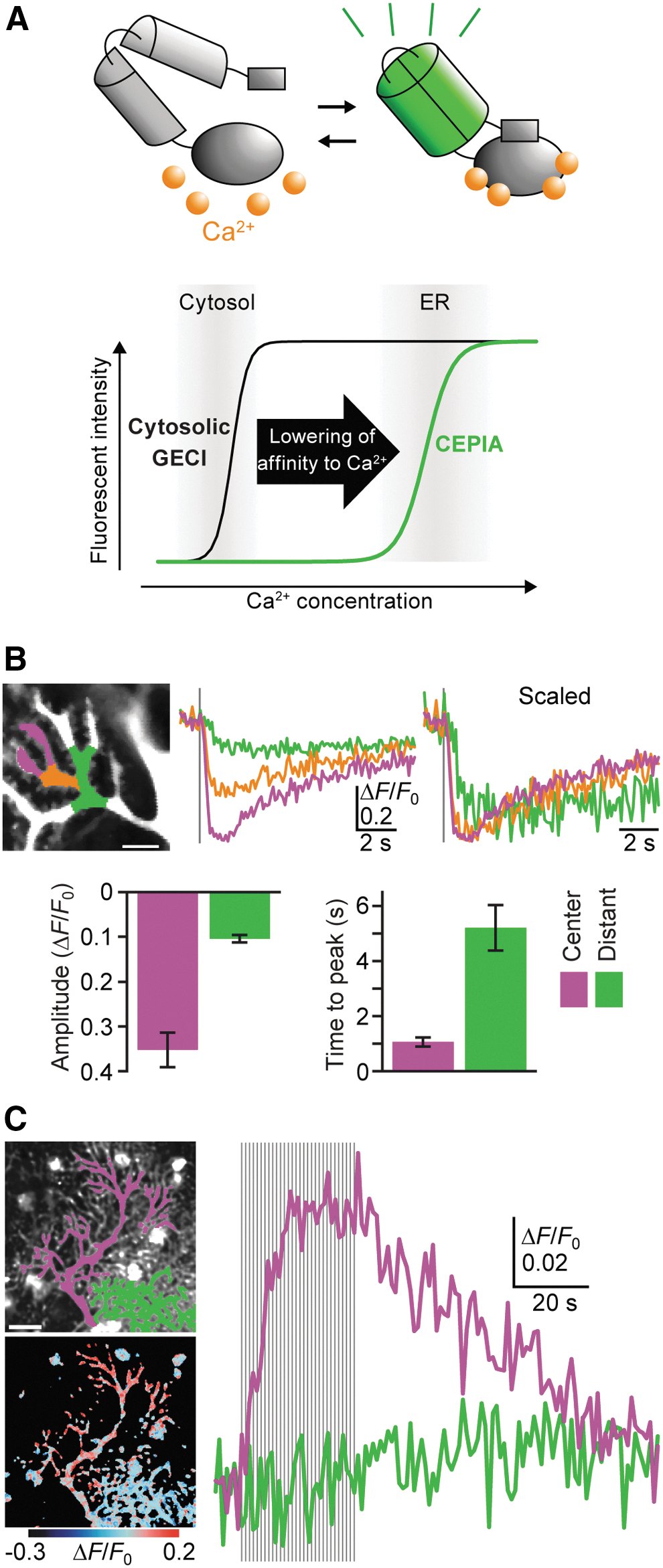
Other high-performance ER Ca2+ indicators have been developed by different groups (50, 84). As in the case of G-CEPIA1er, the low-affinity version of GCaMP6 enables visualization of ER Ca2+ dynamics in the small compartment such as presynaptic boutons (50). A ratiometric Ca2+ sensor of the green fluorescent protein-aequorin protein has an advantage in the quantification of [Ca2+]ER as in the case of other ratiometric indicators such as GEM-CEPIA1er (84). Analysis of ER Ca2+ dynamics under pathological conditions with high spatiotemporal resolution using these newly developed ER Ca2+ indicators may shed new light on the study of neurodegenerative diseases.
Perspectives
In this review, we have summarized current knowledge on involvement of ER Ca2+ signaling in neuronal cell death and neurodegeneration. Accumulating evidence allows us to recognize ER Ca2+ signaling as a promising therapeutic target for neuroprotection during various pathological conditions. The use of genetic methods to inhibit IP3 signaling or new ER Ca2+ indicators may help advance our understanding of the role of ER Ca2+ dynamics in neurodegenerative disease. Such future studies on ER Ca2+ signaling are expected to provide progress for unmet medical needs in neurodegenerative disease.
Footnotes
Acknowledgments
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Nos. JP16K08543 [Y.O.], JP13J00025, and JP15K08227 [Y.M.], JP15H05648 [K.K.], and JP21229004 and JP25221304 [M.I.]), as well as grants from the Tokyo Society of Medical Sciences (Y.O.) and Pharmacological Research Foundation (Y.O.), and project research grants from Toho University (Y.M.).