
Abstract
Significance:
Calcium (Ca2+) hypothesis of Alzheimer's disease (AD) gains popularity. It points to new signaling pathways that may underlie AD pathogenesis. Based on calcium hypothesis, novel targets for the development of potential AD therapies are identified.
Recent Advances:
Recently, the key role of neuronal store-operated calcium entry (nSOCE) in the development of AD has been described. Correct regulation of nSOCE is necessary for the stability of postsynaptic contacts to preserve the memory formation. Molecular identity of hippocampal nSOCE is defined. Perspective nSOCE-activating molecule, prototype of future anti-AD drugs, is described.
Critical Issues:
Endoplasmic reticulum Ca2+ overload happens in many but not in all AD models. The nSOCE targeting therapy described in this review may not be universally applicable.
Future Directions:
There is a need to determine whether AD is a syndrome with one critical signaling pathway that initiates pathology, or it is a disorder with many different signaling pathways that are disrupted simultaneously or one after each other. It is necessary to validate applicability of nSOCE-activating therapy for the development of anti-AD medication. There is an experimental correlation between downregulated nSOCE and disrupted postsynaptic contacts in AD mouse models. Signaling mechanisms downstream of nSOCE which are responsible for the regulation of stability of postsynaptic contacts have to be discovered. That will bring new targets for the development of AD-preventing therapies. Antioxid. Redox Signal. 29, 1176–1188.
Introduction
A
There are many hypotheses of AD pathogenesis: the oldest one is the cholinergic hypothesis (11), the dominant one is the amyloidogenic hypothesis (51), and also popular is the tau hypothesis (67). Recently, amyloidogenic hypothesis has been transformed to the oligomer hypothesis or soluble beta-amyloid (Aβ) hypothesis (41). It differs from the classical amyloid hypothesis by positing that the proximal neurotoxins in AD are soluble oligomers of Aβ, rather than Aβ in the form of amyloid aggregates. However, so far none of these hypotheses has brought successful drugs to prevent the AD pathogenesis. Recently, calcium hypothesis of AD has started to gain popularity. It states that calcium signaling mishandling in neurons occurring at early disease stages is the key event triggering synaptic dysfunction and neurodegeneration (4). Development of new powerful and precise calcium imaging techniques enabled extensive research in this field, and new intriguing data recently appeared. This review is devoted to description of calcium signaling pathways disrupted during AD with particular emphasis on endoplasmic reticulum (ER) calcium channels and store-operated calcium entry. Based on the calcium hypothesis, novel targets for the development of AD-preventing therapies are suggested, and their applicability to the treatment of AD cases is discussed.
Ca2+ hypothesis of AD
The calcium (Ca2+) hypothesis of brain aging was first formulated in 1982. In 1989 and 2017, the hypothesis was revised to introduce new data and outline questions which needed to be answered in the future (4, 63). Calcium hypothesis of AD is connected with other hypotheses in the field since changes in calcium signaling are likely to be secondary to deleterious actions of Aβ oligomers in neurons, disruption of presenilin (PS) functions, defects in mitochondria dysfunction, and aging-related changes.
There is a growing body of evidence that dysregulation in signaling pathways that handle Ca2+ plays a major role in the initiation of AD pathogenesis. Ca2+ is a second messenger that is involved in many if not all cellular processes of neuronal life. Calcium can enter the neuron from extracellular space via membrane-embedded Ca2+-permeable channels. Among them are voltage-gated Ca2+ channels (VGCCs), nonspecific cation channels N-methyl-D-aspartate receptors (NMDARs), and transient receptor potential channels (TRPCs).
Neurons have intracellular Ca2+ stores such as ER and mitochondria. Ca2+ can be released from ER via inositol trisphosphate receptor (InsP3R) and ryanodine receptors (RyanR) (14). Mitochondria can shape intracellular calcium signaling, mainly via Ca2+ sequestering mechanism (97). Ca2+ uptake into mitochondria plays an important role in neuronal physiology by stimulating mitochondrial metabolism and increasing mitochondrial energy production. Excessive Ca2+ entry into mitochondria can lead to opening of a permeability transition pore (PTP) and may lead to apoptosis (111). How these calcium entry pathways affected during AD will be discussed later.
First symptoms start to appear in patients 70–80 years old for SAD. For genetically inherited familiar form of AD, first symptoms may appear already at 50 years of age. The human brain has protective mechanisms that fight with the disease until middle age or later. However, with age the capacitance of such mechanisms gets lower, and at certain moment brain is not able to resist AD anymore. Loss of ability to handle Ca2+ is one of the features of aging neurons. In AD experimental models, Ca2+ is accumulated inside of neurons, and intracellular Ca2+ concentration is increased (4, 15). Elevated calcium levels appear to be toxic to cells and trigger subsequent pathological processes, which drive AD pathogenesis. What are the reasons for the increase of Ca2+ in AD? Is there a main Ca2+ handling mechanism that is dysregulated in AD, or it is a consequence of events that lead to development of the AD? Are there any therapeutic agents that can normalize Ca2+ signaling system in AD? Calcium hypothesis of AD is aimed at answering these and many other related questions.
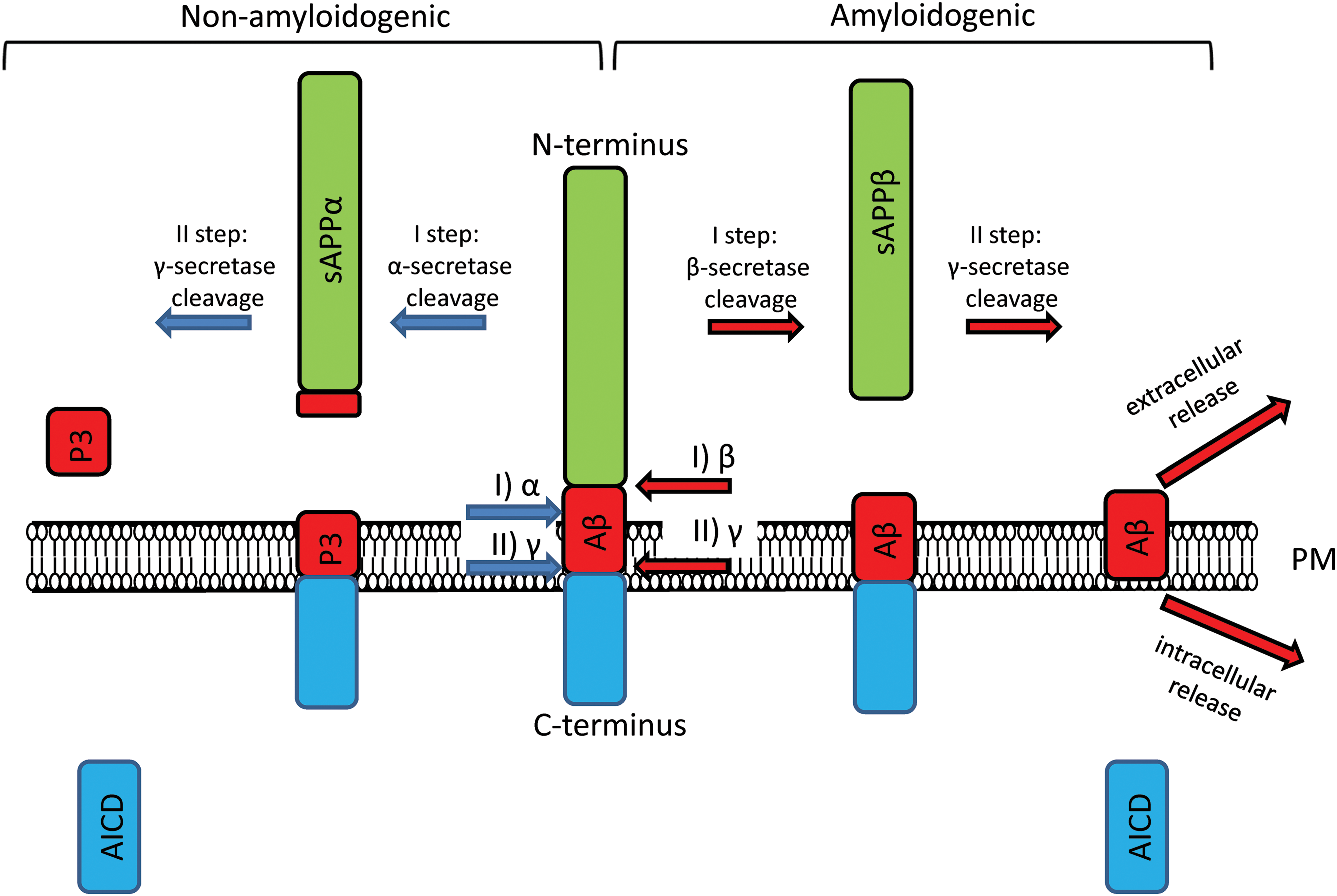
Familial forms of AD are caused by mutations in genes encoding APP, PS1, and PS2 proteins. For a long time, Aβ, the product of proteolytic cleavage of APP, has been considered the initial molecule that triggers AD. While there is a debate on whether the Aβ is a major toxic culpit in AD (55, 80), it plays a major role in the pathogenesis of AD and in calcium dysregulation as well. Other AD-related proteins are PSs, which form the catalytic subunit of gamma secretase. In amyloidogenic pathway (Fig. 1), gamma secretase is responsible for cleavage of APP at its transmembrane domain and produces toxic Aβ (60). In addition to gamma secretase function, PS1 plays the function of passive Ca2+ leak channel (84, 121), which is disrupted by many but not all FAD-associated mutations in PS1. The influence of mentioned proteins on Ca2+ signaling pathways during AD pathology is discussed below.
Aβ and Neuronal Calcium Signaling
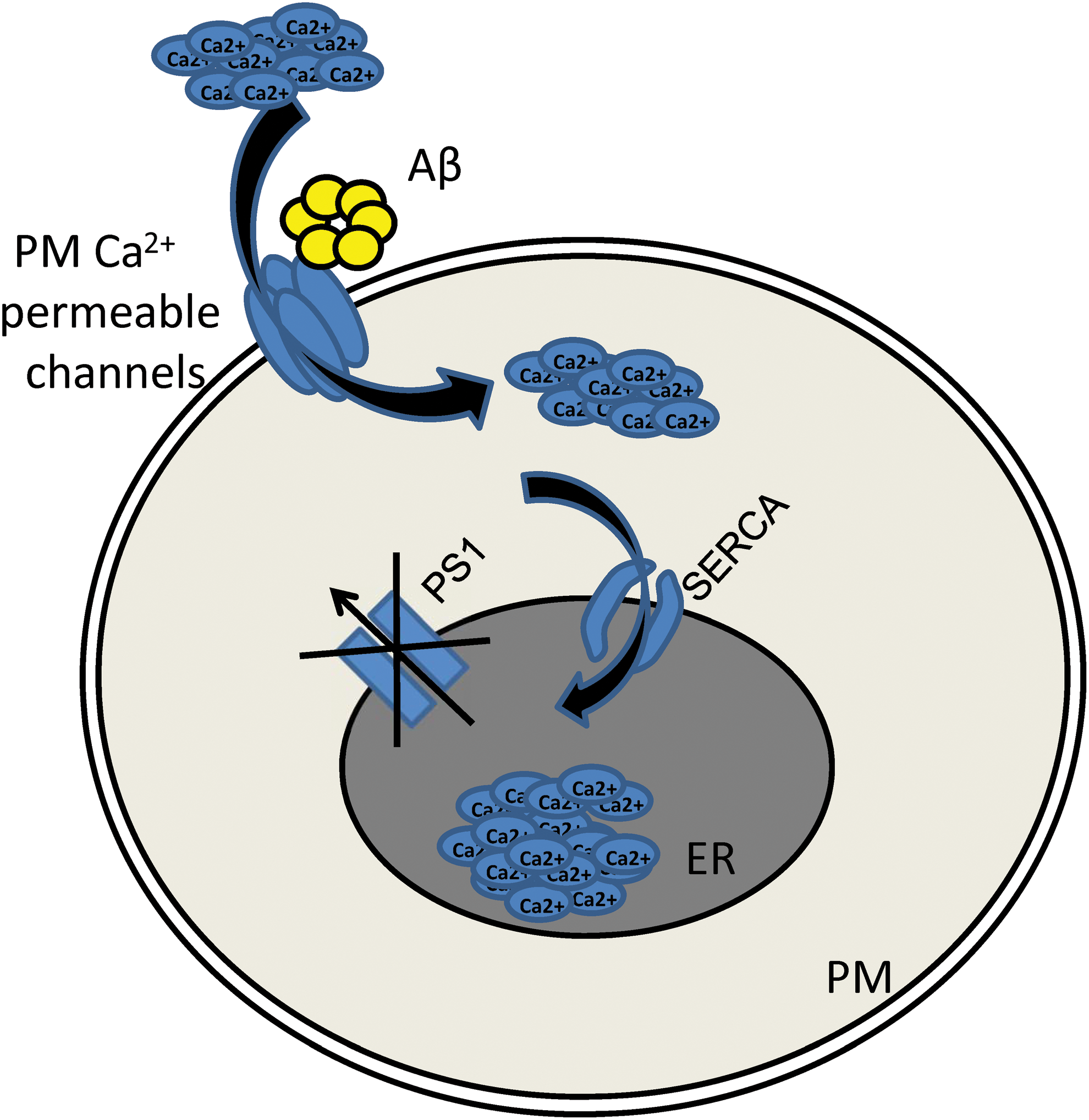
Aβ was initially recognized as the main toxic agent in AD (103). Currently, Aβ theory is under revision (55, 80). It is apparent that Aβ plays an important role in AD pathogenesis, but some other factors also contribute to AD pathology together with Aβ or may even precede the Aβ toxicity. Detrimental effect of Aβ oligomers on neurons has been extensively studied, and many publications demonstrated that Aβ aggregates promote the increase in neuronal cytosolic Ca2+ concentration (16, 34 –37, 46, 68, 107, 124). The exact mechanism of Aβ-mediated disruption of Ca2+ homeostasis is under active investigation. Concerning the role of Aβ in Ca2+ dyshomeostasis during AD, it has been observed that Aβ can make Ca2+-permeable channels in plasma membrane by themselves (7) (Fig. 2).

Probably the most important Aβ targets are NMDA receptors. Activation of NMDA receptors is a key event in long-term potentiation phenomenon, which is thought to be the cellular basis of memory formation process. The effects of Aβ on NMDA receptors were extensively studied (43, 81, 134). Particularly, it has been shown that Aβ is able to increase the vulnerability of neurons to excitotoxicity, which is caused by excessive NMDAR activation with subsequent cell calcium overload (77, 78). Some data indicate that Aβ in its oligomeric form may directly bind and modulate activity of NMDA receptors (30, 69, 108, 117). There is indication that NMDARs are required for synaptic targeting of Aβ oligomers, but they do not appear to comprise the actual binding sites for Aβ oligomers (32). Various deleterious effects of Aβ on NMDAR were reported. It was reported that in early disease stages, Aβ activates NMDAR and induces rapid Ca2+ elevation in neurons (40, 87, 99, 134). Usage of Aβ oligomers at sublethal concentrations induces prolonged Ca2+ signaling via NMDAR. These Ca2+ signals trigger redox-sensitive stimulation of RyanR-mediated Ca2+ release from the ER, decreased RyanR2 protein expression, mitochondrial fragmentation, and prevented RyanR-mediated spine remodeling (89).
Detrimental effect of oligomeric Aβ on RyanR-mediated Ca2+ signaling in ER was also observed in glia, particularly in cultured astrocytes (2). In the study performed by Gavello et al., the oligomeric Aβ42 differently regulated RyanR, NMDAR, and VGCCs by increasing Ca2+ release through RyaRs, and inhibiting Ca2+ influx through NMDARs and VGCCs. According to this study, the overall increased intracellular Ca2+ concentration caused stimulation of K+ current carried by big conductance Ca2+activated potassium (BK) channels and inhibition of hippocampal network firing (44). Application of oligomeric Aβ species in vivo causes fast rise in resting Ca2+ levels which depend on NMDARs activation and triggers dendritic spines loss (6). Treatment with aducanumab (anti-Aβ antibody) restores calcium homeostasis in Tg 2576 mice (61). The treatment effect was connected to restoration of NMDAR function rather than to restoration of intracellular Ca2+ signaling. In addition, it was reported that Aβ induces reduction in NMDAR expression and enhances its endocytosis (109), impairs NMDAR-dependent long-term potentiation (LTP) (29) and reduces NMDAR-mediated calcium influx into active spines (104).
Another calcium-permeable plasma membrane channels are presynaptic VGCC. It has been observed that Aβ oligomers decrease synaptic transmission between hippocampal neurons, most likely via depression of Ca2+ flux through P/Q-type calcium channels (85). However, in HEK293 cells that overexpress recombinant P/Q-type calcium channels the increase in P/Q-type currents by Aβ oligomers has been observed (54). Authors explain such differences by the fact that ion channels can be bidirectionally regulated by the same molecule. For example, potassium channel blocker k-conotoxin PVIIA both enhances and reduces potassium currents depending on its activation state (65). In contrast, the authors report that block of postsynaptic L-type calcium channels by 10 μM nimodipine did not reverse Aβ42-induced deficits, indicating that Aβ oligomer pathology is specifically mediated via presynaptic ion channels. In contrast to the results mentioned above, there are data on age-dependent upregulation of L-type VGCC currents in Cornu Ammonis area 1 region of hippocampus in 3 × TgAD (triple transgenic mouse model of Alzheimer's disease) mice (125). It was reported that antagonists of L-VGCC can protect neurons, and preserve synaptic function in animal models of aging and AD (5, 73, 88, 98, 122).
Beside actions of Aβ on the plasma membrane-embedded calcium channels, it was shown that both extracellular and intracellular Aβ applications alter activity of ER-resident calcium channels—RyanR and InsP3R (42, 57). RyanR- and InsP3R-mediated Ca2+ responses were induced by application of Aβ (25 –35) and Aβ40 on cultured cortical neurons. It was shown that Aβ42-induced Ca2+ release from the ER in intact human neuroblastoma cells was just partially mediated by InsP3R, while the greater part of Ca2+ elevation was induced by an alternative mechanism (57). Interestingly, it was reported that lowering of RyanR-mediated Ca2+ release leads to the reduction of both intracellular and extracellular Aβ load in APP(swe)-expressing (Tg2576) mice (86). According to Briggs et al., it is an example of proposed pathogenic feed-forward cycle in which elevated Ca2+ levels triggered by Aβ further facilitate production of Aβ (17). The link between extracellular Aβ and intracellular channels is still elusive, but several possible mechanisms of action have been proposed. It was shown that Aβ oligomers induce InsP3 production through stimulation and dimerization of synaptic metabotropic glutamate receptor 5 (mGluR5) receptors (96). Another possible link is that in dendritic spines, Ca2+ release by RyanR can be triggered by Aβ-facilitated Ca2+ influx through NMDARs (17, 45, 89). Studies performed by SanMartin et al. demonstrated that Aβ oligomers promote RyanR2-mediated Ca2+ release, mitochondrial Ca2+ entry, ROS generation, and fragmentation of the mitochondrial structural network. It was further shown that RyanR2 knockdown as well as usage of antioxidants reduces Ca2+-mediated noxious effects of Aβ oligomers on mitochondrial function (101, 102). Some AD models demonstrated intracellular Aβ accumulation, which may also take part in ER calcium signaling destabilization (75). Intracellular application of Aβ oligomers into Xenopus oocytes stimulates G-protein-mediated InsP3 production and consequent cytotoxic Ca2+ release from the ERs (35). Another study demonstrated that InsP3Rs were not required for Aβ42-stimulated Ca2+ release from ER in DT40 chicken B-lymphocyte line permeabilized cells, revealing an additional direct effect of Aβ42 upon the ER (57).
Role of PSs in Ca2+ Homeostasis
PSs act as a catalytic subunit of gamma secretase. FAD-associated mutations disrupt gamma secretase function, leading to amyloidogenic processing of APP and production of toxic Aβ species (Fig. 1) (60). Whether FAD mutations cause gain of function or loss of gamma secretase function is the subject of debate (123, 127, 128). Development of gamma secretase modulators as potential anti-AD therapeutics is complicated due to essential role of gamma secretase in Notch processing (31). Calcium signaling effects of Aβ were discussed above. APP intracellular domain also affects ER Ca2+ release by regulating the expression of genes involved in Ca2+ homeostasis (71).
Significant body of research suggests that AD-bearing PS mutants cause Ca2+ dysregulation independently of its gamma secretase function and Aβ accumulation, and due to changes in activity of RyanR and InsP3R (17, 25, 39, 93). Upregulation of RyanR-mediated Ca2+ release and increased levels of RyanR expression among different PS-mutation bearing AD models were reported (17, 21, 23, 26, 33, 45, 113). It was proposed that PSs alter RyanR gating through direct protein–protein interaction mediated by N-terminal cytosolic domain of PSs (52, 91, 100). RyanR gating effects of PS1 and PS2 are isoform specific (91). Increase of the PS2 to PS1 ratio was reported for normal aging mice in both cerebellum and forebrain, which correlates to loss of spatial memory, learning, and motor function (58). Such homologue misbalance is proposed to contribute to age-dependent cytosolic Ca2+ level increase (17). Based on these findings, it was proposed that excessive Ca2+ release from ER and elevated cytosolic Ca2+ concentrations observed during AD may be a result of altered RyanR interaction with PSs (91). Changes in RyanR function have been suggested to be responsible for alterations in synaptic activity induced by PSs (126).
Sensitivity of InsP3R to its agonist InsP3 significantly increased in cell expressing mutant PSs (27, 28). Suppression of InsP3R expression normalized exaggerated Ca2+ signals observed in cortical and hippocampal neurons in PS1-M146 V knock-in and 3 × Tg AD mice models, indicating that it might be a potential therapeutic strategy (106). Recent research using data-based computational modeling provided deeper insight into InsP3R gating in the presence of mutated PSs (76). This model predicted that that the gain-of-function enhancement is sensitive to both InsP3 and Ca2+, and that very small amount of InsP3 is required to stimulate InsP3R channels in the presence of FAD-causing mutant PS. Therefore, significant activity of the InsP3R at resting InsP3 concentration should lead to spontaneous Ca2+ signals in cells (76). Using computational model, the same research group suggested that mutation in PSs increases the open probability of mitochondrial PTP, which in turn triggers pathological processes and may induce cell death (119). It was proposed that mutated PSs enhance Ca2+ release through InsP3R into a cytoplasmic microdomain formed by neighboring cluster of a few InsP3R channels and mitochondria channel uniporter, and therefore facilitate mitochondrial calcium uptake (119). This investigation proposes direct link between Ca2+ disruptions and impaired mitochondrial function, as observed in AD.
Additional gamma secretase-independent function of PSs was suggested. PS1 and PS2 were reported to act as passive ER calcium leak channels (84, 121, 131). This idea was initially controversial (105), but it was supported by unbiased screen for ER Ca2+ leak channels (10). This function of PS1 is altered by many but not all FAD-associated mutations. For example, extensively studied M146 V mutation is a classic example of PS1 mutation that causes disruption of Ca2+ leak function (121). However, the deletion of Exon 9 in PS1 is a pathological mutation that acts as a gain of function for ER Ca2+ leak activity (121). A correlation between patient clinical phenotypes and effects of FAD mutations on ER Ca2+ leak function was observed (82). Site-directed mutagenesis approach was used to map potential ion conduction pore of PS1 (83). It was demonstrated that D385 but not D257 residue is important for channel function of PSs (83, 121). Interestingly, PSs share the fold with chloride channels (118), and the high-resolution crystal structure of archaeal PS homologue, PSH1, has a hole that traverses through the entire protein and is large enough to allow passage of Ca2+ ions (74). This hole was however not apparent when structure of γ-secretase complex was solved (9). Proteolytically cleaved PS does not form ER Ca2+ leak channels (121), which may explain lack of obvious ion conduction pathway in mature γ-secretase complex.
Using molecular dynamics approach, a dynamic all-atom model of mature PS1 embedded into the membrane has been published recently (110). It is important to note that PS1 undergoes post-translational modifications, particularly autoendo-proteolysis. As many other post-translational modifications, autoproteolysis is suggested to be essential for the change of PS1 from inactive to active state (110). Authors have confirmed previously published gating mechanism for PS1 (64). In agreement with previously published data, they have observed that Exon 9 plays a role of a “plug” that closes or opens the “doors” to the catalytic pocket of the PS1 depending on the activation state. Although not modeled in this article, these data suggest that deletion of Exon 9 permanently opens the interior chamber of PS1, consistent with superleaky pore phenotype of PS1ΔE9 mutant (121).
In conclusion, mutations in PSs are shown to enhance calcium release via both ER-resident channels—RyanR and InsP3R. In addition, PSs themselves play a role of ER Ca2+ leak channels. Excessive Ca2+ release from ER contributes to AD pathology. Modifying Ca2+ release from ER is a promising therapeutic strategy to reduce toxic cytosolic calcium elevations.
ER calcium overload in AD hippocampal neurons
ER Ca2+ concentration is increased in experimental models of AD including transgenic mice. It has been observed that InsP3-evoked calcium release from the ER is upregulated in PC12 cells and in fibroblasts that express mutant PS1 (48, 70). Similar effects were observed in neurons in brain slices taken from mutant PS1-M146 V, 3 × TgAD, and APPSweTauP301 L mice (112 –114). Stutzmann et al. suggest that enhanced Ca2+ release from the ER observed in these studies occurs due to upregulation of RyanR function (112 –114).
Another possible mechanism responsible for these effects is that mutations in PS1 disrupt its function as ER calcium leak channel. In addition, it has been suggested that PSs may potentiate the activity of sarco/endoplasmic reticulum Ca2+-adenosine triphosphatase (ATPase) (SERCA) pump via direct protein–protein interactions (46). Aβ can indirectly increase ER calcium content. As discussed above, Aβ potentiates Ca2+ entry via plasma membrane channels. Aβ can also act on SERCA pump that sequesters cytosolic Ca2+ (Fig. 3). To compensate the ER Ca2+ overload, neurons may upregulate the calcium-induced Ca2+ release from the ER via RyanR.
Indeed, changes in expression of RyanR have been described in human AD cases and in patients with mild cognitive impairment (18, 62). It is important to note that there are three subtypes of RyanR—1, 2, and 3. RyanR2 and 3 subtypes are expressed in the brain. It has been observed that RyanR2 is upregulated at early stages and is downregulated in advanced stages of AD in human postmortem samples (18, 62). Concerning RyanR3 subtype, it has been observed that its protein (89) and mRNA expression (18) is upregulated in late stages of the disease, suggesting that upregulation of RyanR3 might be a compensatory response to decreased function of RyanR2. Increase in RyanR2 expression and enhanced Ca2+ release have been reported in presymptomatic AD mice (21, 62, 113, 131). It has been shown that muscle relaxant dantrolene that targets RyanR exerts neuroprotective effects in mouse models of AD (24, 86, 92). Disadvantage of usage of dantrolene in the treatment of AD is that it does not have specificity to neuronal type of RyanR and may lead to side-effects. Moreover, there are data that long-term treatment with dantrolene can worsen AD pathology (131).
Neuronal Store-Operated Calcium Entry Is a Potential Therapeutic Target
Neuronal store-operated calcium entry (nSOCE) is a unique mechanism that refills ER calcium store in response to its depletion (95). For a long time, it has been believed that SOCE exists only in nonexcitable cells where it is the main mechanism to refill intracellular stores (79). However, there is a growing body of evidence that SOCE exists in neurons (8, 13, 47, 66, 94, 115, 129, 130). nSOCE is composed of two parts. The first one is plasma membrane proteins from ORAI and TRPC families that are able to make calcium-permeable channels. Second one is ER membrane protein that has calcium-sensitive domain inside of the ER. There are two ER proteins that participate in functioning of SOCE: STIM1 and STIM2. Stromal interacting molecule (STIM) 2 is predominantly expressed in hippocampus (115, 132). When calcium concentration drops inside of the ER, calcium-sensitive domain sends signal to the STIM to oligomerize. When it is in oligomerized form, it goes to ER–plasma membrane junctions to bind ORAI and TRPC proteins to form nSOC channels (Fig. 4) (132).

Recently, cellular nSOCE-dependent signaling pathway has been described in hippocampal neurons (115). It has been shown that neurons downregulate STIM2 expression in response to ER Ca2+ overload, resulting in drop in the amount of Ca2+ ions that enter neurons via nSOCE channels. STIM2 is downregulated in cultured hippocampal neurons and in hippocampus in animal models of AD, as well as in human AD brain samples (94, 115, 133). Cleavage of STIM proteins by PSs was suggested as a potential mechanism involved in these effects (120). nSOCE channels constitute ternary complex made by STIM2 at the ER part, and calcium release-activated calcium channel protein 2 (ORAI2) and TRPC6 at the plasma membrane part (132) (Fig. 4). In other studies, a role of ORAI1 in supporting SOCE in hippocampal and cortical neurons was demonstrated (47, 66). Knockdown of TRPC6 expression abolished nSOCE in hippocampal neurons. Overexpressed TRPC6 or pharmacological activators of TRPC6 channels restored nSOCE and spine loss in AD neurons (132). The mice that overexpress TRPC6 in the brain display enhanced cognitive performance and increased formation of excitatory synapses (135).
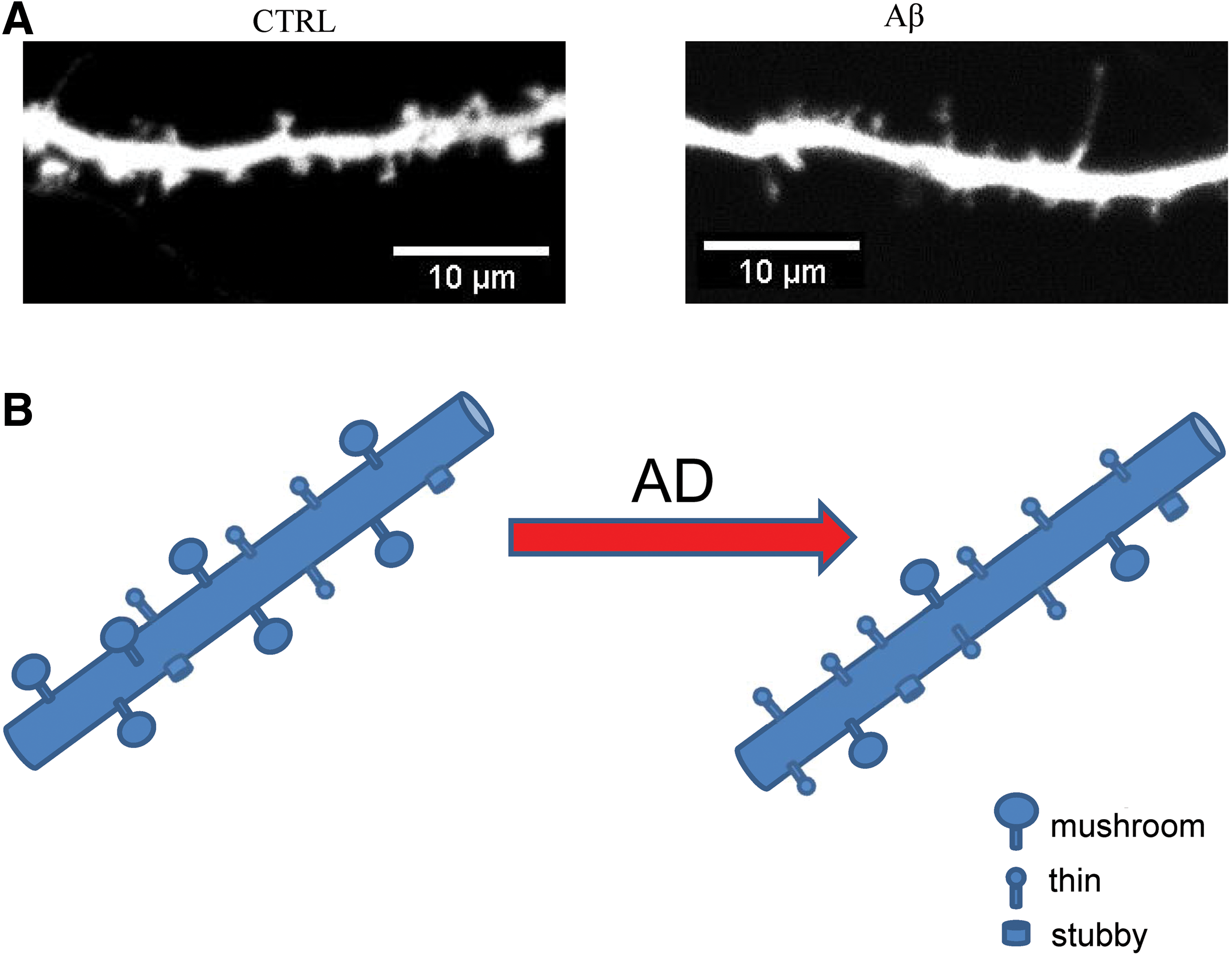
What is a physiological role of nSOCE in hippocampal neurons? It has been shown that nSOCE participates in regulation of stability of mature mushroom spines (Fig. 5) (94, 115, 133). Mushroom spines are sites of strong synapses that are necessary for formation and storage of memories. It has been proposed that downstream target for nSOCE is pCaMKII (phosphorylated calcium/calmodulin-activated protein kinase II), molecule that participates in LTP (Fig. 4). LTP is the best studied physiological mechanism of making participating synapses stronger and is essential for preservation of memories. It has been suggested that nSOCE is active in resting neurons (115), and is the main supplier of Ca2+ ions for CaMKII at rest. CaMKII is necessary for LTP performance. Shifting a balance from CaMKII to CaN is detrimental to synapses, leading to their instability and consequently causing memory dysfunction (93).
From this discussion, TRPC6 appears to be an attractive target for development of AD-preventing therapies. There are two molecules that are able to activate TRPC6 channels—hyperforin and NSN21778 (Fig. 6) (132). Hyperforin is a natural compound that activates TRPC6 channels (72). Beneficial effects of hyperforin and its derivatives in animal models of AD have been demonstrated (20, 38, 56). In double transgenic APPswe/PSEN1DE9 mice, derivative of hyperforin–tetrahydrohyperforin improves memory and prevents the impairment of synaptic plasticity in a dose-dependent manner, inducing a recovery of LTP (56). It has also been reported that tetrahydrohyperforin is able to enhance autophagic clearance of APP (19). In hippocampal neurons, TRPC6-dependent downstream signaling was connected with activation of the RAS/MEK/ERK, PI3K, and CAMKIV pathways (53, 116).

NSN21778 compound was recently discovered as a positive modulator of nSOC (132). It is important to note that NSN21778 is different from hyperforin in the mechanism of TRPC6 activation. It has been shown that hyperforin is a direct activator of TRPC6 while NSN facilitates OAG-induced Ca2+ influx through TRPC6 channels in conditions of partially depleted intracellular stores (132). The neuroprotective mechanism of NSN that is currently proposed is that NSN activates TRPC6 channels in diacylglycerol (DAG)-dependent manner. Following activation of TRPC6 channels, Ca2+ enters spines and activates CaMKII. All these events lead to spine and memory preservation and protection from AD (132) (Fig. 6). Future studies will be needed to establish utility of NSN21778 and its derivatives for treatment of AD.
Conclusions
Calcium hypothesis of AD is gaining popularity (4) since it points to new intracellular signaling pathways that are dysregulated in neurons, and more importantly it brings new targets for the development of AD-preventing therapies. AD is a multifactorial brain disorder (3) that manifests itself as a loss of memory. Modern therapeutical interventions should be based on understanding the mechanisms of memory loss in AD. Multiple lines of evidence suggest that Ca2+ signaling dysregulation plays an important role in synaptic pathology in AD. We propose that downregulation of nSOCE is one of the mechanisms responsible for synaptic and memory loss in AD, and that activators of TRPC6 channels should exert beneficial effects on AD. Future studies will be needed to test these ideas.
Footnotes
Acknowledgments
Ilya Bezprozvanny is a holder of the Carl J. and Hortense M. Thomsen Chair in Alzheimer's Disease Research. This work was supported by the National Institutes of Health Grant R01NS080152 (I.B.) (chapter: Ca2+ hypothesis of Alzheimer disease), Russian Science Foundation Grant 14-25-00024-II (I.B.) (chapter: Beta-amyloid and neuronal calcium signaling), by the state Grant 17.991.2017/