
Abstract
Significance:
The lung is a unique organ, as it is constantly exposed to air, and thus it requires a robust antioxidant defense system to prevent the potential damage from exposure to an array of environmental insults, including oxidants. The peroxiredoxin (PRDX) family plays an important role in scavenging peroxides and is critical to the cellular antioxidant defense system.
Recent Advances:
Exciting discoveries have been made to highlight the key features of PRDXs that regulate the redox tone. PRDXs do not act in isolation as they require the thioredoxin/thioredoxin reductase/NADPH, sulfiredoxin (SRXN1) redox system, and in some cases glutaredoxin/glutathione, for their reduction. Furthermore, the chaperone function of PRDXs, controlled by the oxidation state, demonstrates the versatility in redox regulation and control of cellular biology exerted by this class of proteins.
Critical Issues:
Despite the long-known observations that redox perturbations accompany a number of pulmonary diseases, surprisingly little is known about the role of PRDXs in the etiology of these diseases. In this perspective, we review the studies that have been conducted thus far to address the roles of PRDXs in lung disease, or experimental models used to study these diseases. Intriguing findings, such as the secretion of PRDXs and the formation of autoantibodies, raise a number of questions about the pathways that regulate secretion, redox status, and immune response to PRDXs.
Future Directions:
Further understanding of the mechanisms by which individual PRDXs control lung inflammation, injury, repair, chronic remodeling, and cancer, and the importance of PRDX oxidation state, configuration, and client proteins that govern these processes is needed.
Historical Overview
In 1988, the first peroxiredoxin (PRDX) was described in Saccharomyces cerevisiae as an unknown protein that protected enzymes from thiol oxidative inactivation in the presence of reducing agents such as dithiothreitol or beta-mercaptoethanol (69). The isolated protein did not appear to have any catalase, glutathione peroxidase, or superoxide dismutase activity. Instead it was originally thought to metabolize sulfur radicals and thus was named thiol-specific antioxidant (TSA). It was later discovered that yeast cells lacking TSA were viable under normal conditions; however, if TSA-deficient cells were grown in the presence of various peroxides, they showed a significantly decreased growth rate compared with wild type counterparts (21). In 1993, the sequence of the human TSA analog, TSA protein (PRP, now known as PRDX2), was compared with a human gene sequence database. PRP was found to share homology with several other proteins whose function had not been described, all of which had two highly conserved regions surrounding cysteines (98). In 1994, the sequence of TSA was compared across 11 different organisms from all 5 animal kingdoms, 26 genes were identified with similar conserved regions surrounding 1 of the 2 cysteine residues (23, 24). While the amino-terminal cysteine was conserved in all 26 genes, the carboxy-terminal cysteine residue was not conserved in 6 of the genes. In 1994, the same group published the first article showing that a yeast PRDX homologue was a thioredoxin-dependent peroxidase. It was in this article that the name “peroxiredoxin” was first suggested for this family of proteins, which directly reduces hydrogen peroxide (H2O2) (19). It was clear that PRDXs were thiol peroxidases, but not all members of the family use thioredoxin for regeneration. Therefore, previously suggested names such as thioredoxin peroxidase were not correct. As Rhee et al. stated regarding the creation of the name “peroxiredoxins” in his 2005 review, “peroxi-indicates the nature of the substrate reduced, -redoxin rhymes with thioredoxin and glutaredoxin (GLRX), which also contain redox-sensitive cysteines that undergo oxidation–reduction cycles during protein function (147).” It was postulated that the differences in the sequences of the PRDX family were likely due to differences in the mechanism involved in H2O2 reduction.
The PRDX Catalytic Cycle
Six PRDXs are found in humans and can be subdivided into three distinct classes based on their reaction mechanisms: 2-Cys, atypical 2-Cys, and 1-Cys. Four are 2-Cys PRDXs (PRDX1–4), one is an atypical 2-Cys PRDX (PRDX5), and one is 1-Cys PRDX (PRDX6) (Fig. 1). The 2-Cys PRDXs have conserved amino- and carboxy-terminal cysteines that are separated by 121 amino acids (147) (Fig. 2). The 2-Cys PRDXs reduce H2O2 by donating an electron from the peroxidatic cysteine (amino-terminal) to the H2O2 molecule. This reaction releases water and generates a sulfenic acid intermediate state of the peroxidatic cysteine. The resolving cysteine (carboxy-terminal) of another PRDX monomer then interacts in a head to tail configuration with the oxidized peroxidatic cysteine to form an intermolecular disulfide bond and releases another molecule of water (153). The disulfide bonds between peroxidatic and resolving cysteines of PRDX1–4 are in turn reduced by the thioredoxin/thioredoxin reductase/NADPH (TXN/TXNRD/NADPH) system (Fig. 1A).
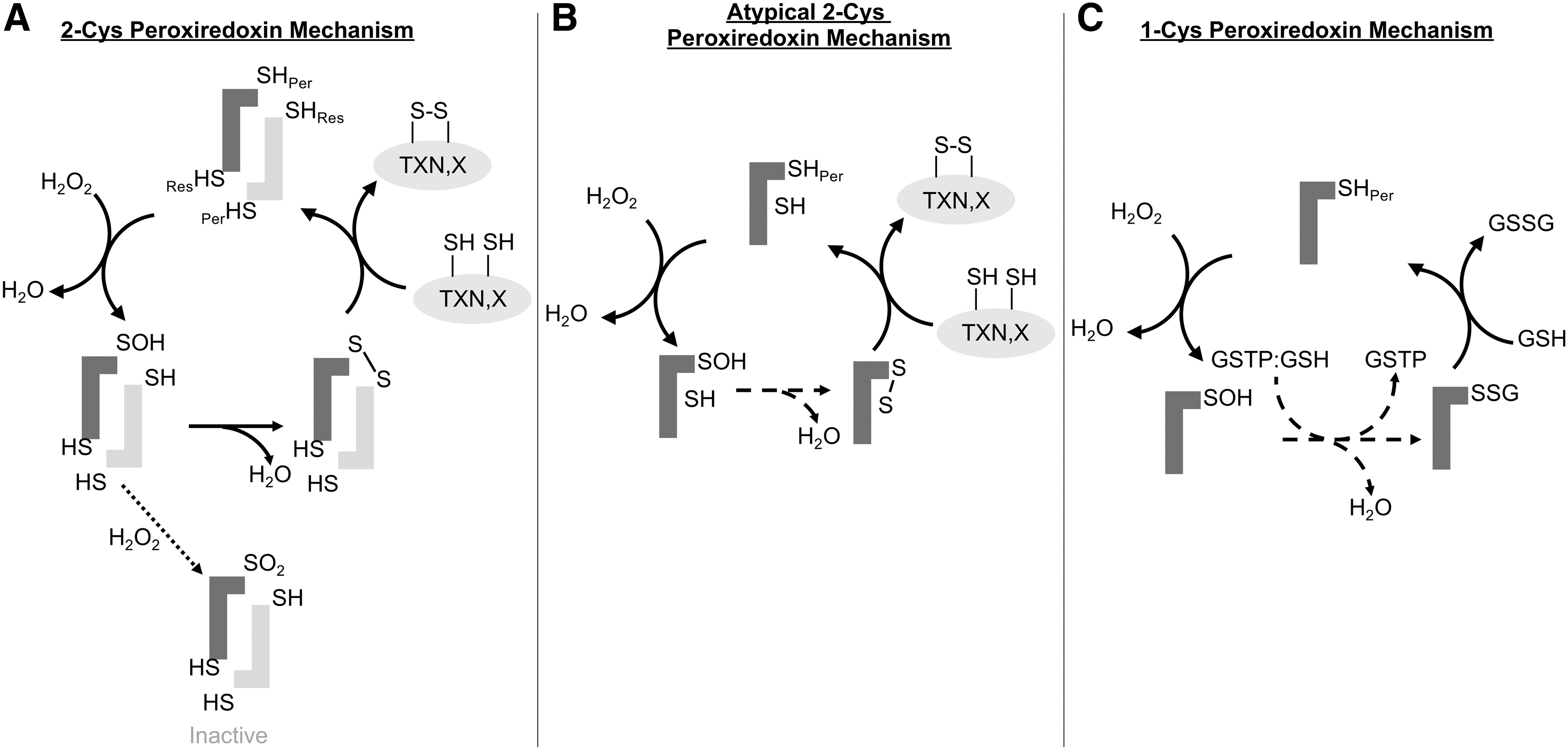

PRDX5 lacks the typical resolving cysteine found in the first four members of the family and functions through an atypical 2-Cys mechanism. PRDX5 has the amino-terminal conserved cysteine at residue 47 and 2 more cysteines, Cys72 and Cys151. It is believed that PRDX5 functions through a unique disulfide shuffling mechanism where after the peroxidatic cysteine (Cys47) reduces H2O2, the resulting sulfenic acid intermediate is resolved by the formation of an intramolecular disulfide bond with Cys151. This disulfide bond is then rearranged to form an intramolecular disulfide bond between Cys47 and Cys72 that can subsequently be reduced by TXN (44) (Fig. 1B).
PRDX6 lacks the conserved carboxy-terminal resolving cysteine found in the 2-Cys PRDXs and is the only 1-Cys PRDX in humans. PRDX6 was found to have phospholipase A2 (PLA2) activity before it was identified as a PRDX (70, 124). Contrasting other PRDXs, PRDX6 can reduce phospholipid hydroperoxides in addition to other organic hydroperoxides and H2O2. Instead of TXN, the electron donor used by PRDX1–5, PRDX6 uses glutathione (GSH) as an electron donor to reduce hydroperoxides (Fig. 1C). The aforementioned phospholipase activity that is unique to PRDX6 plays an important role in lung surfactant homeostasis and degradation of internalized dipalmitoylphosphatidylcholine (156), as further described below. A glutathione-S-transferase Pi (GSTP)-catalyzed reaction is currently believed to catalyze the first step in the reduction of the sulfenylated peroxidatic cysteine of PRDX6 (103). GSTP was shown to bind to the sulfenic acid form of Cys47 (PRDX6-SOH) and in turn S-glutathionylates the active site cysteine (103). Free GSH in turn interacts with the glutathionylated cysteine to regenerate the reduced peroxidatic cysteine, releasing glutathione disulfide (103). Mutation of the residues involved in the GSTP-PRDX6 binding interface reduces peroxidase activity and dimerization of PRDX6 (144, 145, 193), suggesting a role of GSTP-mediated S-glutathionylation in the peroxidatic cycle of PRDX6. Relevant to this perspective on lung disease, it is noteworthy that compared with other PRDXs, PRDX6 expression is highest in lung tissue (78), and PRDX6 expression in lung is higher than other organs (151). Similarly, GSTP expression also is prominent in lung tissue, notably in epithelial cells where PRDX6 also is present (78). A role for S-glutathionylation in the regeneration of other PRDXs (notably PRDX 1 and 2), and their protection against overoxidation, has also been shown (22). The active site Cys residues of PRDX2 can form stable mixed disulfides with GSH, via a sulfenic acid intermediate that is in turn S-glutathionylated, preventing overoxidation (see the PRDX Inactivation section). Reaction with GLRX in turn regenerates the peroxidase activity of PRDX2. Thus, GSH and GLRX provide an alternative mechanism to TXN and TXNRD to regenerate catalytically active PRDX2 (138).
Oligomerization of PRDXs
Originally it was believed that 2-Cys PRDXs formed dimers through the disulfide bond formed between the resolving cysteine of one monomer with the peroxidatic cysteine of a neighboring monomer during the catalytic cycle (25). However, it was later shown that PRDXs form dimers through noncovalent interactions between the monomers (178). The first PRDX decamers were described in 1969 by transmission electron microscopy, where complexes with 10-fold symmetry were observed (48). PRDX1, 2, and 4 tend to form decamers, while mitochondrial PRDX3 forms dodecamers (139) that can self-assemble into high-molecular-weight oligomeric forms (187).
Overoxidation of 2-Cys PRDXs results in the formation of higher order decameric structures, which have been shown to gain molecular chaperone activity, do not require peroxidase activity, and protect protein substrates from thermally induced aggregation, akin to heat shock proteins (56, 96, 119) (Fig. 3). Thus, the cellular protective effects of PRDXs are not limited to the removal of peroxides, as they also directly protect proteins from irreversible denaturation (22). The factors influencing decamer and higher order formations of PRDXs are not fully understood and it is important to note that not all structural transitions under varying peroxide levels are identical between typical 2-cys PRDXs (141). It is believed that PRDXs in vivo cycle between dimer and decamer formations, with decamer formation being favored in the reduced and overoxidized conformations (1, 96, 178) (Fig. 3). The intermolecular disulfide bond between the peroxidatic and resolving cysteine of dimerized monomers requires a conformational unwinding of the active site alpha helix that results in a destabilization of the decamer formation (152). It is possible that the shift from decamer to dimer facilitates the reaction between PRDXs and client proteins, such as TXN, in the catalytic cycle to regenerate reduced PRDX. It has been proposed that the dimer and decamer dynamic is modulated by pH, where at higher pH, PRDXs have an exposed active site loop interfering with decamer formation, while at lower pH, the active site loop is locked down favoring decamer formation (121). S-glutathionylation of PRDX1 at Cys83 converts the decameric PRDX to its dimers with the loss of chaperone activity. Cys83 is located at the putative dimer–dimer interface and it plays a role in stabilizing the hydrophobic interaction required for decamer formation (22). The S-glutathionylation of cysteine residues that are in close proximity to the dimer–dimer interface in the decamer structure suggests a mechanism by which cells regulate the chaperone activity of PRDXs by inhibiting decamer formation via S-glutathionylation.
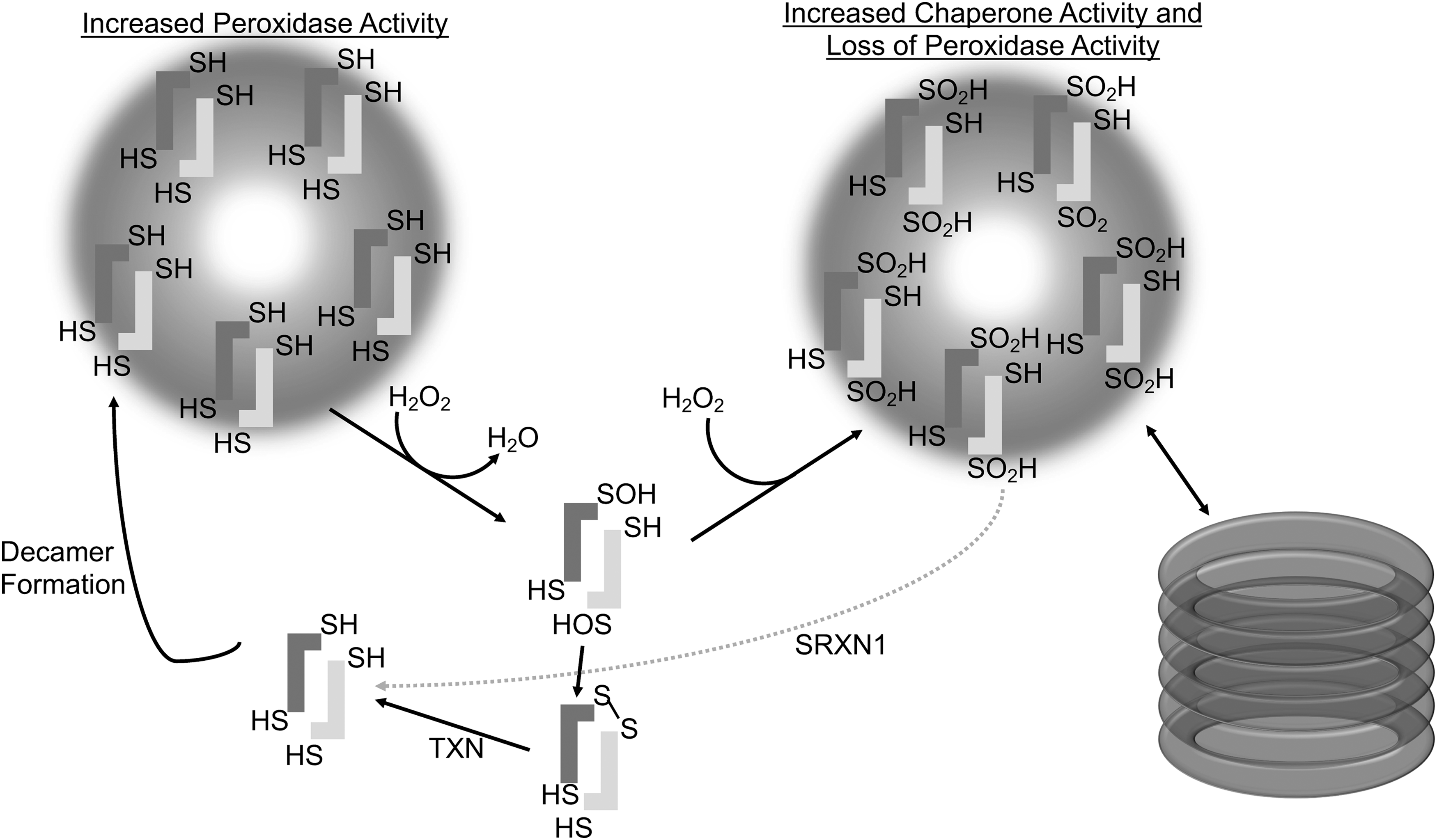
PRDX Inactivation
PRDXs are unusual H2O2 scavenging enzymes in that they are inactivated by high concentrations of their substrates, peroxides. Under conditions of increased H2O2 concentrations, a second H2O2 moiety can interact with the peroxidatic cysteine before it has resolved the sulfenic acid intermediate to an intermolecular disulfide bond. This interaction results in the sulfinic (SO2H) modification of the peroxidatic cysteine (142, 186). The sulfinic acid forms of PRDXs do not have peroxidase activity, as the peroxidatic cysteine in the sulfinic acid state will not react with the resolving cysteine to form a disulfide bond. The inactivation of PRDXs in settings of increased H2O2 seems counterproductive. To explain this phenomenon, the “floodgate theory” was developed in 2003, stating that this inactivation of PRDXs allows H2O2 to accumulate and execute signaling functions (179). It has also been proposed that inactivation of PRDXs frees TXN to interact with other critical substrates that were oxidized during an increased oxidative state (39). PRDXs can also be irreversibly hyperoxidized to the sulfonic acid state (–SO3H); however, this hyperoxidation does not appear to occur spontaneously from the sulfinic acid state (–SO2H) of PRDX (56, 96). Overoxidized PRDXs no longer function as peroxidases, but they have been shown to have greatly increased chaperone activity (96). Of the mammalian 2-Cys PRDXs, PRDX3 has four differing amino acids in the conserved region downstream of the resolving cysteine, which confer a decreased susceptibility to overoxidation (141). When this sequence was cloned into human PRDX2, it significantly decreased the overoxidation of recombinant PRDX2 in response to peroxide treatment (141). This implies that the shift from active to inactive overoxidized forms is regulated and has evolved to be different in each PRDX and suggests a key functional relevance of PRDX overoxidation.
Addition of overoxidized inactivated PRDXs to cell lysates from mammalian cells quickly resulted in reduction of the sulfinic acid modification of the peroxidatic cysteine, regenerating the active PRDX (175). It was later discovered that this process is facilitated by sulfiredoxin (SRXN1), which reduces the sulfinic acid in 2-Cys PRDXs in an ATP-dependent manner, regenerating the active enzyme (9). SRXN1 has different affinities for the 2-Cys PRDXs (172, 176). Using surface plasmon resonance, the PRDX with the highest binding affinity for SRXN1 was shown to be PRDX4 (171). Interestingly, PRDX4 is believed to reside within the endoplasmic reticulum (ER), whereas SRXN1 is a cytosolic protein (171). These observations raise questions about the trafficking of PRDX4 to enable the interaction between ER-located PRDX4 and cytosolic SRXN1. SRXN1 has been shown to translocate to the mitochondria during oxidative stress (127), however, translocation of SRXN1 to the ER has not been shown thus far.
The peroxidase activities of PRDXs can also be affected via phosphorylation. In response to growth factor stimulation, PRDX1, but not PRDX2, was transiently inactivated via an SRC kinase-mediated tyrosine phosphorylation at Tyrosine-194 (97, 177). A role of NOX1 in the activation of SRC kinase and subsequent phosphorylation of PRDX1 was demonstrated (Woo and colleagues). Recent studies from our laboratories demonstrated activation of SRC via sulfenylation of Cys185 and Cys277 in response to NADPH oxidase-dependent signaling (49).These collective findings suggest that H2O2 can promote inactivation of PRDX1 through oxidative activation or SRC, and subsequent phosphorylation of PRDX1, thereby promoting further accumulation of H2O2. H2O2 plays an important role in growth factor signaling, and it is likely that this localized inactivation of PRDX1 is necessary to allow H2O2-mediated oxidation of signaling proteins. During cell division, PRDX1 is phosphorylated at Thr-90 by CDK1 at regions close to the mitotic spindle. The centrosome-bound phosphatase cell division cycle 14B (CDC14B) is susceptible to inactivation through oxidation by H2O2. The local PRDX1 inactivation enables localized H2O2 concentrations to increase and facilitates the oxidative inactivation of CDC14B (97). Later in the cell cycle, centrosome-localized PRDX1 is dephosphorylated and reactivated by okadaic acid-sensitive phosphatases (97). The reactivation of PRDX1 decreases localized H2O2 levels enabling the reactivation of CDC14B. These findings show the spatial and temporal regulation of PRDX activity throughout the cell cycle to propagate H2O2-mediated signaling. PRDX1 can also be phosphorylated by the tumor suppressor, mammalian sterile twenty 1 and 2 (MST1 and MST2) at Thr-90 and Thr-183 leading to its inactivation (146). MST1 can be activated by H2O2 (92), pointing to a feedforward loop between MST1 and PRDX1. Activation of MST1 via oxidation results in inactivation of PRDX1 via phosphorylation, leading to sustained oxidative signaling (146). Contrary to the Thr and Tyr phosphorylation leading to the inhibition of peroxidase activity, Ser32 phosphorylation has been shown to enhance the peroxidase activity of PRDX1 (196).
The 1-Cys PRDX6 can also be phosphorylated (29, 143). PRDX6 is phosphorylated by extracellular signal-regulated kinase (ERK) and P38, causing an increase in PLA2 activity due to conformational change that exposes a hydrophobic residue and enhances phospholipid binding (143, 180). The phosphorylation of PRDX6 also increases its localization to the cell membrane and enhanced PLA2 activity aids in the activation of the NADPH oxidase, NOX2 (29). Similar to observations with NOX2, PRDX6 also promotes activation of NOX1 through interaction of the SH3 domain of NOX activator 1 and requires PRDX6's PLA2 and peroxidase activities (84). These findings demonstrate that PRDXs enhance NOX activities and point to the exquisite feedback regulation through which NOX regulates PRDX, and PRDX in turn promotes NOX activation to sustain redox-based signals.
Cysteines in PRDX1, PRDX2, and PRDX6 can be modified through S-glutathionylation, the addition of the tripeptide moiety GSH to a protein cysteine residue. As discussed earlier, PRDX6 is S-glutathionylated in a GSTP-catalyzed reaction that is part of its catalytic cycle (103, 144). GLRX-mediated deglutathionylation of catalytic site cysteines in PRDX1 and 2 regenerated peroxidase activity (138). Intriguingly, a role of SRXN1 in deglutathionylation of PRDX1 has been shown via the formation of an S-glutathionylated SRXN1 intermediate (132), which in yeast was regenerated via the GLRX/GSH system (10). Besides the roles of GLRX in the regeneration of S-glutathionylated PRDXs and S-glutathionylated SRXN1, GLRX also is important in the regeneration of S-glutathionylated TXN (43). Under conditions of oxidative stress, a two-disulfide form of TXN, containing an active site disulfide (Cys32–Cys35) and a nonactive site disulfide (Cys62–Cys69), was observed. Inactivation of TXN is believed to be important for the transmission of oxidative signals. The nonactive site disulfide of TXN was shown to S-glutathionylated and subsequently could be reduced via the GLRX/GSH system, leading to reactivation of TXN (43). These intriguing observations suggest that the PRDX, TXN, GLRX, and SRXN1 cellular redox systems do not act independently but operate in a highly coordinated manner to control each other's function to restrict or promote redox-based signals.
S-glutathionylation of PRDXs can also occur at cysteine residues outside of the catalytic site (132). As described above, S-glutathionylation of PRDX1 at Cys83 disrupts the formation of decamers and favors dimer formation with concomitant loss of chaperone activity (133). A redox proteomic screen to identify S-glutathionylated proteins secreted following stimulation of macrophages with lipopolysaccharide (LPS) or A549 lung cancer cells with influenza virus identified PRDX1, PRDX2, and TXN as secreted S-glutathionylated proteins (30). The same group also showed that S-glutathionylated PRDX2, along with TXN, can act as a danger signal to enhance inflammation, and suggested that the proinflammatory action of released PRDX2 and TXN involved modification of the redox status of cell surface receptors involved in proinflammatory signaling (149). Toll-like receptor 4 (TLR4) is a putative target in the propagation of proinflammatory signaling, due to the documented binding of PRDX1, 2, and 5 to TLR4 (75, 101, 148), as discussed below.
Comparison of Human PRDX Domains
Each of the six PRDXs has highly conserved regions with a sequence homology of >70% between the first four family members (PRDX1–4). The most highly conserved regions flank the peroxidatic cysteine (amino-terminal), which is conserved in all PRDX members (Fig. 2, “*”). The resolving cysteine (carboxy-terminal) found in PRDX1–4 is equally well conserved among the 2-Cys PRDXs (Fig. 2, “#”).
PRDX1 and 2 are cytosolic proteins, while PRDX3 and 5 are imported into the mitochondrial matrix by a mitochondrial localization signal in their N-terminus comprising alternating acidic and hydrophobic residues (Fig. 2, red). PRDX4 is ER-localized and the membrane signal peptide is located in the large stretch of hydrophobic residues in the N-terminal (Fig. 2, blue). This signal peptide is cleaved once PRDX4 is translated into the ER lumen. PRDX4 does not possess a KDEL sequence for ER retention but is believed to be kept in the ER by chaperone proteins (62, 185); however, PRDX4 can also be secreted (108, 129). It is not currently known what conditions determine whether PRDX4 is retained in the ER, or secreted from the cell, or whether PRDX4 localization is a cell type-dependent attribute. The aforementioned ability of SRXN1, a cytosolic-localized protein, to reduce the sulfinic acid form of PRDX4 also raises questions about the processes and signals that regulate the trafficking of PRDX4. PRDX6 is found predominantly in the cytosol, but a fraction is found in lysosomes, due to its lysosomal targeting sequence (Fig. 2, green) (157).
Signaling via PRDX Relays
Recent studies have illuminated a cardinal role of PRDXs in redox-based signaling through relays (46, 51, 122, 130, 155, 158, 181, 195). Cysteine residues are highly conserved throughout all five kingdoms of species and have a wide range of H2O2 reactivity, depending on solvent exposure, neighboring residues, and protein structure. However, there is no clear consensus about how cysteines in redox-regulated proteins are oxidized by H2O2. Modeling of the oxidized amino acids in transcription factors that are redox reactive predicts a relatively high reaction rate constant (k around 101–102 M −1 s−1) for H2O2 (106). It was hypothesized that there must be specially localized regions of increased H2O2 concentrations where these redox-regulated transcription factors are localized to enable their direct H2O2-mediated oxidation. Regions of high H2O2 likely exist in cells in areas close to NADPH oxidases who produce H2O2 and near aquaporins in the cell membrane where H2O2 is imported into cells; however, mathematical modeling studies do not support the existence of such locations in the cell (163). Alternatively, it was proposed that thiol peroxidases that have a very high reactivity toward H2O2 act as a relay through which they pass their oxidizing equivalents to downstream redox-regulated proteins. The high abundance of PRDXs can make up as much as 1% of soluble total proteins (20), and their high reactivity toward H2O2, k around 105–108 M −1 s−1, makes PRDXs ideal targets for H2O2-mediated redox relays (137, 158, 174). PRDXs are then able to pass their oxidative equivalent to a client protein, typically though disulfide bond formation, thus propagating the redox signaling cascade and regenerating the PRDX to its reduced state.
In 2002, the first mechanism for the thiol oxidation of a target protein by a thiol peroxidase was described in yeast (40). The first indication of redox signaling via PRDXs in mammals was the role of PRDX1 in redox signaling via apoptosis signaling kinase 1 (ASK1) (57). During oxidative stress induced by H2O2, PRDX1 has been shown to catalyze the oxidation of ASK1 to its active state, and to fully activate the ASK1/P38 pathway in response to H2O2 (57). More recently, PRDX1 has been identified in a redox relay regulating the mammalian fork head box transcription factor of the O class 3 (forkhead box O3 [FOXO3]) (51). Under conditions of oxidative stress induced by H2O2, a PRDX1 dimer forms a trimer with FOXO3 through disulfide bond formation. The binding of PRDX1 to FOXO3 inhibits the transcriptional activity of FOXO3 by inhibiting the nuclear translocation. If PRDX1 is overoxidized, then this interaction is inhibited, and FOXO3 can traffic to the nucleus and become activated via MST1, JNK, or p38 (51).
A relay between PRDX2 and signal transducer and activator of transcription 3 (STAT3) also has been documented. PRDX2 modifies the transcription factor STAT3 by catalyzing the addition of an inhibitory disulfide bond (155). On stimulation with H2O2, PRDX2 forms a mixed disulfide intermediate with STAT3, which is then resolved to result in an active reduced PRDX2 and oxidized STAT3 (155). Oxidized STAT3 forms oligomers that attenuate its transcriptional activity. PRDX2 has also been suggested to be involved in a redox relay involving protein deglycase 1 (DJ-1), also known as Parkinson disease protein (7, 46). Under conditions of H2O2 stress, a disulfide link between PRDX2 and DJ-1 is observed before the disulfide bond-dependent dimerization of DJ-1, likely through a thiol/disulfide exchange.
The ER-localized PRDX4 can donate disulfide bonds to naive proteins that are being folded. When ER-oxidoreductin-1 (ERO1) is genetically deleted, thus inhibiting the ERO1-dependent disulfide bond formation via protein disulfide isomerases (PDIs), oxidized PRDX4 can donate its disulfide bond to various PDIs to allow ER protein folding to proceed (195). The ability of PRDX4 to perform thiol/disulfide exchanges with members of the PDI family in the ER is particularly intriguing, because it suggests at an alternate recycling mechanism for PDIs that could be H2O2 dependent (162). PRDX4 has different binding affinities for the various members of the PDI family (150), suggesting that redox-regulated recycling mechanisms could have specificity in protein folding in ER under oxidative stress conditions. PRDX4 can directly catalyze the addition of disulfide bonds to target proteins within the ER, thus linking target protein activity, function, and trafficking to the oxidative state of the ER. The PRDX4-mediated addition of non-native disulfide bonds has been identified for glycerophosphodiester phosphodiesterase 2 (GDE2), also known as GDPD5 (181). Within the ER, PRDX4 forms a non-native disulfide bond in GDE2 which inhibits the trafficking of GDE2 to the cell surface.
Implications of PRDXs in Lung Diseases
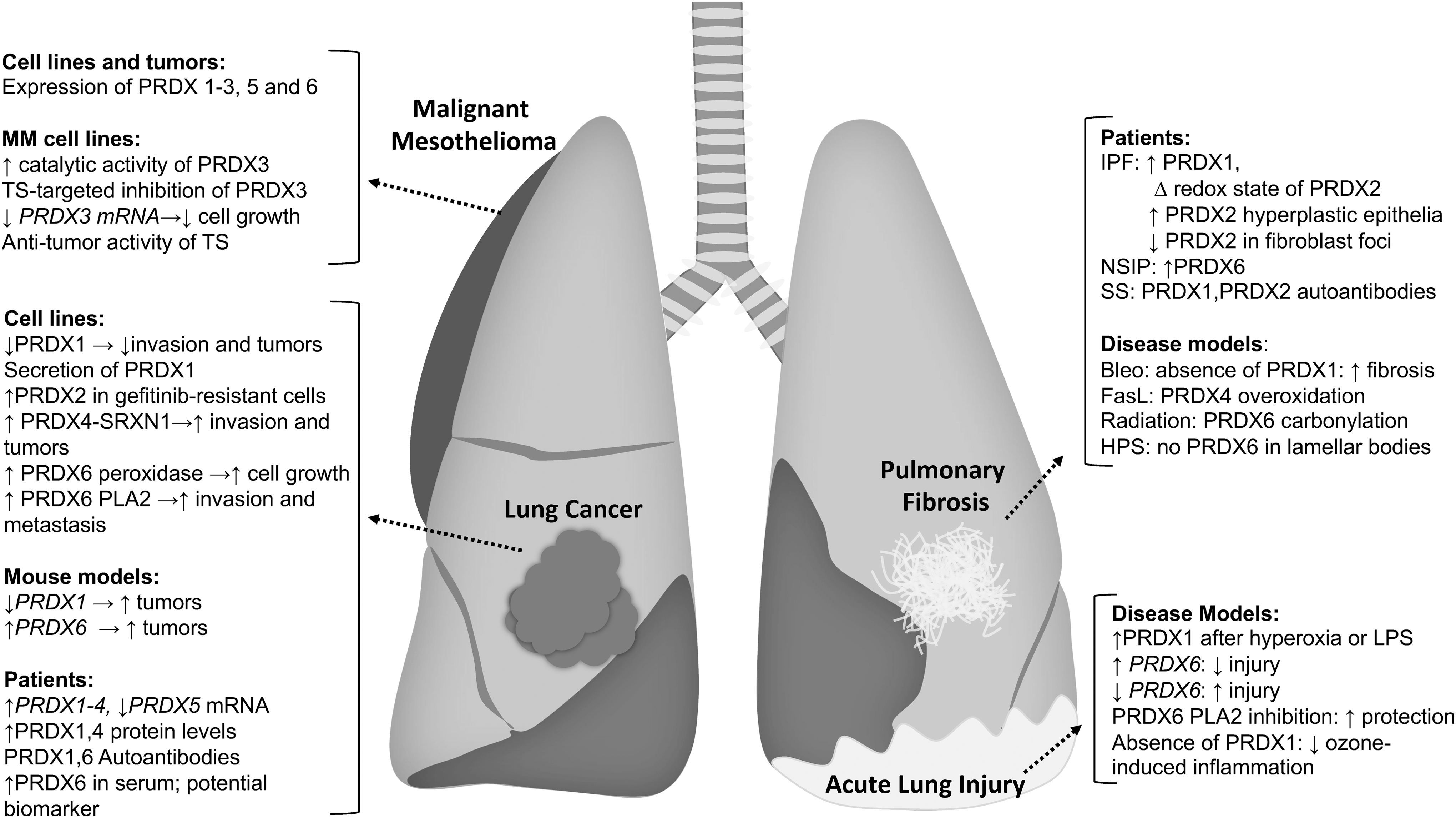
Changes in the redox environment have long been implicated in the pathophysiology of a myriad of lung diseases that include lung cancer, asthma, acute lung injury (ALI), chronic obstructive pulmonary disease, and pulmonary fibrosis. While originally thought to be merely damaging, oxidants are now known to regulate the structure and function of proteins through oxidation of cysteines. Dysregulation of oxidant production, and consequently, protein cysteine oxidations may contribute to the pathogenesis of lung diseases. As described above, PRDXs are major regulators of the cellular redox environment and impact protein cysteine oxidation directly and indirectly. In the next sections we summarize some of the studies conducted thus far that have implicated PRDX in the pathogenesis of lung diseases, emphasizing lung cancer, mesothelioma, pulmonary fibrosis, and ALI, given that most of the studies involving PRDX have been conducted in those diseases and that efforts from our laboratories have focused there.
Lung cancer
Lung cancer is the leading cause of cancer-related mortality in the United States and worldwide. Despite significant progress over the past decade in the early detection and treatment of lung cancer, the 5-year survival rate of patients with advanced disease remains <20% (58). Lung cancer is classified into two major subtypes: small-cell and three types of nonsmall-cell lung cancer (NSCLC), including adenocarcinoma, squamous-cell carcinoma, and large-cell carcinoma (86). Adenocarcinoma is the most common form of lung cancer, and ∼30% of lung adenocarcinoma contains oncogenic single-point mutations of KRAS (115).
A possible contribution of PRDX in lung cancer is not surprising given the increase of oxidative stress in lung tumors, especially H2O2, that was first shown in 1980 (128, 161). Mitochondrial oxidant generation was shown to be increased and essential for KRAS-induced cell proliferation and tumorigenesis (173). The oncogenes, KRAS-G12D, BRAF-V619E, and MYC-ERT2, increased the transcription of nuclear erythroid 2-related factor 2 (NRF2 also known as NFE2L2) to stably elevate the basal NRF2-dependent antioxidant program (41). The transcription factor NRF2 plays a prominent role in lung cancer pathogenesis. Mutations that disrupt the NRF2–KEAP1 interaction to stabilize NRF2 and increase the constitutive transcription of NRF2 target genes have been found, including in patients with NSCLC, where it has been shown that NRF2 activation allows tumors to combat the enhanced oxidative burden of tumor environment (41). A number of PRDX genes, as well as SRXN1 and GSTP that are discussed herein, are transcriptional targets of NRF2 (5, 34, 71, 116, 160).
The first link between PRDX and lung cancer was made in 2001 when a proteomic screen of potential biomarkers for lung cancer showed a significant increase in PRDX1 expression in A549 lung adenocarcinoma cells compared with controls (26). In 2004, a comprehensive analysis of PRDX mRNA and protein levels was conducted in squamous and adenocarcinomas comparing findings with normal control tissue from the same patient. Reverse transcription-polymerase chain reaction showed increases in PRDX1, 2, 4, and 6, whereas Western blot analysis showed increased PRDX1 and 4 protein levels, with variations in expression of PRDXs between tumor subtypes (93). Similarly, assessment of PRDX transcript levels in lung adenocarcinoma or squamous cell carcinoma, and adjacent healthy tissue, using a larger cohort of patients annotated in The Cancer Genome Atlas database (Broad Institute), revealed increases in PRDX1–4 transcripts in the tumors compared with nontumor adjacent tissue, while PRDX5 transcripts decreased and PRDX6 was unchanged (Fig. 4A, B).

PRDX1 was upregulated in NSCLC based on evaluation via immunohistochemistry, Western blot, and proteomic screenings that aimed to identify protein signatures of lung cancer (26, 59, 65, 68, 95, 131, 134, 135). In settings of a KRAS mutation, PRDX1 was upregulated in an NRF2-dependent manner (134). The increased expression of PRDX1 in NSCLC tumors was associated with an increased risk of metastasis and tumor differentiation (95). Using A549 lung adenocarcinoma cells, it was demonstrated that knockdown of PRDX1 resulted in less Matrigel invasion and colony forming ability in soft agar, while overexpression of PRDX1 promoted Matrigel invasion and colony forming ability (59), consistent with a potential protumorigenic role of overexpressed PRDX1.
In contrast to these observations, absence of the PRDX1 gene in mice expressing the KRAS-G12D driver mutation resulted in an increase in tumor number and size, which appeared to be linked to the oxidative activation of the ERK/cyclin D1 pathway (134). Similarly, absence of PRDX1 increased susceptibility to Ras-induced breast cancer (17). Interestingly, A549 lung cancer cells were shown to secrete PRDX1 (27, 28, 52), and autoantibodies against PRDX1 were found in the sera of patients with NSCLC, suggesting that circulating PRDX1 or the autoantibody against PRDX1 is a potential biomarker for lung cancer (27). How the redox state of PRDX1 or its configuration (monomer, dimer, etc.) affects its secretion or antigenicity remains unclear. In addition, the relevance of these autoantibodies in the pathogenesis of lung cancer also requires additional studies.
A number of putative targets of PRDX1-mediated redox regulation potentially relevant to lung cancer have been identified. PRDX1 has been shown to play a role in the regulation of AKT signaling via oxidation of the tumor suppressor, phosphatase and tensin homologue (PTEN). PTEN inactivation has been linked with resistance to epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor therapy and lower survival in NSCLC patients (136). The tumor suppressor, PTEN, is susceptible to reversible inactivation via oxidation by H2O2 (91). Under steady-state conditions, PRDX1 binds to PTEN and prevents the inhibition of PTEN's phosphatase activity by preventing a disulfide bond from forming in the N-terminal region (17). Under conditions of increased oxidative stress, PRDX1 is overoxidized and forms decamers, releasing from PTEN and allowing the inhibition of the active site of PTEN through disulfide bond formation. Inactivation of PTEN leads to increased AKT signaling and oncogenesis (17, 125).
PRDX1 has also been implicated in controlling the activity of dual-specificity phosphatases (DUSP)-1 and -10 (also known as MAP kinase phosphatase 1 and 5, respectively), which have been linked to lung cancer (117). Notably, low DUSP1 levels are associated with a poor clinical outcome in patients with NSCLC (105). Interestingly, overexpression of DUSP1 in gefitinib-resistant NSCLC cells restored gefitinib sensitivity by inhibiting EGFR signaling and inducing apoptosis, whereas its knockdown in sensitive cells conferred gefitinib resistance (99). DUSP1 and 10 can inhibit multiple mitogen-activated protein kinases (MAPKs), including P38, an activator of cellular senescence (165), and ERK. DUSP1-mediated inhibition of ERK has been linked to protection in KRAS-G12V-driven nonsmall-cell lung carcinoma (105). Binding of PRDX1 to DUSP1 or DUSP10 inhibits oligomerization and promotes phosphatase activity toward P38. Overoxidation of the peroxidatic cysteine of PRDX1 leads to a dissociation from DUSP1, while increased binding to DUSP10 (165). Intriguingly, overoxidation of PRDX1-Cys52 resulted in DUSP1 oxidation-induced oligomerization and inactivity toward P38, while overoxidation of PRDX1-Cys52 enhanced the PRDX1:DUSP10 complex that protected DUSP10-mediated inactivation toward P38. The different binding affinities for DUSP1 and DUSP10 that depend on the oxidation state of PRDX1 further highlight that substrate sensitivity of PRDX partners depends on the local redox environment and illustrates the precision through which PRDX oxidations rewire signaling pathways. PRDX1 also is a binding partner for the transcription factor, FOXO3, a tumor suppressor whose deletion has been linked to lung cancer (111). In settings of increased oxidative stress, disulfide-bound heterotrimers linking dimeric PRDX1 to monomeric FOXO3 are enhanced. Absence of PRDX1 enhances FOXO3 nuclear localization and transcription controlled by the presence of Cys31 or Cys150 within FOXO3 (51). It remains unclear precisely how the oxidation state of PRDX1 regulates substrate specificity toward client proteins in lung cancer cells and what the implications are for lung cancer biology. Given that the redox state of PRDX1 regulates its biological functions, targeted drugs to prevent its overoxidation therefore have the potential to elicit biological responses, without affecting the endogenous function of PRDX1. It is not difficult to rationalize the aforementioned contradicting effects of PRDX1 ablation or overexpression in the various lung cancer models, as the substrate targets of PRDX1 may have varied. PRDX2 was remarkably increased only in A549/gefitinib-resistant (GR) cells compared with A549 cells. The elevated expression of PRDX2 resulted in the downregulation of reactive oxygen species (ROS) and cell death and upregulation of cell cycle progression in the A549/GR cells. When PRDX2 mRNA in the A549/GR cells was knocked down, the levels of ROS and apoptosis were significantly recovered to the levels of controls (85).
PRDX3 was also shown to be upregulated in lung adenocarcinoma and this was associated with a loss of expression of Dachshund family transcription factor 1 (DACH1), which has been attributed a tumor suppressor function. Overexpression of DACH1 attenuated PRDX3 expression and decreased colony forming potential of A549 cells, an effect that could be overcome by overexpression of PRDX3 (194).
PRDX4 immunohistochemical staining in 142 patients with stage II NSCLC revealed that positive PRDX4 expression was significantly correlated with recurrence, and shorter disease-free survival in patients with early-stage lung squamous cell carcinoma (54). In contrast, decreased immunohistochemical staining of PRDX4 in patients with primary stage I lung adenocarcinoma was also associated with poor outcomes (154). PRDX4 has also been linked to lung cancer via association with SRXN1 (172). Strong increases in SRXN1 immunoreactivity were found in tissue arrays from patients with squamous and adenocarcinoma (171). The PRDX4-SRXN1 axis in A549 lung cancer cells plays a role in activator protein 1 (AP-1) activation. Knockdown of either PRDX4 or SRXN1 resulted in reduced colony forming units and cell invasion.
Using a tumor xenograft model in SCID mice, knock down of SRXN1 resulted in decreased tumor growth, while overexpression of SRXN1 alone, or SRXN1 in combination with PRDX4, resulted in tumors that grew faster than those from control cells (171). A tumor promoting role of SRXN1 was also shown in studies demonstrating that cigarette smoke promotes the upregulation of SRXN1, and that genetic ablation of SRXN1 attenuated urethane-induced carcinogenesis (114). The sulfiredoxin inhibitor, K27 (N-[7-chloro-2-(4-fluorophenyl)-4-quinazolinyl]-N-(2-phenylethyl)-β-alanine), led to the accumulation of sulfenylated PRDXs, increased mitochondrial oxidants, and preferential death of tumor cells (67), including A549 lung cancer cells (64).
It remains unclear whether PRDX4 was a target of enhanced overoxidation following SRXN1 inactivation. In aggregate, these findings suggest an important role of the SRXN1/PRDX4 inactivation/reactivation pathway in protecting lung cancer cells from oxidative stress, and that inhibition of SRXN1 may constitute a potential therapeutic opportunity for the treatment of lung cancer.
PRDX6 was originally linked to NSCLC when autoantibodies against PRDX6 were found to be increased, which corresponded with an increase in serum PRDX6 levels in patients with squamous-cell carcinoma (184, 192). The role of PRDX6 in lung cancer is complicated by the fact that it has both peroxidase and PLA2 activities, as described earlier. The peroxidase activity of PRDX6 has been linked to A549 cell growth, whereas the PLA2 activity has been linked to increased invasion and metastasis of lung cancer cells (50). The overexpression of PRDX6 in implanted lung tumor cells caused tumors to grow faster than in wild-type control lung cancer cells, and the tumors showed evidence of increased AP-1 and MAPK signaling (60, 188). Furthermore, mice overexpressing PRDX6 show enhanced tumor formation in the urethane model of lung cancer, in association with binding of PRDX6 to JAK2 and activation of the JAK2/STAT3 pathway (189).
A role of PRDX6 in lung cancer development was also suggested in a mouse model lacking presenilin2 (PS2). Presenilins are the enzymatic components of γ-secretase complex that cleaves amyloid precursor proteins, Notch and β-catenin, and are known to have critical roles in cancer development. Mice lacking PS2 were more prone to urethane-induced lung cancer, and this was associated with enhanced expression of PRDX6, increased PLA2 and GSH peroxidase activities, and activation of STAT3. It was speculated that γ-secretase-mediated cleavage at the phospholipase motif site of PLA2 in PRDX6 causes a decrease in PLA2 activity in lung from wild-type PS2 expressing mouse lung, but not in lungs from PS2 knockout mice (190). These findings point to a unique role of the PLA2 activity of PRDX6 in lung tumorigenesis. A recent study using the anti-EGFR therapeutic, gefitinib, in a xenograft model suggested that serum PRDX6 constitutes a potential biomarker of response to anti-EGFR treatment (53), although additional larger scale studies will be required to substantiate that claim.
Malignant mesothelioma
Malignant mesothelioma (MM) is a rare but devastating tumor that develops on the mesothelial cell layer lining the peritoneal and pleural cavities (164). The pathogenesis of MM, which is most often linked to occupational exposure to asbestos, is characterized by loss of tumor suppressor genes, chronic inflammation, and a long latency period. MM is remarkably resistant to common chemotherapies, and average survival times after diagnosis are measured in months, not years. Laboratory studies with animal and cell culture models show that asbestos fibers induce DNA damage, oxidative stress, and chronic inflammation, and antioxidants have been shown to ameliorate many of the effects of asbestos on cells and tissues (7).
MM is considered a “reactive oxygen species (ROS)-driven tumor” due to changes in molecular and cell signaling signatures that support redox-dependent adaptations required for the proliferation and survival of MM tumor cells (31). Cultured MM cells produce increased levels of mitochondrial and cytoplasmic ROS compared with normal mesothelial cells and compounds that perturb cytoplasmic and mitochondrial redox status show antitumor activity in MM cell and animal models, indicating the importance of maintaining a balanced redox environment for survival and proliferation (35, 126). Assessment of PRDX in MM revealed that PRDX1–3, 5, and 6 are expressed at moderate to high levels in MM cells and tumor tissues (73). However, only the mitochondrial TXNRD2–TXN2–PRDX3 axis has been investigated in detail in MM (35 –38, 126).
PRDX3 was identified as a protein of interest in MM when it was discovered to be a redox-dependent target of the anticancer compound thiostrepton (TS). TS was shown to covalently crosslink disulfide-bonded dimers of PRDX3, a catalytic intermediate formed during H2O2 metabolism in the mitochondria, in both MM cells in vitro and human MM xenoplants in vivo (126). Crosslinking of PRDX3 by TS was more abundant in MM cells versus normal mesothelial cells, was potentiated by the addition of the TXN2 inhibitor, gentian violet (191), and was inhibited by pretreatment with the sulfenic acid trapping molecule, dimedone (Fig. 5) (126). In addition, a “PRDX3 turnover assay” consisting of purified recombinant proteins required for PRDX3 oxidation and reduction was used to show that PRDX3 catalytic activity promoted crosslinking by TS (126). These data provide evidence that PRDX3 catalytic activity may be increased in MM tumor cells, and that the disulfide-bonded dimer intermediate of PRDX3 catalysis is the preferred target for TS. This is of considerable importance, as PRDX3 expression in MM tissues is not considered to be an appropriate tumor marker, as it is also expressed in numerous nonmalignant cells (73). Therefore, PRDX3 enzymatic activity may prove to be a more appropriate marker of malignancy, and development of assays to measure PRDX activity in cells and tissues may provide novel diagnostic and prognostic biomarkers.
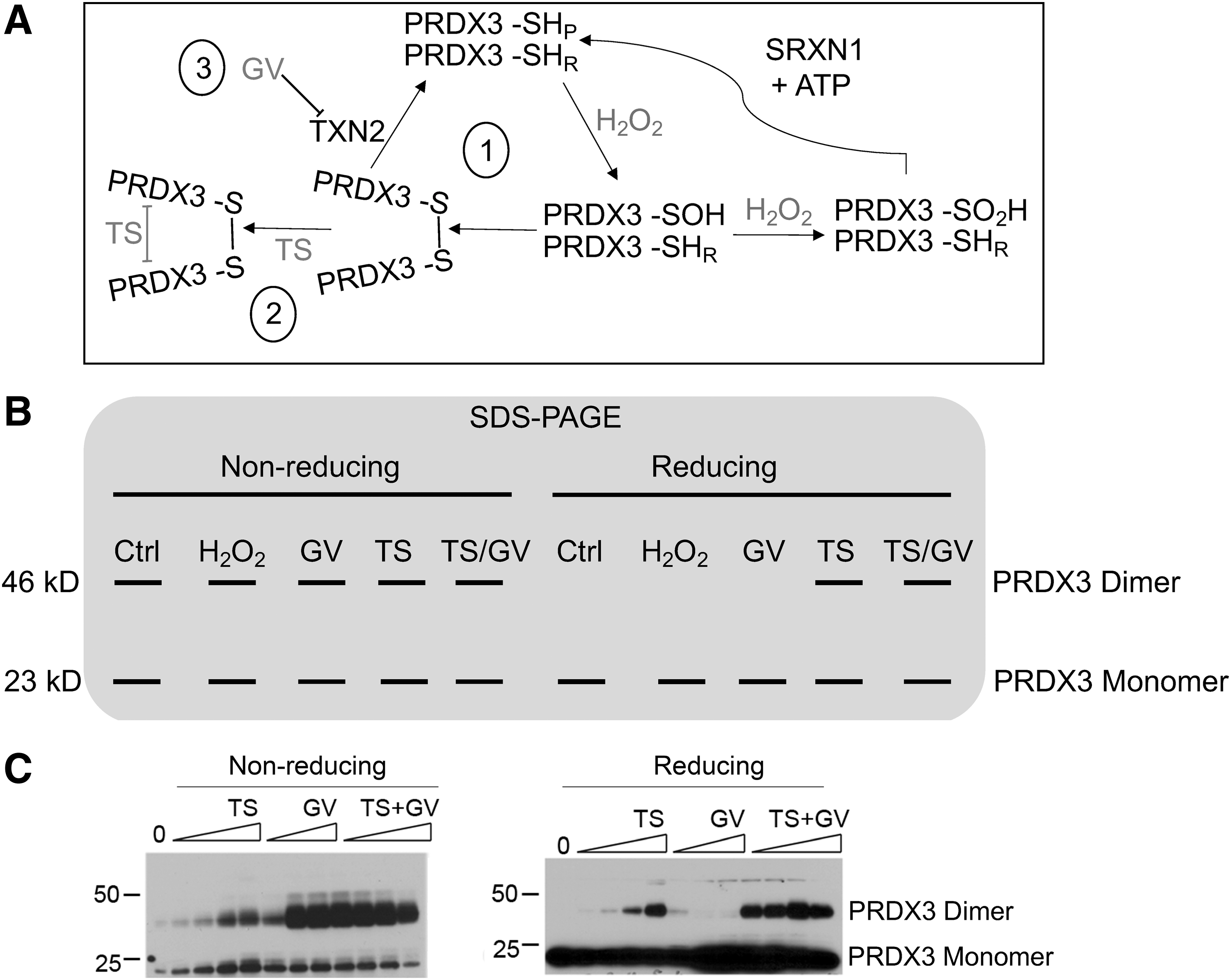
Crosslinking of PRDX3 by TS led to intolerable increases in mitochondrial ROS and MM cell death (36, 126). Furthermore, knockdown of PRDX3 expression by siRNA slowed the growth of MM cells, confirming the importance of PRDX3 in MM cell proliferation. Knockdown of PRDX3 in MM cells also caused cell cycle arrest in the G2/M phase, altered cellular metabolism, and increased mitochondrial fusion, and decreased sensitivity to TS (38), highlighting the relevance of PRDX3 as a therapeutic target in MM (36).
Pulmonary fibrosis
Pulmonary fibrosis is an unrelenting progressive disease, characterized by a loss of normal alveolar architecture, repopulation of alveolar spaces with extracellular matrix, an overpopulation of activated myofibroblasts, and loss of alveolar epithelia. Pulmonary fibrosis is believed to be the outcome of repeated cycles of injury and lack of adequate repair that manifests in individuals in their 50–70s, and affects ∼3 million people worldwide (107). Current therapies have limited effectiveness to halt progression of idiopathic pulmonary fibrosis (IPF), leading to death of patients with IPF within 3–5 years from the time of diagnosis (107).
A number of environmental insults have been shown or speculated to cause pulmonary fibrosis that include inhalation of particulates, such as asbestos or silica, smoking, viral infections, and radiation. In some cases, no clear etiology can be identified in the pathogenesis of fibrosis, leading to the diagnosis of IPF. In cases of familial IPF, fibrosis develops in individuals with germ line mutations in certain genes, including surfactant protein C (SFTPC), and surfactant protein A2 (SFTPA2). These genes encode proteins highly expressed in epithelia, and mutations in these genes result in defects in protein folding, leading to ER stress (80, 87). However, ER stress not only occurs in familial IPF but is now recognized as a common feature of sporadic IPF as well (77).
Besides IPF, a number of other disorders can result in lethal fibrosis, including systemic sclerosis (SS) and Hermansky–Pudlak syndrome (HPS). Numerous studies have linked oxidative stress to the pathogenesis of IPF (15, 16, 32, 72, 74, 83, 109, 118). Perturbations in mitochondria (12, 120) and, as mentioned earlier, the ER (13) have been implicated in the pathogenesis of IPF (88), although the extent to which redox perturbations originating from dysfunctional mitochondria or ER, respectively, contribute to IPF remains unclear. Similarly, the roles of mitochondrially localized PRDX3,5 or ER-localized PRDX4 in lung fibrogenesis remain unclear.
Microarray analysis of 194 samples from patients with interstitial lung disease shows a significant increase in PRDX2,3,4 and a decrease in PRDX6, while chronic obstructive pulmonary disease patients showed significant increases in only PRDX2 and 4, suggesting disease-specific perturbations of PRDXs (Fig. 4C, D). A proteomic screen in lung tissues from control subjects, patients with IPF, or patients with fibrotic nonspecific interstitial pneumonia (NSIP, a fibrotic lung disease with histopathologic features highly distinctive of IPF and better prognosis) revealed alterations in expression of two PRDXs, PRDX1 and PRDX6. PRDX1 expression was increased in lungs from both groups of patients, compared with the control subjects, with higher expression being apparent in NSIP compared with IPF. PRDX6 was increased in lung tissues of NSIP patients but not in patients with IPF, suggesting unique disease-specific modulation of these PRDX proteins.
Immunohistochemical evaluation revealed cell type- and region-specific increases of PRDX1 in settings of fibrosis that occurred in a disease-specific manner (78). The pathways that lead to the cell type- and region-specific increases of PRDX1 and the importance of this upregulation require further investigation. One study investigated the role of PRDX1 in bleomycin-induced fibrosis and showed that mice deficient in PRDX1 were more susceptible to bleomycin-induced mortality, in association with enhanced inflammation and fibrosis. Deficiency in PRDX1 led to increased levels in F2-isoprostanes in lung tissue, indicative of enhanced oxidative stress and increased macrophage migration inhibitory factor (MIF). The worsened phenotype in PRDX1
−/− mice treated with bleomycin could be attenuated by treatment with the low-molecular-weight thiol, N-acetyl-
Immunohistochemical evaluation of PRDX2 in lungs from patients with IPF showed relative increases in expression of PRDX2 in hyperplastic epithelia, while in contrast, PRDX2 immunoreactivity was relatively low in fibroblast foci, the hallmark lesion of IPF (166). Evaluation of PRDX2 expression in lung tissues using Western blots run under reducing and nonreducing conditions revealed diminished immunoreactivity of PRDX2 in lungs from IPF patients, compared with non-IPF subjects, under nonreducing conditions, while differences in PRDX2 expression between the groups were minor, under reducing conditions (166). These observations suggest that the oxidation state of PRDX2 is altered in lung tissues from patients with IPF (166). Further studies will therefore be required to determine the exact redox perturbations of PRDX2 in fibrotic lung tissue. Similar to observations with PRDX1, autoantibodies against PRDX2 were also observed in patients with SS (14) although the implications for PRDX2's function in these patients also require further investigation.
Epithelial cell death and lack of epithelial progenitor function have been recognized as one of the drivers of pulmonary fibrosis, and the death receptor FAS (CD95) plays a critical role in that process (3, 47, 81, 82). Previous work from our laboratories has demonstrated that S-glutathionylation of Fas cell surface death receptor (FAS) amplifies its proapoptotic action, in association with enhanced trafficking to death inducing signaling complexes and binding to FAS ligand (FASL) (2). We also showed that stimulation of cells with FASL induced a rapid overoxidation of PRDX4, suggesting that a redox perturbation in the ER occurs before apoptosis. Indeed, S-glutathionylation of FAS was induced during oxidative processing of a latent pool of FAS in the ER, following protein disulfide isomerase A3-mediated disulfide bridge formation and GSTP-mediated S-glutathionylation (4). Concurrent production of H2O2 was associated with overoxidation of PRDX4. Consequently, overexpression of PRDX4 strongly damped FAS S-glutathionylation, decreased activation of caspases 8 and 3, and increased survival in epithelial cells stimulated with FASL (4). Given the importance of epithelial apoptosis in the pathogenesis of pulmonary fibrosis, these findings point to a putative protective role of PRDX4 in lung fibrogenesis, although additional studies will be required to formally address this scenario. In support of such protective role, transgenic overexpression of PRDX4 diminished liver fibrosis in a model of nonalcoholic steatohepatitis, and type II diabetes, in association with dampened apoptosis of hepatocytes, and decreases in the oxidative stress markers, 8-hydroxy-2′-deoxyguanosine and 4-hydroxy-2-nonenal (123).
As discussed earlier, S-glutathionylation of PRDX and subsequent deglutathionylation by GLRX play a critical role in the reactivation and regulation of higher molecular weight complexes of PRDX (22, 138). Although the exact roles of PRDX in lung fibrosis remain unclear, our laboratories recently illuminated an important role of S-glutathionylation and GLRX in the pathogenesis of lung fibrosis (3). We demonstrated that protein S-glutathionylation was increased in lungs from patients with IPF, compared with non-IPF subjects, and that this correlated with inactivation of GLRX. Furthermore, attenuated GLRX activity and increases in oxidized glutathione inversely correlated with lung function, suggesting a potential contribution of enhanced S-glutathionylation to the pathogenesis of IPF.
In support of this, the global absence of GLRX greatly increased the susceptibility of mice to the development of bleomycin- or adenovirus-expressed active transforming growth factor beta-induced fibrosis. Conversely, transgenic overexpression of GLRX, or direct administration of recombinant GLRX into airways of mice with existing pulmonary fibrosis, reversed the existing increases in collagen content, in association with enhanced collagen degradation, and attenuated apoptosis, in association with diminished S-glutathionylation of FAS (3). Beyond FAS, the exact targets for GLRX-mediated deglutathionylation await further study. For instance, it remains unclear whether GLRX affected the redox homeostasis or chaperone functions of PRDXs, and whether this in turn affected fibrogenesis and/or resolution of disease. In light of the role that GLRX plays in the reduction of S-glutathionylated high molecular weight PRDX complexes, thereby re-establishing active PRDX, it is conceivable that inactivation of GLRX may contribute to increases in inactive PRDXs in settings of lung fibrosis. However, further studies will be required to address the interplay between GLRX and PRDX and implications for fibrogenesis.
As mentioned above, the 1-Cys PRDX, PRDX6, is a bifunctional protein with both PLA2 and peroxidase activities. It remains unclear to this date whether PRDX6 plays a role in the pathogenesis of pulmonary fibrosis. In a gamma-radiation model of pulmonary fibrosis, a carbonylated proteomic screen revealed PRDX6 as one of the target proteins that was carbonylated (6), suggesting potentially compromised function of PRDX6. HPS results from mutations in genes of membrane trafficking complexes that facilitate delivery of cargo to lysosome-related organelles, including lamellar bodies within alveolar type 2 cells in which surfactant components are assembled, modified, and stored (76). HPS can result in lethal lung fibrosis. Using mouse models of HPS, prominent alterations in surfactant were observed in type II alveolar epithelial cells, in conjunction with giant lamellar bodies, early lysosomal stress, late ER stress, and enhanced apoptosis, findings that were confirmed in tissues from patients with HPS (102). As mentioned earlier, ER stress and epithelial apoptosis are prominent features of pulmonary fibrosis, suggesting that pathways that culminate in IPF and HPS may partially overlap. While the role of PRDX6 in fibrosis remains unclear, a role of PRDX6 in HPS has emerged, perhaps not surprising as PRDX6 is found in lamellar bodies where its PLA2 activity plays a key role in phospholipid homeostasis (104, 156). Using a mouse model of HPS type 2 (HPS2) lacking the adaptor protein 3 (AP-3) complex, it was shown that PRDX6 failed to accumulate in lamellar bodies, concomitant with a loss of PLA2 activity in lamellar bodies. AP-3-dependent targeting of PRDX6 to lamellar bodies was shown to require the transmembrane protein LIMP-2/scavenger receptor class B membrane 2 (SCARB2), and a protein/protein interaction between LIMP-2/SCARB2 and PRDX6 was shown to facilitate AP-3-dependent lamellar body trafficking. These findings suggest that the loss of PRDX6 PLA2 activity contributes to the pathogenic changes in lamellar body phospholipid homeostasis found in a subclass of HPS patients (76). The PLA2 activity of PRDX6 was attributed to altered lipid homeostasis in this model of HPS2, although it remains unclear whether its peroxidase activity plays a role in other features of HPS, or other subtypes, inducing enhanced epithelial apoptosis or fibrogenesis. Besides the aforementioned interaction between LIMP-2/SCARB2 and PRDX6, PRDX6 was also shown to bind to the chaperone protein, 14-3-3ϵ, required for targeting of PRDX6 to lamellar bodies (156).
As stated above, the sulfenic acid form of the peroxidatic cysteine of PRDX6 is S-glutathionylated in a reaction catalyzed by GSTP, which is required in the peroxidatic catalytic cycle of PRDX6 (193). Work from our laboratories has illuminated a role of GSTP in pulmonary fibrosis, in association with promotion of epithelial apoptosis. As mentioned earlier, S-glutathionylation of FAS is catalyzed via GSTP-mediated S-glutathionylation, thereby enhancing FASL-induced apoptosis in epithelial cells (4). GSTP expression was prominent in bronchiolar epithelial cells and type II epithelial cells in nondiseased lung tissue, and lungs from patients with IPF, where high immunoreactivity was present in reactive type II epithelial cells that line cysts and in areas of re-bronchiolization. Mice lacking GSTP were partially protected from bleomycin- or adenovirus-encoding active transforming growth factor beta-1 (AdTGFB) -induced pulmonary fibrosis. Direct administration of the clinically relevant GSTP inhibitor, TLK117, into the airways at a time when fibrosis was already apparent, attenuated bleomycin- and AdTGFB-induced remodeling, in association with attenuated S-glutathionylation and dampened epithelial apoptosis (110). Additional studies will be required to elucidate whether oxidized PRDX1 or PRDX6 is a target of GSTP-mediated S-glutathionylation, and whether this impacts alveolar type II cell function and fibrogenesis.
Acute lung injury
ALI is described as acute respiratory failure in association with injury to the vascular endothelium and alveolar epithelium (61). ALI can have many different triggers, which all lead to the activation of the acute inflammatory response in the lung (45). During the inflammatory response, activated neutrophils will pass from the vasculature into the alveolar space and release cytokines and oxidants (33). The increased accumulation of oxidants has been linked to epithelial cell death (11) and progression of ALI (79), and NOX2 has been identified as being a major source of oxidants produced by neutrophils and macrophages during inflammation (42). Despite these observations, apart from PRDX6, the role of PRDXs in ALI remains poorly explored. In a hyperoxic injury model of ALI in mice, PRDX1 mRNA and protein levels were increased (66). In contrast PRDX2 expression was not altered, showing differential regulation between the two cytosolic PRDXs (66). A proteomic screen identified PRDX1 as being elevated in a Pseudomonas aeruginosa model of ALI in rats (101). Furthermore, the proinflammatory mediator, LPS, increased PRDX1 expression in bronchial epithelial cells, which in turn contributed to increased expression of proinflammatory mediators, in association with activation of nuclear factor kappa B (NF-κB) pathway (100).
Other studies have shown that PRDX1 can be secreted from cells in response to distinct stimuli (18, 28), and that PRDX1 in turn can bind to the LPS receptor, TLR4, to enhance production of proinflammatory mediators (148). Similar interactions between PRDX2 or PRDX5 and TLR4 also have been reported (75, 101), suggesting that signals from multiple PRDXs may be transduced via TLR4. In contrast to these stimulatory actions of PRDX on NF-κB activation via TLR4, both PRDX1 and PRDX6 were shown to inhibit NF-κB, in association with inhibition of tumor necrosis factor receptor-associated factor 6 (TRAF6) ubiquitin ligase activity (112, 113). Similar contradicting outcomes of PRDX have been observed in in vivo models of ALI or sepsis. Ozone-induced acute inflammation was attenuated in PRDX1 −/− mice (182), pointing to a proinflammatory role of PRDX1. In contrast, PRDX1−/− mice showed increased susceptibility to LPS-induced lethal shock, in association with enhanced inflammation (159), and similar findings of enhanced injury and/or mortality were reported in mice lacking PRDX2 (183), PRDX3 (94), or SRXN1 (140). Collectively, these findings suggest that the effects of PRDXs on NF-κB activation and subsequent proinflammatory signaling may depend on the location of PRDX (intracellular vs. extracellular) and/or its molecular target(s) (e.g., TLR4 vs. TRAF6), and on the nature and extent of the proinflammatory insult in vivo (an insult to the airway vs. systemic injury elicited by LPS).
A substantial number of studies have been conducted to address the role of PRDX6 in ALI. The overexpression of PRDX6 in mice exposed to hyperoxia attenuated lung injury (169, 170), while knockdown of PRDX6 in the lungs of mice exacerbated hyperoxia- or paraquat-induced lung injury (167, 168). The attenuation of lung injury following overexpression of PRDX6 coincided with less oxidative stress, likely due to the peroxidase activities of PRDX6, reducing lipid peroxides. As previously mentioned, the PLA2 activity of PRDX6 is important in the formation of the active form of NOX2 (29). This is particularly interesting as PRDX6 has two apparently opposing functions: first, promoting the formation of active NOX2, and second, detoxifying lipid hydroperoxides that are increased during ALI. This paradox between enhancing oxidant generation and detoxifying lipid peroxidation makes specific targeting of the PLA2 activity of PRDX6 enticing. Indeed, a PRDX6 transition state analog inhibitor, 1-hexadecyl-3-(trifluoroethyl)-sn-glycero-2-phosphomethanol (MJ33), has been developed to exclusively target the PLA2 activity of PRDX6 while maintaining the peroxidase activity (90). The inhibition of PRDX6 PLA2 activity with MJ33 attenuated the severity of ALI in response to LPS or hyperoxia in mice (8, 89). These important findings suggest that selective manipulation of only distinct functions of PRDX6 has the potential to change the course of disease progression.
Summary and Future Directions
PRDXs are critical regulators of biological functions through their ability to control the redox tone, allowing either the promotion or dampening of signals from H2O2 or related oxidants. The oxidation of PRDXs, and relay of oxidant signals through PRDX-controlled oxidations, controls the outcome of activation of NADPH oxidases or oxidants from other sources, in part, through the oxidation of distinct client proteins. PRDXs do not act in isolation as they require the TXN/TXNRD/NADPH, SRXN1 redox system, and in some cases GLRX/GSH, for their reduction. Furthermore, the chaperone function of PRDXs, controlled by the oxidation state, demonstrates the versatility in redox regulation and control of cellular biology exerted by this class of proteins.
Despite the long-known observations that redox perturbations accompany a number of pulmonary diseases, such as lung cancer, mesothelioma, pulmonary fibrosis and ALI, discussed herein, surprisingly little is known about the role of PRDXs in the etiology of these diseases. Intriguing findings, such as the secretion of PRDXs and the formation of autoantibodies, raise a number of questions about the pathways that regulate secretion, the redox status of PRDXs secreted, implications for the biology of cells secreting PRDXs, and the factors that govern the immune response to PRDXs (Fig. 6).
While client proteins of PRDX-induced oxidation have been identified, and some of these have relevance to lung pathology, these studies have been limited to cell line settings using overexpression strategies to trap proteins bound to individual PRDXs. To date, client proteins for the individual PRDXs in settings of homeostasis or lung diseases remain largely unknown. Some of the studies described herein point to the high level of precision through which oxidation of PRDXs regulates the activity of the repertoire of target proteins.
Similarly, it remains unknown how PRDXs transition from stacked decameric structures that act as chaperones to proteins with redox activities, and which signals govern these conversions. Given the compartmentalization of individual PRDXs, further study of these proteins may provide new insights into redox regulation in subcellular compartments such as the ER, where PRDX4 is localized, and mitochondria where PRDX3 and PRDX5 are found. This exquisite precision of PRDX-mediated control of cellular processes, their high level of expression, and interdependence on the TXN/TXNRD, SRXN1, and GLRX/GSH redox systems make it easy to envision why nontargeted strategies using generic antioxidants have failed in a number of clinical trials and have not yielded new drugs for the management of complex lung diseases.
Insights provided from the biochemical studies conducted thus far offer new insights into potential avenues to target the interface of PRDX with unique client proteins to regulate chaperone activity and control biological oxidations. An exciting example of this precision is the aforementioned work with TS, which covalently crosslinks disulfide-bonded dimers of PRDX3 and preferentially kills MM cells over normal mesothelial cells. These and additional steps in redox drug development will offer the precision required to control oxidant-dependent biological processes with selectivity and specificity currently achieved in other areas of medicinal chemistry. Given the exciting new discoveries that continue to be made in the PRDX field, we have only seen the tip of the iceberg for potential implications of targeting this family or redox enzymes to manage lung diseases.
Footnotes
Acknowledgments
This work was funded by NIH R35HL135828 (Y.M.W.J.-H.), NIH R01HL122383 (V.A.), NIH R01CA219156 (J.v.d.V.), NIH R01HL085646 (A.v.d.V.), NIH R21ESO28857 (A.S.), and T32HL076122 (E.A.E., A.M.). B.C. and N.H.H. are supported through a sponsored research agreement with Paredox Therapeutics, LLC.