
Abstract
Significance:
Cardiovascular disorders are the most important cause of morbidity and mortality in the Western world. Monogenic developmental disorders of the heart and vessels are highly valuable to study the physiological and pathological processes in cardiovascular system homeostasis. The arterial tortuosity syndrome (ATS) is a rare, autosomal recessive connective tissue disorder showing lengthening, tortuosity, and stenosis of the large arteries, with a propensity for aneurysm formation. In histopathology, it associates with fragmentation and disorganization of elastic fibers in several tissues, including the arterial wall. ATS is caused by pathogenic variants in SLC2A10 encoding the facilitative glucose transporter (GLUT)10.
Critical Issues:
Although several hypotheses have been forwarded, the molecular mechanisms linking disrupted GLUT10 activity with arterial malformations are largely unknown.
Recent Advances:
The vascular and systemic manifestations and natural history of ATS patients have been largely delineated. GLUT10 was identified as an intracellular transporter of dehydroascorbic acid, which contributes to collagen and elastin cross-linking in the endoplasmic reticulum, redox homeostasis in the mitochondria, and global and gene-specific methylation/hydroxymethylation affecting epigenetic regulation in the nucleus. We revise here the current knowledge on ATS and the role of GLUT10 within the compartmentalization of ascorbate in physiological and diseased states.
Future Directions:
Centralization of clinical, treatment, and outcome data will enable better management for ATS patients. Establishment of representative animal disease models could facilitate the study of pathomechanisms underlying ATS. This might be relevant for other forms of vascular dysplasia, such as isolated aneurysm formation, hypertensive vasculopathy, and neovascularization. Antioxid. Redox Signal. 34, 875–889.
Introduction
In species having lost the ability to biosynthesize ascorbic acid (AA), such as Homo sapiens, AA sparing is of utmost importance (7, 57). AA-dependent reactions are presumably found in all subcellular compartments of animal cells. Aberrant subcellular compartmentalization of AA has been implicated in both inherited and acquired human diseases, including Werner syndrome (58), cancer (11), and subcellular scurvy (100, 101).
AA has a ubiquitous antioxidant function and acts especially in organelles characterized by oxidative metabolic reactions, such as mitochondria, peroxisomes, and the endoplasmic reticulum (ER). Beyond its general antioxidant properties, AA is required for proper functioning of several enzymes, including AA-dependent mono- and dioxygenases (54, 56, 74) present in various subcellular compartments; for example, hypoxia-inducible factor prolyl hydroxylases in the cytoplasm, collagen prolyl/lysyl hydroxylases in the ER lumen (65, 88), dopamine β-monooxygenase and peptidylglycine α-hydroxylating monooxygenase in chromaffin granules, synaptic and secretory vesicles (27), and histone and DNA demethylases in the nucleoplasm.
Two families of membrane transporters, namely sodium-dependent vitamin C transporters (SVCTs) and facilitative glucose transporters (GLUTs), accommodate for cellular accumulation and the subcellular distribution of AA. Vitamin C can be transported in both a reduced form (AA) and an oxidized form (dehydroascorbic acid [DHA]). The mechanisms involved are summarized in Figure 1.
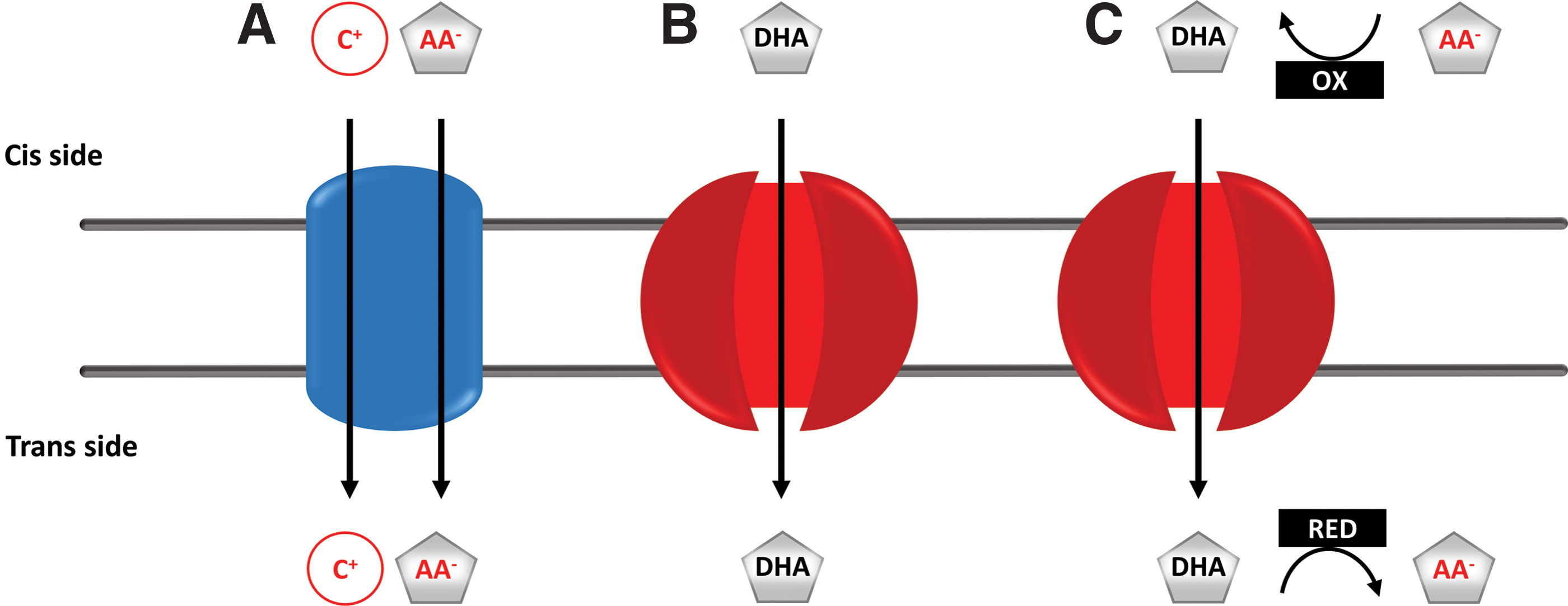
SVCTs are highly specific active transporters of anionic AA (13). SVCT1 (SLC23A1) and SVCT2 (SLC23A2) specifically carry AA across membranes otherwise impermeable for the anion. SVCT1 has a high capacity, but low affinity, for AA and is primarily involved in intestinal absorption (13) and renal reabsorption to reduce urinary loss of AA (59, 81). SVCT2 is a very high-affinity lower-capacity transporter, which is more ubiquitous and responsible for transferring AA into a variety of cells and tissues. This transporter is also present in intracellular membranes, including the inner mitochondrial membrane (5, 64). Thus, SVCT1 enables AA accumulation at the level of the whole organism, while SVCT2 ensures the AA supply of each cell and some of their intracellular compartments.
The oxidized form of vitamin C, DHA, can further be transported by several GLUT family members (SLC2, solute carrier protein 2) via facilitated diffusion [reviewed in (63)]. Under physiological conditions, DHA transport is thought to be less significant since DHA is present at low concentrations in extra- and intracellular compartments due to its unstable nature. DHA is rapidly reduced to AA by thiol-dependent mechanisms on the trans side of the membrane at the expense of nicotinamide adenine dinucleotide phosphate and preserves its antiscorbutic properties. However, GLUT-dependent DHA transport may increase substantially under oxidative stress or by oxidase activity in the vicinity of the membrane (68). GLUT1-4, 8, and 10 are the best-documented, facilitative DHA transporters, which either associate with the plasma membrane or preferentially localize to specific organelles (6).
Overall, the concerted action of both the SVCT and GLUT families results in the intracellular accumulation of tremendous amounts of AA (intracellular: 1–10 mM versus blood plasma: 50–100 μM) and in its distribution between organelles (Fig. 1). Nevertheless, the intracellular distribution of vitamin C and distribution and functioning of AA/DHA transporters remain important, yet incompletely resolved, mechanisms [reviewed in (6)] that are difficult to assess in the absence of in vivo applicable tools for measurement of AA/DHA concentrations in subcellular compartments. A recently proposed approach used high-resolution immunoelectron microscopy and immunogold labeling of AA (99) and demonstrated the presence of AA in organelles of plants at different concentrations that may change under (patho)physiological conditions (45, 84).
Genetic diseases associated with GLUTs are very rare, emphasizing their vital importance and robustness of the family. Mutations in SLC2A1, SLC2A2, and SLC2A10 are, respectively, associated with neurological disorders with variable intellectual disability and epilepsy [OMIM 606777, (83)], Fanconi–Bickel syndrome [a tubulopathy with intellectual disability, OMIM 227810, (79, 89)], and arterial tortuosity syndrome [ATS, OMIM 208050, (26)]. ATS is a rare congenital disorder belonging to the group of cutis laxa syndromes, associating with arterial tortuosity and aneurysm formation (91). The molecular link between mutations in the facilitative glucose transporter GLUT10 (encoded by SLC2A10) and the observed phenotype remains obscure. Hence, it is not surprising that several hypotheses have been forwarded to explain the pathomechanism. We critically review the existing suggestions and draw up a unifying hypothesis.
Clinical Manifestations in ATS
Initial presentation and molecular diagnosis
Most patients present with a cardiac murmur or reduced femoral pulsation. Less frequently, neonatal respiratory failure due to infant respiratory distress syndrome, pulmonary hypertension, or a diaphragmatic hernia may be the initial presentation. A rare initial hint to the diagnosis is a remarkably stretchable or lax skin. Workup with echocardiography reveals kinking of the aortic arch, for which more extensive vascular imaging with magnetic resonance angiography, computed tomography, or percutaneous angiography is initiated. Upon observation of widespread tortuosity of the aorta and middle-sized arteries, genetic analysis is carried out. The identification of biallelic pathogenic variants in SLC2A10 confirms the diagnosis of ATS.
In view of the broad differential diagnosis, most of the patients undergo testing for a panel of genes relevant for arterial aneurysms and tortuosity or undergo targeted exome analysis for a panel of genes related to connective tissue disorders. Of note, a review of evidence for genes that should be included in diagnostic gene panels for heritable thoracic aortic disease does not withhold SLC2A10 (75). Although assigned as a potentially diagnostic gene, evidence is currently limited in the context of heritable thoracic aortic diseases based on its association with a syndromic presentation (rather than isolated vascular disease) and the absence of an unequivocal risk for dissection. Hence, some diagnostic panels for heritable thoracic aortic diseases may not include SLC2A10.
The spectrum of SLC2A10 variants encompasses missense, nonsense, and splice-site mutations, besides large (exonic) and small deletions (4). So far, no genotype–phenotype correlation has been observed, impeding prognosis assessment and disease management by early genotyping (9, 14, 16, 77).
Cardiovascular features and natural history
Generalized tortuosity of the aorta and/or middle-sized arteries is invariably present, sporadically being associated with dilated or slightly tortuous large veins (9, 16, 62). Aortic side branches, including the truncus brachiocephalicus and left subclavian artery, may show an aberrant implantation on the aorta (Fig. 2).
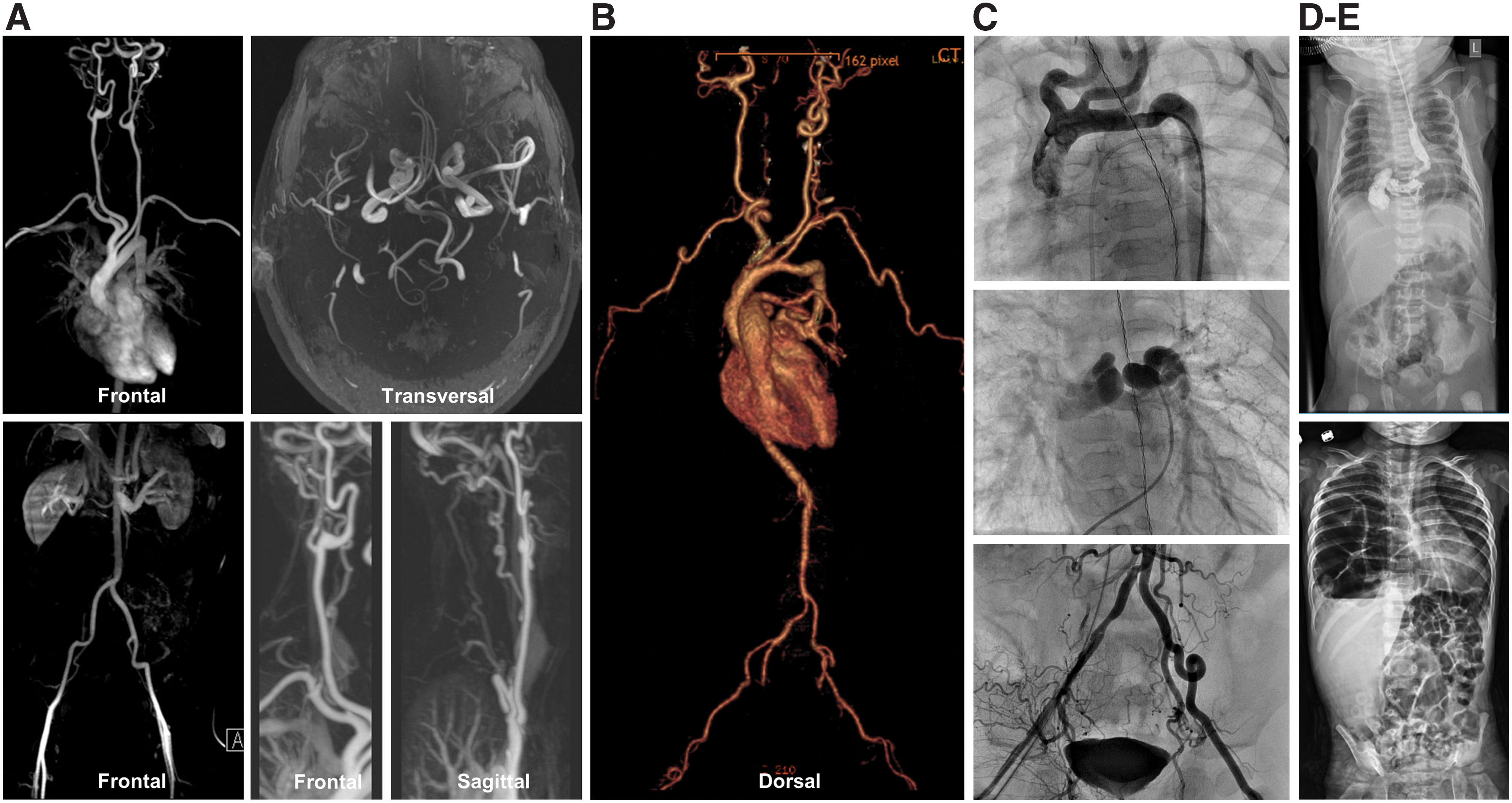
A slight majority of patients (57%) have pulmonary artery stenosis, either of the truncus pulmonalis or the main or peripheral pulmonary branches. Pulmonary hypertension is observed in some patients and may or may not be associated with pulmonary artery stenosis. About one-quarter of the patients have a stenosis of the aorta that may be focal (mostly at the isthmus aortae) or present as a long stenotic stretch or hypoplastic aorta. Other artery stenoses are less common (∼15%), but may involve any middle-sized arteries, including the renal arteries. In addition, severe kinking of arteries may cause functional stenosis (9, 16).
Patients are at risk of developing aneurysms of the aortic root (commonly at the sinuses of Valsalva). Some patients have presented with aggressive aortic root dilatation before the age of 4 years, requiring urgent surgery. Others have shown slowly progressive aortic root dilatation in young adulthood. Nevertheless, no aortic dissections have been unequivocally recorded to date (9, 16). Mitral valve prolapse and cardiomyopathy have been reported (9). It is unclear if cardiomyopathy is due to secondary remodeling or a primary event (as in the related Marfan syndrome) (18).
Ischemic events (9, 16, 19, 33), including stroke, are a concern. Of note, initial reports have described organ infarctions as a cause of death in young children clinically diagnosed with ATS (8, 71, 93), some of whom were confirmed molecularly afterward.
While initial mortality rates were reported up to 40% before the age of 4 (93), larger cohorts of patients with a molecularly confirmed diagnosis revealed a milder disease course (9, 16).
Pulmonary manifestations
In a recent cohort (9), infant respiratory distress syndrome was a relatively frequent observation that may relate to impaired lung maturation, diaphragmatic hernia, or pulmonary hypertension. Obstructive sleep apnea may be observed in older children and adults and may relate to hypotonia of the pharyngeal musculature and retrognathia. It remains to be established if patients are at risk to develop pulmonary emphysema, as has been observed in other elastinopathies (24).
Ocular manifestations
Corneal thinning and pellucid corneas are recently described manifestations in several ATS patients (43, 44). This may result in irregular astigmatism, keratoconus, and keratoglobus (16, 43). It may also associate with corneal opacities. Myopia is often present, although it is unclear if it is more frequently observed than in the general population. Laser-assisted in situ keratomileusis surgery is a risk factor for developing corneal complications (43).
Connective tissue and craniofacial features
ATS patients may have some distinctive craniofacial features that become more prominent with age (Fig. 3). Frequently reported features include a long face, hypertelorism, downslanting palpebral fissures, epicanthal folds, periorbital fullness, sagging cheeks, large ears, a highly arched palate, and retrognathia (9, 16). Bifid uvula or a cleft palate seems not to be associated with the disease, which may differentiate it from the Loeys–Dietz syndrome.
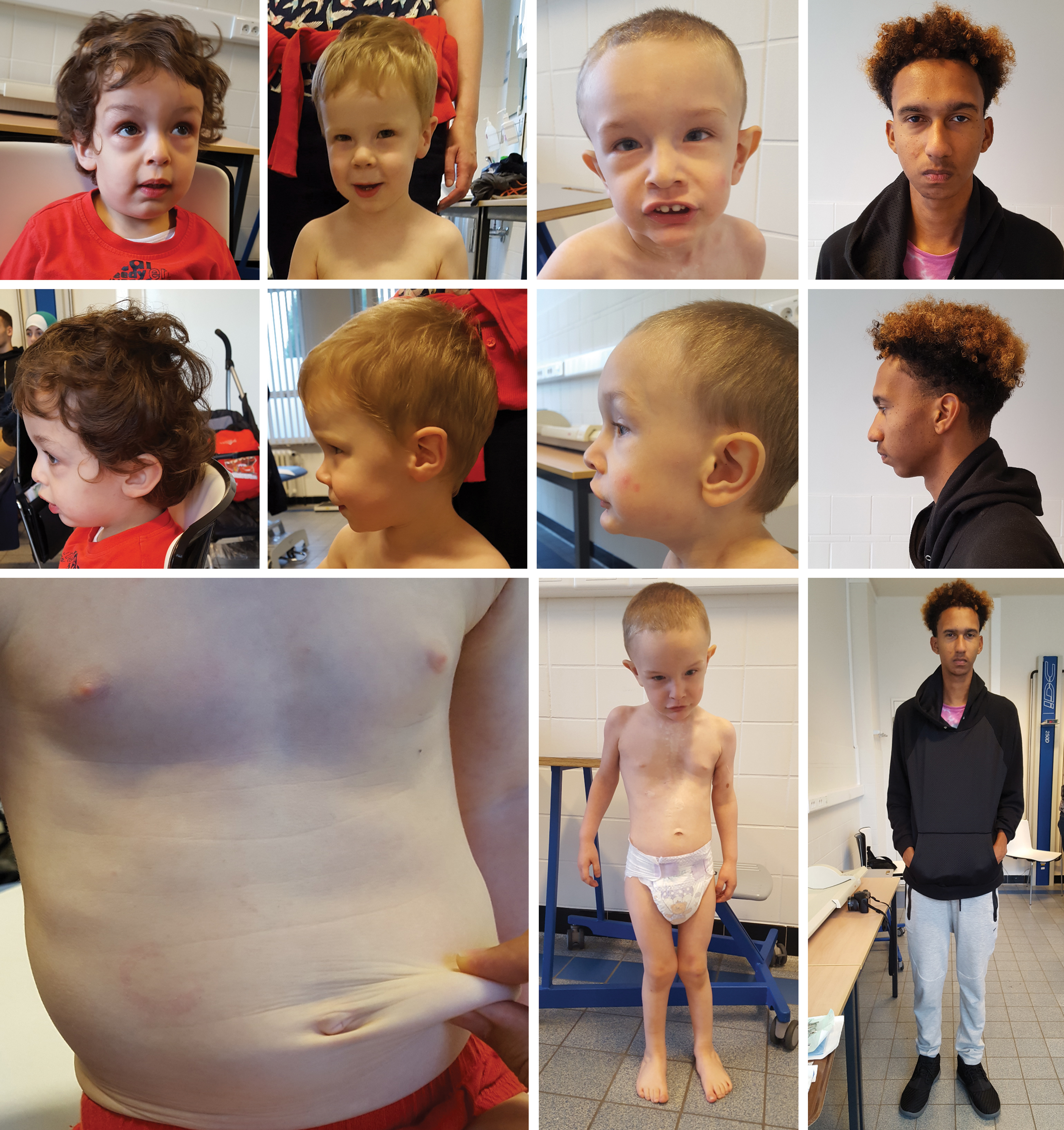
Overgrowth results in arachnodactyly and pectus deformity, while dolichostenomelia is not routinely observed. Hypermobility of small and large joints is frequently present and hypotonia may cause isolated motor delay (9, 16). Skin anomalies range from a thin hyperextensible skin with a velvety texture (often described as soft and doughy) to loose skin folds (cutis laxa) (Fig. 3). Wound healing is normal. Umbilical and inguinal hernias may occur (9) as well as pelvic organ prolapse in women (especially after child bearing) (20).
Other manifestations
Hiatal hernia and diaphragmatic hernia may be present in up to 30% of the patients and their presentation may vary from asymptomatic over postprandial discomfort to neonatal respiratory failure (Fig. 2) (9, 98). About one-third require surgical correction. Pyloric stenosis seems more prevalent (10%) as well as urinary tract anomalies with dilatation of the pyelocaliceal system (9). Finally, some patients may have lengthening of the gastrointestinal tract, resulting in esophageal tortuosity or dolichocolon. Several patients reported symptoms of autonomic dysfunction such as obstipation (that may partly be due to a dolichocolon), Raynaud phenomenon, slow eye accommodation, and orthostatic hypotension.
Many patients have psychological burdens related to both self-esteem and anxiety and distress for an uncertain vascular prognosis (Callewaert, personal observation).
A summary of the clinical characteristics and presentation rates in a series of 50 novel patients and 52 previously reported patients with a confirmed molecular diagnosis was recently presented by Beyens et al. (9).
Histopathology
Vascular tissue histology shows fragmentation of the inner elastic membrane and the elastic lamellae in the tunica media of the aortic wall. The elastic fibers are short, thickened, and disorganized, an observation made as well in other heritable disorders of the connective tissue with cardiovascular manifestations (1, 8, 9, 34, 39, 71). Cultured fibroblasts show disorganization of the actin cytoskeleton and multiple extracellular matrix (ECM) components, including fibronectin, fibrillin, type 3 collagen, type 5 collagen, and decorin (9, 26, 41, 102).
Transmission electron microscopy on skin biopsies of ATS patients reveals a poorly organized elastin assembly at the periphery of the elastic fiber, accompanied by infiltration of microfibrils in an interrupted elastin core (9).
Patient Management
Given the systemic manifestations associated with ATS, a coordinated and multidisciplinary approach for management and treatment is required. Treatment is largely supportive and symptomatic.
Baseline magnetic resonance arteriography from head to pelvis is required upon diagnosis and should be repeated every 3 years. During the first 5 years of life, echocardiographic follow-up every 3 months is necessary to detect aggressive aneurysm formation or evaluate pulmonary hypertension. Afterward, yearly follow-up suffices if measurements remain within normal limits (9). In case of aneurysm formation, surgical guidelines for Marfan syndrome can be applied as the risk for acute dissections on small aortic diameters seems relatively small (9). Pulmonary artery stenosis can be corrected by percutaneous surgical or hybrid approaches (80, 92). Systemic stenosis—if focal—may require excision and end-to-end anastomoses (2, 3, 12, 22, 28, 35, 46, 49, 80, 92). Currently, the experience is limited and is based on expert opinion and on personal experience of the thoracovascular surgeon (14).
In analogy with treatment strategies applied for other heritable disorders of connective tissue, such as Marfan syndrome and Loeys–Dietz syndrome, agents that reduce hemodynamic stress on the arterial wall, including beta-adrenergic blockers, angiotensin-converting enzyme inhibitors, and angiotensin II receptor 1 antagonists, such as losartan, can be considered. The experience is limited and in case of aortic hypoplasia/stenosis and/or renal stenosis, caution is necessary because of the risk of renal failure. Similarly, caution is needed to use nonsteroidal anti-inflammatory agents (9, 14). Pulmonary hypertension may be severe and related to pulmonary artery stenosis (causing a ventilation–perfusion mismatch), increased vascular wall tension, or lung hypoplasia due to diaphragmatic hernia. Treatment depends on the cause and may require percutaneous angioplasty and/or surgery or medical treatment (e.g., with sildenafil).
Regular orthopedic (joint laxity and scoliosis), orthodontic (highly arched palate and dental crowding), pulmonary (obstructive sleep apnea syndrome, also postoperatively), and ophthalmological (keratometry and myopia) assessments are advised. Corneal cross-linking has been done in two ATS patients with corneal thinning, but long-term effectiveness needs to be established (Callewaert, personal communication). Early rehabilitation may improve motor function in case of joint hyperlaxity and hypotonia. Above all, patients should remain active in moderation (aerobic joint-gentle activities, such as swimming), as advised for other connective tissue disorders.
Differential diagnosis
ATS clinically shows some overlapping vasculopathy with other heritable disorders of the connective tissue. More specifically, autosomal recessive cutis laxa type 1B (ARCL type 1B) (76) caused by mutations in fibulin-4, encoding for the elastin-binding protein fibulin-4, is also associated with arterial tortuosity, arterial aneurysms, and stenosis, although ARCL type 1B more commonly leads to focal stenosis at the aortic isthmus and a more aggressive arterial phenotype. Furthermore, Loeys–Dietz syndrome, caused by heterozygous pathogenic variants in genes involved in transforming growth factor beta (TGFβ) pathway signaling, such as TGFβ cytokines (TGFB2 and TGFB3), TGFβ receptors (TGFBR1 and TGFBR2) and signal transducers (SMAD2 and SMAD3), is characterized by cerebral, thoracic, and abdominal arterial aneurysms and/or dissections (55).
Occipital horn syndrome, an X-linked disorder caused by mutations in ATPase copper-transporting alpha (ATP7A), presents not only with intracranial vascular tortuosity but also with distinctive skeletal and urogenital features (10). Loose skin may be reminiscent of different types of cutis laxa, including ARCL type 1A, type 1B, type 1C, and ARCL type 2. When the skin is rather hyperextensible, it should be differentiated from the Ehlers–Danlos syndrome, hypermobility and vascular type (14).
Phenotypic Analysis of Animal Models for ATS
To date, two mouse models have been described for ATS. These models were generated through N-ethyl-N-nitrosourea mutagenesis in mice with a C3HeB/FeJ background. Two lines harboring missense mutations in Slc2a10 (c.383G>A—p.G128E and c.449C>T—p.S150F) were recovered, and these nucleotide alterations were predicted to be deleterious (15).
Callewaert et al. executed comparative studies on wild-type, heterozygous, and homozygous mice at 5 months of age, including ultrasound analysis of the abdominal aorta, whole-animal vascular corrosion casting, and histological analysis of the tail and popliteal arteries. For both lines, none of the examinations revealed ATS-related phenotypic abnormalities. Suggested explanations for an absent phenotype include (i) several GLUT family members may show functional redundancy, thus compensating for the loss of GLUT10; (ii) phenotypic penetrance is hindered by the used genetic background (C3HeB/FeJ); and (iii) GLUT10 does not contribute to vasculogenesis in mice (15).
Utilizing the same mouse models, Cheng et al. executed a series of additional experiments (21). Again, echocardiograms did not reveal any abnormalities, and using brain magnetic resonance angiography, architectural anomalies in the cerebral arteries such as tortuosity, stenosis, or aneurysms could not be detected at 10 months of age. Histopathological analysis, however, disclosed a mild vascular phenotype, which was more severe in mutant mice carrying the p.G128E mutation (in comparison with the p.S150F mutant) and in older mice (10 months of age vs. 6 months of age). Older mutant mice had a higher blood pressure and displayed a thickened and irregularly shaped vessel wall, fragmented elastic fibers with a disruption of the internal elastic lamina, and hypertrophy of endothelial cells in the tunica intima (21).
In 2012, Willaert et al. published a report on an slc2a10 zebrafish knockdown model (95). Morpholino-injected embryos (morphants) showed several cardiovascular abnormalities: cardiac edema, a reduced heart rate and blood flow, and an incomplete and irregular patterning of the vasculature, resulting in disrupted blood flow in the sinus venosus. These anomalies possibly represent developmental precursors to vascular anomalies, as observed in ATS patients.
Molecular Background
ATS exhibits autosomal recessive inheritance and approximately a decade ago, its causal gene, SLC2A10, was identified (26). The protein it encodes (GLUT10) is a member of the facilitative GLUTs, which are involved in assisting the transport of monosaccharides, polyols, and other small carbon compounds across eukaryotic cell membranes (63). The GLUT family consists of 14 proteins, which are categorized into three classes based on sequence similarity: class 1 (GLUTs 1–4 and 14), class 2 (GLUTs 5, 7, 9, and 11) and class 3 (GLUTs 6, 8, 10, 12, and H+/myo-inositol cotransporter). The general structure of each GLUT protein is similar: 12 transmembrane domains connected by linker domains, cytoplasm-pointing amino and carboxy termini, and a unique N-linked oligosaccharide side chain, positioned in the first (classes 1 and 2) or fifth (class 3) exofacial linker domain (63).
In GLUT10, this particular extracellular loop is distinctively larger in size compared with the other GLUT family members. Some authors have speculated that this loop may attribute alternative functions to GLUT10 (60). However, none of the 34 known mutations causing ATS reside in this exofacial linker domain (9), possibly reflecting a less critical role of this domain.
Hypotheses for the Pathophysiology of ATS
GLUT10 is widely expressed, but is most abundant in smooth muscle-rich organs (97), which correlates with its importance in blood vessel development and homeostasis. Other organs showing expression are the heart, placenta, lung, liver, skeletal muscle, pancreas, brain, kidney (32, 60), and adipose tissue (96).
On a subcellular level, GLUT10 has been localized to the nuclear envelope (
Inhibition of glucose-mediated transcriptional regulation in the nucleus
Coucke et al. localized GLUT10 in the perinuclear region and identified TGFβ pathway upregulation in ATS patients (26). According to the hypothesis, deficiency of GLUT10 would result in reduced nuclear glucose levels, downregulating transcription of genes with glucose-responsive elements in their promoter regions. One such gene is DCN encoding the proteoglycan, decorin. Decorin sequesters TGFβ (37) and its expression was reduced in cultured vascular smooth muscle cells (SMCs) of ATS patients. Consequently, TGFβ-driven expression of the proteoglycan, versican, was increased (26). Versican has an inhibitory role on elastic fiber assembly (94) and its increased expression could explain failed elastogenesis in ATS (Fig. 4).
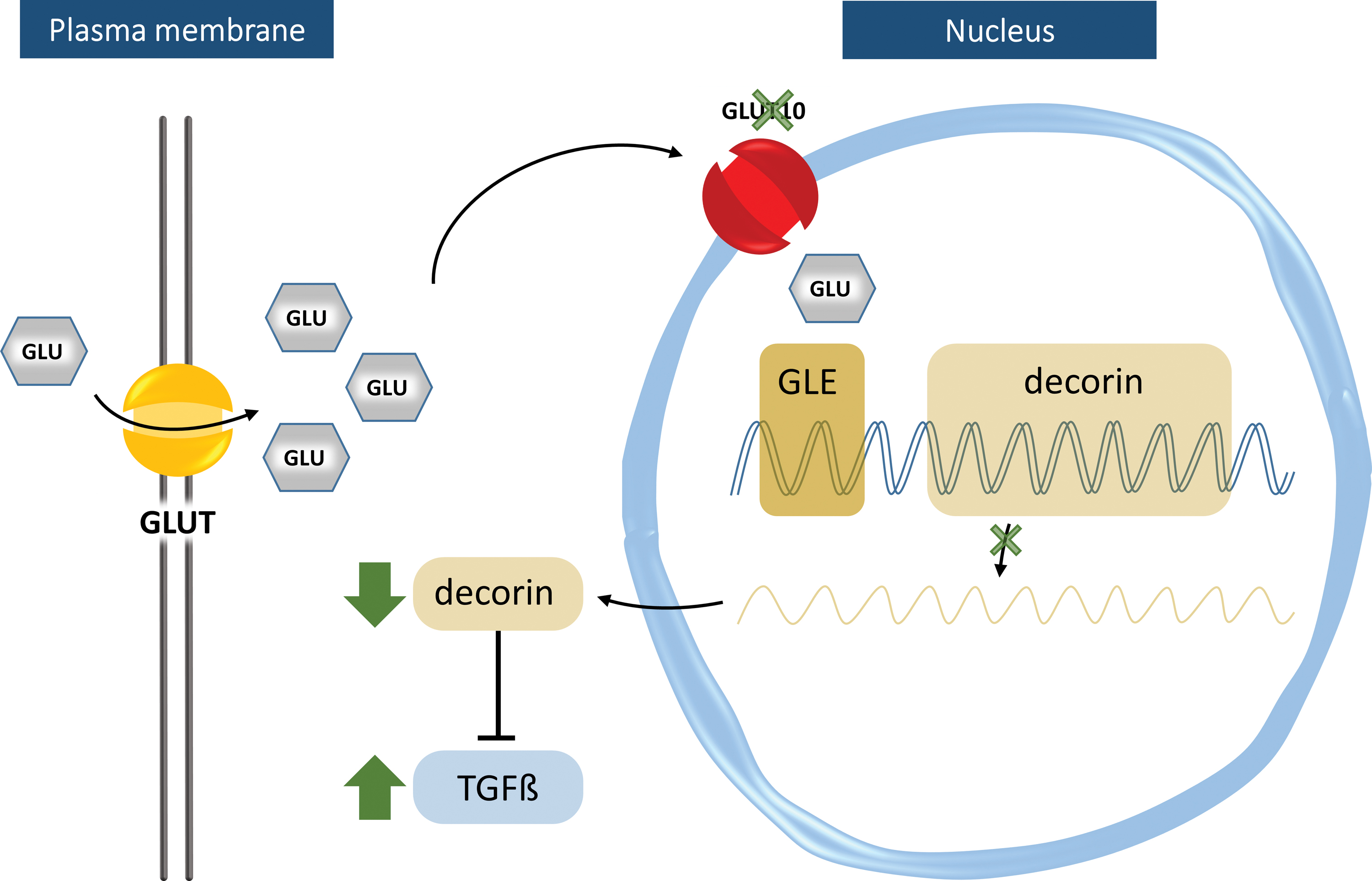
Nevertheless, the role of GLUT10 as a nuclear GLUT is questionable based on the following: (i) small polar molecules such as glucose are able to enter the nucleus via diffusion through the nuclear pore, hence not requiring specialized transporters, and (ii) glucose-dependent transcription is not directly mediated by intranuclear glucose, but rather by indirect mechanisms transducing the glucose signal to the genome (82).
Oxidative stress
Several studies, described in detail below, pointed toward a contribution of oxidative stress in the pathogenesis of ATS. Disturbed redox status and increased reactive oxygen species (ROS) production, promoting protein oxidative modifications, can modulate signaling molecule activities and dysregulate key pathological cellular processes, such as apoptosis, migration, proliferation, inflammation, and ECM component degradation, accumulating in vascular remodeling as observed in ATS (38).
A first study, performed by Lee et al., confirms that GLUT10 operates as a DHA transporter (53). They observed that exogenous tagged GLUT10 localized primarily to the mitochondria in rat A10 SMCs and insulin-stimulated mouse 3T3-L1 adipocytes, while the protein localized to the Golgi apparatus in mouse NIH3T3 fibroblast cells. Mitochondrial import of DHA increased in a dose-dependent manner and following oxidative stress upon H2O2 administration.
These findings prompted a second hypothesis (Fig. 5) where GLUT10 has a major role in replenishing mitochondrial antioxidative capacities through DHA transport. Under normal conditions, both DHA and AA are transported into the cytoplasm, where most DHA is reduced to AA. Under oxidative conditions, DHA accumulates in the cytoplasm and is transported to the mitochondria in a GLUT10-dependent manner, followed by its regeneration to AA. There, AA exerts its function as an antioxidant, quenching ROS molecules and free radicals, protecting the cell from damage due to oxidative stress. A GLUT10 defect would result in accumulation of ROS molecules, which might trigger development of ATS-related anomalies (53).
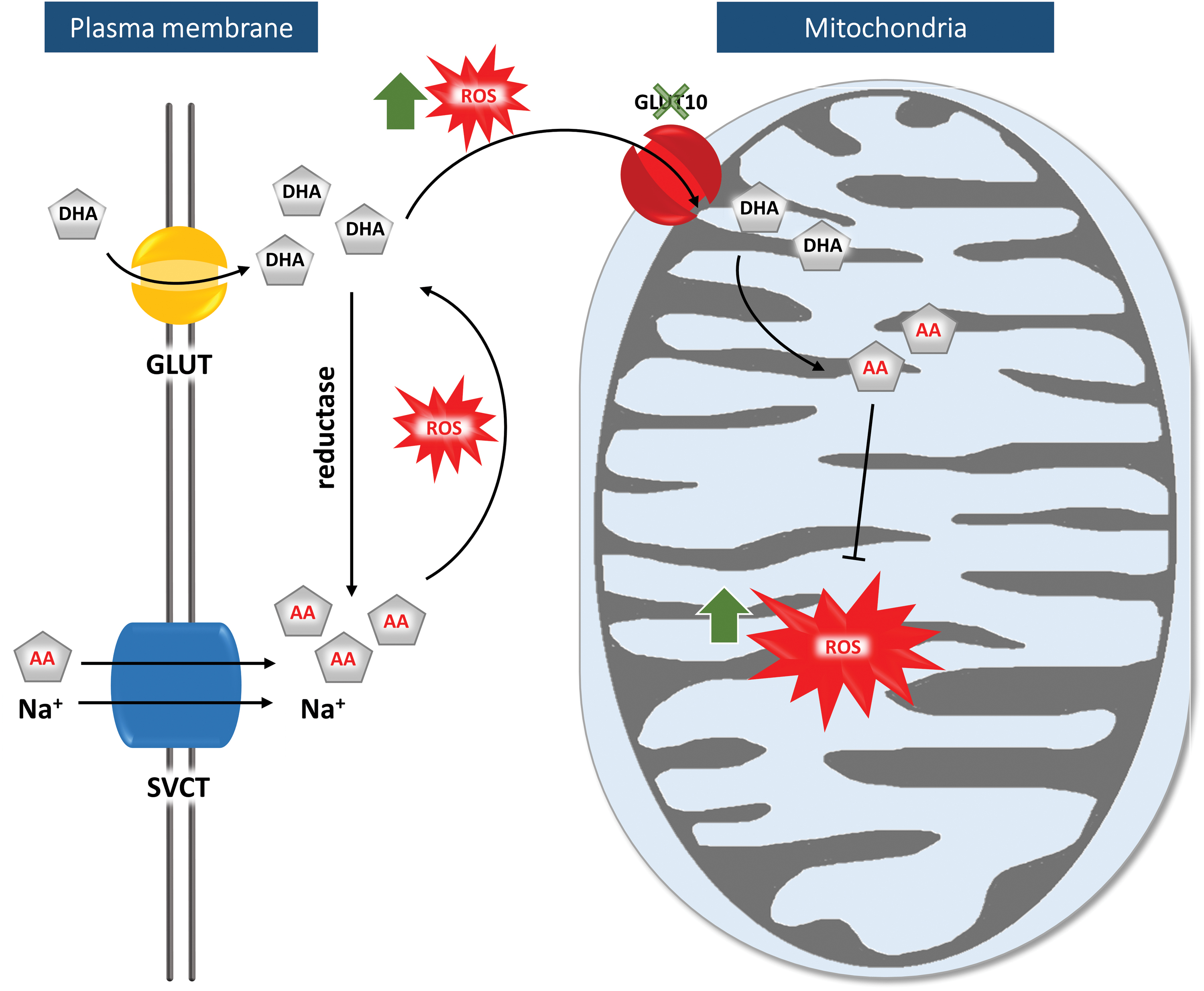
A couple of arguments, however, dispute this pathogenesis model. A recent in silico study identified GLUT10 as one of the transporters with the lowest predicted mitochondrial localization score (87). In addition, the sodium-coupled ascorbic acid transporter-2 (SVCT2) has already been identified as a mitochondrial AA transporter, questioning the necessity of GLUT10 for mitochondrial DHA transport (5, 64).
This hypothesis was nonetheless reinforced by the same laboratory with additional data obtained on rat, mouse, and human SMCs. This study revealed that stress and aging conditions increased intracellular and mitochondrial ROS levels and promoted targeting of GLUT10 to the mitochondria, increasing mitochondrial DHA uptake. In addition, GLUT10 knockdown and overexpression models in mouse MOVAS SMCs, respectively decreased or increased the inner mitochondrial membrane potential and oxygen consumption rate (86). Compared with control SMC, SMCs from GLUT10G128E mice showed higher ROS levels, both intracellularly and in the mitochondria, a reduced oxygen consumption rate, and altered mitochondrial morphology and density. These features were also confirmed in aortic tissue of 15-month-old GLUT10G128E mice. (86).
Comparative transcriptome analysis at 48 h postfertilization between slc2a10 morphants (following morpholino-based slc2a10 knockdown) and control embryos revealed altered expression of genes involved in oxidative phosphorylation, reactive oxygen production, the Szent–Györgyi–Krebs cycle, the glycolysis/gluconeogenesis pathway, and glycogen metabolism. These findings suggested mitochondrial dysfunction as a pathomechanism, which was further validated by impaired oxygen consumption rates in morphants. In contrast to observations made in the ATS mouse model, electron microscopy analysis of slc2a10 morphants did not reveal any changes in mitochondrial morphology (95).
Disturbed post-translational modification in the ER
In silico tools predict GLUT10 localization in the ER with high probability (40) because of the presence of an ER retention signal YXXI/V (48) and a Lys-Arg-Arg (KRR) motif (42) in the C-terminus of the protein. Since ATS patients show a remarkable clinical overlap with heritable disorders of the connective tissue with abnormal collagen and/or elastin homeostasis, Segade hypothesized that GLUT10 operates as a transporter of AA, a necessary component for post-translational modification of collagen and elastin molecules in the ER (82).
AA—being a cofactor of prolyl and lysyl hydroxylases—catalyzes the hydroxylation of proline and lysine residues in elastin and collagen, thereby facilitating cross-link formation. Lowered AA levels in the ER would result in defective and immature collagen and elastin molecules, contributing to histopathology observed in collagen- and elastin-rich tissues in ATS patients (Fig. 6). In this scenario, the observed upregulation of the TGFβ pathway in ATS patients would result from weakening of the ECM and increased activation of latent TGFβ molecules (82).

This hypothesis has been experimentally supported by identification of GLUT10 as a DHA transporter in the ER. Transient transfection of GLUT10-green fluorescent protein constructs in rat SMCs confirmed the presence of GLUT10 in the ER (82). Furthermore, immunocytochemistry-supported localization experiments, either employing ATS patient cells or exogenous tagged GLUT10, showed a reticular distribution and perinuclear abundance of the protein in fibroblast cells (40, 102), and GLUT10 was found to be present in microsomal (ER-derived) fractions obtained from human fibroblasts and liver tissue (40).
Incorporation of in vitro-produced GLUT10 into liposomes resulted in efficient transport of DHA in a concentration-dependent manner. Moreover, selective permeabilization experiments in fibroblasts showed that the transport of DHA through endomembranes was markedly reduced in ATS patient cells when compared with control fibroblasts (66).
Epigenetic modulation through AA deficiency in the nucleoplasm
Recently, nucleoplasmic JmjC domain-containing demethylases (50, 90) and ten-eleven translocation methylcytosine dioxygenases (47, 51), all members of the AA-dependent Fe2+/2-oxoglutarate-dependent dioxygenases, gained attention since they promote demethylation of histones and DNA, respectively. Both mechanisms are involved in epigenetic regulation of transcription (17).
It has recently been shown that by applying high-resolution immunoelectron microscopy and immunogold labeling of AA (99), AA accumulation in the nucleoplasm was diminished in ATS fibroblasts (67). Since the syndrome is due to mutations in GLUT10, it can be supposed that GLUT10 is required for the proper AA concentration in the nucleus.
Because the membranes of the ER and the NE are continuous, DHA uptake by GLUT10 in the nucleoplasm can theoretically follow two routes. DHA can be transferred from the ER across the inner membrane of the NE to the nucleoplasm. Alternatively, DHA could be transported directly through the membranes of the NE (Fig. 6). As a consequence, altered DNA hydroxymethylation patterns at both the global level and at specific gene regions (including PPARG encoding peroxisome proliferator-activated receptor [PPAR]γ) were found in ATS patient fibroblasts (67), which suggests an epigenetic role of AA transport in the ATS pathomechanism.
Discussion
Vitamin C compartmentalization: unifying the different disease mechanisms?
Scurvy, a generalized AA deficiency due to insufficient AA intake, has been known long before the discovery of AA and causes muscular weakness, pain, generalized bleeding, and impaired wound healing. Scurvy at the cellular level can manifest with normal intake and serum concentrations of AA. This latent or tissue scurvy may contribute to complications of insulin-dependent diabetes mellitus, such as endothelial dysfunction and atherosclerosis (73), due to competition between glucose and DHA for GLUTs during hyperglycemia (29, 72).
In a series of elegant experiments, it has been demonstrated that subcellular scurvy may also result from either increased consumption of AA in a cellular compartment or defective transport into a specific organelle. Zito et al. studied the effect of the combined loss-of-function mutations in genes encoding the ER thiol oxidases ERO1α, ERO1β, and peroxiredoxin 4 in a murine model. These enzymes catalyze electron transfers implicated in oxidative protein folding (101). Surprisingly, only minor alterations in disulfide bond formation were found, suggesting the presence of alternative electron transfer routes. However, low tissue AA concentrations and decreased 4-hydroxyproline content of procollagen were observed.
Hence, in the absence of ER thiol oxidases, cysteinyl thiol groups are oxidized to sulfenic acid through an alternative hydrogen peroxide-dependent way, consuming AA as a reductant (100). This competes with prolyl hydroxylases for AA in the ER lumen, resulting in impaired procollagen hydroxylation, and impairs ECM homeostasis, causing scurvy-like disease. Thus, neither AA synthesis (mice are able to produce AA) nor AA transport could keep pace with the increased consumption.
Due to the presence of transporters, competition may arise between AA-requiring reactions of two (or more) compartments as well. For instance, in pluripotent stem cells, increased prolyl-4-hydroxylase activity in the ER hampers 5-methylcytosine demethylation in the nucleoplasm, affecting epigenetic regulation, and vice versa (30). Genetic ablation of prolyl-4-hydroxylase subunit alpha-2 or its pharmacological inhibition reverted epigenetic changes and antagonized cell state transition. Thus, it can be hypothesized that AA consumption in the ER by collagen hydroxylation reduces AA availability for DNA and histone demethylases in the nucleoplasm, preventing mesenchymal transition.
Finally, mislocalization or translocation of AA/DHA transporters can also influence vitamin C distribution between two neighboring compartments. Indeed, Pena et al. demonstrated that normal and cancer cells handle AA differently (69). Human breast cancer tissues expressed SVCT2, but were not able to take up AA. This was explained by the fact that this form of SVCT2 was absent from the plasma membrane and expressed in mitochondria of breast cancer cells (78). Augmented expression of the SVCT2 mitochondrial form and mitochondrial sequestration of AA might profoundly alter AA-dependent mechanisms in other subcellular compartments and might represent an adaptation mechanism to the special metabolic features in cancer cells.
These mechanisms balancing transport and consumption within and between cellular compartments may participate in the pathogenesis of ATS, although still several aspects remain to be addressed.
Pathophysiological mechanisms related to oxidative stress
Different lines of evidence support a role for GLUT10 in DHA transport through the ER-NE continuum or the contribution of oxidative stress to the ATS pathomechanisms. The complementarity and hierarchy of these identified disease mechanisms need further investigation, as well as the exact role of GLUT10 herein. However, molecular alterations linked to ATS pathogenesis were rescued upon reexpression of GLUT10 in ATS patient fibroblasts (under low, nonphysiological AA concentrations), suggesting that GLUT10 might also harbor transport-independent functions (102, 103).
Comparable with the zebrafish morpholino model, transcriptome analysis comparing ATS patient and control cells identified affected redox homeostasis, energy and lipid metabolism, TGFβ signaling, and ECM architecture (102). Of note, the most upregulated gene was ALDH1A1, encoding a member of the aldehyde dehydrogenases (ALDHs). ALDHs act in detoxification induced by sustained presence of lipid peroxidation (LPO)-derived molecules that were found to be increased in ATS fibroblasts. Elevated ROS levels in ATS fibroblasts were suggested to initiate the LPO mechanism by interaction with polyunsaturated fatty acids (PUFAs).
Furthermore, PPARγ activity is boosted by peroxidized PUFAs and an altered cellular energy metabolism. PPARγ is a master regulator in multiple (patho)physiological processes and influences transcription and post-translational modification events. Oxidative stress in ATS might depend on a PPARγ-dependent mechanism through a maladaptive feedback, upregulating β-oxidation, and PUFA metabolism, establishing a vicious circle of oxidative stress induction. ROS directly promote ECM protein fragmentation and induce LPO-derived molecules that could hamper the secretion and organization of collagen and elastin molecules in the ECM, causing ER stress (102).
TGFβ signaling: a delicate balance at the crossroads of oxidative stress, defective ECM production, and epigenetic modification
A key pathway in multiple heritable disorders of connective tissue with cardiovascular implications is TGFβ signaling. Initial immunostaining experiments for connective tissue growth factor and pSMAD2 on arterial tissue of one ATS patient demonstrated an extreme increase in signal intensity in comparison with control tissue (26). Similar analyses in skin and artery tissue in a subsequent study could not confirm this observation (9). These observations likely point to variability in spatiotemporal TGFβ signaling and add to the uncertainty regarding the (primary) role of TGFβ signaling in the disease mechanisms underlying elastic fiber diseases.
TGFβ is secreted in the ECM in its latent form, called the large latent complex (LLC), comprising TGFβ, latency-associated peptide (LAP), and latent TGFβ-binding proteins, which are covalently cross-linked to ECM components. TGFβ can be released from this complex through LAP proteolysis, interaction with αv-containing integrins such as αvβ3 integrin, or ROS-induced mechanisms.
Disturbed TGFβ signaling has been claimed to be a primary actor in ATS pathogenesis (26), or to result secondarily from increased release of latent TGFβ from a damaged ECM (82), due to ineffective hydroxylation of collagen and elastin. Furthermore, epigenetic inhibition of PPARγ will enhance TGFβ signaling, and ROS contribute to the release of active TGFβ from the LLC.
Finally, Zoppi et al. found a key role for αvβ3 integrin, cross talking with noncanonical TGFβ signaling. More specifically, αvβ3 integrin-dependent release of TGFβ from the LLC induces αvβ3 integrin association with TGFβ receptor II and noncanonical TGFβ pathway stimulation through p38 MAPK (102). This is supported by an increased presence of TGFβ receptor II, αvβ3, integrin, and p38 MAPK, but lowered pSMAD2, a biomarker for canonical TGFβ signaling, in mock-transfected ATS patient fibroblasts compared with GLUT10 transfected ATS cells. Although p38 MAPK is a negative regulator of cellular stress, it does not seem to correct ECM organization, as demonstrated by staining of several ECM components in ATS patient fibroblasts.
Future Perspectives
The different hypotheses provide paradigms to understand ATS pathophysiology and developmental biology of the arteries, but none of them are currently supported by sufficient evidence to be regarded as the sole, or even main, mechanism. Nevertheless, the formulated hypotheses are not mutually exclusive.
Indeed, GLUT10 may translocate to different organelles depending on the cell state (25, 40, 53, 82, 102). Moreover, intracellular membranes as well as the plasma membrane are no longer regarded as static structures, but instead are attributed to plasticity, interchanging phospholipids, and membrane-bound (or enclosed) molecules through physical links and/or transport mechanisms. Of note, a complex, ER-mitochondria encounter structure, brings ER and mitochondrial membranes in close proximity and is necessary for cell survival (52). In a related disorder, occipital horn syndrome caused by deficiency of the copper ATPase, encoded by ATP7A, it has been shown that the molecule translocates from the cell membrane to trans- and post-Golgi compartments depending on copper concentration (70).
Finally, as discussed above, it is crucial to understand the balance between AA use, import, and transport between organelles both under physiological and GLUT10-deficient states. Accumulating evidence shows that local subcellular concentrations of vitamin C are equally or even more important than the global cellular or blood levels. Mapping of subcellular AA concentrations, determination of the redox state of the AA/DHA couple in the different cellular compartments, and exploration of AA/DHA transport mechanisms between the cytoplasm and various organelles, both under physiological and diverse pathological situations, are future challenges.
This knowledge will lead to a more detailed understanding of known and yet unknown functions of AA in the different cellular compartments in health and disease. Knowing the importance of AA in vascular development and homeostasis (as evidenced by, respectively, ATS and scurvy), this knowledge will contribute to understanding neovascularization both in desirable (wound healing, neovascularization after vascular injury, or thrombosis) or undesirable (tumor formation) states.
Furthermore, we should keep in mind that in vivo versus in vitro experiments may reveal completely different biological outcomes, as has been observed for the Loeys–Dietz syndrome, caused by mutations in TGFBR2 (31, 61). In addition, observed pathophysiological aberrations in postdevelopmental life may be protective in early development (when the arterial system is being developed) (23). Similarly, the damaged arterial wall may behave differently on blood pressure when still developing or in postnatal life (as hypothesized for intracranial tortuosity in autosomal dominant cutis laxa due to ALDH18A1 defects) (36, 85). Finally, it may prove difficult to distinguish primary versus secondary effects. Indeed, while increased TGFβ signaling seems a common final pathway in many vascular disorders, it remains to be resolved whether it precedes ECM remodeling or whether it is a secondary effect of altered ECM homeostasis.
Animal models could be extremely helpful to test the different hypotheses. Unfortunately, the reported mouse models with missense mutations showed extremely mild phenotypes, not reminiscent of the severe cardiovascular anomalies encountered in ATS patients. Therefore, it is questionable if the mild phenotypic differences observed in older GLUT10G128E mice are relevant to study ATS pathogenesis. Genetic compensation might be at play to counteract the mechanisms induced upon Slc2a10 mutations. Moreover, since mice are able to synthesize AA, it can be anticipated that the abovementioned results should be interpreted with caution. It is possible that mice will present with a more relevant ATS-related phenotype if made incapable of synthesizing AA (82). Alternatively, CRISPR/Cas9 knockout and knockin approaches in zebrafish might be a good alternative, especially since results on the morphants were motivating (95).
Footnotes
Acknowledgments
B.C. is a senior clinical investigator of the Research Foundation–Flanders (FWO). É.M. was supported by the János Bolyai Research Scholarship from the Hungarian Academy of Sciences and by the New National Excellence Program from the Ministry of Human Capacities.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research is supported by FWO funds VS.080.16N, FWOOPR2013025301, and by the Hungarian National Research, Development and Innovation Office (NKFIH grant number: FK124442).