
Abstract
Significance:
Mitochondria undergo constant morphological changes through fusion, fission, and mitophagy. As the key organelle in cells, mitochondria are responsible for numerous essential cellular functions such as metabolism, regulation of calcium (Ca2+), generation of reactive oxygen species, and initiation of apoptosis. Unsurprisingly, mitochondrial dysfunctions underlie many pathologies including cancer.
Recent Advances:
Currently, the gold standard for cancer treatment is chemotherapy, radiation, and surgery. However, the efficacy of these treatments varies across different cancer cells. It has been suggested that mitochondria may be at the center of these diverse responses. In the past decade, significant advances have been made in understanding distinct types of mitochondrial dysfunctions in cancer. Through investigations of underlying mechanisms, more effective treatment options are developed.
Critical Issues:
We summarize various mitochondria dysfunctions in cancer progression that have led to the development of therapeutic options. Current mitochondrial-targeted therapies and challenges are discussed.
Future Directions:
To address the “root” of cancer, utilization of mitochondrial-targeted therapy to target cancer stem cells may be valuable. Investigation of other areas such as mitochondrial trafficking may offer new insights into cancer therapy. Moreover, common antibiotics could be explored as mitocans, and synthetic lethality screens can be utilized to overcome the plasticity of cancer cells.
Introduction
Mitochondria, commonly known as the “powerhouse of the cell,” are responsible for many essential cellular functions. The mitochondrial network undergoes dynamic changes such as biogenesis, fusion, fission, and mitophagy. Key pathways that are regulated by mitochondria include bioenergetics, mitochondrial reactive oxygen species (mROS) generation, mitochondrial calcium (Ca2+) regulation, and apoptosis. These multifaceted functions of mitochondria make them important for sensing cellular stress and adaption to the environment (Fig. 1).
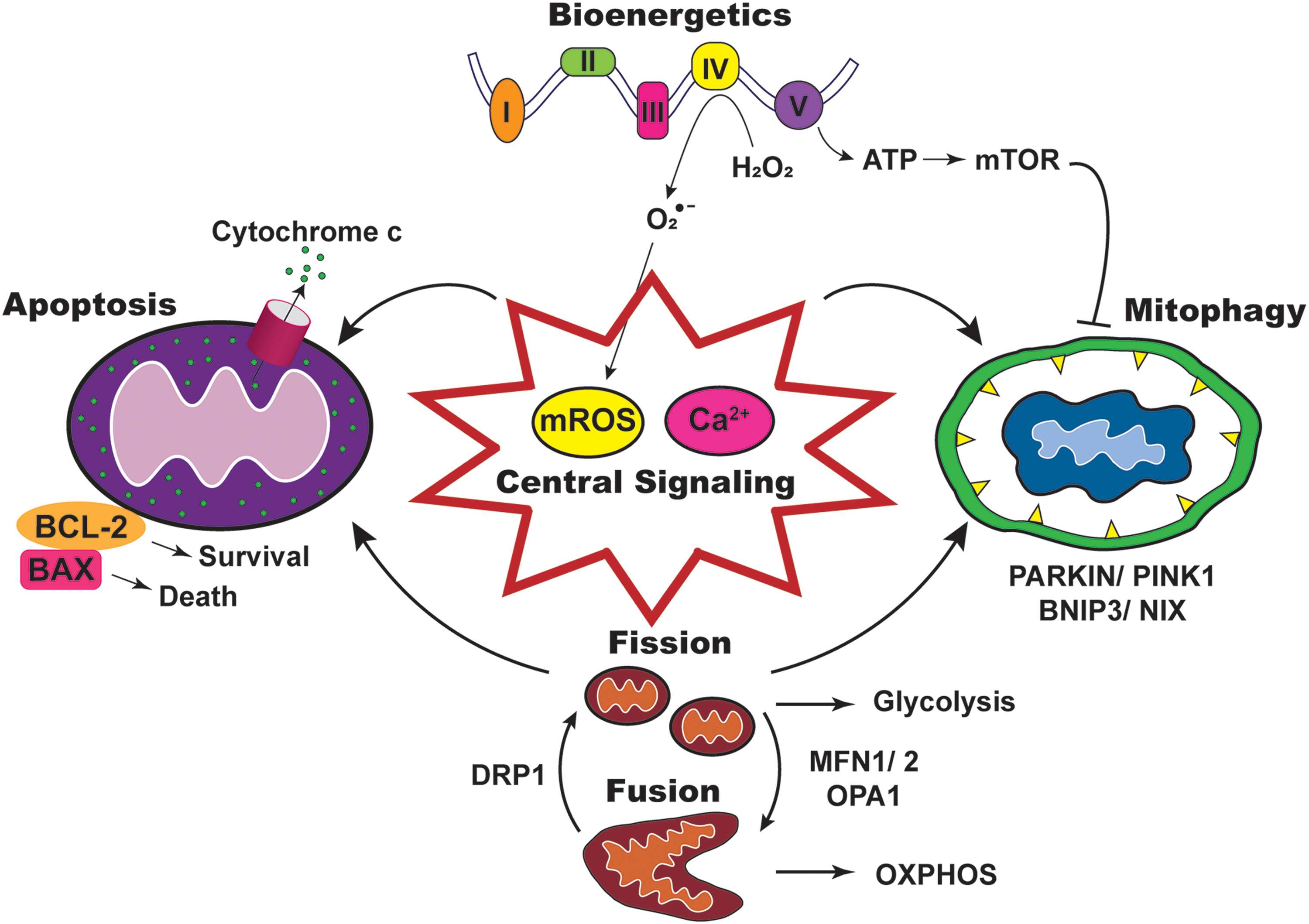
Mitochondrial reprogramming or mutations that disrupt function are associated with many pathologies, including cancer, which contributes to approximately one in six deaths worldwide (108). Although mitochondrial dysfunction may not be the driver of cancer, dysregulated mitochondrial mass due to an imbalance in biogenesis and mitophagy is central to oncogenic signaling pathways. For instance, many genes involved in mitochondrial biogenesis are downstream targets of c-MYC, a potent oncogene resulting in upregulation of mitochondrial metabolism to promote tumor progression (162). Imbalanced fission and fusion dynamics have also been reported in various cancers, which are associated with energy production and apoptosis (142).
A hallmark of cancer cells is the reprogramming of metabolism from oxidative phosphorylation (OXPHOS) to oxidative glycolysis, also known as the Warburg effect. More importantly, the flexibility in adjusting metabolism enables cancer cells to meet the increased cellular energy requirements for tumor progression. This energy mainly comes from the production of adenosine triphosphate (ATP) through mitochondrial respiration. Mitochondrial respiration involves the Krebs cycle and electron transport chain (ETC) to convert glucose into usable ATP. Mitochondrial function in promoting proliferation, migration, invasion, and evasion from apoptosis requires large amounts of energy from ATP, and mitochondrial Ca2+ is an important factor in this process. To promote proliferation and migration, cancer cells increase the uptake of nonessential amino acid glutamine to feed the Krebs cycle and increase ATP production. This increase in ATP is only possible with the presence of three enzymes, namely alpha-ketoglutarate dehydrogenase (α-KGDH), isocitrate dehydrogenase, and pyruvate dehydrogenase. Since Ca2+ is a cofactor for these enzymes, the increase in mitochondrial Ca2+ upregulates their enzymatic activity and thereby ATP production.
Disrupted reactive oxygen species (ROS) homeostasis is another hallmark of cancer. ROS is a common by-product of metabolic processes, and is produced in many cellular organelles such as endoplasmic reticulum (ER), peroxisomes, and mitochondria (162). However, the main bulk of ROS is produced in the mitochondria, also known as mROS. mROS is the by-product of mitochondrial respiration as a result of electron leak during OXPHOS. Increased mROS aids tumor progression by promoting genomic instability and metastatic potential. Dysregulated mitochondrial Ca2+ is also commonly observed in cancers and promotes oncogenic phenotypes. For instance, lower basal mitochondrial Ca2+ reduces the possibility of mitochondrial permeability transition pore (mPTP) opening and activates apoptosis.
In view of the pivotal role of mitochondria in tumor progression, this review summarizes major mitochondrial dysfunctions, including altered dynamics, bioenergetics, signaling, and apoptosis in various cancers (Fig. 1). Therapeutic approaches based on the various dysfunctions are discussed. In addition, based on recent findings, new avenues for mitochondrial-targeted cancer therapies are examined.
Mitochondrial Dynamics in Cancer
Fission and fusion
Continuous cycles of fission and fusion that maintain the dynamic morphology of mitochondria under physiological conditions are disrupted in cancer, often with a shift toward fission, to promote tumorigenesis and metastasis (100, 148). Mitochondrial fission and fusion are mediated by guanosine triphosphatases (GTPases) present on the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM) (Table 1). When a cell is dividing or undergoing severe stress (i.e., oxidative or hypoxic stress), mitochondrial fission is induced by dynamin-related protein 1 (DRP1), which is activated post-translationally by Serine 616 (S616) phosphorylation (14, 150) (Fig. 2). In contrast, mitochondrial fusion is elevated to create an extended mitochondria network for increased energy production via OXPHOS. Mitofusin 1 (MFN1) and 2 (MFN2) mediate OMM fusion, while optic atrophy 1 modulates IMM fusion (Fig. 2). These molecules are predominantly regulated transcriptionally (84).

Summary of Proteins Involved in Mitochondria Dynamics and Their Cellular Functions
BCL-2, B-cell lymphoma 2; BH3, BCL-2 homology domain 3; LC3, microtubule-associated protein 1A/1B-light chain 3; LIR, LC3 interacting regions.
In multiple cancers, mitochondria are highly fragmented, accompanied by increased activation of DRP1 via S616 phosphorylation (61, 130, 163, 187). This may result from calcium signaling, as increased cytosolic Ca2+ upregulates DRP1 expression (58). Mitochondrial fission in turn elevates cytosolic Ca2+, forming a positive feedback loop between Ca2+ and fission (58). Mitochondrial fragmentation may also be induced by oxidative stress, as high mROS can inhibit fusion and promote fission in cancer cells (70, 173).
Mitogen-activated protein kinase signaling also promotes mitochondrial fission. Specifically, in oncogenic KRAS-driven pancreatic cancer cells, ERK2 phosphorylates DRP1 on S616 and promotes DRP1 activity (69). Inhibition of DRP1 reduces the ability of cells to generate tumors, indicating the importance of mitochondrial fission in KRAS-driven malignant transformation and proliferation. In contrast to fission, increased mitochondrial fusion appears to suppress growth in several cancer cell lines, thus assuming an antitumorigenic role (130).
In addition to transformation, increased fission may support metastasis. In glioblastoma, DRP1 inhibition significantly reduced invasiveness (163). Similarly, in metastatic breast cancer cells, DRP1 inhibition reduced invasion without affecting cell viability (187). On the contrary, mitochondrial fusion inhibits invasive properties of breast and lung cancer cells (177).
A recent study proposed that mitochondrial fission is crucial for maintenance of cancer stem cells (CSCs) (176). Fragmented mitochondria and hyperactivated DRP1 were observed in patient-derived brain tumor initiating cells (BTICs). As expected, inhibition of DRP1 hindered growth and self-renewal of BTIC. More importantly, the overexpression of hyperactive DRP1 in non-BTIC induced the expression of core stem cell (SC) regulators and repressed differentiation markers, suggesting the importance of DRP1-mediated fission in BTICs. Interestingly, increased mitochondria fission is also discovered in prostate CSCs (193), raising the tantalizing possibility that elevated fission could be a common feature across all CSCs (16).
Despite strong connections between fission and tumorigenesis, mitochondria fusion can also be upregulated in certain cancers. Specifically, the MYC oncogene preferentially activates fusion to promote OXPHOS and drive cell growth (37, 49). Since MYC is known to be highly expressed in multiple cancers, including triple-negative breast cancer, mitochondrial fusion may assume a protumorigenic role in the context of these tumors. These findings suggest that the restoration of fusion–fission balance in cancer cells could be a potential therapeutic strategy.
Mitophagy
Increased fission promotes mitophagy by generating depolarized and hyperpolarized mitochondria (155). Such asymmetric fission enables the removal of depolarized mitochondria via mitophagy, a form of autophagy that selectively degrades defective mitochondria to maintain cellular fitness (11). Due to its cytoprotective role, mitophagy is dysregulated in different cancer stages (35, 76). On the one hand, mitophagy is downregulated to promote malignant transformation during cancer initiation. On the other hand, mitophagy is upregulated to promote survival under stress during cancer progression (Fig. 3). Thus, depending on the cancer type and stage, mitophagy fulfills different roles.

Mitophagy is induced by stress signals such as mitochondrial depolarization and hypoxia. Depending on the signals, mitophagy can be activated by several pathways (Table 1). The phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1) is activated by mitochondrial depolarization to induce mitophagy. At the OMM, PINK1 phosphorylates and activates E3 ubiquitin ligase PARKIN, which proceeds to ubiquitinylate other OMM proteins. The ubiquitin chains on these proteins are further phosphorylated by PINK1, resulting in an amplification of signals to undergo mitophagy (Fig. 3).
Mitophagy can also be activated by hypoxia through the B-cell lymphoma 2 (BCL-2) interacting protein 3 (BNIP3)/NIX pathway. Both BNIP3 and NIX have conserved microtubule-associated protein 1A/1B-light-chain 3 interacting regions capable of recruiting autophagosomes for mitophagy (Fig. 3). Under hypoxic conditions, the hypoxia-inducible factor 1 transcriptionally upregulates BNIP3 and NIX, leading to increased mitophagy.
These key pathways are commonly dysregulated in tumors. Specifically, downregulation of PARKIN and PINK1, either via mutation or deletion, appears to promote tumorigenesis. For instance, loss of PARKIN has been identified in multiple human cancers, including glioblastoma (157). Loss of PARKIN promotes genomic instability, increases cancer cell proliferation and resistance to apoptosis (44, 57, 83). PARKIN may also be linked to cancer metastasis, as its downregulation promotes cell migration and invasion in breast cancer cells (85). Similarly, loss of PINK1 occurs in multiple human cancers, including glioblastoma and neuroblastoma (1, 124). It is plausible that downregulation of PINK1/PARKIN-mediated mitophagy decreases OXPHOS, increases mROS production and glycolysis, thereby contributing to the Warburg effect and consequently tumorigenesis (184).
In contrast to PINK1/PARKIN, BNIP3 may serve as a tumor suppressor or oncogene. Loss of BNIP3 is associated with tumor progression and metastasis in triple-negative breast cancer (21), and contributes to chemoresistance in pancreatic cancer (2). However, its expression is upregulated in another subset of cancers (18, 147). In nonsmall-cell lung cancer and uveal melanoma, high BNIP3 expression correlates with poor patient prognosis (48, 64). Further, BNIP3 upregulation has been reported to promote cell migration and invasion (91, 171). These contradicting findings on BNIP3 may be attributed to the preferential expression of BNIP3 splice variants, as full length BNIP3 promotes cell death while spliced BNIP3 favors survival.
Mitophagy also plays a part in chemoresistance (19). In colorectal CSCs, doxorubicin treatment induced NIX expression and promoted chemoresistance (178). NIX inhibition significantly reduced mitophagy and resensitized cells to doxorubicin. Similarly, ariadne RBR E3 ubiquitin protein ligase 1 (ARIH1)/PINK1-mediated mitophagy was upregulated in cisplatin-treated lung cancer cells and ARIH1 knockdown resensitized the cells to cisplatin-induced death (161). These findings collectively suggest that mitophagy inhibition may be a potential adjuvant therapy to overcome chemoresistance and improve therapeutic outcomes in cancer.
Interestingly, some cancer cells exploit mitophagy to fulfill their bioenergetics needs (39). Several studies observed that OXPHOS-dependent cancer cells release ROS to mimic hypoxia and induce mitophagy in cancer-associated fibroblasts. Due to mitophagy, these fibroblasts switch to glycolysis to produce lactate and pyruvate, which are then taken up by cancer cells for OXPHOS (96, 97). Thus, cotargeting mitophagy and OXPHOS may be a more effective path in cancer therapy.
Mitochondrial Bioenergetics in Cancer
As the cellular powerhouse, mitochondria's primary task is to generate sufficient ATP to fulfill the cell's bioenergetic needs. Mitochondria generate ATP via OXPHOS, a process that couples proton pumping with ATP synthesis. Due to the Warburg effect, it was initially assumed that OXPHOS is universally defective in cancer cells. Subsequent studies revealed that tumors preferentially switch between OXPHOS and glycolysis under different microenvironmental conditions. Specifically, metabolic rewiring to OXPHOS provides cancer cells with greater independence from external nutrient supply conferring selective advantage under nutrient deprivation. Further, cancer cells can generate more ATP via OXPHOS to drive proliferation and metastasis, making OXPHOS inhibition an attractive strategy in cancer therapy (6, 167).
Mitochondrial OXPHOS generates ATP by transferring electrons through a series of transmembrane protein complexes in the IMM, known as the ETC. When OXPHOS is active, nicotinamide adenine dinucleotide and flavin adenine dinucleotide donate electrons to complexes I and II, respectively. The transfer of electrons down the ETC is accompanied by pumping of protons into the mitochondrial intermembrane space, generating a proton gradient across the IMM. These protons flow back to the mitochondrial matrix via complex V (also known as ATP synthase), which harness the proton motive force to synthesize ATP. At the end of the ETC, oxygen acts as the final electron acceptor, thereby producing mROS as by-products of OXPHOS (Fig. 4).
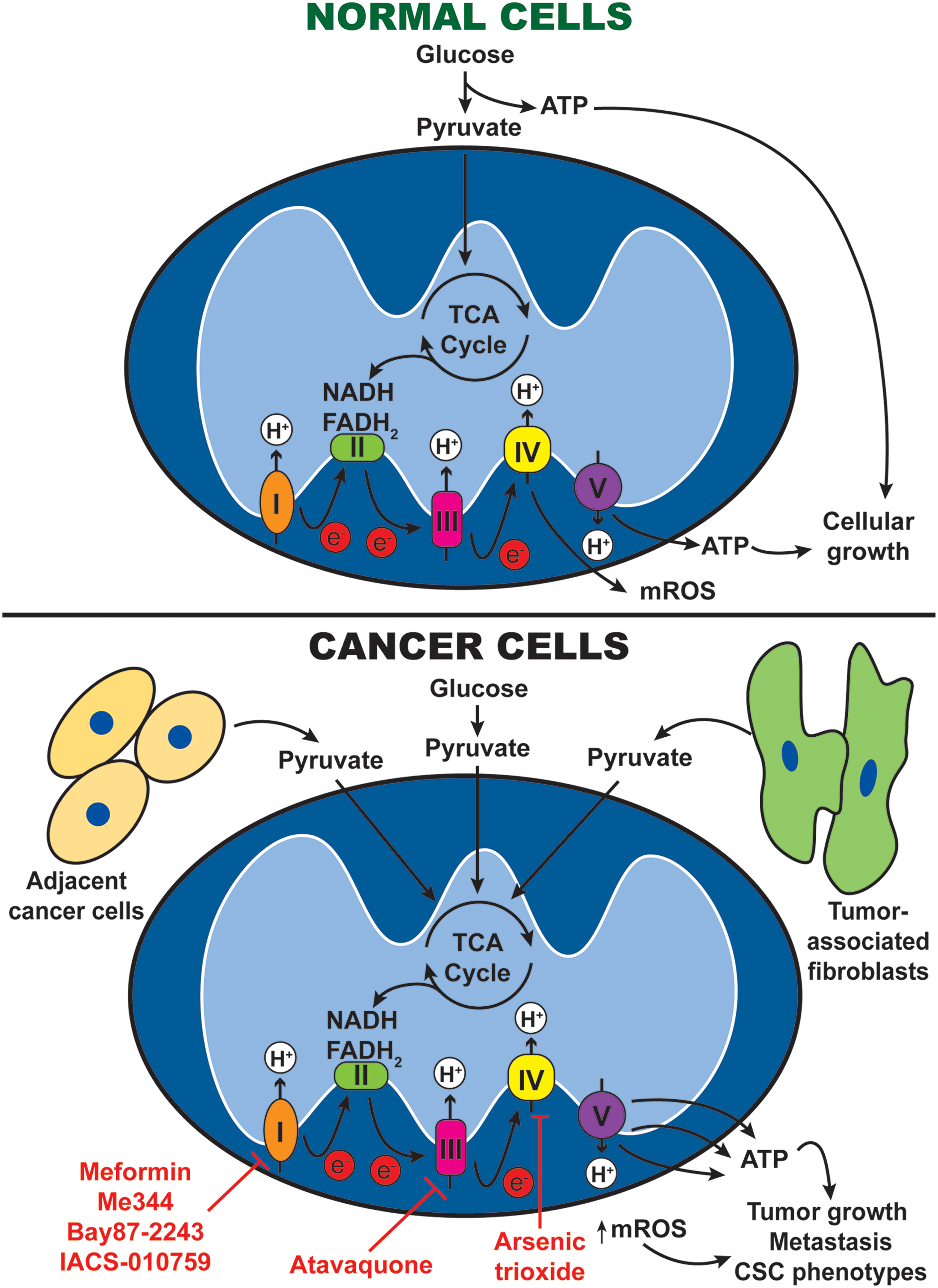
OXPHOS upregulation has been shown to be important in several cancer types. In human breast cancer, upregulation of ETC proteins and increased OXPHOS activities were observed (169), while OXPHOS inhibition by tigecycline significantly impaired growth of breast cancer xenografts (65). Similarly, in Hodgkin's lymphoma, OXPHOS upregulation is evident from an increase in ETC protein expression and decrease in lactate production, while OXPHOS inhibition led to apoptosis and reduced cell growth (10).
Some CSCs also preferentially upregulate OXPHOS, including the sphere-forming CD133-positive glioblastoma and pancreatic ductal adenocarcinoma (PDAC) SCs (63, 135, 136). Such a preference could be mediated by peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), which is crucial for both OXPHOS and CSC phenotypes (136). RAS oncogene could also underlie OXPHOS dependence, as PDAC cells harboring oncogenic KRAS are highly reliant on OXPHOS and displayed CSC properties (160). Consequently, OXPHOS inhibition resulted in tumor regression and improved survival in the KRAS mutant mice, highlighting the potential application of OXPHOS inhibitors in targeting KRAS-driven pancreatic cancers (160).
OXPHOS is also upregulated as a result of chemotherapy (104). Upregulated OXPHOS may support chemoresistance by producing sufficient ATP for the removal of drugs via multidrug transporters, since increased intracellular ATP levels directly correlate with chemoresistance in colon cancer (191). Oxaliplatin and fluorouracil (5-FU) treatment induced colon cancer cells to upregulate OXPHOS via sirtuin 1 (SIRT1) and PGC-1α (158). Moreover, reduction of OXPHOS via SIRT1/PGC-1α knockdown resensitized cells, suggesting that OXPHOS inhibition may well be another attractive choice as adjuvant therapy.
Despite the supportive roles, certain cancers downregulate OXPHOS and switch to glycolysis. Metagenomic analysis revealed deleterious, tumor-specific mitochondrial DNA mutations across multiple cancer types (81). These mutations affected genes encoding for mitochondria ETC proteins and potentially lead to reduced OXPHOS. Importantly, OXPHOS downregulation is associated with poor clinical outcomes and invasive phenotypes, suggesting that its reduction may be linked to cancer metastasis (46).
More recently, it was proposed that metabolically distinct cancer cells may form symbiotic relationship with one another in the tumor microenvironment to support growth (Fig. 4). Specifically, glycolysis-dependent cancer cells produce lactate and pyruvate, which serve as OXPHOS metabolites in OXPHOS-dependent cancer cells (109). Similarly, cancer-associated fibroblasts may preferentially perform glycolysis to produce lactate to fuel OXPHOS in neighboring cancer cells (40, 95). Such observations support the combined inhibition of OXPHOS and glycolysis to effectively eradicate cancer cells (9), although the resulting toxicity to noncancerous cells must be considered.
Mitochondrial Signaling in Cancer
Reactive oxygen species
Cellular ROS is a common by-product of oxidative metabolism, and mitochondria contributes to ∼80% of all cellular ROS produced (77). mROS are generated largely through ETC and participate in various signaling pathways affecting cellular processes (38). Common ROS includes superoxide (O2 •−), hydrogen peroxide (H2O2), hydroxy radical, and reactive nitrogen species. O2 •− are produced during oxidative respiration due to partial reduction of oxygen, which is then transformed into H2O2 by matrix antioxidant defense enzymes such as superoxide dismutase. mROS generation is mainly affected by OXPHOS, and mitochondrial Ca2+ plays an important role in influencing mROS generation both directly and indirectly. First, mitochondrial Ca2+ fuels the Krebs cycle and production of ATP and in this process, mROS are generated as by-products of ETC. Second, mROS can be generated directly through enzymes such as glycerol phosphate and α-KGDH upon activation by mitochondrial Ca2+. Third, mitochondrial Ca2+ can activate nitric oxide synthase and block complex IV through formation of nitrogen oxide. This leads to excessive mROS generation. Finally, prolonged mitochondrial Ca2+ overload in the matrix triggers the opening of mPTP, which leads to permanent mitochondrial membrane depolarization and apoptosis. In this process, crista of the IMM unfolds and reverse electron transport is activated, which causes significant amount of mROS to be generated (38).
Redox has been proven to modulate a wide range of receptors and signaling pathways, which may have direct or indirect effect on mitochondrial Ca2+ homeostasis. For instance, mROS promote mitochondrial calcium uniporter (MCU) oxidation and cause a conformational change in the MCU complex, which results in increased mitochondrial Ca2+ uptake (33). The rise in mitochondrial Ca2+ in turn generates even more mROS and sensitizes cells toward cell death, illustrating the link between mROS and mitochondrial Ca2+. When mitochondrial Ca2+ homeostasis is maintained, a balance between mROS production and elimination is achieved to ensure proper cellular functions such as autophagy, adaptation to hypoxic environment, and mitochondria bioenergetics (38). However, when this balance is disrupted and there is excess mROS generation, cell death and cellular damage occur. Prolonged disruption of mROS homeostasis leads to pathologies like cancer (119).
Generally, cancer cells exhibit elevated mROS levels, and this can have both pro- or antitumor effects depending on the range of elevation as well as downstream pathways. High mROS is often associated with increased apoptotic stimuli through opening of mPTP, which has an anticancer effect. However, elevated mROS can also favor tumorigenesis by promoting genome instability, DNA mutations, and DNA damage. Some of the mROS may also travel to nucleus and stimulate epigenetic changes such as hypermethylation and silencing of tumor suppressor genes (119). Moreover, elevated mROS may be accompanied by upregulation of antioxidant system such as nuclear factor erythroid 2-related factor 2 and peroxiredoxin. Hence, mROS may only be elevated slightly to promote cancer progression signaling pathways. For instance, mROS has been shown to inhibit redox-sensitive PTEN and activate oncoproteins such as protein kinase B (AKT) (119). Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and ERK are also activated by mROS to promote tumor growth.
The crosstalk between mitochondrial Ca2+ and mROS is evident in cancer. MCU overexpression, which results in elevated mitochondrial Ca2+, is associated with poor prognosis in breast cancer (152). In triple-negative breast cancer, MCU silencing showed a significant reduction in mitochondrial Ca2+ concentration and mROS production. Correspondingly, reduction in tumor growth, migration, and invasive capacity was observed. Since, crosstalk between AKT-phosphoinositide 3-kinases and mROS has been well established (75), phosphorylation of mitochondrial calcium uptake 1 (MICU1) by AKT at the N-terminus was found to increase mitochondrial Ca2+ uptake, mROS production, and promote cancer progression (94).
Mitochondrial calcium
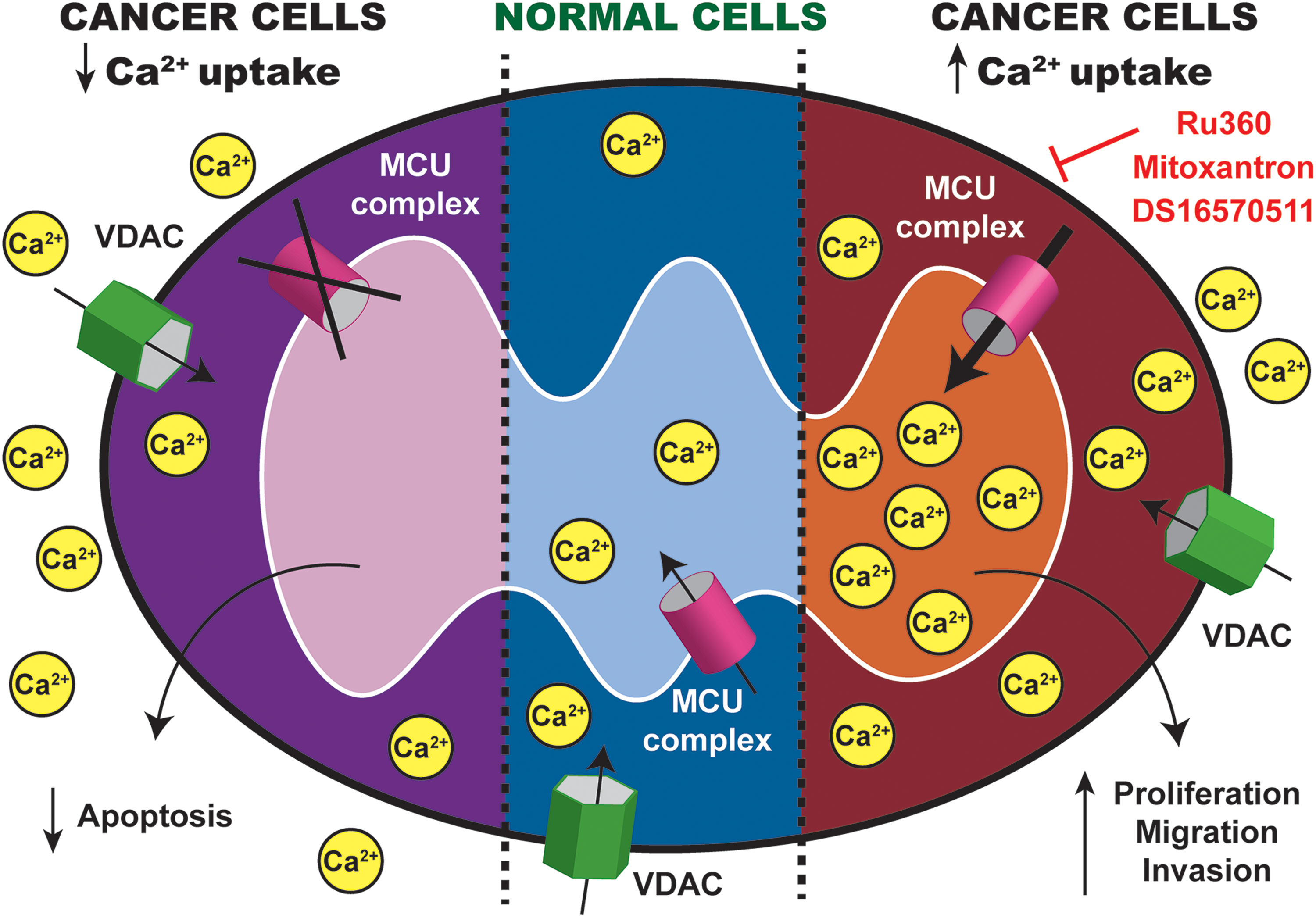
Ca2+ ions are important secondary messengers that extensively regulate mitochondrial functions in mROS generation, cell death, metabolism, and energy homeostasis. MCU complex, the main mitochondrial Ca2+ influx channel, plays a fundamental role in regulating global calcium signaling, aerobic metabolism, and inducing apoptosis. The main channel protein, MCU, is essential for efficient mitochondrial calcium uptake, and its loss inhibits mitochondrial calcium uptake by ∼70%. MCU regulators include MICU1, mitochondrial calcium uptake 2, and mitochondrial calcium uniporter regulator 1. MICU1 is the gatekeeper and prevents mitochondrial calcium overload under basal cytosolic calcium condition. MICU1 acts to determine the threshold of MCU opening and cooperates to activate the channel under high calcium concentration. MICU1 is therefore important in determining the sigmoidal response of mitochondrial calcium uptake in response to calcium transients in the cytosol.
Since MCU and MICU1 regulate mitochondrial calcium uptake, their deregulation has been reported in several cancers. Overexpression of MCU was reported in breast cancer, and its expression correlated with tumor size and poor prognosis (152). Conversely, loss of MCU expression reduced invasiveness, metastatic potential, and tumor growth. Likewise, deregulation of MICU1 has also been shown in ovarian cancer. Thus, some cancers upregulate MCU complex to increase mitochondrial Ca2+ and promote tumor progression, whereas other cancers downregulate it to decrease mitochondrial Ca2+ and evade apoptosis (Fig. 5).
While it was thought that the mitochondria can accumulate high amount of Ca2+, this idea seems counterintuitive due to the low affinity of MCU complex for Ca2+ uptake. Under physiological condition, cytosolic Ca2+ has a resting concentration of ∼100 nM, which is too low to allow for Ca2+ uptake into mitochondria (129). The discovery of contact sites between ER and mitochondria provides an explanation. High-resolution electron microscopy and fluorescence microscopy identified the contact points to be ∼10–30 nm wide (174) and relatively stable. These contact sites named mitochondrial-associated ER membranes (MAMs) are critical for Ca2+ uptake into mitochondria since Ca2+ concentration at these regions can be as high as 10 μM (129). The distance between ER and mitochondria, together with the size of MAMs, has been shown to greatly influence Ca2+ transfer into mitochondria. MAMs are enriched in calcium transport channels and proteins encoded by both oncogenes and tumor suppressors that regulate various cellular signaling pathways. Several proteins such as MFN2 (129), thioredoxin-related transmembrane protein 1, and calnexin are associated with MAMs (32); however, their exact functions are still widely debated and await proteomic analysis for characterization of the dynamic environment at MAMs.
Uptake of Ca2+ into mitochondria mainly originates from ER, which is the main intracellular Ca2+ storage organelle. The transfer of Ca2+ at MAMs mainly depends on two proteins: the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) and inositol 1,4,5-triphosphate receptors (IP3R) (32). SERCA is responsible for pumping Ca2+ from the cytosol into the ER to create a Ca2+ gradient between the cytosol and ER. IP3R is the main intracellular Ca2+ release channel and interacts physically with voltage-dependent anion channel 1 (VDAC1) at MAMs to facilitate Ca2+ uptake into mitochondria. Ca2+ released from IP3R crosses the OMM through VDAC and accumulates in the IMM forming microdomain of high Ca2+ concentration. This in turn allows for efficient Ca2+ uptake from IMM into mitochondrial matrix through MCU complex (32, 105).
Since ER–mitochondria transfer accounts largely for mitochondrial Ca2+ uptake and its alterations play a crucial role in cancer progression, cancer cells often exploit this system to their advantage. Several cancers alter the activity of IP3R through phosphorylation by AKT, which decreases Ca2+ transfer from ER into the mitochondria and protects cancer cells from apoptosis. Other cancers downregulate tumor suppressor such as breast cancer type 1 susceptibility protein and promyelocytic leukemia protein, which stabilize IP3R and promote Ca2+ transfer, thereby conferring resistance to apoptotic stimuli. Besides apoptosis, the regulation of mitochondrial Ca2+ by MAMs affects mitochondrial ROS production, autophagy, and mitochondrial dynamics. Consequently, many therapeutic options have since been devised to target ER–mitochondria Ca2+ transfer and downstream mitochondrial functions to improve treatment outcomes.
Mitochondria-Mediated Apoptosis in Cancer
Under physiological conditions, apoptosis is tightly regulated by mitochondria to maintain tissue homeostasis. The ability to inhibit apoptosis and resist cell death is one of the well-established hallmarks of cancer (52).
Apoptosis is activated by cell death inducers such as Ca2+ and mROS (33, 122). Upon activation, the antiapoptotic BCL-2 family proteins such as BCL-2 and B-cell lymphoma-extra large (BCL-XL) are displaced, permitting the proapoptotic BCL-2-associated X protein (BAX) and BCL-2 antagonist/killer 1 (BAK) to translocate to the OMM and mediate mitochondrial outer membrane permeabilization (MOMP). Specifically, BAX and BAK form oligomers in the OMM, thereby promoting pore formation and the release of proapoptotic agents from the intermembrane space, including cytochrome c, apoptosis-inducing factor (AIF), second mitochondrial-derived activator of caspases (SMAC)/Diablo homolog (DIABLO), and endonuclease G (66) (Fig. 6). These agents initiate apoptosis via several signaling pathways: (i) cytochrome c and apoptosis protease activating factor 1 form the apoptosome to activate the effector caspases (98); (ii) Smac/DIABLO antagonize the inhibitors of caspases and other apoptosis proteins (159); and (iii) AIF and endonuclease G promote chromatin condensation and DNA degradation (117).
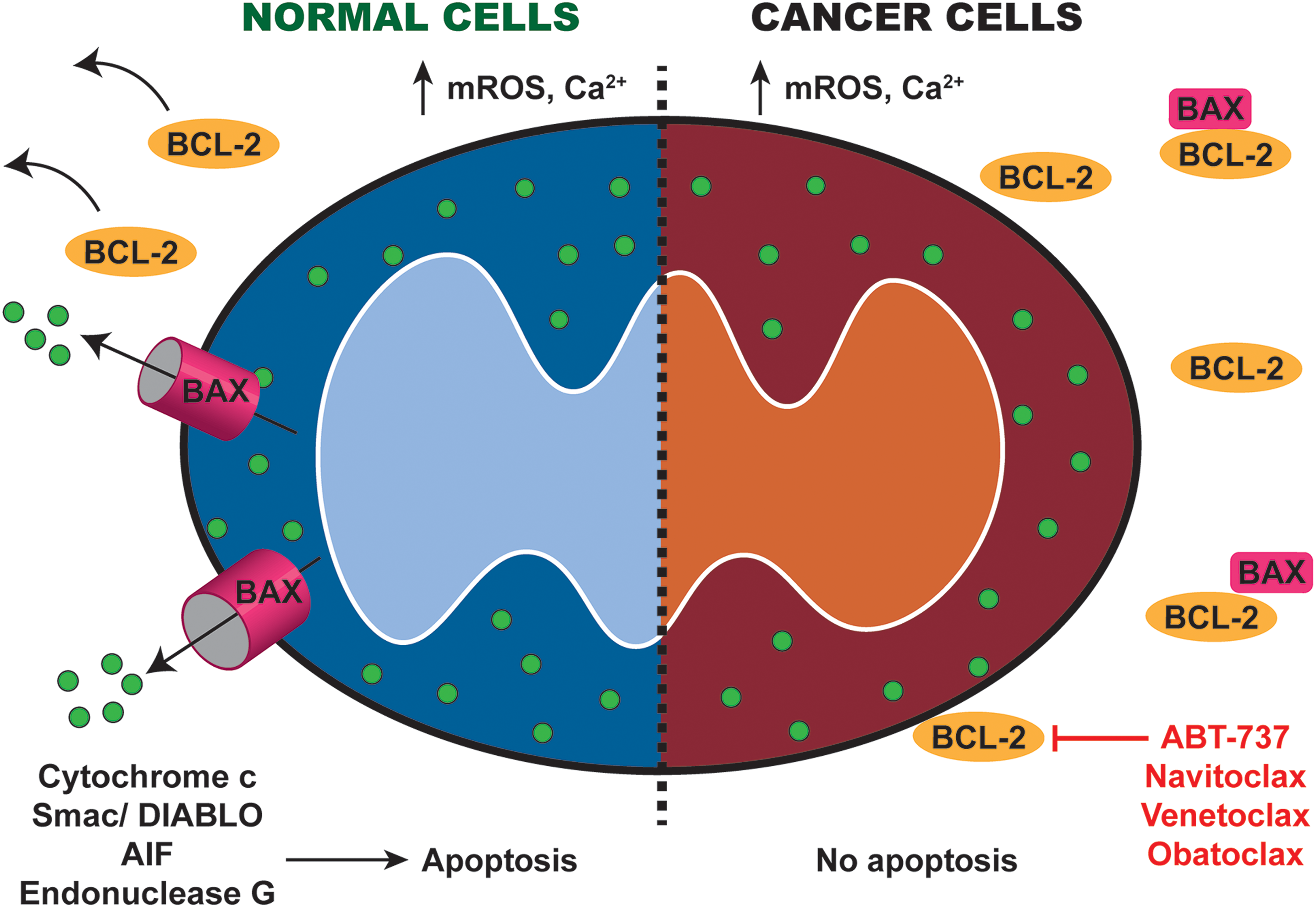
The balance between pro- and antiapoptotic BCL-2 family proteins is critical for activation of apoptosis. Expression of the antiapoptotic gene BCL-2 is commonly upregulated in many cancer types as a result of gene amplification, transcriptional upregulation, or downregulation of BCL-2 targeting microRNAs. Apart from antagonizing the death-inducing actions of BAX, BCL-2 interacts with IP3Rs at MAMs to inhibit ER Ca2+ release, thereby preventing mitochondrial Ca2+ overload and apoptosis in cancer cells (134). Thus, increased BCL-2 expression significantly enhances tumor onset, especially when combined with growth promoting oncogenes such as MYC. Upregulated BCL-2 and BCL-XL expressions also confer therapeutic resistance in multiple cancer models. Similarly, downregulation or deletion of proapoptotic BAX and BAK has also been identified in various cancers, and may be crucial for oncogenic transformation and therapy resistance.
A balance between fusion and fission also dictates apoptotic susceptibility (162). Phosphorylated MFN1 inhibits mitochondria fusion and interacts with BAK to promote its oligomerization for MOMP (126). On the contrary, loss of MFN1 results in resistance to apoptosis as mitochondrial hyperfragmentation prevents BAX from interacting at the OMM (131). Restoration of mitochondria dynamics via DRP1 inhibition resensitized cells to apoptosis, indicating that balanced mitochondrial fission–fusion dynamics is crucial for mitochondrial shape to support BAX/BAK-mediated apoptosis.
Mitochondrial-Targeted Therapeutic Strategies
Distinct aspects of mitochondrial dysfunctions have roles in different cancer stages. Mitophagy and mROS play essential roles in inducing genomic instability and malignant transformation during cancer initiation. Downregulation of mitophagy promotes accumulation of defective mitochondria and production of mROS. As the by-product of OXPHOS, mROS epigenetically alters expression of multiple oncogenes and tumor suppressors (57). During cancer progression, uncontrolled proliferation of cancer cells is largely mediated by elevated fusion and metabolic rewiring to OXPHOS. Increased mitochondria fusion promotes ATP generation via OXPHOS, allowing the cancer cells to sustain continuous cell divisions without having energy constraints. On the contrary, cellular survival is strongly promoted by increased mitophagy and evasion of mitochondria-mediated apoptosis. Elevated mitophagy promotes clearance of dysfunctional mitochondria and protects cancer cells from excessive accumulation of mROS. Imbalance in the pro- and antiapoptotic BCL-2 family proteins further protects the cell from death-inducing stimuli. Such potent prosurvival signals inevitably lay the foundation for therapeutic resistance and subsequent tumor relapse. Increased mitochondrial OXPHOS and fission stimulate metastasis to secondary sites.
Since mitochondrial dysregulation plays a role in cancer, we evaluate current strategies aimed at selectively reversing distinct types of dysfunctions (Table 2).
Summary of Mitochondrial-Targeted Therapeutic Compounds and Their Current Status
ATP, adenosine triphosphate; BCL-XL, B-cell lymphoma-extra large; BIRD-2, BCL-2-IP3R disruptor 2; Ca2+, calcium; DHE, dihydroergotamine tartrate; DRP1, dynamin-related protein 1; ER, endoplasmic reticulum; FDA, Food and Drug Administration; GPX, glutathione peroxidase; GSH, glutathione; HER2, human epidermal growth factor receptor 2; MCU, mitochondrial calcium uniporter; MDIVI-1, mitochondrial division inhibitor 1; OXPHOS, oxidative phosphorylation; PEITC, β-phenylethylisothiocyanates; PINK1, PTEN-induced kinase 1; ROS, reactive oxygen species; SERCA, sarco/endoplasmic reticulum Ca2+ ATPase.
Targeting mitochondrial fusion and fission
Inhibition of fission is promising since it plays extensive roles in cancer development (Fig. 2). The DRP-1 inhibitor mitochondrial division inhibitor 1 (MDIVI-1) displayed synergy with cisplatin to overcome resistance and promote cell death in several cancer cell lines (127). Similarly, a combination of MDIVI-1 and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) enhanced TRAIL-induced death in ovarian cancer cells, implying MDIVI-1's potential as an adjuvant to conventional cancer drugs (164). No other fission inhibitors have been tested, presenting an open door for future development in cancer treatment.
In contrast, induction of mitochondrial fission also demonstrated anticancer properties. The combination of saikosaponin and cisplatin resensitized cisplatin-resistant ovarian cells to apoptosis via DRP-1 upregulation (154). In this context, cisplatin resistance was mainly driven by increased fusion due to p53 mutation. By inducing fission, the apoptotic effects of cisplatin were restored by saikosaponin.
Similarly, resveratrol may induce cancer cell death via upregulation of DRP-1-mediated fission. In human cervical, colon, breast, and liver cancers, resveratrol was found to promote mitochondria fragmentation, leading to cell death (172). However, since resveratrol is capable of regulating multiple cellular processes, the death-inducing effects of resveratrol are unlikely to be solely fission dependent.
Due to the context-dependent roles of mitochondrial fission and fusion in cancer, limited therapies are developed to target these processes. In particular, the challenge lies in how to induce mitochondrial fragmentation or to suppress mitochondrial fusion, and to what extent.
Targeting mitophagy
Similar to fission, both the inhibition and induction of mitophagy have shown to be effective anticancer strategies (Fig. 3). Specifically, compounds that activate mitophagy inhibited growth and induced death in several cancer cell lines. For instance, dihydroergotamine tartrate (DHE), a common drug for migraine, has recently been shown to inhibit lung cancer cell growth by inducing mitophagy (15). DHE induced PINK1/PARKIN activation for mitophagy-induced cell death, indicating its potential to be repurposed for cancer treatment. Similarly, CerS1/C18 pyridinium-ceramide treatment potently induces lethal mitophagy and abrogated growth of tumor xenograft (143). LCL-461, a mitochondrial-targeted ceramide analog, also demonstrated efficacy in treating crenolanib-resistant acute myeloid leukemia (AML) cells through lethal mitophagy (25). It will be interesting to evaluate the impact of mitophagy-inducing compounds across different cancers. Inhibitors of mitophagy are poorly studied in cancer. Liensinine, a universal autophagy inhibitor, prevents autophagosome–lysosome fusion and resensitized breast cancer cells to chemotherapy (186, 189). Liensinine also synergized with other classical chemotherapy drugs, demonstrating its potential as an adjuvant therapy (19).
Targeting mitochondrial bioenergetics
OXPHOS inhibitors are promising in targeting OXPHOS-dependent tumors and also to alleviate tumor hypoxia—a condition correlated with resistance and poor clinical outcomes (Fig. 4). One of the most promising OXPHOS inhibitors is metformin, a Food and Drug Administration (FDA)-approved diabetic drug repurposed for cancer treatment. By inhibiting ETC complex I, metformin reduces OXPHOS and tumor growth in multiple cancer models (87, 140, 168). In addition, metformin alleviates hypoxia in tumor xenografts and improves sensitivity to radiotherapy (182). Metformin is currently under clinical trials to assess its efficacy as an antitumor agent. Despite its remarkable safety profile, the bioavailability of metformin in tumors is not robust, thus compromising its antitumor effects. To overcome this, triphenylphosphonium-conjugated metformin has been developed to specifically target tumor mitochondria (20). Other biguanides with higher potency are also being studied as an alternative to metformin. Phenformin is readily transported into cancer cells and exhibits higher affinity to mitochondria membrane than metformin (4). However, clinical usage of phenformin leads to side-effects such as lactic acidosis, thus dosage has to be carefully monitored.
Atovaquone is a U.S. FDA-approved malaria drug that is recently repurposed for cancer treatment. It exerts antitumor effects by inhibiting ETC complexes III and by alleviating tumor hypoxia at pharmacologically achievable concentrations (43). It is now under clinical trial for the treatment of AML. As for ETC complex IV inhibitors, arsenic trioxide is FDA approved for promyelocytic leukemia and is being investigated for other cancers (30).
Since drug treatment often induces OXPHOS upregulation, OXPHOS inhibitors are now tested as adjuvants to conventional chemotherapy. The ETC complex I inhibitor ME344 is now under clinical trial for human epidermal growth factor receptor 2-negative breast cancer in combination with bevacizumab. ME344 has also been shown to synergize with other tyrosine kinase inhibitors to control spontaneous breast cancer models. Similarly, phenformin also synergizes with BRAF inhibitors to promote tumor regression in melanoma mouse models, further highlighting the potential of OXPHOS inhibitors as adjuvant therapy (181).
In addition, OXPHOS inhibitors could be a viable approach to overcome chemoresistance that arose from metabolic reprogramming. Recently, IACS-010759, a small-molecule inhibitor of ETC complex I, has shown promising targeting effects in OXPHOS-dependent ibrutinib-resistant mantle cell lymphoma model (185). This compound also potently inhibited tumor growth in brain cancer and AML models at well-tolerated doses, and is currently under phase I clinical trial for refractory AML and solid tumors (102). These exciting preclinical data strongly support efforts to develop OXPHOS inhibitors for cancer treatment.
Targeting ROS
Most current therapeutic drugs exert anticancer effects by tipping the balance between ROS production and removal (by dedicated antioxidant system) in favor of ROS generation, leading to apoptosis. Common drugs that increase ROS include anthracyclines (e.g., doxorubicin, epirubicin) (101), platinum coordination complexes, topoisomerase inhibitors (e.g., topoisomerase II inhibitors), and alkylating agents. Antioxidant system can be inhibited by arsenic agents (e.g., arsenic trioxide) (30), imexon or curcuminoids (179). Another category of chemotherapeutic drugs including taxanes and vinca alkaloids (179) decreases ROS to inhibit cancer progression by decreased growth rate and invasiveness. However, these may target ROS-independent pathways such as DNA damage and disrupt protein synthesis as well.
Elesclomol sodium (STA-4783) is a novel potent drug that has shown promising efficacy in AML in preclinical trials (54). It functions through inhibition of ETC flux and enhances mROS production. Since cancer cells generally have elevated basal mROS, STA-4783 can induce cytotoxicity and apoptotic cell death in tumor cells while sparing healthy cells. STA-4783 has been tested alone or in combination with paclitaxel in both phase I and II trials in patients with adult AML and solid tumors and shown promising results (47). However, a phase III trial in melanoma patients showed differential result among patients with different lactate dehydrogenase (LDH) levels (54, 113, 125). Since patients with high LDH showed decreased survival rate, this trial was discontinued in 2013 (113). Another group investigated STA-4783 as a single agent in relapse and refractory AML patients. While STA-4783 had good safety profile, clinical efficacy would need further investigation given the small sample size of 9 patients (54). A phase II trial on patients with recurrent ovarian cancers with the combination of STA-4783 and paclitaxel is ongoing, but good tolerance of this combinatorial treatment has been reported (103).
Another promising mROS targeting compound is β-phenylethylisothiocyanates (PEITC), a thiol modifier that activates apoptosis (13, 125, 175). In combination with metformin, PEITC exhibited promising antiproliferative and cytotoxic effect in ovarian cancers (13). A phase II clinical trial completed in 2017 on effect of PEITC to prevent lung cancer in smokers showed modest but encouraging results (55, 180). A novel analog LBL21 exhibited superior anticancer effect and improved potency as a mROS modulator (165). Preclinical trials in vitro and in vivo demonstrated enhanced cell death and decreased proliferation in lung cancer cell lines and animal models (180).
Targeting mitochondrial Ca2+
Since the discovery of MCU, immense efforts have been put into developing effective pharmacological inhibitors/activators. Direct manipulation of mitochondrial Ca2+ through MCU complex has been challenging. Ruthenium red and Ru360 are direct inhibitors of MCU. However, they are not cell permeable and affect a wide range of ion channels. This limits their usage mainly to in vitro analysis on isolated mitochondria. Interestingly, mitoxantrone, a topoisomerase type II inhibitor, which disrupts DNA synthesis and repair mechanism, has been found to inhibit MCU activity directly through a yeast-based chemical screening (5). However, the precise mechanism of action requires further validation. Moreover, the use of mitoxantrone as MCU inhibitor in mammalian cell line has yet to be verified. However, as a commonly prescribed chemotherapeutic drug in AML and breast cancer, mitoxantrone shows great potential to be used as MCU inhibitor in mitochondrial-targeted therapy. Another potential small-molecule MCU inhibitor compound is DS16570511 (72, 82), which shows improved specificity for MCU and is cell permeant. However, the mechanism of action remains unclear (72), and off-target effects on mitochondrial membrane potential have been reported recently (120).
Another approach for intervention is by targeting MAMs that form the interface between ER and mitochondria. The BCL-2-IP3R disruptor 2 triggers cancer cell apoptosis by promoting mitochondria Ca2+ overload through IP3R and BCL-2 (3, 188). Cancer cell death can also be triggered by SERCA inhibition. Thapsigargin induces ER stress-mediated apoptosis in cancer cells through SERCA inhibition (93). As thapsigargin can induce toxicity in all cell types, its specificity is further improved by combining with a targeting peptide Mipsagargin G-202 (29). G-202 is currently in clinical trials for several cancers (31). Similarly, resveratrol and its derivative picaetannol can induce cancer cell death by inhibiting SERCA and enhancing ER stress (90). Given that prolonged ER stress can also increase tumorigenic and metastatic potential of cancer cells, one must exercise caution in the use of these ER stress agents (24). Another potential drug is PS89, a small molecule, which has been identified as potent chemosensitizing drug that inhibits protein disulfide isomerase (PDI). PDI is important for maintaining ER homeostasis, and inhibition by PS89 affects crosstalk at MAMs and induces apoptotic cascades (71).
Targeting mitochondria-mediated apoptosis
Owing to the precise understanding of mitochondria-mediated apoptosis, multiple drugs have been developed to target this process (Fig. 6). ABT-737, a BCL-2 homology domain 3 (BH3) mimetic that antagonizes antiapoptotic BCL-2 and BCL-XL, kills cancer cells via mitochondria-mediated apoptosis, and is ineffective in BAX- and BAK-negative cells (17). In multiple cancer models, ABT-737 has demonstrated potent anticancer activities as monotherapy or in combination with chemotherapy or radiotherapy (78, 99, 149). When used in combination therapy, ABT-737 reverses therapeutic resistance and resensitizes cancer cells to drug-induced death (53, 78). Due to poor bioavailability of ABT-737, ABT-263 (Navitoclax) was developed as a more soluble and orally available alternative to ABT-737. Specifically, navitoclax is modified at three chemical sites along the backbone of ABT-737 to maximize the pharmacokinetic/pharmacodynamic relationship between oral exposure in animals and efficacy in human tumor cell lines (153). Navitoclax mimics the effects of ABT-737 in promoting cancer cell death, but presents side-effects of thrombocytopenia in clinical trials possibly due to BCL-XL inhibition. A third BH3 mimetic ABT-199 (Venetoclax) was chemically modified at several sites to enhance its potency and specificity against BCL-2, but not BCL-XL (146). Venetoclax demonstrated promising outcomes in clinical trials and is recently approved by FDA for treating chronic lymphocytic leukemia in combination with obinutuzuma.
Other than BH3 mimetics, small-molecule GX15-070 (Obatoclax) induces cancer cell death by antagonizing BCL-2 and BCL-XL (111). In preclinical trials, obatoclax showed synergy with ABT-737 to induce apoptosis in AML (73). Despite passing phase I clinical trials, obatoclax displayed limited clinical activity and prominent side-effects in phase II clinical trials in cancer patients.
G3139 (Oblimersen) is another BCL-2 targeting drug that stalled at phase III clinical trials and failed to receive FDA approval for cancer treatment. As an oligonucleotide, oblimersen specifically targets the first six codons of BCL-2 mRNA to induce apoptosis. In early stages of clinical trials, oblimersen demonstrated promising effects when combined with multiple chemotherapeutic agents, including doxorubicin, docetaxel, fludarabine, and cyclophosphamide (107, 132). Yet, it was eventually rejected due to insignificant clinical activity in a phase III trial with multiple melanomas. Clearly, BCL-2 inhibitors show great potential as anticancer drugs if one can overcome the challenges associated with current BCL-2 targeting compounds.
Antibiotics to target mitochondria in cancer cells
The use of readily available antibiotics to target mitochondria in CSCs was suggested in 2015 (79). Considering the endosymbiotic bacterial origin of mitochondria (133), screening and repurposing antibiotics originally developed to kill bacteria or other prokaryotes may uncover new approaches to target mitochondria in cancer. Various commonly prescribed antibiotics that inhibit bacterial protein synthesis such as doxycycline and azithromycin have been examined for their effects in targeting biogenesis of mitochondria in CSCs (79). Five different classes of FDA-approved antibiotics were examined across numerous cell lines and tumor types (36, 79). There are several classes of drugs that target prokaryotic ribosomes, which share homology with mitochondrial ribosomes. Indeed, these antibiotics showed reduced tumorsphere formation across different cell types (16, 79). Moreover, as these antibiotics are commonly used for treatment of bacterial infections, they have minimal side-effects and cytotoxicity, and are nontoxic to normal cells. Other antibiotics that have been shown to be effective in targeting mitochondria in cancer include bedaquiline (TMC207), which acts on ATP synthase (42, 145). Although more clinical trials are warranted to test the efficacy of these antibiotics in cancer therapy, using currently FDA-approved drugs not only benefits from the already established safety profiles but also accelerates new cancer drug development greatly.
Synthetic lethality
Synthetic lethal interaction was proposed >20 years ago to identify new anticancer drug targets, but only one made it successfully to clinical use (114, 183). This is due to the difficulty in identification of robust and clinically relevant interactions (114). The metabolic reprogramming and plasticity in mitochondria of cancer cells has made exploring metabolic synthetic lethality attractive. Succinate dehydrogenase (SDH) is made up of several subunits such as SDHA, SDHB, and SDHC, and they were found to be commonly mutated in cancers such as gastrointestinal stromal tumors, renal cell carcinoma, and neuroblastoma, which led to SDH deficiency in cancer (41, 62, 156, 183). Interestingly, both Lussey-Lepoutre et al. (89) and Cardaci et al. (12) reported preferential use of pyruvate carboxylase (PC) to generate oxaloacetate in SDHB-deficient cells. Hence, PC and SDH form synthetic lethal interactions, and silencing of PC in SDH-deficient cancer cells appears to affect proliferation and tumor progression (12, 183). Recently, proline dehydrogenase (PRODH), which is a p53-inducible IMM flavoprotein linked to ETC and ATP production, was reported to have synthetic lethal interaction with glutaminase (GLS1) (141). Inhibiting PRODH alone with S-5-oxo or N-PPG resulted in a metabolic shift and did not yield favorable results. However, in combination with GLS1 inhibitors such as CB-839, synergistic loss of growth and viability was observed in several breast cancer cell lines (141). Common to many cancer therapeutics and chemotherapeutic drugs, single agent often results in minimal efficacy and resistance due to plasticity of cancer cells. Therefore, a more effective anticancer strategy may be identification of synthetic lethal interactions and treatment combinations. Since mitochondrial dysfunctions contribute greatly to onset and progression of cancers, investigating possible synthetic lethal targets relevant to mitochondria may yield promising results.
Novel Approaches to Mitochondrial-Targeted Therapy
In addition to approaches aimed at restoring mitochondrial function described above, which have shown promising outcomes, several novel avenues are under investigation.
Mitochondrial trafficking
Mitochondrial trafficking has been reported in neurons and motor neuron diseases (45). Mitochondrial subcellular localization and movement is crucial for maintaining cell polarity, morphology, and cell homeostasis (45). Although the precise mechanism is unknown, the relocation of mitochondria to the periphery is important for cellular movement (16, 115). Indeed, decreased migration in multiple cancer types (glioma, breast, and pancreatic) with loss of NF-κB-inducing kinase caused aggregation of mitochondria at perinuclear regions (67). The pathway of mitochondrial aggregation is ADP-ribosylation factor 6-ARF-GTPase activating protein dependent and induces detrimental ROS generation (115). This study provides an explanation for the improved tolerance of mROS in cancer cells due to enhanced trafficking of mitochondria to the periphery to minimize regions of mROS to prevent activation of apoptotic pathway. Mitochondria at the periphery can also facilitate movement and invasiveness by providing localized ATP. Drugs inhibiting this anterograde trafficking pathway could be developed to inhibit localized mROS production at the periphery for cellular movements. The number of mitochondria has been associated with how cancer responds to drug therapy (137). Using a mathematical framework DEPICTIVE, a difference in mitochondria number was found to account for up to 30% of the varied response to BCL-2 inhibitors.
Mitochondrial dysfunction in CSCs
CSCs (80) are a subpopulation of cancer cells that are capable of perpetuating themselves through self-renewal and undergo cellular differentiation. CSCs are gaining attention in cancer therapy as they contribute to tumor heterogeneity and therapy resistance. However, developing effective anti-CSC therapy is challenging due to the plasticity of CSCs and a high degree of commonality in SC maintenance mechanisms between CSCs and normal SCs. For instance, CSCs possess higher levels of short fragmented mitochondria than non-CSCs, similar to SCs (121, 145, 176, 190). Downregulation of mitochondrial fission proteins such as DRP1 has been shown to decrease sphere-forming capacity and tumorigenicity in CSCs of glioblastoma (176) and pancreatic adenocarcinoma (69). On the contrary, upregulation of fusion proteins such as MFN2 showed reduced tumor growth (16, 144, 145). Interestingly, decreased proliferation in only CSCs and not in glioblastoma non-CSCs upon DRP1 knockdown was reported (176). Some possible mitocans include Mdivi-1 (DRP1-specific inhibitor) and P110, which had decreased off-target effects compared with Mdivi-1. The activity of DRP1 can also be inhibited by targeting COX-2 with inhibitors, including resveratrol and celecoxib.
Balanced mitophagy and biogenesis are also important for regulation of CSC phenotypes (145). An increase in mitophagy with carbonyl cyanide m-chlorophenyl hydrazone increases the association of p53 with mitochondria and induces mitophagy-dependent degradation of p53 (16, 86). This prevents binding of p53 to NANOG promoter and increases NANOG expression. Since NANOG maintains pluripotency, this results in increased stemness and self-renewal property. Mitochondrial biogenesis is closely associated with CSC maintenance through prominent oncogenes such as c-MYC and pathways such as WNT signaling (16). Moreover, inhibition of mitochondrial biogenesis through PGC1α/estrogen-related receptor α signaling by XCT790 significantly reduces sphere-forming ability of breast carcinoma cells (16, 27).
Mitochondrial bioenergetics is another important factor affecting stemness. Although SCs commonly obtain source of energy from glycolysis and shift to OXPHOS upon differentiation, CSCs do not fully recapitulate this phenomenon. In fact, CSCs of different cancer types depend highly on both glycolysis and OXPHOS for energy (26, 135, 145). Rather than the utilization of certain metabolic profile, it is the plasticity and ability to switch readily between glycolysis and OXPHOS that is important in maintaining stemness in CSCs (88, 135, 145). Thus, to target CSCs through metabolic targeting drugs, it will be more effective to have a combinatorial treatment, of which one drug inhibits OXPHOS and sensitizes CSCs to second drug that targets glycolysis. For instance, when multikinase inhibitors are used as monotherapy to downregulate glycolysis, the effect is very minimal due to a switch to OXPHOS and results in tumor resistance. However, when used in combination with OXPHOS inhibitors such as phenformin or ME344, very potent synergistic effect on tumor growth reduction in vivo was observed (7, 110). Besides using one drug to sensitize CSCs toward a second drug, additional drugs such as 5′ AMP-activated protein kinase inhibitors can be used to prevent activation of mitophagy or fatty acid oxidation, which can serve as compensatory pathways to provide energy for CSCs survival (145).
Finally, other mitochondria-related functions such as apoptosis, de novo pyrimidine biosynthesis, ATP-binding cassette transporters, and iron metabolism may also play a part in maintaining CSC phenotypes. In essence, mitochondria play a crucial role in the induction and maintenance of CSCs; therefore, targeting CSCs through mitochondrial-targeted therapy may increase efficacy of cancer therapy.
Summary and Future Directions
Significant advances have been made in delineating mitochondrial dysfunctions that occur in cancer. Nevertheless, several questions need to be addressed: Is it possible to differentiate mitochondrial dysfunction in CSCs and cancer cells? What are the interactions between mitochondria and key oncogenic pathways in cancer? Given that cancer cells readily undergo metabolic reprogramming, is OXPHOS and/or glycolytic inhibition sufficient to prevent tumor relapse? Is it possible to make use of elevated mitochondrial membrane potential for enhanced uptake of drugs to selectively target cancer cells? What are potential approaches to improve delivery of chemotherapeutic drugs to the mitochondria?
Future studies in these directions will aid understanding of tumor adaptation and therapeutic failure, and provide a new axis for mitochondrial therapeutic intervention.
Footnotes
Funding Information
Work in the R.T. laboratory is supported by grants from the National Medical Research Council (NMRC/OFIRG/0073/2018) and the Ministry of Education (MOE2019-T2-1-024).