
Abstract
Significance:
Selenoproteins incorporate the essential nutrient selenium into their polypeptide chain. Seven members of this family reside in the endoplasmic reticulum (ER), the exact function of most of which is poorly understood. Especially, how ER-resident selenoproteins control the ER redox and ionic environment is largely unknown. Since alteration of ER function is observed in many diseases, the elucidation of the role of selenoproteins could enhance our understanding of the mechanisms involved in ER homeostasis.
Recent Advances:
Among selenoproteins, selenoprotein T (SELENOT) is remarkable as the most evolutionarily conserved and the only ER-resident selenoprotein whose gene knockout in mouse is lethal. Recent data indicate that SELENOT contributes to ER homeostasis: reduced expression of SELENOT in transgenic cell and animal models promotes accumulation of reactive oxygen and nitrogen species, depletion of calcium stores, activation of the unfolded protein response and impaired hormone secretion.
Critical Issues:
SELENOT is anchored to the ER membrane and associated with the oligosaccharyltransferase complex, suggesting that it regulates the early steps of N-glycosylation. Furthermore, it exerts a selenosulfide oxidoreductase activity carried by its thioredoxin-like domain. However, the physiological role of the redox activity of SELENOT is not fully understood. Likewise, the nature of its redox partners needs to be further characterized.
Future Directions:
Given the impact of ER stress in pathologies such as neurodegenerative, cardiovascular, metabolic and immune diseases, understanding the role of SELENOT and developing derived therapeutic tools such as selenopeptides to improve ER proteostasis and prevent ER stress could contribute to a better management of these diseases.
Introduction and Scope
Reactive oxygen species (ROS) are radical or molecular species derived from molecular oxygen. Under physiological conditions, ROS are generated during aerobic respiration as a by-product of the mitochondrial electron transport chain. The incomplete reduction of oxygen to superoxide anions (O2 −) is the first step of ROS formation (106, 135). The forms of ROS that are relevant in the biological systems comprise free radicals such as O2 − and hydroxyl radical (OH•), and also nonradical forms able to generate free radicals such as hydrogen peroxide (H2O2).
Although mitochondria are the main site of O2 − and H2O2 production, the endoplasmic reticulum (ER) may be the most significant ROS producer in some cell types, in addition to peroxisomes. Importantly, the ER forms membrane contact sites with mitochondria (mitochondria-associated membrane [MAM]) where redox-sensitive proteins are enriched. The latter include redox-sensitive calcium (Ca2+)-handling proteins regulating ER–mitochondria Ca2+ flux and, consequently, redox-dependent mitochondrial metabolism. ROS rapidly diffuse across membranes through aquaporins and specific channels, and the ER has emerged as the most important contributor (60%) to cytosolic ROS in certain conditions (133).
The ER is involved in the synthesis of lipids, membrane proteins, and proteins destined for secretion, which represent up to one-third of total cellular proteins. It is also the main organelle participating in the regulation of Ca2+ homeostasis. In the ER, disulfide bonds formation is catalyzed by protein disulfide isomerases (PDIs), which act to stabilize the tertiary and quaternary structures of proteins. Two electrons are exchanged between a cysteine (Cys) residue of a substrate and PDI. Then, to restore PDI and start another cycle, two electrons are transferred from PDI to ER oxidoreductin 1 (ERO1), resulting in H2O2 production (Fig. 1) (103, 122). One PDI, designated ERp57, mediates protein folding by promoting the formation of disulfide bonds in its newly synthesized glycoprotein substrates thanks to its interaction with the ER lectins calnexin (CANX) and calreticulin (CALR).

The CANX/CALR cycle ensures the folding and the quality control sorting of folded glycoproteins from terminally misfolded glycoproteins destined to degradation (22, 62). When the ER protein-folding capacity is overwhelmed, cells have to minimize the effects of oxidative damage resulting from ERO1 activity. To restore ER homeostasis, cells activate an evolutionarily conserved signal transduction pathway called UPR (unfolded protein response), consisting in three mechanistically distinct branches, namely serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1 α (IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6), whose main purpose is to repress protein translation, to increase protein folding, and to enhance misfolded protein degradation via the ER-associated protein degradation (ERAD) pathway (Fig. 1) (45, 46, 127).
In conditions that alter ER functions, principally overwhelmed protein-folding capacity and oxidative stress, N-glycan synthesis inhibition (23), and Ca2+ depletion, cells undergo a condition termed ER stress. If ER stress is not resolved, accumulation of unfolded proteins and increased oxidative folding further enhance the production of ROS first in the ER, and then in the whole cell, thus irreversibly damaging biomolecules (25, 51, 103) and ultimately leading to ER stress-mediated apoptotic cell death.
Although low ROS production is essential to maintain physiological functions (proliferation, signal transduction, host defense, gene expression), high levels of ROS are toxic for the cell (20, 41, 44, 51, 91, 93). Consequently, ROS production, diffusion, and transorganellar transport are highly regulated. Most organelles are equipped with their own defense system. Among the most important are catalase and Cu/Zn-superoxide dismutase (Cu/Zn-SOD, SOD1) in peroxisomes, manganese superoxide dismutase (Mn-SOD, SOD2), and glutathione peroxidases (GPx)1 and GPx4 in the mitochondria, and peroxiredoxin 4 (PRX4), GPx7 and GPx8 in the ER (133). Both PRXs and GPxs protect the organism from oxidative damage through the reduction of H2O2 in H2O.
It is worth noting that certain GPx (GPx1, GPx2, GPx3, GPx4, and GPx6) contain the essential nutrient selenium (Se) and belong to the selenoprotein family. Cytosolic GPx1 was the first identified selenoprotein. Other important members of this family include TrxR1 and TrxR2, which constitute with NADPH and thioredoxin a major disulfide reductase system that controls cellular redox status (57, 60). Selenoproteins incorporate Se in the form of one or more selenocysteine (Sec, the 21st amino acid) which, compared with Cys, has a slightly higher nucleophilicity, making it very efficiently targeted by electrophiles, and providing a higher resistance to irreversible inactivation by overoxidation (10, 50, 99).
During atypical selenoprotein synthesis, the Sec residue is synthesized on its own tRNA, the tRNA[Ser]Sec encoded by the Trsp gene, and incorporated in an unconventional manner during translation. Selenoprotein transcripts harbor in their 3′-untranslated region a SECIS (selenocysteine insertion sequence) element, which is a 60-nucleotide-length RNA sequence that adopts a stem-loop structure. The SECIS element recruits a protein complex composed of SBP2 (SECIS binding protein 2), eEFSec (eukaryotic elongation factor, selenocysteine-tRNA specific), and other factors with the purpose to site specifically deliver the Sec-tRNASec at defined stop UGA codons in selenoprotein mRNA, thus incorporating Sec into the protein elongating chain (Fig. 2) (113).

The number of genes coding for selenoproteins in eukaryotes is highly variable: 2 in worms, 3 in fruit flies, 24 in rodents, 25 in humans, and >30 in fish (17, 57, 70, 74). Nevertheless, selenoproteins are not present in all forms of life (10). This is the case in higher plants, some fungal species, certain insects and yeast. In these organisms, Cys variants of some selenoproteins are found (70, 73). There is a possibility that these organisms never made selenoproteins, or alternatively that they have lost the Sec insertion machinery during evolution. Less costly energetically, Cys-containing protein orthologs would thus be maintained in certain genomes (in drosophila for instance) in place of the Sec-containing forms.
Inasmuch as Se is essential for life, the significance of selenoproteins for human health is increasingly recognized through the identification of patients with mutations in genes involved in the selenoprotein biosynthetic pathway (111). Mutations in selenocysteinyl-tRNA(Sec) synthase (SEPSECS) lead to neurodevelopmental disorders, and diseases associated with SBP2 alteration are characterized by impaired metabolism and action of thyroid hormones, bone growth and maturation deficits, azoospermia and other defects related to nervous system development or muscle, inner ear, skin and immune system function.
Mutations in individual selenoproteins are also associated with diseases, including selenoprotein N (SEPN1, SELN, SELENON)-related myopathy, GPx4-related Sedagathian-type spondylometaphyseal chondrodysplasia characterized by respiratory failure and bone defects, TrxR2-related dilated cardiomyopathy, and familial glucocorticoid deficiency (60, 107, 108, 116, 134). Se deficiency or excess is also associated with health risk in humans (94), although long-term Se deficiency could be viewed as a mimic of calorie restriction, a condition known to increase life span (131).
Meanwhile, the role of about half of selenoproteins, which were first described in silico by the Gladyshev laboratory, is still unclear. This is the case for selenoproteins localized in the ER, which have for some of them a key role in the regulation of the ER redox balance and efficient quality control of glycoproteins. In this review, we summarize the current knowledge on ER selenoproteins, focusing especially on selenoprotein T (SELENOT) whose function is essential to maintain ROS and ER homeostasis.
ER-Resident Selenoproteins
Seven selenoproteins have been shown to reside in the ER. The luminal ones include selenoprotein F (SEP15, SELENOF) and selenoprotein M (SEPM, SELM, SELENOM). Others are associated with the membrane. The best characterized of them is type 2 iodothyronine deiodinase (DIO2), which catalyzes the conversion of prohormone thyroxine (3,5,3′,5′-tetraiodothyronine, T4) to the bioactive thyroid hormone (3,5,3′-triiodothyronine, T3). The others are selenoprotein K (SELK, SELENOK), SELENON, selenoprotein S (SELS, VIMP, SEPS, SELENOS), and SELENOT.
All of them are indisputably important to maintain ER homeostasis (67, 88, 99), and their function is just beginning to be understood. Several of them have a role in the regulation of Ca2+ homeostasis. For instance, SELENOK is associated through Src-homology 3 (SH3)/SH3 interactions with DHHC6 (single letter symbols representing Asp-His-His-Cys amino acid sequence), which is an ER membrane enzyme catalyzing protein palmitoylation. SELENOK possibly serves as a cofactor, stabilizing the acyl-DHHC6 intermediate and reducing hydrolyzation of the thioester bond until transfer of the palmitoyl group on the target protein can occur (29). It has been shown that the DHHC6/SELENOK complex is required for the palmitoylation of certain proteins such as the inositol 1,4,5-trisphosphate receptor (IP3R), an essential step for the activation of this Ca2+ channel (30, 126).
The ryanodine receptor (RyR), another Ca2+ channel present in the sarcoplamic reticulum, is a “redox sensor” regulated by a number of associated proteins, including SELENON, with which it is physically associated, at least in zebrafish (54). SELENON is also associated with the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump, reducing its luminal Cys that are hyperoxidized by ERO1-generated peroxides (71). This oxidoreductase is considered as a prominent MAM-localized regulator of ER–mitochondria crosstalk, limiting Ca2+ efflux from the ER to mitochondria through the activation of SERCA by a reduction of its oxidation state (65, 71, 133). Mutations in SELENON gene lead to myopathies (18, 54, 87), although its precise contribution to muscle development is not yet understood.
SELENOM also influences Ca2+ homeostasis, but in contrast to SELENON and SELENOK the underlying molecular mechanism is totally unknown. It has probably a major role in energy metabolism since its gene knockout (KO) in mouse leads to an obese phenotype (89, 95).
Other ER selenoproteins, such as SELENOF, SELENOK, and SELENOS, are involved in the folding and quality control of glycoproteins. SELENOF, which is ancestrally linked to SELENOM, directly participates in protein quality control through its binding to UDP-glucose:glycoprotein glucosyltransferase (UGGT), an enzyme that specifically reglucosylates unfolded proteins to be recognized by CALR for recycling to the ER (132). SELENOS, in association with SELENOK, participates in the protein quality control process via its interaction with valosin-containing protein, an ATPase present at the ER membrane, which is associated with the ERAD complex (64).
Finally, compelling data on SELENOT, that is, sequence data, genetic information, in particular phenotypes of KO animals, subcellular location, and functional studies, also indicate a vital function in relation with its presence in the ER. The rest of this review will be focused on SELENOT for which major breakthroughs have been recently achieved.
SELENOT, an Essential Gene Conserved Across Organisms
SELENOT was initially identified in silico using the computer program SECISearch, based on the recognition of the SECIS element and thermodynamic characteristics (59). Different Sec- and Cys-containing SELENOT orthologs have now been identified in numerous eukaryotes, including plantae, metazoans, and some protists such as ciliates (Fig. 3 and Supplementary Data) (72), but not in yeast and bacteria where apparently no homolog exists. The Cys substitution is present in the two Caenorhabditis elegans SELENOT isoforms (SelT1.1 and SelT1.2), in several insects (Drosophila melanogaster, Drosophila willistoni, and Culex quinquefasciatus), land plants (Arabidopsis thaliana, Oryza sativa japonica, and Zea mays), and in Kinetoplastida (Trypanosoma and Leishmania) (69, 79).
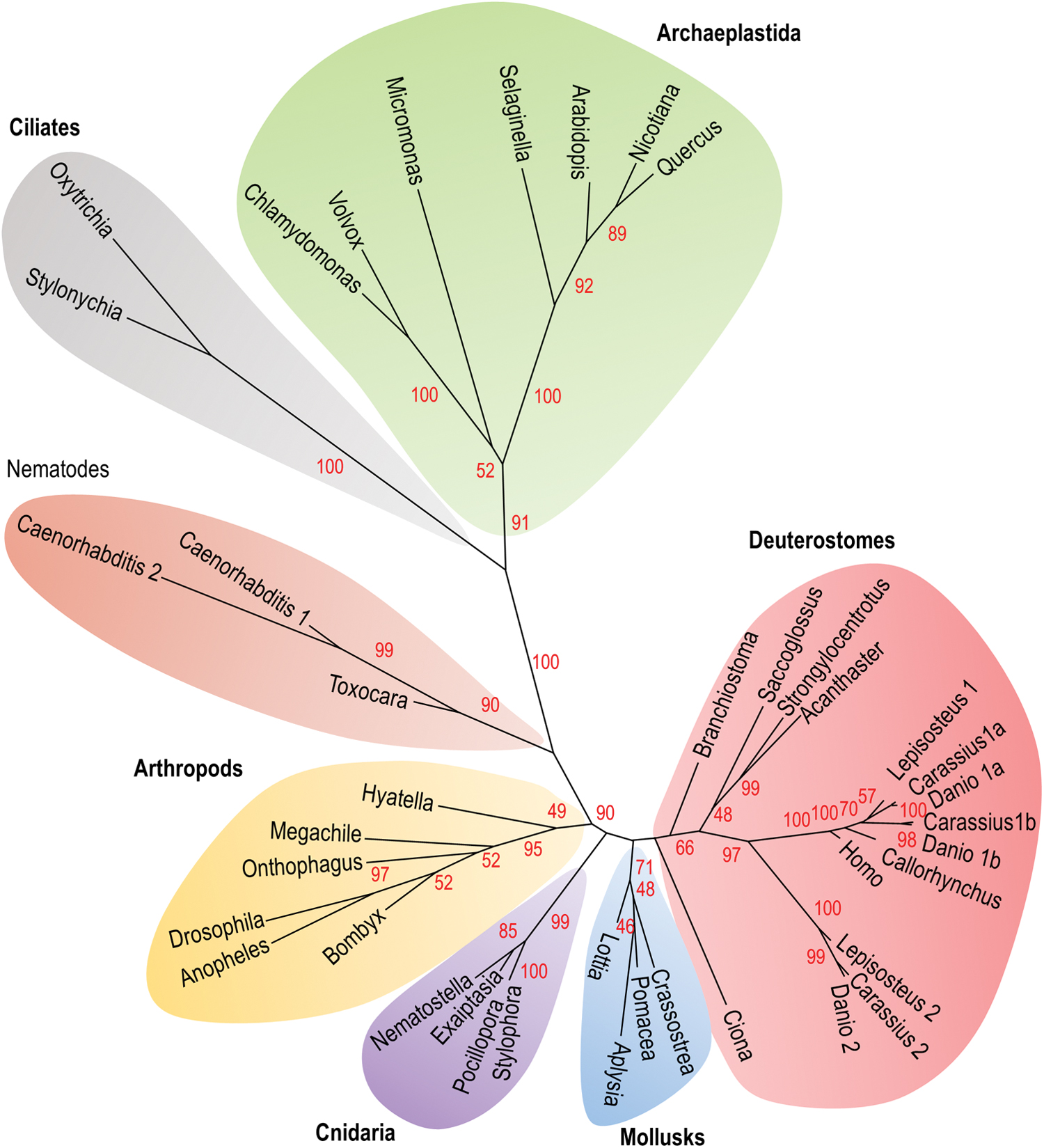
Based on our phylogenetical analysis, it is noteworthy that in certain groups such as arthropods and archaeplastida, the most basal species (e.g., Hyatella and Chlamydomonas, respectively) have a SELENOT-like isoform with a Sec in the redox center, while in the others (e.g., Arabidospsis and Drosophila, respectively), the Sec residue has been substituted by a Cys residue. These data strongly suggest that Sec to Cys substitution would be a recent acquisition, in these groups at least. They also show that SELENOT exists in the form of Sec or Cys variants, and that the Cys isoforms appear as closely evolutionary related to Sec-containing SELENOT.
The SELENOT gene family has been strongly shaped during evolution. In humans, the gene is localized on chromosome 3 at position 3q25. It includes six exons with the Sec-encoding TGA codon in exon 2, the TAG stop codon in exon 5, and the SECIS element in exon 6 (8, 38, 79). Two processed pseudogenes, SELENOTP1 (ENSG00000233846) and SELENOTP2 (ENST00000603971.1), are also present in the human genome at positions 9p24 and 5q23, respectively. Interestingly, SELENOP1 is a nearly complete copy of the mRNA transcribed from the functional SELENOT gene, including the SECIS sequence, thus raising the interesting possibility that it might be functional. Like in humans, most species possess a single SELENOT gene although with exceptions. Nematodes for instance possess a single SELENOT gene, but as mentioned above, some taxa may have up to two (C. elegans) or three (Caenorhabditis brenneri) copies of the SELENOT gene (100). Likewise, three SELENOT-related genes, called SelT1a, SelT1b, and SelT2, are present in some teleost species such as zebrafish (58) and goldfish (21), while two SELENOT genes, SelT1 and SelT2, are present in spotted gar, a nonteleost ray-finned fish.
Up to now, the phylogenetic status of the different SELENOT-related genes (SelT1a, SelT1b, and SelT2) found in ray-finned fish was unknown. As shown in Figure 4, synteny analysis shows that all these genes belong to the same paralogon; that is, a set of paralogous chromosomal regions derived from a common ancestral region. According to Nakatani et al. (83), this paralogon was already present in the last common vertebrate ancestor and was probably generated in 2R, the two whole-genome duplication events that took place early during the vertebrate history. From these data, we can speculate that up to three SELENOT-related genes could have been present in the vertebrate ancestor. Among these three genes, only two, SelT1 and SelT2, are still present in bony fish, and one, SelT1, in tetrapods, including human. Synteny analysis also suggests that the two copies of SelT1, SelT1a, and SelT1b, present in zebrafish and goldfish, arose through 3R, the teleost fish-specific genome doubling.

The conservation of gene sequence during evolution is indicative of strong functional constraints leading to a very selective pressure, which is stronger when genes are essential. According to gene prediction and phylogenetic reconstruction methods, SELENOT, selenoprotein, and nonselenoprotein orthologs included, is the most conserved selenoprotein in vertebrates, with 95% amino acid identity between mammalian and chicken SELENOT for instance. There is also an impressive identity across all mammals even at the nucleotide sequence level, thus supporting an essential function (27, 72, 79). This seems indeed to be the case since SELENOT gene disruption is embryonic-lethal in mouse. No alive embryo could be found in the uterus of pregnant mice at the earliest embryonic stage analyzed; that is, embryonic day 8 (14). However, the precise stage of pregnancy at which embryos die, that is, before or after implantation, is not known yet and remains to be determined.
In humans, the dispensability of selenoprotein genes has recently been assessed by quantifying the natural occurrence of loss-of-function (LoF) variants. The intolerance of LoF between human and mice for the SELENOT gene is matching, indicating that SELENOT may be essential in humans as in mice. These findings do not support the possibility that SELENOP1 pseudogene would be functional in humans. It should be noted that at least three other mammalian selenoproteins, namely TrxR1, TrxR2, and GPx4, are also essential in mice. Among these, only TrxR1 seems essential in humans as SELENOT (102).
Further emphasizing the functional importance of SELENOT, its cellular levels are marginally impacted by Se deficiency (86, 118). In addition, gene invalidation of SELENOP, which transports Se throughout the body for its use in the synthesis of other selenoproteins, has a moderate impact on SELENOT expression, except in the testis. Finally, the levels of SELENOT mRNA are significantly upregulated between control and Se-supplemented conditions in HEK293 cells, whereas the levels of 19 other selenoprotein mRNAs remained insensitive to Se-level variation (121). Taken together, these data show that in the hierarchy of selenoprotein production, SELENOT is among the highest priority selenoproteins, along with TrxR1, and that SELENOT expression cannot drop below a critical threshold.
SELENOT, an Oxidoreductase Associated with the ER Membrane
Based on its structure, SELENOT is expected to be a selenosulfide/disulfide oxidoreductase, like all selenoproteins that have been functionally characterized. In addition to a 19-amino acid N-terminal signal peptide, it possesses six α-helices and four β-sheets with the β1-α1-β2-α2-α3-α4-α5-β3-β4-α6 typical arrangement (Fig. 5) (8, 27, 79). The N-terminal redox motif, Cys-Val-Ser-Sec/Cys (CVSU/C) in most species, is located in a loop between the β1-sheet and the α1-helix, thus forming a typical domain characteristic of the thioredoxin-fold proteins. By analogy to other thioredoxin-fold proteins such as thioredoxins, glutaredoxins, and PDI, the redox center is presumed to confer to SELENOT an oxidoreductase activity. Using an assay based on the reduction of 5,5′-dithiobis(2-nitrobenzoic) acid (DTNB) with NADPH, a TrxR-like activity of recombinant SELENOT was shown, which depends on Cys/Sec residues of the thioredoxin-like fold (14), although such activity remains to be established in a more physiological context.
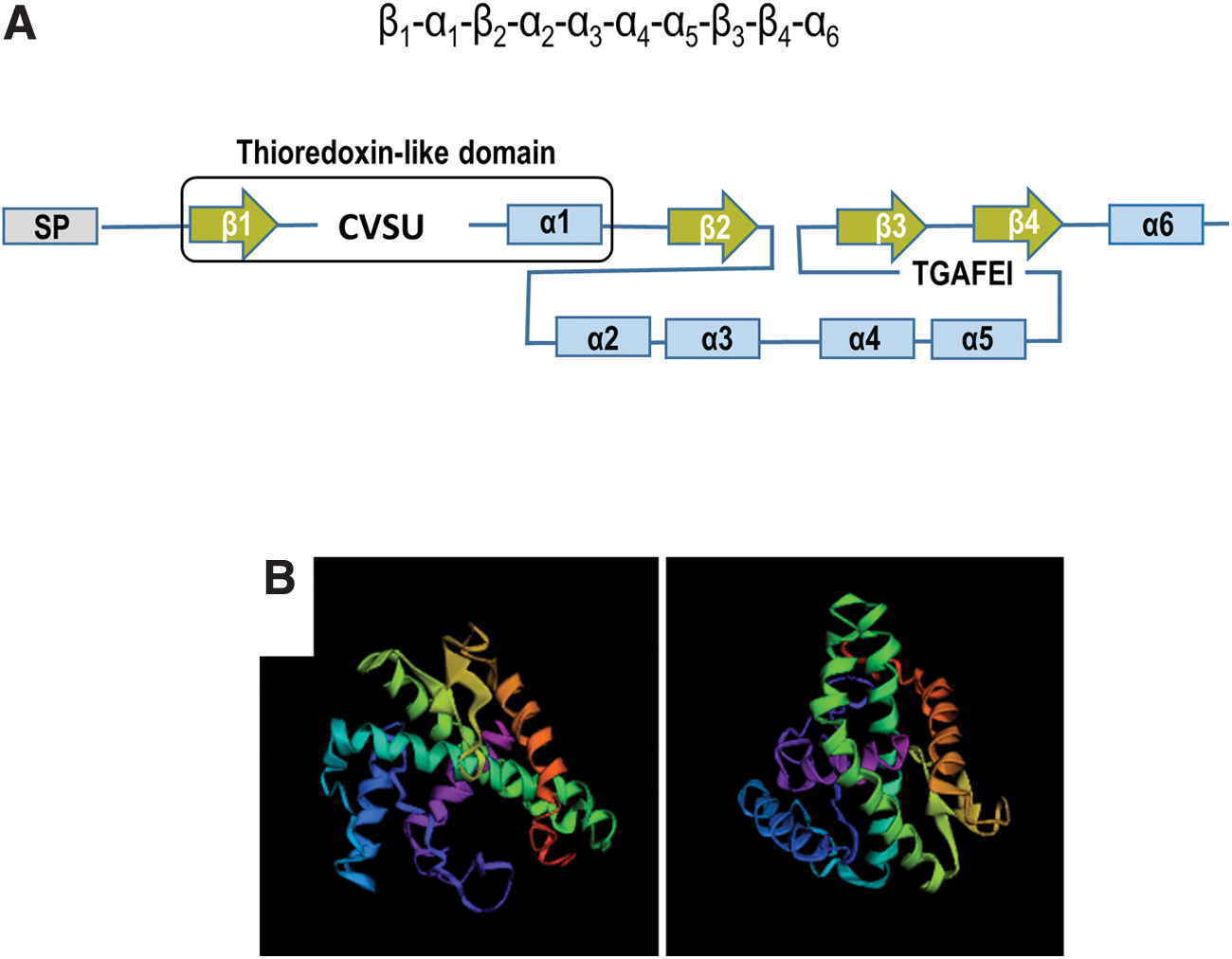
In contrast to many other thioredoxin-fold proteins, an additional feature of SELENOT is a conserved tGxFEI (Thr-Gly-X-Phe-Glu-Ile, X is any aminoacid, conserved residues in capital letters, residues most often observed in the indicated position in small letters) stretch of amino acids in the C terminus, which is also present in selenoprotein W (SELENOW), selenoprotein V (SELENOV), selenoprotein H (SELENOH) and Rdx12, in mammals (27). This motif and the thioredoxin-fold domain constitute the genetic signature of a new family of thioredoxin-type selenoproteins named redoxins (Rdx), as defined by Dikiy et al. (27). In this family, SELENOT is the only member localized in the ER.
The ER localization of SELENOT has been established in plant, invertebrate, and vertebrate species: in A. thaliana, a classical model of higher plants, the SELENOT-like protein with the Sec/Cys substitution was identified in the ER fractions by mass spectrometry (85); in C. elegans, the two SELENOT isoforms also carrying the Sec/Cys substitution were detected in the ER by immunofluorescence (100); in a nonmammalian vertebrate, the goldfish Carassius auratus, genuine SELENOT was characterized in the cardiac sarcoplasmic reticulum (76); in mammals, SELENOT was found to be colocalized with several ER proteins, including an ER-targeted green fluorescent protein (GFP), the ER-resident protein BiP, which is also referred to as 78-kDa glucose-regulated protein, and keratinocyte-associated protein 2 (KCP2) (38, 40, 92, 96, 120).
How does SELENOT achieve its localization in the ER is still an open question. Intriguingly, a SELENOT lacking the N terminus exhibited a cytoplasmic localization that did not virtually differ from that of the full-length protein (27, 38), suggesting that other sequences are required for the targeting of SELENOT to the ER. Some membrane proteins may be addressed to the ER by their membrane domains (6, 12). Accordingly, SELENOT, unlike the structurally close SELENOW, harbors two hydrophobic amino acid stretches at positions 87–102 and 125–144, inserted between β2 and β3 sheets, which could allow a membrane insertion (27, 38, 79). Modeling studies suggest that the first hydrophobic segment likely contains one long amphipathic helix, which forms extensive contacts with the lipid bilayer, probably promoting monotopic attachment (Fig. 6) (67). A recombinant SELENOT devoid of the 87–102 hydrophobic region is no more confined to the ER, suggesting that at least this domain is required for ER localization (38).

Other observations reinforce the idea that SELENOT is anchored to the ER membrane. SELENOT is detected in microsomal fractions prepared from mouse endocrine cells, although it is technically challenging to find it systematically (40). In addition, immunogold staining data show a proximity of SELENOT signal with the ER membrane in rat anterior pituitary cells (40). Interestingly, a proportion of SELENOT is also observed in the cytoplasm with this approach, supporting the possibility of a partial cleavage of the signal peptide and a possible distribution of the protein between the ER and the cytosol.
Several mechanisms might participate in SELENOT retention at the level of the membrane, including static retention and protein–protein interactions with other resident proteins (82, 125). An important issue is whether SELENOT leaves the ER, since SELENOT does not display the Lys-Asp-Glu-Leu tetrapeptide necessary for ER retention. This question cannot be answered yet based on the currently available data. Indeed, SELENOT has been described in the Golgi apparatus in one report (27), but SELENOT was not found in secretory granules by immunogold staining (40).
Together, these observations indicate that SELENOT is an atypical member of the Rdx family exhibiting membrane association and a main ER localization. Other locations for SELENOT have also been suggested but remain to be more accurately characterized, as the Golgi apparatus or the cytoplasm.
SELENOT Serves to Maintain ER Redox Homeostasis
The induction of the luminal chaperone BiP, due to its ability to alleviate proteotoxicity, is considered as a marker of ER stress (11, 91). Overexpression of BiP in AtT20 cells that have been depleted in SELENOT is indicative of a stress in the ER, also visualized by an enlargement of the ER volume (40). The transcripts of atf6, choP, and atf4 (two effector genes of the UPR), and that of the short form of Xbp1 (X-box binding protein 1), were also significantly enhanced in line with the activation of the IRE1, PERK, ATF6 branches of the UPR (Fig. 1) (45, 46). Unexpectedly, a lower degradation of misfolded proteins was observed, congruent with an inhibition of the ERAD pathway at the transcriptional level (40).
Consistent with UPR activation (Fig. 1), oxidative stress and decreased cell survival were reported after SELENOT gene knockdown in dopaminergic neurons, in cortical neuroblasts, and in PC12 cells, indicating that oxidative stress could not be resolved despite UPR activation, at least in these cells (3, 14, 19). Also consistent with SELENOT redox function, its depletion in murine fibroblasts enhances the expression of several oxidoreductase genes, including Cbr3 (carbonyl reductase 3) a member of the short-chain dehydrogenase/reductase family of proteins (112).
In brain conditional SELENOT-deficient mice (Nes-Cre/SelTfl/fl ), oxidative stress was detected in SELENOT-depleted immature neuroblasts, resulting in the loss of immature neurons but not glial cells, through apoptotic cell death (19). In addition, disruption of the SELENOT gene in these mice provoked early lethality and higher vulnerability to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a neurotoxin used to generate animal models of Parkinson's disease (PD). In the nigrostriatal system of these mice, a marked oxidant/nitrosative stress was observed, associated with neurodegeneration, and decreased tyrosine hydroxylase activity and dopamine levels (14). Conversely, SELENOT expression limited the oxidative stress and neurodegeneration in the nigrostriatal pathway of wild-type mice after treatment with MPTP or rotenone, another neurotoxin used to model PD. Likewise, SELENOT also protects the heart against oxidative stress and ischemia, after an ischemic-reperfusion episode in rat (98).
Some Cys residues present in Ca2+-handling proteins are located in the ER lumen, regulating in a redox-dependent manner the activity of RyR, IP3R, and SERCA (9). The crosstalk between Ca2+ and redox signaling is an important process, which determines the amount of Ca2+ available in the cytoplasm and the mitochondria, and consequently the cell fate. Indeed, preventing IP3R-dependent Ca2+ leakage from the ER and calcium/calmodulin-dependent protein kinase type II (CaMKII) activation in the cytosol is sufficient to prevent the activation of proapoptotic pathways (45, 119).
SELENOT gene silencing in PC12 cells, in addition to disturbing the redox homeostasis, actually alters Ca2+ flux, and in these conditions, the pituitary adenylate cyclase-activating polypeptide (PACAP)-induced increase in [Ca2+]i is abolished. This has also been demonstrated in rat primary gastric smooth muscle cells in which SELENOT knockdown reduces their Ca2+ content, myosin light chain kinase activity, and the subsequent 20 kDa myosin light chain phosphorylation, two key elements in smooth muscle contraction (66). Noteworthily, the redox center in SELENOT is necessary to preserve Ca2+ homeostasis, although its precise contribution to the regulation of Ca2+ flux has not yet been elucidated (38).
Unlike ERp57 and SELENON, which modulate SERCA activity, SELENOT ablation did not result in SERCA pump inhibition in PC12 cells (38), and the possibility that SELENOT interacts with IP3R, like ERO1, is not excluded (9). Alternatively, oxidative stress and UPR activation in SELENOT-deficient cells may be sufficient to promote Ca2+ leakage through the translocon (124).
SELENOT Plays a Role in N-Glycosylation
To identify SELENOT partners and better understand the mechanisms underlying the function of SELENOT in ER homoeostasis, a modified split-ubiquitin system for in vivo detection of interactions between transmembrane proteins in yeast has been used (40). Among the subset of putative candidates identified was KCP2, an ER-resident transmembrane protein described as a subunit of the oligosaccharyltransferase (OST) complex (96, 97).
OST catalyzes the transfer “en bloc” of the high-mannose oligosaccharide Glc3Man9GlcNAc2 (Glc, glucose; Man, mannose; GlcNAc, N-acetylglucosamine) from the ER membrane onto Asn residues of a specific N-glycosylation acceptor sequence (Asn-X-Ser/Thr/Cys) in a neosynthesized polypeptide (4). Mammalian OST is a hetero-oligomeric complex, which exists in two paralogous forms containing either the catalytic subunit STT3A or STT3B (Fig. 6). The STT3A isoform is stably integrated into the translocon and catalyzes cotranslational N-glycosylation, while the STT3B complex acts post-translationally as a proofreader for sites missed by STT3A (15, 101, 114, 115). Importantly, KCP2 is a specific subunit of the STT3A complex.
Coimmunoprecipitation experiments have shown that native SELENOT does interact with KCP2, as well as with STT3A and OST48, a common subunit of the STT3A- and B-types OST complexes, but not with STT3B, suggesting that KCP2 likely provides the binding site of SELENOT to the STT3A complex (115). The levels of STT3A, OST48, and KCP2 are significantly reduced in a corticotrope cell line depleted in SELENOT, thus implicating SELENOT in the stability of the OST complex or its binding to the ribosome. Consistently, the association with the translocon is disrupted in the STT3A complex devoid of KCP2 or DC2, another specific subunit of the STT3A complex, which interacts with KCP2 (115).
It is not yet established how SELENOT interacts with the STT3A complex. The seleno/disulfide oxidoreductase activity may be required for SELENOT/STT3A interaction. Indeed, redox interactions have been demonstrated between ER SELENOF and UGGT, and cytosolic SELENOW, which is distantly related to SELENOT, with 14-3-3 protein. In the latter case, the interaction with SELENOW interrupts 14-3-3 binding to Rictor, thus leading to the activation of the mTORC2/Akt pathway (1, 27, 53).
SELENOT itself is not N-glycosylated (40). This excludes the possibility that it physically interacts with the STT3A complex as a substrate, but rather as a modulator of N-glycosylation in the same way as KCP2 (97). Consistent with this hypothesis, SELENOT levels, like those of STT3A and KCP2, and to a lesser extent STT3B, are higher in professionally secreting cells and tissues (13, 96).
SELENOT gene silencing in endocrine cells leads to an alteration of N-glycan occupancy on specific substrates, including proopiomelanocortin (POMC), a 235-amino acid prohormone precursor harboring two N-glycosylation sites at positions 91 and 152, and a modified GFP engineered to detect impaired N-glycosylation (40). The issue of substrate specificity of SELENOT is not solved yet, but it is quite remarkable that SELENOT expression coincides with that of a number of glycosylated hormones or polypeptide precursors with disulfide bonds such as POMC, somatostatin, FSH, TSH, and insulin (40, 92, 120). This observation has led us to speculate that SELENOT, owing to its oxidoreductase activity, might ensure the redox quality control of glycoproteins with disulfide bonds, like the other oxidoreductases TUSC3/N33 (tumor suppressor candidate 3) and MAGT1/IAP (magnesium transporter protein 1) in the STT3B-type OST (78), and their orthologs OST3/OST6 in the yeast OST complex (109). These oxidoreductases form transient mixed disulfide complexes with a set of substrates to slow glycoprotein folding and increase glycosylation efficiency.
Since OST3/6, MagT1/IAP, or TUSC33/N33 have no homolog in the STT3A complexes, it is tempting to speculate that SELENOT can exert the same function in the STT3A complex, provided that the SELENOT N-terminal redox CVSU motif faces the ER lumen (Fig. 6) (24). Remarkably, SELENOT, along with other factors of the glycoprotein quality control cycle (CANX, for instance), was detected in haploid genetic screens designed to identify factors involved in glycosylphosphatidylinositol (GPI) deacylation (68). This step, which is N-glycan dependent, enables GPI-anchored proteins (GPI-APs) to be transported from the ER to the Golgi and to be anchored to the plasma membrane. This finding suggests that GPI-APs are, in addition to glycohormones, other SELENOT targets. GPI-APs group comprises ∼150 proteins, involved in diverse biological processes such as immune recognition, cell adhesion, and vertebrate organ development, a number of processes where SELENOT has also been implicated (19, 100, 112).
So far, SELENOT is the only ER selenoprotein for which the physical association with the STT3A complex has been demonstrated. It is now necessary and urgent to identify SELENOT redox partners, using thiol-trapping methods for example, to better understand at the molecular level the role of SELENOT in N-glycosylation. This essential co- or post-translational modification contributes to protein folding, stability, and transport, and mediates cell adhesion and repulsion, protein dimerization properties, and other processes required when cells communicate with each other. A role of SELENOT in N-glycosylation could in fact provide the basis to understand a number of alterations in cell adhesion, differentiation, and secretion, observed in genetically engineered animals and cell models with SELENOT depletion (3, 14, 19, 40, 68, 92, 112).
SELENOT Couples Metabolic Activity with Differentiation and Secretion
Initially, SELENOT was characterized as a gene overexpressed in PC12 cells exposed to PACAP (38), a model of neuroendocrine differentiation. When SELENOT gene expression was silenced, PACAP-induced neuritogenesis was inhibited, and this was associated with oxidative stress and decreased cell viability. PACAP-induced SELENOT gene expression was found to rely in part on the activation of the canonical cAMP/protein kinase A (PKA) pathway and to be Ca2+ dependent (38), which fits well with the trophic effects of the neuropeptide and its known transduction pathways in neuroendocrine cells. However, when the SELENOT gene promoter was characterized, additional signaling components impacting SELENOT expression were highlighted (3).
Using comparative bioinformatic analysis, several binding sites for nuclear respiratory factor 1 (NRF-1) could be identified in the SELENOT promoter, which are conserved in evolutionary distant species (from human to chicken). Chromatin immunoprecipitation experiments and promoter studies revealed that NRF-1 governs both basal and PACAP-induced SELENOT gene expression. NRF-1 is a basic region-leucine zipper transcription factor that activates the expression of nuclear genes essential for mitochondrial biogenesis and function, and some key metabolic genes regulating cellular growth (104, 105).
In line with these activities, NRF-1-dependent SELENOT expression was found to rely in part on the upstream activation of AMP-activated protein kinase (AMPK), a key kinase that acts as a fuel gauge regulating energy metabolism by switching on catabolic pathways while switching off anabolic pathways and stimulating mitochondrial biogenesis (42, 43). This cascade was also dependent on peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), which is a transcriptional coactivator of NRF-1 and CREB, and which links external physiological stimuli to the regulation of mitochondrial biogenesis (130).
Altogether, these observations indicate that PACAP reprograms cell metabolism via the activation of the AMPK/PGC-1α/NRF-1 cascade and the concomitant stimulation of SELENOT expression. Such a signaling cascade could allow cells to cross a critical metabolic checkpoint (32) with a tight control of ER and oxidative stress, and to proceed successfully toward differentiation. Of note, PACAP also stimulates catecholamine secretion from PC12 cells, a process that may also require SELENOT as previously reported, linking SELENOT effects in secretion and differentiation (38, 40, 92). Overall, these observations suggest that SELENOT exerts a pivotal role in cell metabolism that could impinge various cellular functions.
Responsible for the correct folding, secretion, and membrane insertion of proteins representing a third of the proteome in eukaryotic cells, the ER is crucial to achieve the extensive protein synthesis required to undergo normal development and maintenance of tissue homeostasis (47, 61, 110). ER structure is modified to adapt lipid and protein synthesis and Ca2+ storage.
SELENOT is in fact widely and abundantly expressed during this period, in line with its role in cell metabolism and ER proteostasis (100, 120). The SELENOT mRNA is detected at embryonic day 7 in rat embryo and placenta (38). A strong expression is observed at embryonic day 20 in many developing organs (brain, thymus, liver, kidney, testis, adrenal), whereas other organs like the gastrointestinal tract or the skeletal muscle showed a less intense expression (38, 120). In the developing brain, a strong SELENOT signal is found in neural progenitors located in the subventricular zone, the intermediate zone, and the cortical plate, and in neurons of the external germinative layer of the cerebellar cortex (120). This pattern is conserved during evolution, since in C. elegans the SelT1.1 isoform is expressed throughout development in most cells from prebean embryonic stages at the epidermal enclosure stage to the adult stage (100).
When the SELENOT gene was specifically disrupted in neural precursors (mutant Nes-Cre/SelTfl/fl mice), several brain structures including the hippocampus, cerebral cortex, and cerebellum showed a significant growth retardation during the first postnatal week (19). Their volume reduction was associated with an increase in neuroblast death by apoptosis, which could be linked to an elevation of ROS levels. Despite a morphological compensation in adulthood, mutant mice exhibit hyperactive and anxiety-like behaviors, raising the possibility that SELENOT is also required for neural network maturation. Disturbances in ontogenesis are also observed in pancreatic islets of SELENOT-insKO mice, as morphometric analyses showed that the islets were smaller and their number increased in mutant mice compared with controls (92).
In line with these developmental abnormalities, some data support a crucial role of SELENOT in adhesion processes. Hence, cell adhesion is impaired in murine fibroblasts with decreased SELENOT levels (112), and HEK293 cells depleted in SELENOT have a deficit in the maturation of GPI-APs, a class of proteins comprising adhesion molecules such as semaphorins, ephrins, and neural cell adhesion molecule 1 (56, 123). These processes being fundamental during implantation and placentation, they may explain the embryonic lethality observed as early as embryonic day 8 in SELENOT-KO mice and which may occur earlier (14).
As mentioned above, SELENOT expression at adulthood is restricted to different endocrine cell types. It is highly abundant in the spermatogenic and the testosterone-producing Leydig cells of the testis (120), in lactotrope, gonadotrope, thyreotrope, corticotrope, and somatotrope cells of the pituitary (40), or in β and δ cells of the pancreas (92), suggesting its implication in major functions, including growth, reproduction, and energy metabolism.
Endocrine cells are devoted to the regulation of body homeostasis, and selenoproteins such as SELENOT could play a key role in this vital process. For instance, the hypothalamo-pituitary axis is responsible for the neuroendocrine adaptation to the stress response. This response is characterized by the hypothalamic release of corticotropin-releasing factor (CRF), which binds to CRF receptors on the anterior pituitary gland and stimulates adrenocorticotropic hormone (ACTH) secretion.
CRF was able to stimulate SELENOT expression in pituitary corticotrope tumor cells (AtT20) (40). In addition, CRF-induced ACTH secretion was abolished after SELENOT gene knockdown, indicating that SELENOT is essential to adapt secretion to environmental cues. Supporting this idea, SELENOT levels are not homogeneous between pituitary cells of the same population, which may be related to their distinct secretory status, a phenomenon well described for different endocrine cell types (36, 37, 81). In the periphery, SELENOT was detected in β-pancreatic cells, and it has been shown that glucose tolerance after an oral administration is altered in conditional β-pancreatic cell SELENOT-KO mice, suggesting that SELENOT is essential to adapt insulin production/secretion to changing glucose levels (92). Further supporting a role in β cell function, SELENOT was also able to potentiate PACAP-induced insulin secretion.
In summary, SELENOT, as a target of the trophic AMPc/PKA and metabolic AMPK/PGC-1α/NRF-1 signaling pathways, profoundly impinges ontogenesis, cell differentiation, and secretion. These functional studies corroborate its implication in N-glycosylation and ER homeostasis, being overexpressed when the folding and quality control machinery has to adapt to the massive protein synthesis required to undergo normal development and maintenance of endocrine/neuroendocrine tissue homeostasis (47).
SELENOT Promotes Cell Survival in Pathophysiological Stressful Conditions
SELENOT expression has been shown to be upregulated after exposure to a number of noxious pathophysiological signals, including oxidative stress, ER stress, inflammation, heavy metal, and neurotoxin exposure.
In vitro exposure to oxidative stress (H2O2) or ER stress (tunicamycin or dithiothreitol) inducers leads to an increase in SELENOT levels, and cells in which SELENOT levels have been reduced experimentally were less resistant to all these stressors (3, 14, 19, 40). SELENOT was hardly detected in the normal brain, but its expression increased in the rodent brain in hypoxic conditions (49) and in neurons and astrocytes of the nigrostriatal pathway in the mouse MPTP model of PD (14). These observations are consistent with data obtained in humans; that is, SELENOT overexpression in the caudate putamen of postmortem PD patients (14). When Nes-Cre/SelTfl/fl mice were exposed to MPTP, the neurotoxin effect resulted in the death of the majority of KO animals within 2 h after injection, in contrast to wild-type animals, which survived normally in these conditions (14).
Likewise, SELENOT expression was undetectable in the adult heart, but cardiac SELENOT expression was induced after ischemic injury (98, 99). SELENOT levels increased in liver and spleen in fish (C. auratus) after exposure to different stressors, such as cadmium or H2O2 (21). Increased SELENOT expression was also observed in spleen and thymus during lipopolysaccharide-induced inflammatory response in pig (117). Furthermore, SELENOT was upregulated in the mouse liver during its regeneration after hepatectomy (120).
It should be mentioned that SELENOT mRNA levels may vary oppositely according to the nature of the stress. For example, isoproterenol and T3 both induce cardiac hypertrophy, but SELENOT levels are either down- or upregulated by these agents, respectively (48). Likewise, in cancer cells, SELENOT is overexpressed in the hepatocarcinoma HepG2 and HUh7 cell lines (39, 136), while it is reduced in paracarcinoma and in gastric cancer, compared with controls (63).
Hence, these studies point to an important role of SELENOT in pathophysiological stressful situations. Together with the high SELENOT expression in endocrine cells, which have to face a physiological stress during sustained secretion, these observations highlight the role of SELENOT in the ER, allowing different cells to cope with various stressors.
Conclusions and Future Directions
The ER is the principal subcellular compartment involved in the quality control and folding of secretory and membrane proteins. Protein folding in the ER is a complex and error-prone process. When misfolded proteins accumulate in the ER, ROS increase and Ca2+ leaks from the ER, thus compromising cell viability if the stress is not resolved (20, 22, 25, 41, 91, 129). ER stress actually contributes to the etiology of many human diseases. For instance, the accumulation of misfolded and aggregated proteins is a common characteristic of neurodegenerative diseases (106). It is also the hallmark of cancer, diabetes, metabolic syndromes, cardiovascular and inflammatory/immune diseases, many diseases for which the restoration of ER homeostasis is proposed as a new potential therapeutic target (31, 34, 55, 77, 128). Because UPR activation is inextricably linked to the maintenance of ER and cell homeostasis, finding UPR biomarkers and targets is a major therapeutic issue (7).
Major breakthroughs have recently been made on the function of SELENOT, showing that it is essential to life and to cope with cellular stress situations, making it one of the most important selenoproteins. Even if the precise function of this selenoprotein remains to be further investigated, recent data identified SELENOT as an ER-resident protein and a partner of KCP2, a subunit of the STT3A-type OST complex involved in cotranslational N-glycosylation. Through its involvement in N-glycosylation, a process that is essential for glycoprotein quality control system, it is very likely that SELENOT plays a crucial role to maintain ER homeostasis when cellular metabolic activity increases, such as in secreting or in differentiating cells, to ultimately ensure that its protein-folding capacity is not overwhelmed (Fig. 7A, B).

Owing to the important biological functions of oligosaccharides in glycoproteins, incorrect synthesis of these compounds results in diverse human diseases with up to 100 inherited disorders, named congenital disorders of glycosylation (CDG) (52). SELENOT could be a candidate gene in these diseases, but the possibility of a genetic alteration in SELENOT has not yet been documented, neither in these diseases nor in any other pathology, while mutations or polymorphisms in two other ER-resident selenoproteins, SELENON and SELENOS, are associated with human pathophysiological conditions, such as myopathies and inflammation, respectively (26, 87). The increase in SELENOT expression found in the caudate putamen of PD patients and in different pathophysiological animal models also suggests that SELENOT could be a new biomarker of these disorders (14).
Mainly expressed during embryogenesis, SELENOT contributes to the development of many organs, including the brain and the pancreas. The studies performed with the mutant Nes-Cre/SelTfl/fl mice provide valuable clues regarding the role of SELENOT during neurogenesis. Indeed, the volume reduction of several brain structures (neocortex, cerebellum, and hippocampus) observed during the first postnatal week and the hyperactive behavior observed in this mutant at adulthood indicate that SELENOT is also critical for the formation of functional neural circuits.
The recent finding of SELENOT requirement for the N-glycan-dependent maturation of GPI-anchored protein is highly relevant in this regard. Indeed, it is now well established that a subset of GPI-APs, as cell adhesion molecules, have an impact on brain development and function through their involvement in neuritogenesis and synaptogenesis (75, 84). Also, a number of neurodevelopmental disorders, such as autism spectrum disorder, are associated with mutations of several GPI-APs, including contactin-4, -5, -6 (90) and Ntm (16). Further studies are warranted to confirm in Nes-Cre/SelTfl/fl model the influence of SELENOT on GPI synthesis.
Altogether, these observations suggest that SELENOT participates in various physiological processes, including hormone/peptide secretion, growth, neuroprotection/regeneration and inflammation, or pathological conditions such as cancer, which are associated with an increase in energy demand and stress. In particular, altered N-glycosylation and ER stress, which are observed in the hypothalamus and pancreas, may lead to metabolic perturbations, and in fine, to obesity, diabetes, and the metabolic syndrome, as recently reported for the ER-resident SELENOM-deficient mice (33).
A better understanding of the mechanisms linking ER selenoproteins and protein folding and redox homeostasis regulation might lead to the development of novel therapeutic targets for different human diseases.
In support of this notion, it is interesting to note that a SELENOT-derived peptide, SELENOT43–52 (PSELT), encompassing the Sec-containing redox motif, inhibits oxidative and nitrosative stress and exerts a potent pharmacological postconditioning protective effect in the heart after ischemic injury (98). PSELT-dependent cardioprotection is accompanied by a significant increase in phosphorylated Akt, Erk-1/2, and Gsk3α-β and a reduction of apoptosis. Another brain-penetrant selenopeptide-TAT (SelPep), composed of a TAT peptide attached to a stretch of amino acids derived from the C terminus of SELENOP, has been designed to deliver Se into the cells. SelPep possesses a wide therapeutic window in vitro and in vivo for hemorrhagic stroke and possibly other central nervous system (CNS) and non-CNS conditions associated with ferroptotic death. It drives a TFPA2c and Sp1-dependent transcriptional response, including genes of the selenome, to counteract reactive lipids and cell death (5).
Our preliminary results indicate that PSELT could exert protective effects in various diseases, associated with cellular stress and degeneration. This opens up a new avenue in clinical research, especially for different degenerative and metabolic diseases associated with oxidative and ER stress, for which no preventive or curative therapies are currently available.
Footnotes
Funding Information
This work was supported by Inserm, Rouen Normandie University, Conseil Régional de Normandie, the FEDER program of the European Union (ERDF) (PACT-CBS 2016–2019 and PHEDERCPG 2017–2019 programs). Europe gets involved in Normandie with ERDF. Hugo Pothion was supported by a fellowship from the Federation Hospitalo-Universitaire “SURFACE.”
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Figure S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.