
Abstract
Significance:
Alongside well-characterized permanent multimeric enzymes and multienzyme complexes, relatively unstable transient enzyme–enzyme assemblies, including metabolons, provide an important mechanism for the regulation of energy and redox metabolism.
Critical Issues:
Despite the fact that enzyme–enzyme assemblies have been proposed for many decades and experimentally analyzed for at least 40 years, there are very few pathways for which unequivocal evidence for the presence of metabolite channeling, the most frequently evoked reason for their formation, has been provided. Further, in contrast to the stronger, permanent interactions for which a deep understanding of the subunit interface exists, the mechanism(s) underlying transient enzyme–enzyme interactions remain poorly studied.
Recent Advances:
The widespread adoption of proteomic and cell biological approaches to characterize protein–protein interaction is defining an ever-increasing number of enzyme–enzyme assemblies as well as enzyme–protein interactions that likely identify factors which stabilize such complexes. Moreover, the use of microfluidic technologies provided compelling support of a role for substrate-specific chemotaxis in complex assemblies.
Future Directions:
Embracing current and developing technologies should render the delineation of metabolons from other enzyme–enzyme complexes more facile. In parallel, attempts to confirm that the findings reported in microfluidic systems are, indeed, representative of the cellular situation will be critical to understanding the physiological circumstances requiring and evoking dynamic changes in the levels of the various transient enzyme–enzyme assemblies of the cell. Antioxid. Redox Signal. 35, 788–807.
Introduction
Protein interactions play an important role in the proper functioning of living cells, with broad-scale screens in humans having proposed the presence of ∼130 000 binary protein–protein interactions at any point in time (18, 52), thereby underlining the commonality of such interactions. Different categories of protein–protein interactions have been defined in former studies (97, 105). These definitions include whether a complex is obligate or nonobligate (105). In obligate complexes, the promoters of the participating proteins are not found as stable structures on their own, unlike the situation observed in nonobligate complexes. Obligate complexes mostly reflect the need for stability or the evolution of a function that requires both promoter and inter-subunit active sites, which means the protein–protein interfaces are generally larger and more hydrophobic than those associated with nonobligate associations.
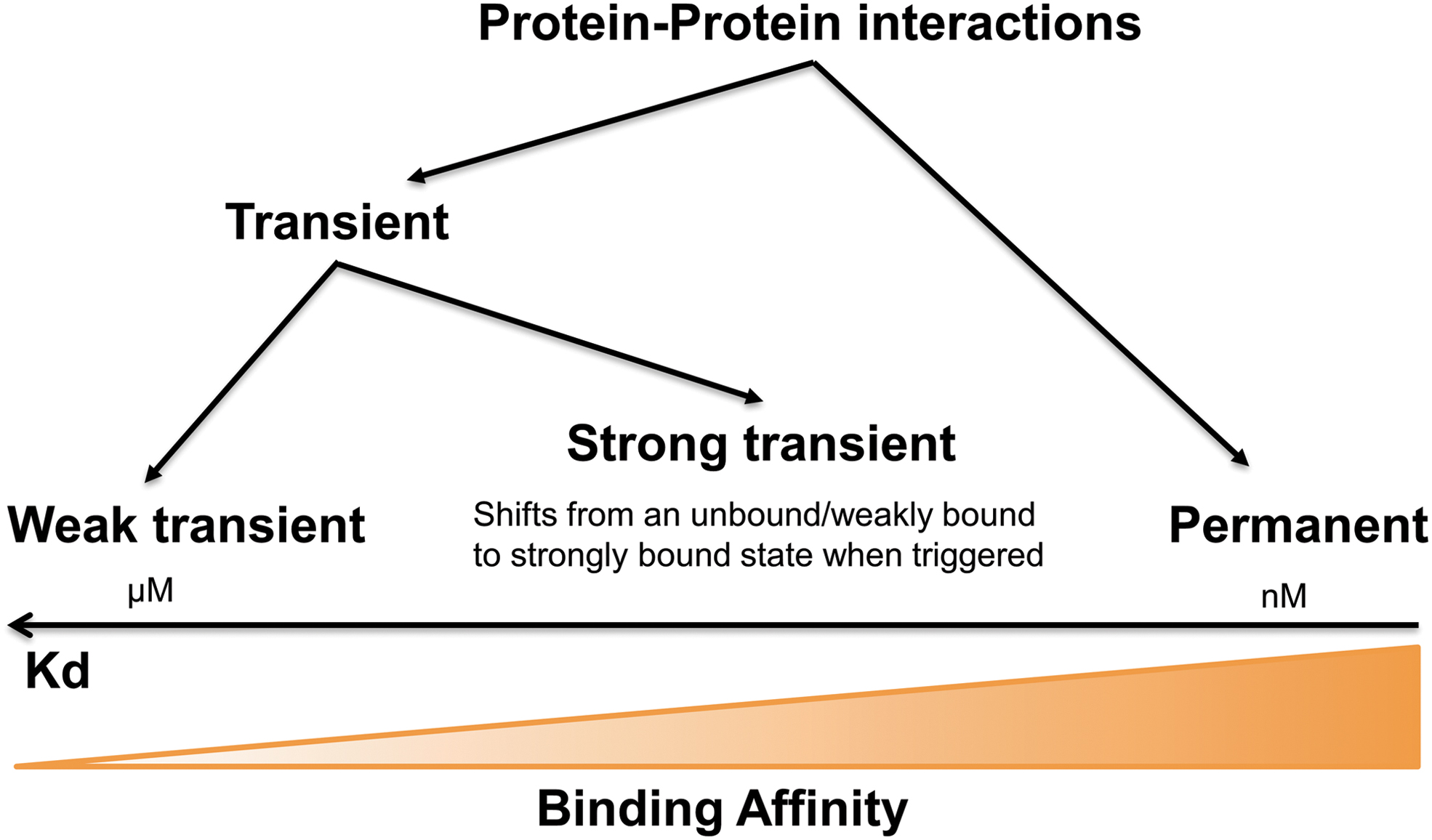
In addition, nonobligate complexes can exist independently as hetero-oligomeric structures such as antibody–antigen, receptor–ligand, and enzyme–inhibitor complexes (97, 105). Complexes can additionally be either homomeric or heteromeric in nature (105). A further distinction that is often made is that between permanent and transient complexes, with the basis of this distinction being subunit affinity. Permanent protein–protein interactions are irreversible and strong, whereas the interaction qualifies as transient if it freely undergoes changes in its oligomeric state (105). These types of interactions can easily be distinguished from one another biochemically on the basis of their binding affinities that are inversely related to the dissociation constant (Koff/Kon) KD. Although as illustrated in the cartoon of Figure 1 in nature these values form a continuum, permanent interactions usually display strong binding affinities (in the nM range) and weak transient interactions show rapid bound to unbound equilibrium with KD values in the μM range. Such a range is in keeping with recorded local concentrations (105), underscoring the biological/physiological relevance of such interactions. Such low KD values do, however, render their study difficult, necessitating the use of several strategies as described next.
In the major pathways involved in energy and redox metabolism in plants (44, 112), microbes (33, 145), and mammals (126), both obligate and nonobligate, homomeric and heteromeric, and permanent and transient enzyme–enzyme, enzyme–protein, and protein–protein interactions have been characterized (145). We will discuss these later in detail in the designated sections. However, before doing so, we feel it important to provide background into (i) how these different types of interactions occur, (ii) how they are experimentally detected, (iii) how they evolved, and (iv) their functional significance. Although we will describe all types of interactions, our major focus will be on metabolons—temporary enzyme–enzyme assemblies that form between sequential enzymes of a metabolic pathway—that are involved in substrate channeling (40, 166). Cellular processes have been suggested to be organized via spatial micro-compartmentalization and the assembly of multienzyme complexes in metabolons (17, 101). The term “metabolon” is much overused and erroneously often simply taken to be synonymous with an enzyme–enzyme assembly; as previously stated (40, 141), a metabolon is a subclass of enzyme–enzyme assemblies that acts to physically channel metabolites from the active site of one enzyme to that of another without exposure to the bulk solvent. The components of a metabolon can be specific to a single metabolon or dynamically shared between metabolons for swift adaptation of metabolism to environmental challenges and cellular needs (40, 101, 141). Such an organization of metabolic pathways at the molecular level has been postulated to have several advantages, including (i) increasing local concentrations of the enzymes and their substrates, (ii) improving channeling of intermediates into specific subpathways, (iii) increasing metabolic fluxes, and (iv) sequestration of reactive intermediates (61, 71, 109). However, the evidence in support of these postulates varies depending on the metabolon in question and as previously stated (141). Indeed, a more likely universal reason for their formation is to ensure that metabolic fluxes are regulated in a manner befitting the cellular circumstance. Before discussing both stable and transient complexes in specific pathways, we felt it useful to provide a brief historical overview of their study. Our main focus will be on enzymes; however, we will also mention both members of electron transport chains and transporter proteins where these have a strong influence on metabolism. Throughout the article, we adopt the terms “enzyme–enzyme assembly” for enzymes that are merely demonstrated to interact and we reserved the term “metabolon” for the subset of enzyme–enzyme assemblies for which substantial evidence of metabolite or substrate channeling exists. We intend to provide case study analyses of five key pathways of energy and redox metabolism, namely glycolysis, the tricarboxylic acid cycle (TCA cycle), the mitochondrial electron transport chain, the Calvin-Benson cycle, and photorespiration, before attempting to define general features of the enzyme assemblies in these pathways and to interpret why at certain steps of the pathway stable as opposed to transient interactions have evolved and vice versa. Finally, we review exciting pertinent developments emanating from cell-free systems. While stressing that proof for their physiological relevance is currently lacking, we expound on the potential importance of these findings. Wherever possible, we will review the available data at a cross-kingdom level.
Types of Enzyme–Enzyme Interactions
Multimeric enzyme complexes
There are a staggering number of enzymes that only function or only optimally function as multimeric enzyme complexes characterized by permanent interactions; examples of such enzymes are aldolases, oxidases, catalases, many galactosidases, and many dehydrogenases (38, 40). With regard to the pathways, this review is concerned with a complete list of multimeric enzyme complexes that is provided in Table 1. In cases where proteins are multimeric, attachment of one enzyme to several scaffold proteins could allow crosslinking to form larger structural protein complexes, which often possess low stability (96, 125). In addition, the stabilization of multimeric proteins relates to the dissociation of the enzyme subunits or the loss of their correct assembly structure (38). Thus, examining multimeric enzyme complexes depends on experimental conditions that mimic the enzyme's native environment (presence of ions, polymers, etc.), the existence of inter-subunit disulfide bridges, or chemical- or physical-crosslinking with poly-ionic polymers (38). Given that there is a vast literature regarding the study of such multimeric enzyme complexes (38, 96, 125, 127) and the fact that they appear to be universally characterized by co-evolution of the many residues that mediate their binding at the subunit interface (38), we will not extensively discuss such interactions here with the exception of a brief comparative evaluation of the evolution of permanent and transient enzyme complexes in The Evolution of Protein Complexes section.
Comparison of the Different Types of Enzyme–Enzyme Assembly
Multienzyme complexes
Several copies of one or several enzymes (polypeptide chains) are stably associated by strong permanent noncovalent interactions in multienzyme complexes, which carry out a single or a series of biochemical reactions such as the pyruvate dehydrogenase (PDH), 2-oxoglutarate dehydrogenase (OGDH), glycine cleavage system, fatty acid synthase, glutamine synthase, proteasome, and Rubisco complexes (118). As discussed earlier, with regard to the pathways this review is concerned with a complete list of multienzyme complexes that is provided in Table 1. These multienzyme complexes have been studied to varying levels; although for some the subunits are unique to the complex, for others the subunits are shared between complexes, for example mitochondrial dihydrolipoyl dehydrogenase is shared as an integral component of several multienzyme systems, including the TCA cycle (PDH complex, OGDH), photorespiration (glycine decarboxylase complex [GDC]), and the degradation of branched-chain α-ketoacids (branched-chain 2-oxoacid dehydrogenase complex, BCDHC) (98, 143). The reaction intermediates of the multienzyme complex are entirely enclosed in internal chambers within the protein complex, which directly renders diffusion between active sites of the component enzymes or uses swing arms to physically mobilize intermediates or cofactors (141). Multienzyme complexes can dramatically improve the catalytic efficiency of the reaction chains in question, having even been argued to have reached catalytic perfection (63, 135). Given that channeling clearly exists in these complexes, their comparison with metabolons is particularly apt and we also attempt this in The Evolution of Protein Complexes section. Similar to the multimeric enzymes described earlier, the mechanisms underlying the interaction are largely understood, although the presence of subunits in multiple different protein complexes is probably more common for this type of interaction, thus rendering the evolutionary pressure that, at least some of, the subunits receive unequal. Unfortunately, due to space constraints, we cannot comprehensively review this type of permanent enzyme complex here, so we rather refer the reviewer to a number of excellent recent reviews on this subject (104, 123, 125, 149).
Transient enzyme–enzyme assemblies
Transient enzyme interactions include protein interactions that are formed and broken easily, that is, relatively unstable complexes that play an important role in many aspects of cellular function (3, 105, 145). Transient interactions are characterized by dissociation constants (koff/kon)—KD—in the micromolar range and lifetimes of seconds (Fig. 1), whereas strong transient interactions may have lower KD's in the nanomolar range and longer lifetimes. Commonly weak transient complexes are represented by dynamic mixtures such as protein phosphorylation or dephosphorylation in vivo, whereas strong transient complexes change their quaternary state, for example when triggered to do so on ligands. Although transient protein interactions play an important role in biological systems, notably in regulating the dynamics of biological networks, their experimental detection is not easy (3). In addition, the transient interactions of sequential enzymes of several metabolic pathways may well participate in the substrate channeling formation inside living cells. As such, metabolons represent a subset of transient assemblies, relying on delicate local changes in solutes, structural elements, and possibly scaffolding proteins to allow their operation (61). Given the importance of this subset of assemblies, we will discuss them next in detail.
The metabolon
The concept of physical enzyme–enzyme complexes was initially conceived in 1970 by A.M. Kuzin of the USSR Academy of Sciences (68), and it was subsequently adopted in 1972 by Paul A. Srere of the University of Texas for the enzymes of the citric acid cycle (130). The hypothesis that the structural assembly of enzymes was common and explained many of the inconsistencies between theoretical expectations and biological observations was well accepted in the former USSR and further developed in B.I. Kurganov and A.E. Lyubarev's finding of complexes of glycolytic enzymes (66, 67). In the mid-1970s, the group of F.M. Clarke at the University of Queensland, Australia also provided considerable support for the concept (24). The name “metabolon” was first coined in 1985 by Paul Srere during a lecture in Debrecen, Hungary (113), who went on to provide the first detailed experimental study of the phenomenon while working on the mitochondrial TCA cycle (131, 166). From these studies, he notes that “through the dynamic association and/or dissociation, transient complexes allow the regulation of metabolic pathway flux” (17, 40, 108, 148). As mentioned earlier, metabolons mediate “substrate channeling” (also known as metabolic channeling) wherein reaction intermediates are isolated from the bulk environment surrounding them. Various advantages of substrate channeling have been postulated, including isolation of intermediates from competing reactions, local enrichment of metabolite concentrations to achieve high reaction rates, sequestration of cytotoxic metabolites, and protection of unstable intermediates (129, 131, 154). Further, during the functioning of metabolons, the amount of water needed to hydrate the enzymes is decreased and enzyme activity is enhanced. A vast number of metabolons have been proposed to mediate substrate channeling in various organisms, including those proposed to operate in branched-chain amino acid metabolism in human mitochondria (56); the glycolytic pathways of mammals, yeast, and plants (9, 48, 88, 107); and a wide variety of specialized metabolic pathways, including polyamine (103), isoprenoid (74), alkaloid (155), and a number of different phenylpropanoid pathways [including lignin, carotenoid, flavonoid, isoflavonoid, and cyanogenic glucoside synthesis in plants (14, 42, 71, 72, 124)]. That said, strictly speaking, metabolite channeling must be observed to claim the presence of a metabolon and not all enzyme–enzyme assemblies are metabolons. Based on this requirement, there is relatively limited evidence for the presence of metabolons. Indeed, to our knowledge in plants: only glycolysis (48, 88), the TCA cycle (166), and the cyanogenic glucoside biosynthetic pathway (71) have been demonstrated to operate in this way, with all other structures that were described as metabolons to date being more accurately described, on the basis of cumulative evidence, merely to be enzyme–enzyme assemblies. This is not said to diminish the importance of the interactions in question, merely to reinforce a more rigorous definition of the term “metabolon” (40, 108, 141).
Experimental evaluation of enzyme–enzyme interactions and substrate channeling
We have earlier discussed how protein–protein interactions could drive both metabolic and regulatory events and how large macromolecular complexes are highly stable and even permanent, whereas dynamic and transient interactions are key components with regard to signaling, regulatory networks, and the formation of substrate channels (3, 105). Given the instability of transient complex formation, it is technically difficult to study and harder to detect weak protein–protein interactions, compared with the more stable interactions exhibited by multimeric enzymes and multienzyme complexes. That said, high-throughput yeast two-hybrid (Y2H) screens are able to detect binary transient interactions (105). However, similar to other experimental detection methods of protein–protein interaction, Y2H has its own weaknesses, including a high rate of false-positive detections with attempts to ameliorate this problem, often resulting in a reduced overall ability of the method to detect transient interactions (20). Despite this fact, it is still the popular method for large-scale detection of protein–protein interactions, owing to its scalability and accessibility (20). Further, several recent developments to this approach enabled the detection of additional types of interactions, including those involving cytosolic, extracellular, or membrane-bound proteins (71, 171). These developments, notwithstanding proof of complex formation, are now almost universally required to not be based on a single technique alone and, as such, Y2H is usually used in tandem with another technique or in teaming with multiple techniques. In keeping with this development, several research efforts have focused on bioinformatics integrating different protein–protein interaction methods to gain reliable interaction lists (142). For instance, the recent database STRING V11 collects, scores, and integrates all publicly available experimental sources of protein–protein interaction information, and it allows comparison of these with computational predictions (142). In addition to this, considerable research works have focused on decreasing the problem of false-negative protein–protein interactions by improving the protein interaction method. For example, two recently developed approaches, namely bioluminescence resonance energy transfer (BRET) (82) and fluorescence/Foerster resonance energy transfer (FRET) (50), were developed to detect protein interactions in living cells. For this purpose, one protein is fused to NanoLuc luciferase or mCitrine and the other protein to Halotag and mCherry. After introducing the fusion proteins via transgenes, BRET and FRET between the two proteins are measured (50, 82). These approaches were demonstrated to have the additional benefit of highlighting transient, membrane-bound, and extracellular protein interactions, since they detect the interactions on the basis of physical distance (3). Measurements yielded by these methods thereby supply a noninvasive means by which to visualize the spatiotemporal dynamics of interactions between protein partners in vivo. In addition, use of confocal laser scanning microscopy may enable the direct imaging of substrate channels after the transient or stable expression of target proteins as immunologically tagged proteins or as fluorescent-fusion proteins (61, 154). Direct visualization of the substrate channel, allowing assessment of their association by sequential incorporation of their component polypeptides, can now also be achieved by atomic force microscopy or the fluorescence lifetime imaging microscopy-based FRET (71). Moreover, new methodologies such as the use of Clear-Native-polyacrylamide gel electrophoresis, crosslinked mass spectroscopy, proximity-dependent biotin identification (BioID), and ascorbic acid peroxidase enable the isolation and characterization of enzyme–enzyme assemblies in which adherence between the different subunits is weak (62, 114, 156). Another well-developed high-throughput protein–protein interaction detection method is affinity purification followed with mass spectroscopy (AP-MS) (163, 164), which allows rapid purification protein complex under native conditions, even when expressed at their native level. As several washing steps are required to remove nonspecific binding, this method has traditionally been unable to detect the transient interaction. However, with the use of in vivo chemical crosslinking, it has become possible to detect transiently formed complexes by inducing covalent-bond formation between interacting proteins (73). In addition, technologies subsequently allow the preservation of the crosslink during the washing phase of affinity purification (73, 157).
Despite advances in the implementation of the techniques described earlier, a functionally important subset of transient interactions is dependent on post-translation modification events. Such interactions are often missed when protein–protein interaction screening is performed in Y2H (73). This limitation has been tackled in different ways, including the use of native cell culture systems, an approach that remains constrained by the difficulty of transforming either mammalian or plant cell cultures. Among other methods that feature the use of endogenous cell systems for protein–protein interaction screening are bimolecular fluorescence complementation (BiFC) (65, 159, 163) and the split-luciferase assays (147), both of which are able to detect transient (as well as permanent) protein–protein interactions in vivo, thereby eliminating the need for high-throughput purification studies. The characteristics of the various experimental techniques to identify transient protein–protein interactions are summarized in Figure 2.
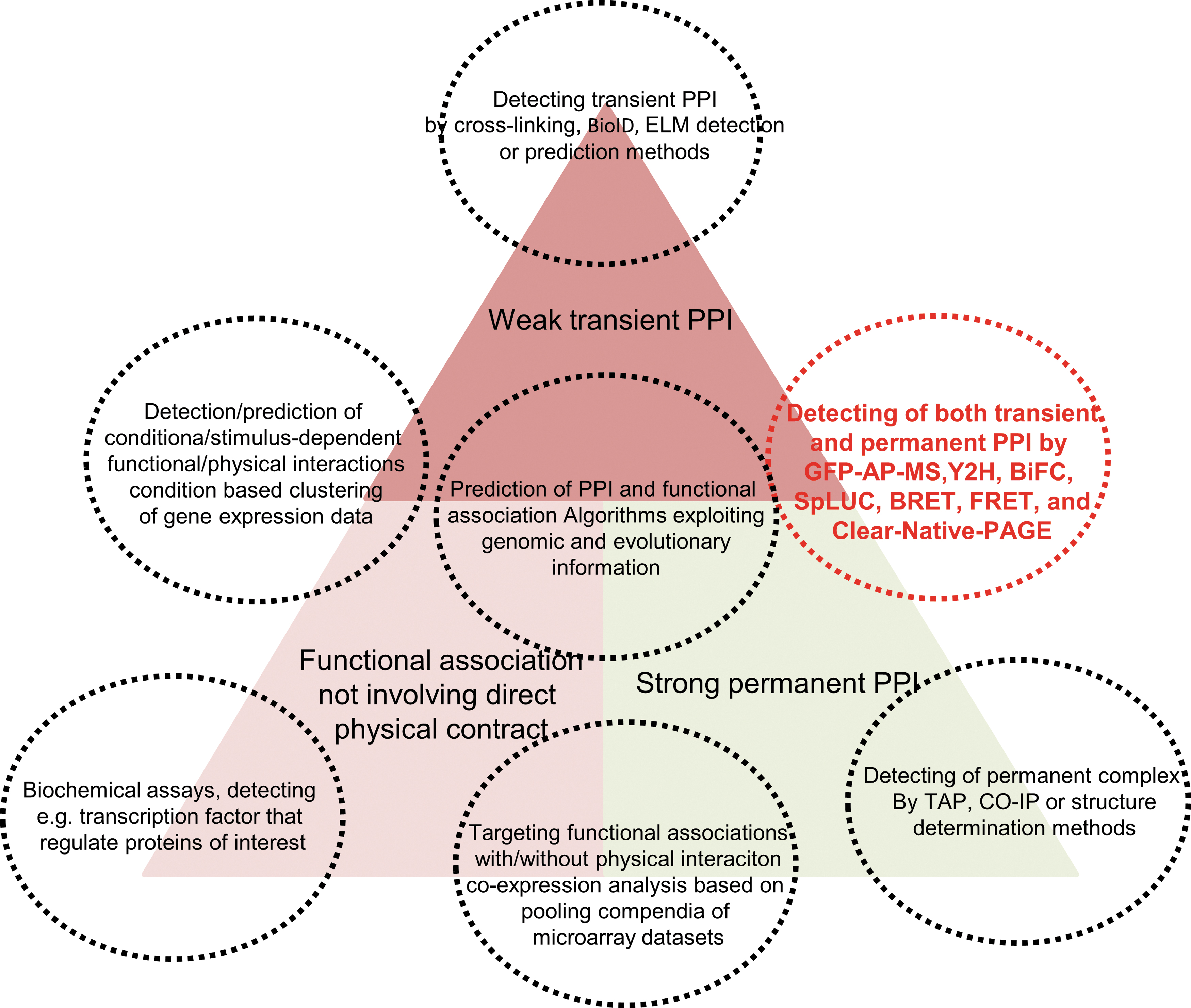
Recently developed approaches give us various options to test protein–protein interactions (69, 159). Indeed, the reports discussed here utilize various techniques to investigate protein complex formation as well as the interactions between specific enzyme pairs (Fig. 2). However, there is still no perfect approach that can study protein–protein interactions without any false-negative and -positive detection. This renders the validation and screening of novel enzyme–enzyme assemblies complicated. The Y2H assays, co-localization of fluorescently tagged proteins, BiFC, and co-purification are commonly used to identify enzyme–enzyme complexes (65, 69, 141, 159, 164). All these approaches tend to generate a lot of false-positive results and have to be conducted with special care, paying particular regard to the use of proper negative controls (65, 159). In addition, given the relatively high false-positive and -negative rates of these assays, evidence from multiple independent approaches is required to accurately call the presence of enzyme and enzyme assemblies (166). The use of multiple techniques greatly aids in the reduction of false-positive results, thereby improving the reliability of the data. However, these techniques require special equipment and expertise and their use is often limited to highly specialized research groups. When the purpose of research comes to the screening of protein complexes within a large set of enzymes or interacting protein pairs, it becomes increasingly complicated to gain reliable results. The use of multiple techniques is, therefore, essential to avoid not only false-positives but also false-negatives. Given the transient nature of nonpermanent enzyme–enzyme assemblies, their associations are most likely dependent on the microenvironments that the enzymes find themselves in. As such, use of a single approach may provide an improper microenvironment, resulting in false-negative results. Conversely, problems arise to integrate the results from multiple techniques and in setting an appropriate threshold at which to declare a protein pair or multiple proteins as interacting with one another. In the screening of protein–protein interactions within the plant TCA cycle, a combination of three rather conventional methods, split luciferase complementation, Y2H, and AP-MS, was used (165, 166). All three of these approaches provide information on in vivo protein–protein interactions; however, the principle of detection in all instances differs from one another. Importantly, however, they generate semiquantitative scores that can be statistically combined to produce a single reliability score. This method successfully identified expected and novel elements of plant TCA cycle metabolon (77) and, as such, provides a robust framework for the screening of physiologically occurring enzyme–enzyme assemblies.
The techniques described earlier represent the state-of-the-art in detecting enzyme–enzyme assemblies, whereas the specific identification of metabolons requires stricter proof than is commonly provided (40, 141, 166, 170). The identification of enzyme–enzyme assemblies that include consecutive enzymes certainly represents good candidates for complexes that may contain metabolons. That said earlier, describing such assemblies as metabolons or even metabolon containing proof of substrate channeling is required. As mentioned earlier, this has been achieved for a subset of the reactions comprising the glycolytic and TCA cycle pathways (45, 48, 88, 157, 166). Such proof is nontrivial. Given that it is debated in length in a recent review (141), we will not reiterate the arguments made there merely to briefly state that both isotope labeling (48, 57, 141) and non-isotope labeling approaches (100, 125, 151) have been used with those of greatest relevance to the pathways covered in this review being summarized in Figures 3 and 4.


The Evolution of Protein Complexes
A relatively recently expanding research field concerned with enzyme interactions concerns the evolutionary histories of stable enzyme multimers, multienzyme complexes, and dynamic assemblies of enzymes (141, 166). In this vein, considerably more evolutionary-based research has been implemented on the former—with many permanent interactions being highly conserved throughout evolution. For example, pioneering research proposed that the α-ketoacid dehydrogenase complexes appeared very early in evolution, being found in aerobic members of the bacteria, archaea, and eukaryotes (93). Another well-studied multienzyme complex is the fatty acid synthase complex (FAS)—of which two amazingly different types have evolved in eukaryotes: the fungal and metazoan FAS (21). The fungal FAS is associated with a 2.6-MDa assembly comprising 48 functional domains and, alongside the homologous CMN-FAS recently described in Corynebacterium, Mycobacterium, and Nocardia (43), constitutes one of the most complex biochemical protein machineries uncovered to date (49); whereas the metazoan FAS is a 540-kDa homodimer that shares a common architecture with bacterial polyketide synthases (84). A recent study determinates trans-acting polyketide enoyl reductases and non-canonical bacterial fatty acid synthases as potential ancestors of the scaffolding regions with a striking conservation of insertions to scaffolding components despite minimal sequence identity (21). In contrast, many bacteria and plants harbor only monofunctional FASs (22). Another classical example of a multienzyme complex is the penta-functional Arom complex of shikimate biosynthesis, which appears likely to have evolved via gene fusion with the enzymes of most other species being monofunctional (146). A final multienzyme complex that merits discussion is that of tryptophan synthase (75) for which a plausible model by which the sophisticated multienzyme complex evolved via stepwise association of protein subunits has been proposed (28, 75). Research into the evolution of protein complexes is currently largely focused on residues at the interface of the subunits and, as such, on their co-evolution (29, 102).
The evolution of metabolons (and indeed of other transient enzyme–enzyme assemblies) is far less studied. However, as presented in Table 1, these demonstrate a similar range of conservation throughout evolution as the stable multienzyme complexes. In particular, metabolons within glycolysis and the TCA cycle are highly conserved. Others, such as the urea/polyamine substrate channel, are found, at least in part, in several species. However, other substrate channels, especially those involved in fungal and plant specialized metabolism, are far more limited in their species range. That said, despite the widespread occurrence of metabolons (and other types of dynamic enzyme assemblies) and stable multienzyme complexes, there is no reason to suspect that the evolution of the former is an obligatory step toward the evolution of the latter. Indeed, it is likely that the two types of enzyme assemblies have evolved to play quite distinct functions from one another (141). What is almost certain, however, is that less conservation at the interface of the interacting proteins will be apparent in metabolons and other transient enzyme–enzyme assemblies. Moreover, in philosophizing about the role of these transient structures, one could conceivably argue that the flexibility afforded by their transient nature is likely to provide an advantage, rather than a compromise, to fitness. Since this clearly was not the case for multimeric enzymes, multienzyme complexes raise the following fascinating questions: What properties render a certain reaction in an enzyme cascade, or alternatively the protein that catalyzes it, a good candidate to stabilize, and which characteristics rather lead to the maintenance of (relative) plasticity?
We will return to the questions just raised later. On a more general note, the presence of protein aggregates has been argued to be under negative selection (166). However, at the same time, protein aggregation may be an unavoidable consequence of the limited solvency capacity of the cell in relation to the total protein concentration, rendering, as such, functional organization around such aggregates an evolutionary compromise (99). Given that a wider range of oligomeric proteins can adopt multiple structural conformations, there may be unique opportunities for mutations to lead to novel protein–protein contact points (166). Although in many cases novel aggregations will be deleterious, some will be neutral and may even ultimately prove beneficial to the cell (99), in which case it would seem reasonable to anticipate their stabilization. However, recent studies of protein interaction data in the context of evolution have left several unanswered questions (41). Most important of these are: (i) to what extent do protein interactions act as constraints during the evolution of the protein sequence, (ii) what role do transient or obligate interactions play in these constraints, and (iii) are mutations in the binding site of an interacting protein correlated to those in the binding site of its partner? (166). Fascinatingly, studies aimed at answering these questions observed that amino acid residues at the interfaces of obligate complexes evolved at a relatively slower rate, allowing them to co-evolve with their interacting partners (29, 91, 102). By contrast, when the protein–protein interactions are transient, the rate of residue substitution is considerably higher (91). These observations are thus in keeping with our suggestion that multienzyme complexes and metabolons evolved independently from one another. As such, these result in distinct repurposing of the ancestral enzymes. We have previously proposed that the key functional discriminator is the requirement for access of competing enzymes to the channeled metabolite so that metabolic branch points can operate. This would require disassembly of the enzyme complex that supports substrate channeling and would provide a selective pressure against the evolution of a stable multienzyme complex for such a set of reactions (166). Put more simply, we believe that stable multienzyme complexes can only evolve where there are no other cellular reactions that require access to the channeled metabolite; in the absence of such conditions, there is no selective advantage in conservation of residues that improve interface binding and, as such, the complex process of co-evolution is unnecessary (166). To date, very few studies have been carried out looking at the evolution of metabolons but following the assumptions stated earlier we would anticipate much higher conservations of the residues involved in hollow-core or electro-static channeling of metabolites than at the protein–protein interaction interface (59). The results for enzyme–enzyme interactions that do not lead to substrate channeling are, however, much harder to predict.
Protein Complexes in Respiratory and Photosynthetic Metabolism
Respiratory metabolism
Respiratory metabolism, including the pathways of glycolysis, the TCA cycle, and the mitochondrial electron transport chain, provides carbon skeletons for biosynthesis of a series of key metabolites and, as such, is a central feature of metabolic networks across all kingdoms of life and the core of biochemical energy transformation (40). The basic electrochemistry and chemistry of the pathways are highly conserved (39, 121), with the exception of energy parasites such as chlamidiae, mistletoe, and diatoms, which have reduced respiratory function (12, 83, 120, 144). Despite the fact that the pathways operate in widely different physiological settings, a degree of metabolite channeling—the promoted transfer of the metabolite from one enzyme to the sequential enzyme in the metabolic pathway without the intermediates equilibrating with the large amount of aqueous solvent (141)—appears to be a common feature of these pathways across all kingdoms of life. Here, we will direct our attention toward this aspect in respiratory metabolism, describing the nature of metabolite channeling for three metabolic pathways. However, we will additionally provide details of other enzyme–enzyme interactions, that is, those that do not, or at least have not yet been demonstrated to result in channeling. Further, we will discuss these transient interactions alongside discussion of the cardinal multienzyme (and multiprotein) complexes that operate within these pathways.
Glycolysis
The observation of transient assemblies of glycolytic enzymes has an extensive history. Initial, although controversial and indirect evidence, came from the evidence of independent pools of glycolytic metabolites in rats and Escherichia coli (27, 81). Similar evidence of sub-compartmentation of metabolites within the cytosol has subsequently been provided by high-resolution nonaqueous fractionation in the model plant Arabidopsis thaliana (64). However, considerably stronger evidence for assemblies of glycolytic enzymes has been provided, such as direct proof of specific protein–protein interactions between constituent pairs of glycolytic enzymes (40, 141), association of enzymes of glycolysis with actin (86, 94) or red blood cell membranes (27, 86), and complexes of all glycolytic enzymes (27, 86). There is an ample body of research that glycolytic enzymes are associated with red blood cell membranes (34) with an environmentally dependent binding affinity (26). Several glycolytic enzymes (glyceraldehyde phosphate dehydrogenase, aldolase, pyruvate kinase, and lactate dehydrogenase) were proved to bind to an immobilized F-actin-tropomyosin complex (36). However, as a recurrently observed pairing, the specific interaction between constituent enzymes of aldolase and glyceraldehyde phosphate dehydrogenase is the real breakthrough in building the occurrence of glycolytic enzyme associations (132). These interactions were revealed by a diverse range of experimental approaches, including polarization of fluorescence, kinetic studies, affinity electrophoresis (16), and active enzyme centrifugation. However, intriguingly a simple gel filtration of cell extracts of Saccharomyces cerevisiae (132) and E. coli (95) revealed only a small proportion of the enzymes to be in complex with one another. There is, in addition, a large body of evidence that proposes an interaction of glycolytic enzymes with mitochondria. For example, a study of Tetrahymena pyriformis reported that 100% of lactate dehydrogenase, 75% of phosphofructokinase, and 50% of glyceraldehyde phosphate dehydrogenase are associated to mitochondria (26, 34, 36); whereas the association of hexokinase to the mitochondrial outer membrane was earlier believed to be the general nature of eukaryotes (132). It has been postulated that a mitochondrial location of hexokinase was beneficial in terms of energy efficiency, providing ready access to the ATP being generated by the mitochondria and leading to the suggestion that metabolic channeling occurs, with associated kinetic and regulatory benefits (132) (Fig. 5). We have previously questioned this interpretation (40); however, notwithstanding this fact, these studies nicely provide quantification of the amount of assembled as opposed to non-assembled activities—something that unfortunately is rarely provided.
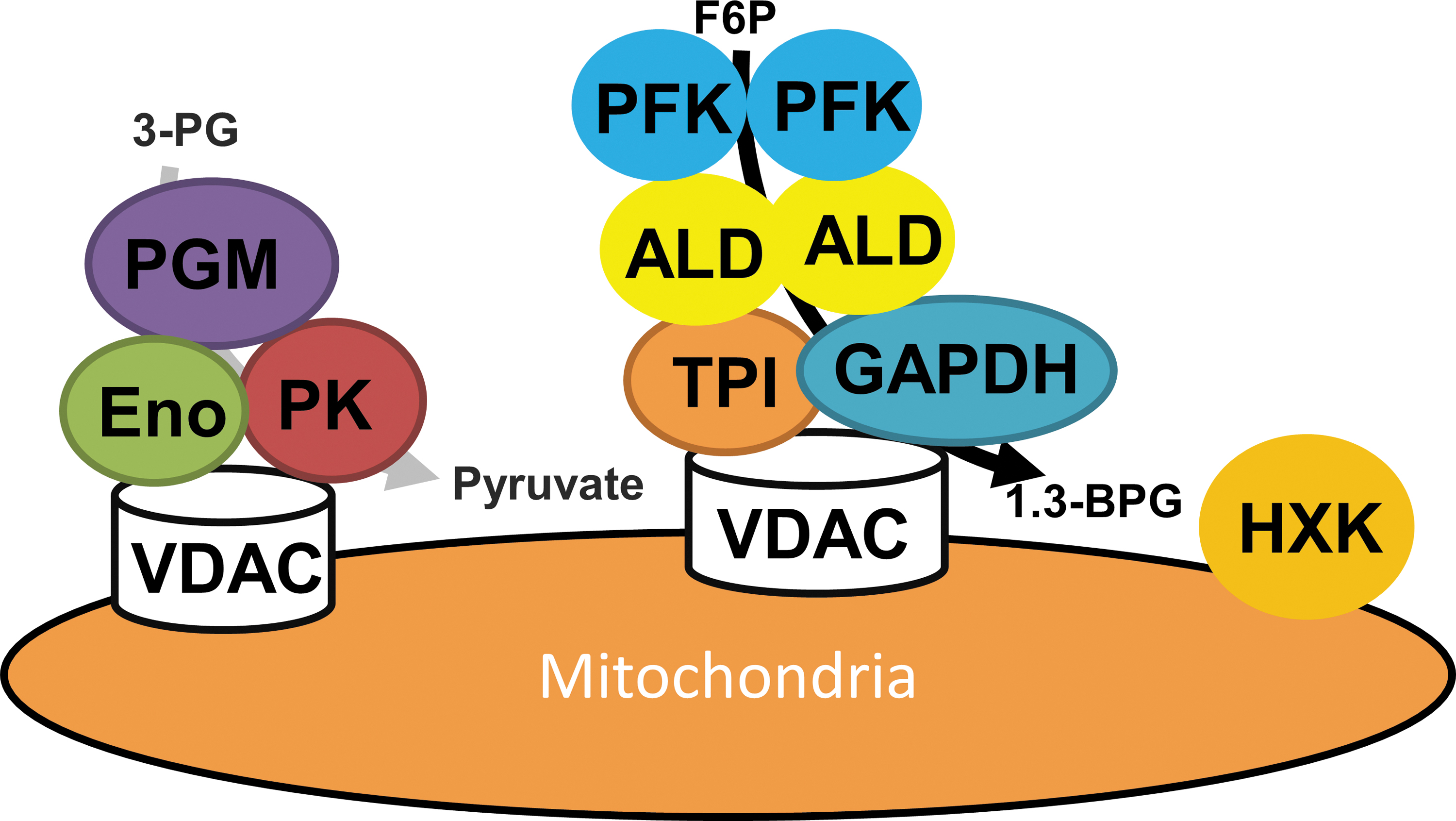
The evidence described earlier for glycolytic complex formation was considerably boosted by a wealth of cross-kingdom evidence during this century (87, 141). A handful of proteomics studies have suggested the presence of the enzymes of glycolysis in isolated mitochondrial fractions from Arabidopsis (45, 48, 88), humans (107), and yeast (9). Studies employing both blue native sodium dodecyl sulphate–polyacrylamide gel electrophoresis and enzyme assays as well as co-immunoprecipitation of proteins with anti-enolase antibodies demonstrated that enolase is a member of a large macromolecular protein complex assembled to the mitochondria and also including enzymes of the TCA cycle and mitochondrial membrane carriers in yeast (19). In Arabidopsis, protease treatments were used to demonstrate that these enzymes were mainly localized on the cytosolic face of the outer mitochondrial membrane (45). Subsequent experiments in which the glycolytic substrates 13C-glucose and 1-13C fructose-1,6-bisphosphate were supplied to isolated mitochondria demonstrated that the complete glycolytic sequence was present and active in this fraction. Further research—this time working on potato mitochondria—showed that this association was dynamic in that inhibition of respiration by KCN led to a proportional decrease in the association of glycolytic enzymes with mitochondria whereas stimulation of respiration enhanced this association. A detailed study suggested that this association is mediated by VDAC, which anchors the glycolytic enzymes to the membrane and is the outer mitochondrial membrane protein (48, 88). Importantly, this research also provided indirect evidence for the channeling of the glycolytic metabolites, with the labeling patterns of glycolytic metabolites being followed by nuclear magnetic resonance proposing a leaky channeling of glucose 6-phosphate and fructose 6-phosphate but a tighter channeling of intermediates from fructose 1,6-bisphosphate onward (48). Channeling. although to a lesser extent, has also been demonstrated for mammalian glycolysis (25), and even in species, such as E. coli, which lack mitochondria (122).
Two approaches have proven particularly informative in assessing the presence of metabolite channeling in glycolysis, namely analysis of modeling of 13C isotopic labeling studies and NADH channeling (Fig. 4). The first of these was an early experiment by Srivastava and Bernhard that demonstrated the direct transfer of NADH from liver glyceraldehyde phosphate dehydrogenase to alcohol dehydrogenase (134). Further work from this lab has demonstrated that a series of such transfers can occur between dehydrogenases and that in every case the transfer occurs between dehydrogenases with alternate stereospecificity for NADH. The second method was, in addition, able to indicate that modeling data acquired from isotope labeling assays in a pattern that consists of channeled glycolysis provides a better fit to the result than when channeling of this pathway is not considered (135, 136). There are six further recent publications regarding glycolytic assemblies—a review on the activity of glycolytic metabolons in muscle (87), three cell biology studies on glycolytic assemblies in animals (58, 63, 107), a study of the glycolysis actin interaction in yeast (9), and finally an example of chemotactic-driven enzyme assembly using glycolytic enzymes and intermediates in a cell-free system (169). Given that we reviewed these recently (40), we will only detail the last of these here. This study reported the fascinating recent finding that using microfluidic and fluorescent spectroscopic techniques it was possible to demonstrate that the first four sequential enzymes of the glycolytic pathway each independently abides by its own specific substrate gradient. As such, this study provided considerable insight into one potential mechanism for the assembly of transient enzyme complexes (169). Indeed, this finding opens up an additionally possible physical route, alongside the earlier described cytoskeletal and membrane platforms, by which enzyme–enzyme interactions can nucleate. Indeed, there is an ever-increasing body of evidence, as detailed in the Structural Support and Nucleation Points for Enzyme–Enzyme Assemblies section, that this mechanism is one that can be demonstrated, at least in vitro, to occur in a range of pathways.
The TCA cycle
The TCA cycle is one of the iconic pathways in metabolism that was elucidated by Hans Krebs in 1940 (153), although some cross-kingdom variation in this pathway has been observed (167). The recent elucidation of an alternative route from 2-oxoglutarate to succinyl–coenzyme A (162) means that these differences are more subtle than initially thought. Indeed, several different bypaths of the TCA cycle exist, ranging from the nearly universal GABA shunt to more taxonomically restricted bypasses such as malate and acetate shunts (167). Apart from these minor differences, the TCA cycle is, however, remarkably conserved.
As for glycolysis, the search for dynamic enzyme assemblies in the TCA cycle was largely driven by the need to explain kinetic observations that did not fall under the conventionally accepted view of a well-mixed metabolism (132). Srere et al. further proved that an immobilized pairing of malate dehydrogenase and citrate synthase had a kinetic benefit over the diffusion enzymes (133). A variety of other early experiments using a wide range of diverse approaches provided further support for the organization of the TCA cycle enzymes that Srere first termed the “metabolon” (40, 131). Since these pioneering studies, dynamic aggregation of consecutive enzymes of glycolysis has been observed across the kingdoms of life (40).
Over a period of 25 years of studies, either by or inspired by Srere and Sugemi and their co-workers, applying gel filtration and precipitation in polyethylene glycol discovered physical protein interactions between six of the eight enzymes of the pathway (132). The importance of these researchers cannot be underestimated since their research cannons additionally include the first structural model of the malate-dehydrogenase-citrate synthase-aconitase complex (148) and the determination of dissociation contents of the complex after the incubation of a fluorescein isothiocyanate labeled citrate synthase with malate dehydrogenase in pig heart and a series of intermediates intimately related to the TCA cycle. Interestingly, 2-oxoglutarate increased the dissociation constant whereas NADH lowered it. The effect of 2-oxoglutarate remains difficult to understand; however, NADH is a major determinant of the energy-generating flux of the mitochondria (40).
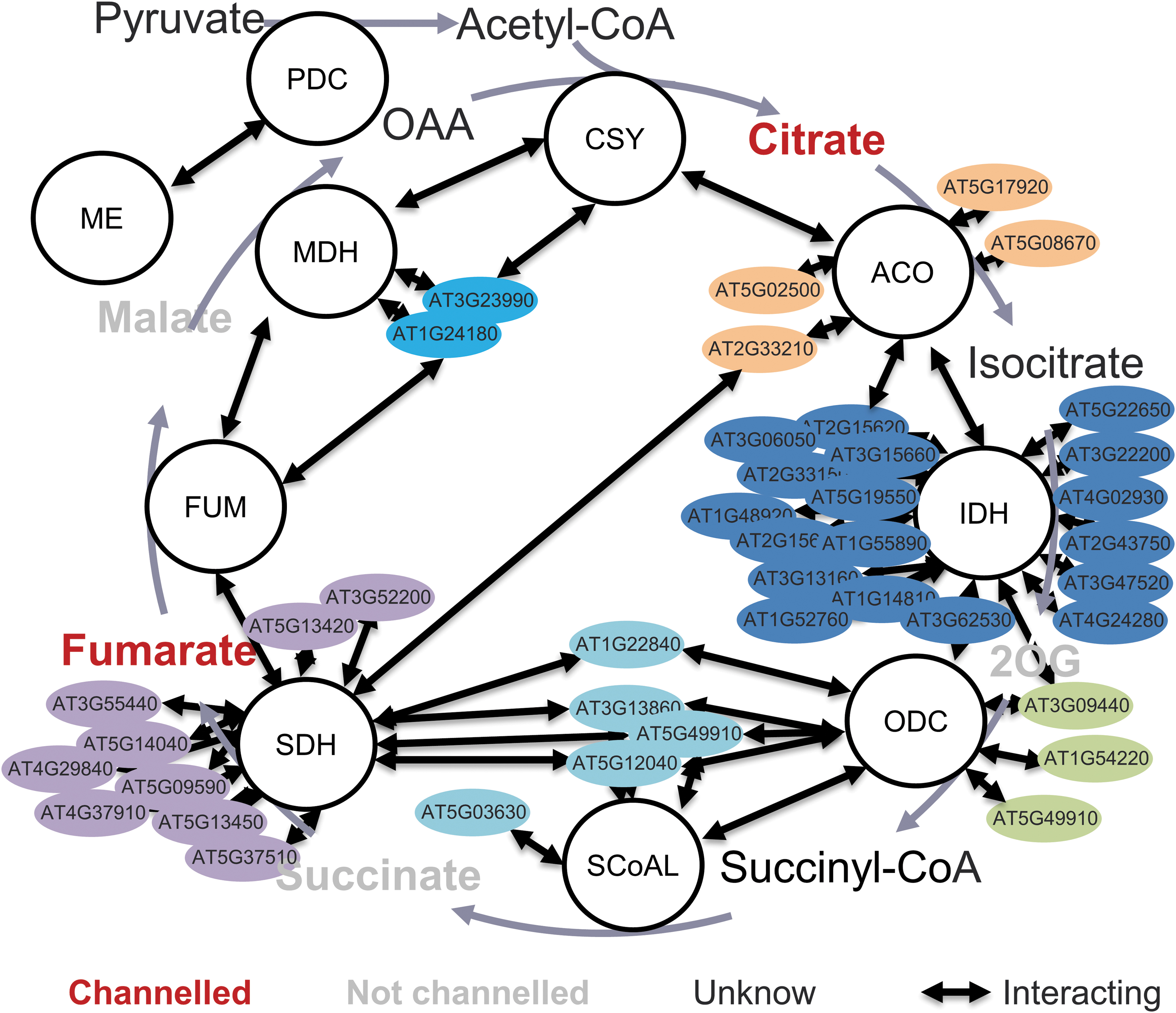
Again, as demonstrated for glycolysis earlier, the advent of fluorescence-based cell biology and proteomics facilitated the study of pathway-wide interactions of the TCA cycle. For example, a comprehensive characterization of the interactions between Bacillus subtilis enzymes revealed interactions between six sequential enzymes of the TCA cycle. Moreover, the TCA cycle enzyme complexes' structural models are consistent with metabolite channeling via electrostatic retention of the channeled metabolite on charged domains of the enzyme surfaces (157, 158). Similarly, as already described, a comprehensive range of techniques, including affinity purification mass spectrometry, Y2H assays, and split-luciferase, have been used to demonstrate protein–protein interactions in the plant TCA cycle (166). One surprising feature of wide-scale protein interaction studies is the substantial number of interactions between nonconsecutive pathway enzymes. It is conceivable that many of these interactions, which occur largely between regulatory subunits of one enzyme and the catalytic subunit of another, act as nucleation points that aid in the formation of metabolons (166) (Fig. 6). In a recent study, the extra-pathway interactions of the plant TCA cycle enzymes were revealed, with 125 interactions being identified that highlighted many novel interactions, including the amino acid metabolism, mitochondrial electron transport chain complexes (mETC), ATP synthesis, signaling, lipid metabolism, nitrogen metabolism, and redox stress (165) (Fig. 6). Interesting, the ATP synthase interacted with the succinate dehydrogenase (10), and it was proposed to organize the super-complex with TCA cycle metabolon in the inner membrane of mitochondria (119). In addition, several subunits of succinate dehydrogenase were found to interact with the other complexes of the mETC. These results also support the postulated super-complex of the mitochondrial electron transfer chain complex (76).
In a more recent study, Wu and Minteer (157) coupled in vivo crosslinking and mass spectrometry to build a low-resolution model of the malate dehydrogenase-citrate synthase-aconitase complex. Interestingly, this revealed protein interactions between all the TCA cycle enzymes. Employing distance constraints derived from the crosslinking, two possible structure models of the malate dehydrogenase-citrate synthase-aconitase protein complex were suggested. The average distances of aconitase-citrate synthase and malate dehydrogenase-citrate synthase between the active site are 50 Angstrom and 35 Angstrom, respectively, in the model based on the most prevalent protein structure.
A theoretical research by Elcock and McCammon (35) suggested that the existence of electrostatic interactions could highly increase substrate-transport efficiency. Moreover, recent research, from the same lab, suggests that, as demonstrated for glycolytic enzymes earlier, metabolite-level gradients increase association of the TCA cycle metabolon (157).
As recently reviewed (40), a variety of isotope labeling methods have also been used to provide evidence for metabolic channeling in the TCA cycle. Arguably more importantly, labeling methods have also been employed as a more common test of the existence of channeling. For instance, the “isotope dilution” approach uses the range of dilution of labeled intermediate pools after the addition of amounts of unlabeled metabolites as a measure of metabolite channeling. For instance, Zhang et al. fed purified mitochondria of potato tuber with 13C-labeled glutamate or pyruvate until accumulation of 13C in the next TCA cycle intermediates achieved an isotopic steady state (166). Then, unlabeled TCA cycle intermediates were supplied and the “dilution” effect on labeling was followed over time. To avoid the complications in interpretation of labeling patterns caused by multiple turns of the cycle, the TCA cycle was linearized by inhibiting succinate dehydrogenase with malonate in the pyruvate feeding assay and by inhibiting aconitase with fluoroacetate in the glutamate assay. These experiments exposed dilution in 2-oxoglutarate, malate, and succinate but none in fumarate or citrate, indicating that the latter metabolites are channeled. Intriguingly, the data from a metabolic flux investigation due to 13C-label redistribution in heterotrophic Arabidopsis cell culture suggested channeled flux from fumarate to malate but no channeled flux from 2-oxoglutarate (2OG) or succinate to citrate (152). More detailed studies in potato tuber mitochondria after 13C-labeling demonstrated channeling of both fumarate and citrate (166).
An alternative nonstructural approach that warrants discussion is the so-called enzyme buffering approach that has been extensively used in studies of NADH channeling by NADH dehydrogenases. In this method, the question addressed is whether the second enzyme (E2) uses the enzyme 1 (E1) bound form of the common intermediate as well as the free form. Given that the dissociation constant KD of E1-NADH is ∼1 mM in the presence of excess E1, it is possible to decrease [NADH]r to a value well under its Km for E2. The major arrow in Figure 4 suggests that for such a binding equilibrium NADH is more than 99% in its E1 bond form. Therefore, if the experimental tested reaction rate is highly in excess of that which could be reached from free NADH alone, then this provides evidence that the NADH is channeled between the sequential enzymes (136). Intriguingly, NADH channeling has only been found in consecutive enzymes of opposite chirality but clear cut results have been reported for several such enzyme pairs (136) By contrast, if NADH is not channeled then E1 is simply buffering NADH to a low [NADH]f (129). Although currently only reported for NADH-containing reactions, this method still has broad utility given the high number of dehydrogenase reactions in the cell and their importance in energy and redox metabolism.
The Mitochondrial Electron Transport Chain
The mitochondrial electron transport chain is more divergent than the other respiratory pathways, with considerable divergences in alternative respiratory pathways and the sizes of the respiratory complexes being proposed in plants (30). It has, however, been studied for at least as long as glycolysis and the TCA cycle (89). More recently, our understanding of the process has been advanced by molecular-level information regarding the structures of the protein complex and electron-transport complexes that catalyze oxidative phosphorylation and constitute the respiratory chain (80, 89). These studies resulted in our current understanding of the three proton translocating complexes, complex I, III, and IV, as well as the mobile electron carriers cytochrome c and ubiquinone (40, 80), with the mechanism underlying the proton-motive force required for ATP synthesis now being very well investigated (Fig. 7).
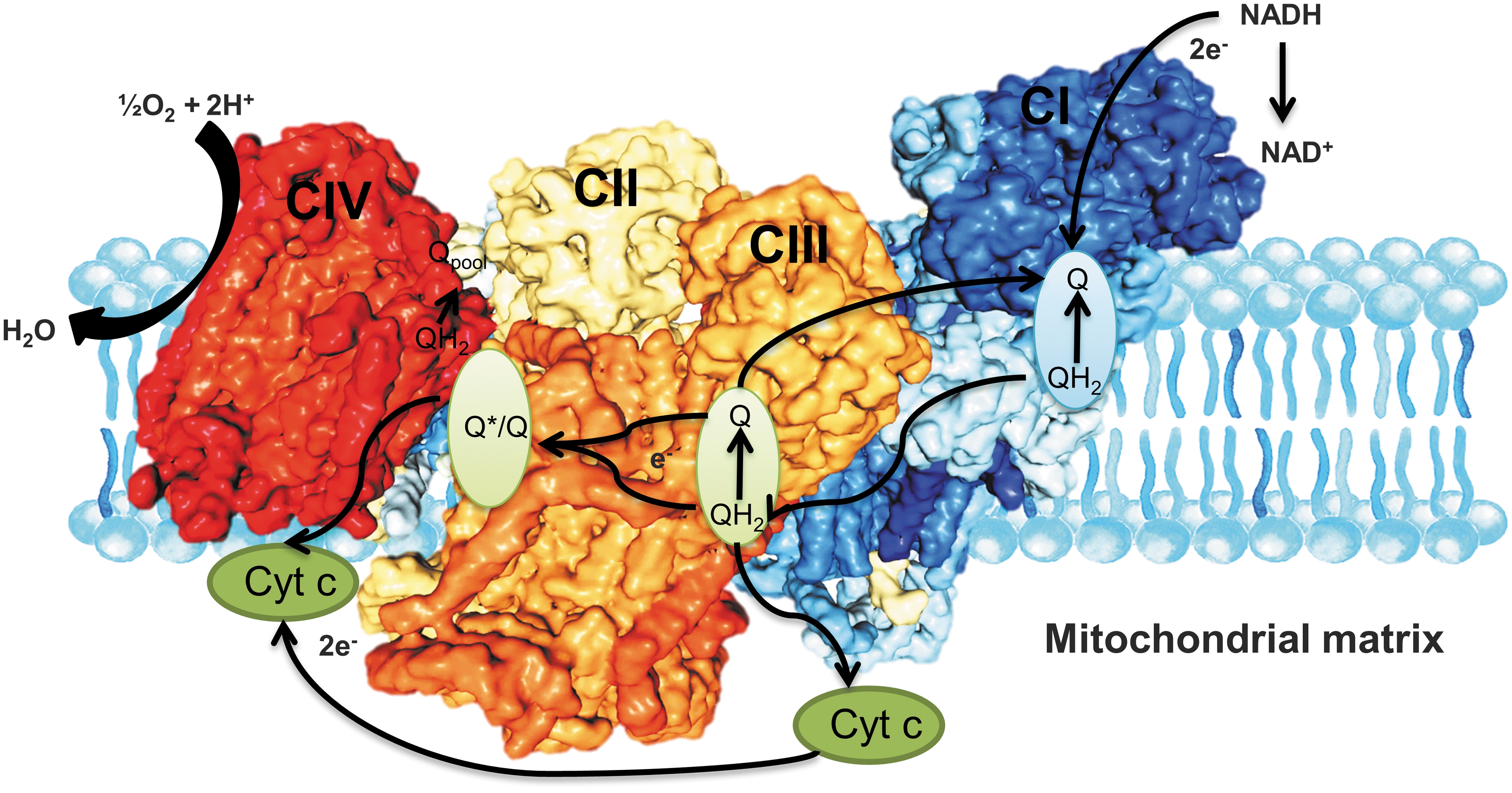
The respiratory complexes are well-determined, stable multi-subunit protein complexes that have been subjected to substantial research programs given the significance of respiratory chain dysfunction in disease and aging (89). Indeed, the separate respiratory protein complexes could be dynamically assembled into super-complexes as observed in plants (37, 110), mammals (70, 115, 116), yeast (115), and even some bacteria such as Paracoccus denitrificans (40, 137). However, some species-specific variations between these super-complexes have been observed (Fig. 7). For instance, patches of identical complexes are suggested in different parts of its cell membrane in E. coli (79), whereas Blue native–polyacrylamide gel electrophoresis has proved that super-complexes of varying stoichiometries with complexes I and III are far more similar to be discovered in a super-complex than complex IV (116). In addition, only simpler super-assemblies of complexes III and IV were observed in S. cerevisiae, which does not contain complex I (53, 90). That said, despite initial skepticism, respiratory super-complexes are now firmly accepted (40). However, the question as to whether they confer any function advantage and the reasons for their existence remain much debated (89). Indeed, by contrast to the glycolytic TCA cycle pathways, evidence that the super-complexes perform metabolite channeling remains considerably contended with researchers debating whether structural proof is consistent with the presence of substrate channeling or not (5, 31, 76, 128). If this conclusion is correct, the question of the physiological purpose of super-complex formation becomes the key. Alternative suggestions include: (i) modulation of cristae morphology regulation of respiratory chain activity, (ii) stabilization of the individual complexes, and (iii) the prevention of protein aggregation in the highly crowded mitochondrial membrane (89). That said, considerable further research is required to assess the relative likelihood of these hypotheses.
Photosynthesis
Unlike heterotrophic systems, photosynthetic tissue captures light energy (47). In the two steps of photosynthesis, light-dependent and light-independent (i.e., the Calvin–Benson cycle) reactions depend on several enzyme complexes. In addition, the process of photorespiration—a metabolic repair cycle—returns photosynthesis to the Calvin–Benson cycle after the oxygenation, as opposed to carboxylation, of ribulose bisphosphate (15). Despite the fact that the pathways function in widely different physiological settings, the emergence of new kinetic and regulatory properties as a consequence of protein–protein interactions will be discussed to aid an understanding into how flux is regulated through these pathways. Our discussion of these pathways will be shorter than the previous ones, not only because they are taxonomically narrower but also given that less evidence is currently available regarding enzyme–enzyme assemblies in this pathway.
The Calvin–Benson cycle
In the micro-compartment of the chloroplast stroma, the enzymes involved in the Calvin–Benson cycle pathway are not randomly distributed, but they interact with the multienzyme complex (47). For example, a potential multienzyme complex that comprised five pathway enzymes, ribose-phosphate isomerase (RPI), phosphoribulokinase (PRK), ribulose-bisphosphate carboxylase/oxygenase (RuBPCase), phosphoglycerate kinase (PGK), and glyceraldehyde-phosphate dehydrogenase (GAPDH), has been proposed (47, 138, 160). In a further experiment, the 900 kDa stable super-complex of the Calvin–Benson cycle was organized by enzymes RPI, ribulose-5-phosphate kinase (Ru-5-P-K), RuBPCase, GAPDH, sedoheptulose-1,7-bisphosphatase (Sed-1,7-bPase), and the electron transport protein ferredoxin-NADP+ reductase (FNR) (138). Immunoelectron microscopy revealed that Ru-5-P-K, GAPDH, Sed-1,7-bPase, and FNR are bound to stroma-faced thylakoid membranes in situ, whereas rubisco and rubisco activase are randomly distributed throughout chloroplasts. The interaction between GAPDH and aldolase was demonstrated by several methods in the pea chloroplast (Pisum sativum L.) (8); whereas multienzyme complexes of varying compositions of the Calvin–Benson cycle enzymes have been isolated from pea and spinach, with the most predominant being the PRK/GAPDH complex (117, 150, 160). This complex may, therefore, correspond to the core complex of a super-complex involved in CO2 assimilation. In addition, a small protein CP12 has also been identified in most of these complexes (47, 150). As the formation of this supramolecular complex occurs under oxidizing conditions, the GAPDH/CP12/PRK complex is disassembly by reducing agents such as DTT, which facilitates the purification of isolated PRK and a subcomplex of GAPDH and CP12, also known as native GAPDH (13, 85). The interaction between oxidized CP12 and GAPDH provides full protection from oxidative damage (85). In higher plants, chloroplast GAPDH forms an A2B2 tetramer. The B subunit holds a C-terminal extension that is responsible for the oligomerization of higher plant chloroplast GAPDH into an A8B8 regulatory form (78, 85). In the algae Chlamydomonas reinhardtii, the PRI, PRK, PGK, GAPDH, and fructose bisphosphatase (FBPase) co-localize and are located near the thylakoid membrane by the immunolocalization studies. As the Calvin–Benson cycle of higher plants, these enzymes in the C. reinhardtii are also assembled as the supramolecular complexes (139) (Fig. 8). In addition, the GAPDH is only present as an A4 homotetramer in the C. reinhardtii and Scenedesmus obliquus. Interestingly, the sequence similarity of CP12 with the C-terminal extension of the B subunit of GAPDH, which contains two cysteine residues, is believed to be involved in the redox regulation of this enzyme (106). However, the experimental evidence for substrate channeling in the Calvin–Benson cycle is lacking, despite the fact that the formation of supercomplexes is proposed to be related to a regulatory mechanism for CO2 assimilation.
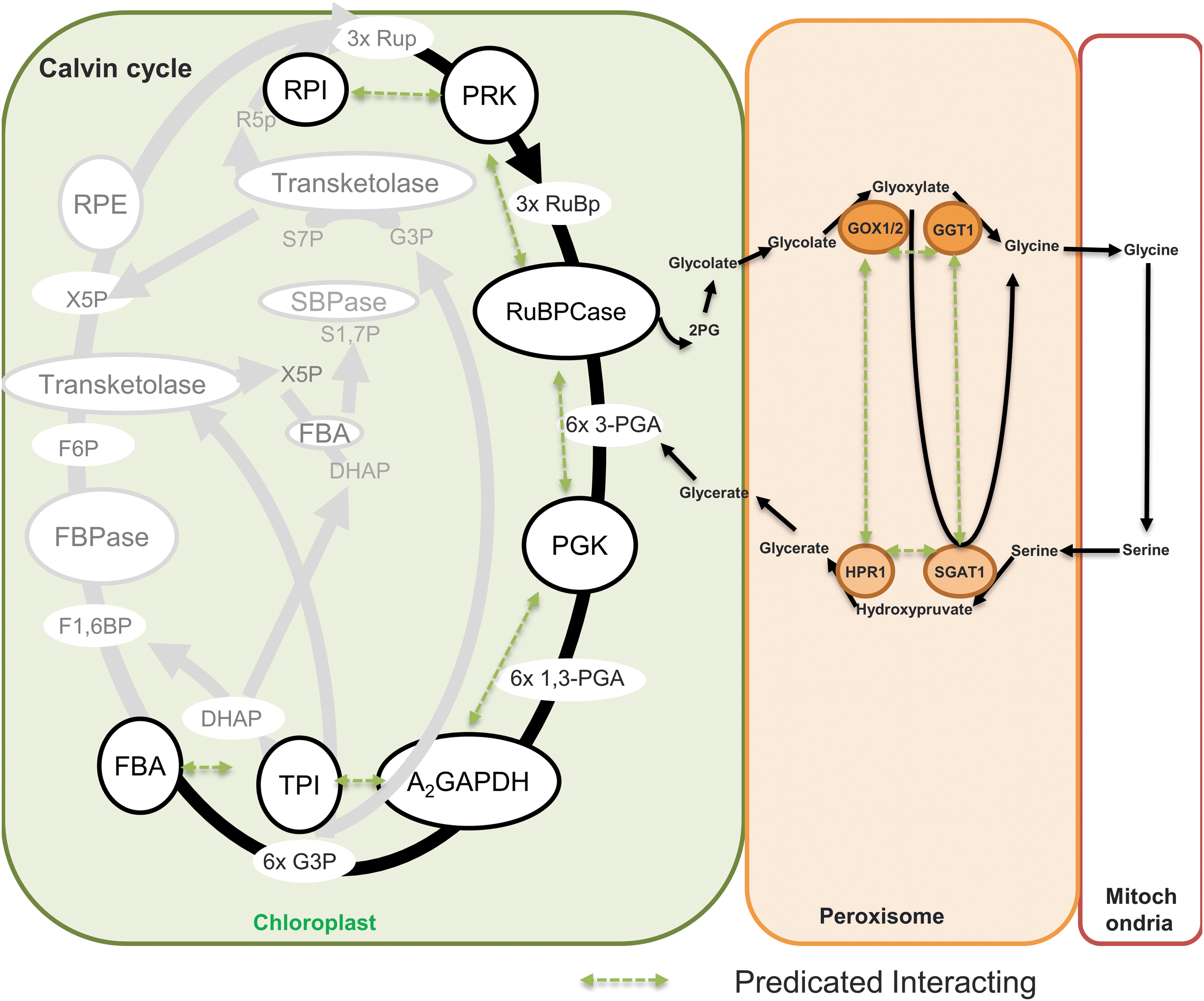
Photorespiration
In the central photorespiratory pathway of leaf peroxisomes, the inner membrane microcompartment of peroxisomes is the site of the possible interaction of the glycolate oxidase (GOX), catalase, Ser-glyoxylate and Glu-glyoxylate aminotransferase, hydroxypyruvate reductase (HPR), and malate dehydrogense (MDH) (Fig. 8). Moreover, the complex formation and possibility metabolite channeling among the components of the glycine decarboxylase system remains unclear. The association–disassociation of GOX and catalase depends on dynamic changes in hydrogen peroxide (H2O2) levels (168). In the isolated spinach leaf peroxisomes, the glycerate was produced at the same rate with and without an intact membrane and, in both cases, the intermediates glyoxylate and hydroxypyruvate are not detected in the suspension medium, indicating the possible substrate channeling in the photorespiratory pathway of leaf peroxisomes (54). Assembly and dynamics of these multienzyme complexes in vivo in peroxisomes may reveal the additional regulatory mechanisms for photorespiration (125). In addition, the channeling of tetrahydrofolate between mitochondrial glycine decarboxylase system T protein and serine hydroxymethyl transferase has been expected due to the sensitivity of tetrahydrofolate to oxidation; however, this was not proved by the in vitro enzyme kinetics (111). To conclude, although enzyme–enzyme assemblies clearly appear to be prominent in the twin pathways of photosynthesis and photorespiration considerably more study is required to better understand their form and function.
Structural Support and Nucleation Points for Enzyme–Enzyme Assemblies
Unlike oligomeric protein assemblies and stable multienzyme complexes, an understanding of the regulation of transient enzyme–enzyme assemblies, that is, factors that promote their association/dissociation is currently relatively poor. Indeed, until recently, it has largely been confined to the demonstrations that lipid microdomains (23, 32, 61, 161), cytoskeletal elements (9), and carrier proteins (7) could form structural support for enzyme–enzyme assemblies. As described earlier, the latter two mechanisms have been demonstrated to be involved in enzyme–enzyme assemblies that are engaged in energy and redox metabolism. Similarly, a wide range of phenylpropanoids metabolons have been proposed to be stabilized at the ER membrane (61, 71). A further recent hypothesis emanated from the comparative study of cell-free and E. coli glycolytic pathways that identified the phosphotransfer system of E. coli as a possible nucleation point for the glycolytic metabolon in this species (1, 2). Both in vitro and in vivo experiments proved that the Embden-Meyerhof-Parnas flux between isozymes of phosphofructokinase and fructose-bisphosphate aldolase are channeled and the phosphotransfer system may be an anchor point to initiate enzyme assemblies (2). In addition to the TCA cycle, the interactions between regulatory subunits and catalytic subunits of different proteins act as nucleation points (141) that help in the formation of the specific interaction between the sequential enzymes (166). Thus, the sequential enzymes of the metabolic pathway formed the metabolon with the help of both protein–protein interactions and by structural elements of the cell (integral membrane proteins and proteins of the cytoskeleton).
Although reports have suggested that phosphorylation disrupts the human carbonic anhydrase metabolon (6), large-scale structure–function studies have yet to be carried out to ascertain whether this is a common mechanism for the regulation of enzyme–enzyme assemblies. An intriguing recent hypothesis that is receiving growing theoretical and experimental support is that the metabolic intermediates of the pathways themselves are determinants of the complex formation. An early example of this concerns the metabolon formation that was observed after the conformational change in cytochrome P450 enzymes on substrate binding that mediates the attachment of cytochrome 450 reductase (61, 154). It seems likely that advances in crystallography and electron microscopy will render such discoveries more common in the near future. Chemists and physicists have recently developed experimental systems for testing enzyme–enzyme assemblies that are based on cell-free systems that either replicate molecular crowding (11) or represent simple systems in which to evaluate enzyme chemotaxis (92). Although the former will, undoubtedly, provide new insight into the dynamics of cellular metabolism, for reasons described earlier (140) we do not believe that such simulated cells will prove great insight into enzyme–enzyme assemblies. As described earlier, the enzyme chemotaxis studies, on the other hand, provide a novel mechanism by which metabolic intermediates may govern the formation of enzyme–enzyme assemblies; this has been uncovered by in vitro experiments of the effect of metabolite gradients on complex formation. These studies suggest that enzymes show chemotactic movement along their substrate gradient in a manner that could drive their co-localization (4, 55, 157, 169). This is typically followed by using fluorophore-tagged enzymes to follow movement in microfluidic devices (125); however, the conclusions made have been critically discussed due to issues in the interpretation of the fluorescence correlation spectra (51). A further issue is whether these devices adequately reflect how the enzymes behave in a cellular system. As such, developments of approaches to test this are an utmost priority. Notwithstanding this criticism, these findings have inspired chemists and physicists alike to assess these phenomena with many further recent articles being published in this area that are worthy of mention.
The results of two recent microfluidic experiments are particularly interesting. The first of these used super-resolution fluorescence measurements performed across four orders of magnitude of substrate concentration demonstrated that this dramatically boosted motion, providing demonstration that catalytic activity is linked to chemotactic and anti-chemotactic properties (60). The second study used 13C-labeling in cell-free systems to investigate channeling in glycolysis and compare this with in vivo studies, evidencing considerable advantage in the more channeled system arising in the cell-free system (2).
Modeling studies have also focused on the chemotactic movements exhibited by catalytic enzymes and proposed potential mechanisms by which this is achieved with a wide range of possibilities being explored, including self-thermophoresis, boosting of kinetic energy, stochastic swimming, and collective heating theories being proposed (4). As yet, there does not appear to be a consensus regarding the most likely mechanism; however only modeling a combination of stochastic swimming and collective heating has yet been demonstrated to reproduce changes in diffusion that are consistent with experimental evidence (46). The picture is complicated yet further by hydrodynamic interactions between the enzyme and the nonuniform substrate with which it interacts (4). However, given the vast interest in these phenomena, it would appear likely that considerable advances in this area will be made within the next decade.
Conclusions
Although there is, admittedly, on the basis of dissociation constants, no clear distinction between stable multienzyme complexes and transient enzyme–enzyme assemblies, these complexes would appear to have evolved under different selective pressures to take different roles in metabolic regulation. Although there is a huge literature underlying the structure–function relationships of multimeric enzymes as well as multienzyme complexes that has led to a detailed understanding of the mechanisms that confer their stability, much less is known regarding this feature for transient enzyme–enzyme assemblies (including metabolons). That said, there is growing support that cytoskeletal or membranous structures provide support or nucleation points for transient enzyme–enzyme assemblies. The nucleation points of the metabolon formation also provide more information on the in vivo enzyme cluster engineering in synthetic biology. Moreover, work in the past few years has led to the fascinating hypothesis that such assemblies form due to the presence of concentration gradients of their substrates. Although questions remain regarding the interpretation of the fluorescence signals that these conclusions are based on and the occurrence of such behavior in vivo, this is a highly exciting idea that has brought fresh impetus to the study of transient enzyme–enzyme complexes. Provision of in vivo evidence of such behavior is clearly a pressing challenge for future research. However, once this is achieved, alongside structure–function studies, this will represent a fantastic opportunity to gain deeper understanding of the stability of transient enzyme–enzyme complexes, alongside properties of their association and dissociation.
Footnotes
Funding Information
This work was supported by funding from the Max-Planck Society (Y.Z. and A.R.F.); A.R.F and Y.Z. would like to thank the European Union's Horizon 2020 research and innovation programme, project PlantaSYST (SGA-CSA Nos. 664621 and 739582 under FPA No. 664620) for supporting their research.