
Abstract
Significance:
The management of cancer patients is frequently complicated by the occurrence of a complex syndrome known as cachexia. It is mainly characterized by muscle wasting, a condition that associates with enhanced protein breakdown and with negative energy balance. While the mechanisms underlying cachexia have been only partially elucidated, understanding the pathogenesis of muscle wasting in cancer hosts is mandatory to design new targeted therapeutic strategies. Indeed, most of cancer patients will experience cachexia during the course of their disease, and about 25% of cancer-related deaths are due to this syndrome, rather than to the tumor itself.
Recent Advances:
Compelling evidence suggests that an altered redox homeostasis likely contributes to cancer-induced muscle protein depletion, directly or indirectly activating the intracellular degradative pathways. In addition, oxidative stress impinges on both mitochondrial number and function; the other way round, altered mitochondria lead to enhanced redox imbalance, creating a vicious loop that eventually results in negative energy metabolism.
Critical Issues:
The present review focuses on the possibility that pharmacological and nonpharmacological strategies able to restore a physiologic redox balance could be useful components of treatment schedules aimed at counteracting cancer-induced muscle wasting.
Future Directions:
Exercise and the use of exercise mimetic drugs represent the most promising approaches capable of reinforcing the muscle antioxidant defenses of cancer patients. The results from ongoing and new clinical trials are needed to validate the preclinical studies and provide effective therapies for cancer cachexia. Antioxid. Redox Signal. 33, 542–558.
Introduction
Cachexia is a severe comorbidity of cancer that significantly complicates patient management, reducing the tolerance to treatments, worsening the quality of life, and resulting in about 20% of cancer deaths (4). The consensus reached some years ago on the definition of cachexia (83) indicates two main factors as hallmarks of this multiorgan syndrome: body weight loss, mainly due to fat and muscle wasting, and chronic inflammation.
The causative mechanisms in the onset and progression of cancer cachexia are still largely unknown, although in the past decades, the different contributions of nutrition, reduced anabolism, hypercatabolism, and altered energy metabolism have been shown to play a role (Fig. 1). This picture is further complicated by the need to treat cancer patients in the attempt to get rid of the tumor; such treatments are generally very aggressive and by themselves pose several limitations to patient management, being endowed with significant toxicity (100).

The present review takes into consideration the possible role played by alterations of the oxidative/reductive (redox) homeostasis in the pathogenesis of cancer-induced muscle wasting, as well as the possibility that targeting the redox balance could be useful to manage this wasting syndrome.
Mechanisms of Cancer-Induced Muscle Wasting
The depletion of muscle proteins is one of the main features of cachexia. It is mainly associated with enhanced protein degradation rates that can be coupled or not with reduced protein synthesis. This hypercatabolic response is mediated by malnutrition and humoral factors, such as proinflammatory cytokines and hormones, as well as by alterations of the energy balance (4). In addition to play a role in asthenia and weakness, the loss of muscle mass also contributes to the reduced treatment tolerance displayed by many cancer patients, including the recently achieved immunotherapy by checkpoint inhibitors (27).
Being so relevant in terms of patient management, quality of life, and survival, the evaluation of muscle mass depletion should be part of the standard protocols in oncology. Despite this evidence, however, patients do receive a cancer diagnosis and staging, but very little is done to assess if muscle wasting is occurring. Indeed, the vast majority of patients are recorded for body weight or body mass index, without taking into account body composition and muscle cross-sectional area.
The skeletal muscle comprises the majority of body proteins. In the adult, muscle mass is maintained fairly constant through a convergent regulation of protein synthesis and breakdown rates that in healthy individuals allows the achievement of a new steady state when the balance is altered. By contrast, during aging or chronic diseases, breakdown rates generally exceed those of synthesis, resulting in muscle hypotrophy (91).
All the intracellular proteolytic pathways operating in the skeletal muscle (proteasome, lysosomes, caspases, and calpains) have been shown to be activated above physiological levels in cancer cachexia (91). In particular, proteasome and lysosomes appear those mainly in charge of degrading short-lived and regulatory proteins, dysfunctional organelles, and structural proteins (91).
As for lysosomes, their contribution to cancer-induced muscle wasting mainly reflects the supraphysiological activation of autophagy, a process by which intracellular material is sequestered into vacuoles and subsequently degraded upon vacuole fusion with lysosomes. It occurs physiologically but is rapidly enhanced by stress conditions such as lack of nutrients/energy and/or organelle damage. While initially described as an aspecific degradative process, autophagy has been shown to be finely tuned to ensure the disposal of specific cargos such as mitochondria (mitophagy), peroxisomes (pexophagy), bacteria (xenophagy), and lipid droplets (lipophagy) (72).
The possibility to follow the activation of the different types of autophagy has become feasible due to the discovery of molecular markers such as Beclin-1, LC3B, p62, Bnip3, Parkin, and Pink1, the latter three used to detect mitophagy. Increased autophagic rates have been shown to contribute to muscle wasting in fasting, aging, denervation, disuse, and neurodegenerative diseases (91). Similarly, muscle autophagic rates above normal have been reported in experimental cachexia as well as in cancer patients (8, 95, 97, 132). However, autophagy induction in cancer cachexia is peculiar in comparison with other muscle wasting-associated states: instead of being degraded, autophagosomes do accumulate in the tissue, probably reflecting lysosomal engulfment and protease activity exhaustion (95). Consistently with these observations, both inhibition and overstimulation of autophagy are detrimental in tumor-bearing mice (92, 95).
The second relevant proteolytic system in the muscle is that dependent on proteasome. This mainly acts on regulatory and/or damaged proteins targeted for degradation by ubiquitylation, operated by three different classes of enzymes, namely E1, E2, and E3. In particular, the E1 group includes enzymes able to activate ubiquitin, and E2s conjugate ubiquitin to the protein substrate, while E3s are ubiquitin ligases. The latter are endowed with tissue/substrate specificity. Along this line, there are several muscle-specific E3s: atrogin-1 and MuRF1, the first to be identified, MUSA1,TRIM32, TNF receptor-associated factor (TRAF)6, and SMART (63). Muscle-specific E3s are currently used as molecular markers of proteasome-dependent proteolysis. In this regard, their increased expression has been shown in several conditions associated with muscle wasting, including cancer cachexia (4), although contradictory results have been reported (88, 132).
Despite lysosomes and proteasome appear overactivated in conditions associated with muscle wasting, they are unable to directly break down myofibrillar proteins. To become available for degradation, the latter should undergo a preventive cleavage likely operated by calpains and/or caspases (91), whose activity in the muscle is indeed upregulated in both tumor-bearing animals (29, 71, 101) and cancer patients (124).
The enhanced protein turnover that occurs in muscle wasting can also reflect reduced protein synthesis rates. However, in cancer cachexia at least, this is not a definite finding, since muscle protein synthesis has been reported to increase, decrease, or remain unchanged (4). Data obtained studying experimental model systems have provided contrasting information: reduced protein synthesis has been observed in mice hosting the C26 or the Lewis lung carcinomas (LLC) (9, 87, 136), while normal anabolic potential has been shown by rats bearing the Yoshida AH-130 hepatoma (30). The situation is far from being clear also in cancer patients. While there are studies reporting a reduction of muscle protein synthesis (42, 55), improvement of baseline synthetic rates has been shown to occur in pancreatic cancer patients (36).
Another relevant mechanism impinging on muscle wasting in cancer cachexia is the early alteration of energy metabolism. In this regard, most of the cancer patients are characterized by resting energy expenditure (REE) increased in comparison with the normal population (141). Of interest, a significant number of patients considered hypermetabolic do not report loss of body weight and are characterized by good performance status (141). The lack of correlation between body weight loss and hypermetabolism is also confirmed by another study performed on bladder cancer patients, showing that the recovery after surgery is faster for metabolic rates than for body weight loss (135).
Hypermetabolism in cancer patients is likely driven by molecules released by the tumor and/or by the organism in response to tumor growth. The possibility that large tumors drain energy, producing a sort of stripper action against the organism, is not particularly sounding. Indeed, tumor mass in cancer patients is generally low in comparison with body weight (106).
Alterations in thermogenesis, the process in charge of heat production, also contribute to increase REE in cancer patients. It mainly occurs in the skeletal muscle (shivering thermogenesis) and in the brown adipose tissue (BAT; nonshivering thermogenesis). The former modality of heat production is characterized by low metabolic efficiency and is generally rapidly activated when the organism faces heat loss due to cold exposure. By contrast, the adaptation to a cold environment is more effectively achieved by activating the nonshivering thermogenesis. In addition to cold exposure, also meal ingestion stimulates thermogenesis in the BAT at an extent that depends on meal size and composition, as well as on the action of the sympathetic nervous system, mainly exerted by norepinephrine release. Both skeletal muscle and BAT are characterized by high expression of uncoupling proteins (UCPs), which are involved in the dissipation of the proton gradient in mitochondria, resulting in energy loss that is used to produce heat rather than ATP. Consistently with the occurrence of hypermetabolism, increased expression of UCPs with respect to control values has been reported in both tumor-bearing animals and cancer patients (14, 26).
The presence of the tumor is also associated with a phenomenon defined as “browning,” characterized by the conversion of white adipocytes into brown adipocyte-like cells. This process contributes to cancer-induced fat depletion and increased REE by shifting energy from ATP to heat production (4). Of interest, both browning and increased REE have been associated with increased expression of the tumor-derived parathyroid-hormone (PTH)-related protein, whose knockdown confers resistance to cancer-induced cachexia in experimental models (65, 66). These observations confirm the existence of a cross talk between fat and muscle (4). Consistently, genetic deletion of the adipose triglyceride lipase (33) or treatment with the fatty acid oxidation inhibitor etomoxir (48) has been shown to prevent cancer-induced muscle wasting in mice.
The increased REE in cancer patients is associated with decreased energy production that mainly derives from reduced food intake and from the activation of futile metabolic cycles that divert energy from storage to wasting. In addition to these rather obvious explanations, in the last decade, the idea that something wrong likely occurs to the energy production machinery in mitochondria is gaining a growing consensus.
Along this line, ultrastructural and functional changes have been observed in muscle mitochondria in tumor-bearing animals (47, 99, 122), ultimately resulting in impaired oxidative capacity (61, 138) and, by activating mitophagy, in reduced mitochondrial number. Other studies have reported the occurrence of alterations in mitochondrial fusion, fission, and biogenesis (see also the Mitochondria as Pivotal Elements in Muscle Oxidative Stress section), frequently associated with a proinflammatory environment (9, 148). Cancer-related effects on muscle mitochondria also affect the ability of the cell to maintain the physiological redox balance that is frequently disrupted in favor of the oxidative arm (see Role of Oxidative Stress in Muscle Wasting and Dysfunction section).
Role of Oxidative Stress in Muscle Wasting and Dysfunction
Oxidative stress is caused by the accumulation of the physiologically produced reactive oxygen or nitrogen species (ROS/RNS). ROS increase can derive from increased production or decreased antioxidant capacity, due to low concentration of molecules endowed with antioxidant properties and/or impaired activity of the antioxidant enzymes (3). At physiological concentrations, ROS regulate distinct signaling pathways, mainly activating mitogen-activated protein kinases (MAPKs) such as ERK and JNK and inhibiting the phosphatase calcineurin, leading to the nuclear translocation of transcription factors involved in cell proliferation, differentiation, survival, or in the regulation of intracellular protein turnover (102). On the contrary, excessive ROS/RNS levels result in modification of DNA, proteins, and lipids such as nitration, nitrosylation, carbonylation, and glycation, causing cellular dysfunction and potentially leading to cell death, according to the magnitude of the oxidative insult.
The bimodal response to ROS fits with the hormesis theory, based on the concept that the exposure to low levels of a damaging agent may induce an adaptive response able to “train” the cell to properly respond to a subsequent damaging stimulus of enhanced intensity (59). Adapting the concept to the redox status, a transient oxidative stress reflects a potentially beneficial adaptation process, while an uncontrolled accumulation of ROS/RNS leads to pathological cellular damage.
The importance of oxidative stress in modulating muscle mass and function was highlighted many years ago studying age-related sarcopenia. Indeed, the activity of antioxidant enzymes decreases with age and similar results have been obtained in several chronic pathological conditions (81). As any other cell type, muscle fibers counteract ROS activity at different levels: (i) prevention of ROS-generating mitochondrial electron leakage; (ii) ROS scavenging by enzymes of the detoxification pathway, including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPX), and peroxiredoxin; and (iii) ROS neutralization with antioxidant molecules such as reduced glutathione and vitamins C and E.
The cellular antioxidant capacity is stimulated by low ROS levels that promote an adaptation mechanism leading to increased expression of the detoxifying enzymes and accumulation of antioxidant molecules (see the Exercise Counteracts Cachexia Modulating the Redox Homeostasis section for further details). Among the signaling regulatory factors that sense intracellular ROS, the nuclear factor-kappaB (NF-κB), the activator protein-1 (AP-1), the heat shock transcriptional factor-1 (HSF-1), and the nuclear respiratory factor (Nrf)2 regulate several muscle homeostatic processes and trigger the increased expression of antioxidant enzymes (84).
In cancer-induced muscle wasting, the status of the antioxidant defense is not univocally defined. The reduction of most antioxidant enzyme expression and activity coupled with increased superoxide levels has been reported in the muscle of cachectic mice bearing the MAC16 tumor (130). Similar observations have been confirmed in patients affected by distinct cancers, either weight stable or weight losing, suggesting that the loss of the antioxidant defense is more related to the presence of a tumor rather than to the loss of muscle mass (19).
By contrast, a preclinical study has shown that cachexia induced in the host rat by the AH-130 tumor leads to distinct responses in locomotor and respiratory muscles, the former being more susceptible to oxidative stress than the latter, despite the increase of the endogenous antioxidant defense (115), likely due to an ineffective adaptive response. Similarly, increased pro-oxidant species coupled with the activation of the antioxidant defense has been reported in the muscle of C26-bearing mice (6, 12). These observations are supported by data obtained in the quadriceps muscle of lung cancer patients, suggesting that tumor growth does not halt the adaptive antioxidant muscle response (104), although it might be insufficient to cope with the magnitude of the oxidative insult.
When ROS accumulation exceeds the adaptation capacity of the cell, several molecular pathways are activated, eventually leading to muscle atrophy and/or dysfunction (Fig. 2). As described above (see the Mechanisms of Cancer-Induced Muscle Wasting section), muscle wasting results from the imbalance between protein synthesis and degradation, the latter overpassing the former. High oxidative stress can directly or indirectly modulate both the protein synthesis machinery and the degradation pathways. Indeed, ROS are known modulators of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt/mTORC1 pathway, the main intracellular regulator of protein synthesis (131).
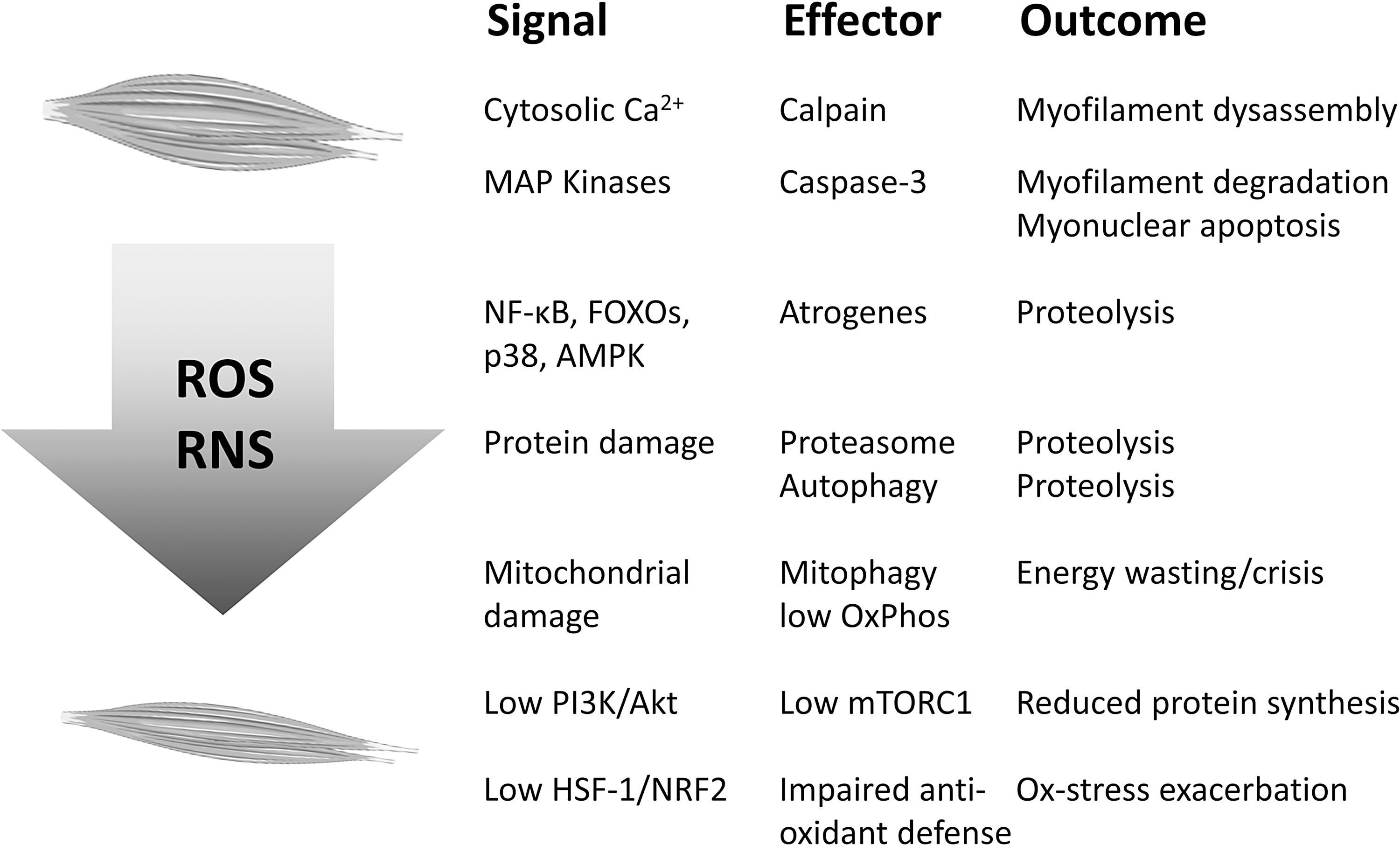
As for the protein degradation machinery, ROS excess can alter calcium homeostasis, which in turn activates the cysteine protease calpain leading to myofilament release (77). In parallel, calcium dysregulation might promote aberrant NF-κB activity triggering the simultaneous activation of distinct proteolytic systems (118). Beyond the calpain system, severe muscle wasting is generally associated with the hyperactivation of the degradation systems responsible for the disposal of bulk proteins and organelles, namely the ubiquitin/proteasome and the autophagy systems. As for the former, a direct link with the oxidative stress has been suggested by in vitro observations on C2C12 myotubes, where the exposure to H2O2 results in increased enzymatic activity and expression of proteasome components (51).
In addition, a recent study has shown that diaphragm atrophy occurring during mechanical ventilation depends on ROS-induced proteasome and autophagy activation (80). In this regard, the occurrence of a cross talk between oxidative stress and autophagy has been previously suggested by observations reporting that blocking mitochondrial ROS production using the antioxidant SS-31 in rats exposed to mechanical ventilation improves both increased autophagy and muscle wasting (126). Beyond inactivation of the PI3K/Akt/mTORC1 and activation of the FOXO pathways, ROS-induced autophagic degradation in the skeletal muscle was proposed to rely on both p38 MAPK and the energy sensor AMP-activated protein kinase (AMPK) (109).
In addition to proteasome and autophagy, oxidative stress also results in activation of caspases. Indeed, caspase-3 has been reported to be activated by ROS in the diaphragm upon mechanical ventilation (76, 126). Caspase-3 has been linked to protein depletion in cancer cachexia (123), despite its activation being generally associated with apoptotic cell death. In this regard, ROS-induced loss of myogenic cells or cellular senescence has been suggested to be relevant in the onset of aging-related sarcopenia, such mechanism being also ROS and autophagy dependent (49, 127). Dysregulation of myogenesis has been reported to contribute to the onset of cancer-induced muscle wasting (56, 94), with a mechanism that involves excessive ERK or NF-κB activation. Of interest, both signaling pathways could be stimulated by local oxidative stress, although such hypothesis has not been tested, so far.
Finally, it is worthy mentioning that, beyond muscle quantity, oxidative stress might also affect muscle quality, that is, the ability to produce workload. Consistently, quality of life in cancer patients is frequently worsened by the occurrence of fatigue. From the cellular and molecular point of view, most of the ROS-induced alterations affecting muscle quality have been studied in aged individuals or rodents, suggesting that ROS interfere with the neuromuscular junction functionality, with the excitation/contraction (EC) coupling at the ryanodine receptor (RyR), and with the cross-bridge cycling in the sarcomere (13). The oxidative stress-induced degeneration of the neuromuscular junction was demonstrated in mice lacking the antioxidant enzyme SOD (58) and is now considered a relevant alteration associated with muscle wasting (113), linking oxidative stress, muscle wasting, and functional impairment.
Mitochondria as Pivotal Elements in Muscle Oxidative Stress
The skeletal muscle accounts for about 40% of the body mass. It requires large amounts of oxygen for contraction, but even at rest it is responsible for more than 20% of body oxygen consumption (153). Such elevated respiration implies that the muscle can generate a relevant quantity of ROS, however, the magnitude of ROS production only partly relies on the amount of oxygen consumed, being highly dependent on mitochondrial “health” or quality. Mitochondria are an interconnected and very dynamic network able to adapt to the metabolic requirement of the cell/tissue. When a damaging insult occurs, the mitochondrial network undergoes degeneration, eventually leading to loss of mitochondrial ATP production and to dramatic increase of ROS release (68).
The maintenance of mitochondrial quality requires a class of proteins involved in the so-called mitochondrial quality control, taking care of the mitochondrial network health (112). Briefly, these proteins are involved in mitochondrial turnover, on one hand regulating the biogenesis of new mitochondria and their fusion to incorporate them in the elongating network, and on the other hand, the fission of the dysfunctional mitochondrial domains from the network and the degradation through the autophagy system, specifically defined “mitophagy.” The altered expression of the proteins involved in each of these processes has been causally linked to the regulation of oxidative stress and muscle mass (Fig. 3).
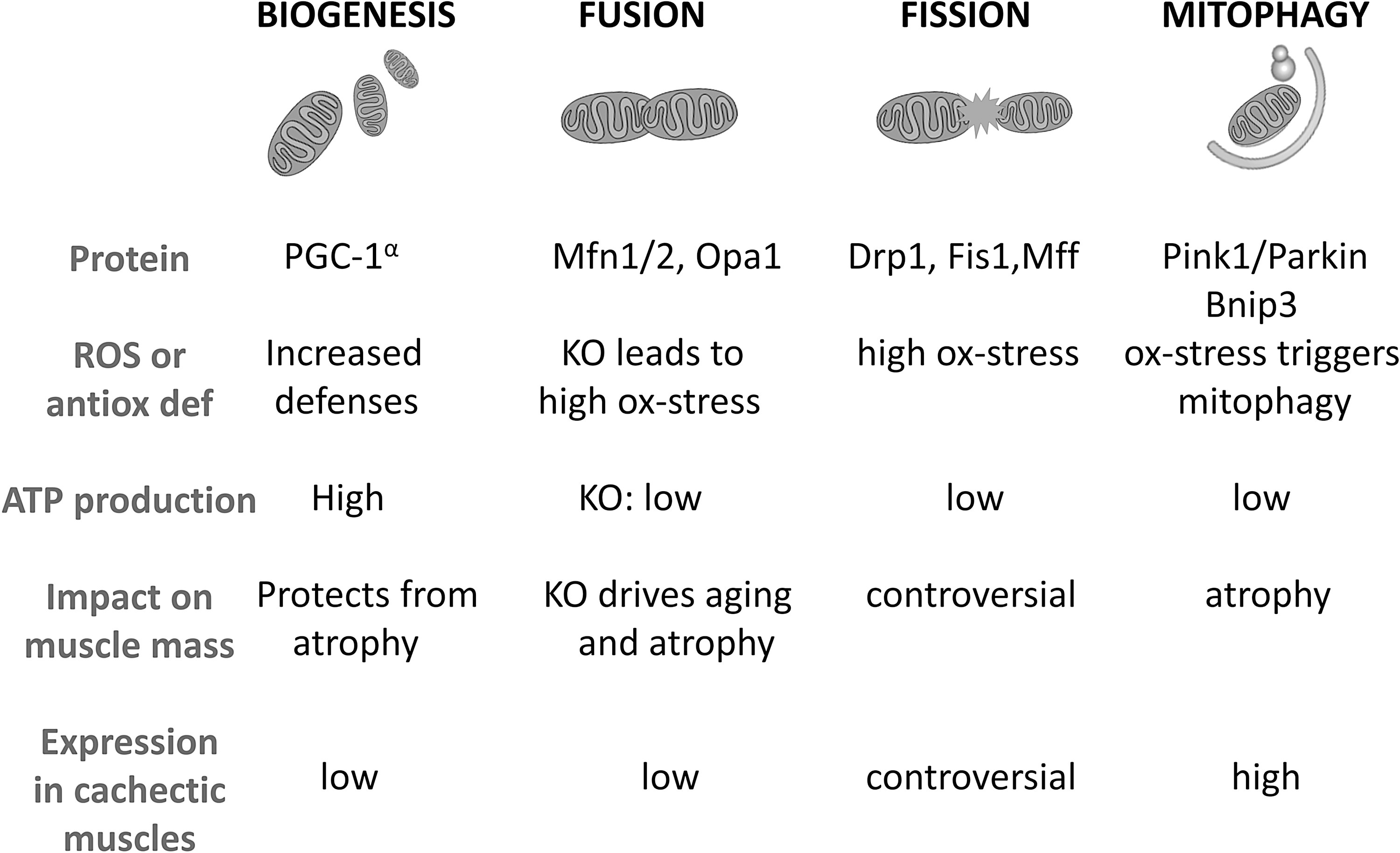
Mitochondrial biogenesis mainly depends on the activity of the transcriptional coactivator PGC-1α that promotes the activity of transcription factors critical for the synthesis of mitochondrial components (105). In the heart, PGC-1α is required for the induction of the antioxidant defense, triggering the expression of SOD2 and thioredoxin (73). In addition, PGC-1α muscle expression is impaired in tumor-bearing animals and further reduced upon chemotherapy administration (9). The same work has demonstrated that muscle PGC-1α overexpression is not able to spare cancer- and chemotherapy-induced muscle depletion, distinctly from denervation and fasting where PGC-1α has proven sufficient to rescue muscle mass (117). The higher mitochondrial content and PGC-1α expression of oxidative fibers (70) associate with increased antioxidant defenses when compared with glycolytic fibers, having a lower mitochondrial load (22). The observation that oxidative fibers are less susceptible to cancer-induced atrophy (25) points to a relationship among fiber metabolism, oxidative stress, and wasting, suggesting that the antioxidant and metabolic properties of oxidative fibers should be preserved to spare muscle mass.
Beyond mitochondrial biogenesis, also the mitochondrial fusion process, which along with fission is generally described as mitochondrial dynamics, has been recently involved in the regulation of muscle mass. The fusion machinery is composed by mitofusin (Mfn)1 and 2, involved in the fusion of the outer mitochondrial membrane and optic atrophy protein 1 (Opa1), required for the fusion of the inner membrane (112). Muscle-specific Mfn2 ablation causes the appearance of an aged phenotype characterized by massive ROS accumulation and impaired muscle mass and function (120). Consistently, Mfn2 deficiency was described in experimental (9, 150) and human cancer cachexia (75, 150). The overexpression of Mfn2 was sufficient to counteract skeletal muscle wasting in mice bearing the HCT116 tumor (150). Similarly to Mfn2, muscle Opa1 loss leads to severe disruption of the mitochondrial network with marked muscle atrophy and an aged phenotype (110, 133).
Opposite to fusion, fission produces a fragmented mitochondrial network. Contributing to the latter, the dynamin-related protein 1 (Drp1) interacts with protein receptors bound to the outer mitochondrial membrane, namely Fis1 and Mff (112). Regarding the role of mitochondrial fission in the onset of muscle atrophy, there are scanty and conflicting data. Fis1 protein levels have been reported to be upregulated in cachectic cancer patients (75), while both Fis1 and Drp1 gene expressions are downregulated in mice bearing the C26 tumor (9, 92). Also, the genetic manipulation of fission-related genes has produced contrasting data. On one hand, muscle-specific Drp1 deletion has been associated with muscle wasting and weakness (44), while Fis1 knockdown is protective against fasting-induced myofiber atrophy (111). These data could be reconciled keeping in mind the hormesis theory, thus considering that basal fission activity is essential for the maintenance of muscle health, while excessive fission induces muscle wasting.
The final step of mitochondrial turnover is degradation through mitophagy. Dysfunctional or damaged mitochondria are recognized by the lysosomal/autophagic system when tagged with specific proteins, such as the Pink1/Parkin complex that triggers the ubiquitylation of several mitochondrial substrates, including Mfn2 (24) or Bnip3 that binds to the autophagosome membrane protein LC3B (40). Keeping in mind that oxidative stress is highly dependent from mitochondrial health, an increased ROS production from mitochondria also turns into a mitochondrial damaging insult, enhancing the oxidative stress and triggering mitophagy. In this regard, in cachectic muscles, a feed-forward mechanism has been postulated, in which oxidative stress-induced mitochondrial damage triggers mitophagy, impairing mitochondrial activity and abundance, eventually activating the catabolic pathways responsible for muscle wasting (144). Such mechanism finds confirmation from recent works showing that oxidative stress and mitochondrial degeneration are an early event that precedes muscle wasting in mice bearing the LLC tumor (18). Similarly, mitophagy induced by C26 tumor growth impairs mitochondrial respiratory function and forcing autophagy in cachectic mice exacerbates mitophagy and consequently muscle protein depletion (92). Consistently, increased expression of markers of autophagy and mitophagy has been detected in muscle biopsies from cachectic cancer patients (8).
Mitochondrial oxidative stress, leading to impaired mitochondrial function and abundance, does not only imply the activation of muscle protein catabolism but even more importantly affects the metabolic properties of the muscle as well, producing a systemic energy failure and worsening patient fatigue. The impairment of mitochondrial oxidative capacity triggers a metabolic shift of the myofibers from oxidative to glycolytic (99), reducing the ability to produce ATP (9, 138). Both cancer and chemotherapy were shown to interfere with muscle oxidative metabolism. Indeed in C26-bearing mice or in mice exposed to chemotherapy, severe oxidative stress was coupled with a dramatic reduction of the tricarboxylic acid cycle and β-oxidation flux, producing an increased glucose demand that requires both glucose and glycogen mobilizations from the liver (69, 98).
Inflammation and Oxidative Stress Cross Talk in the Muscle and Beyond
As reported above, chronic inflammation is one of the hallmarks of cancer cachexia. In this regard, high circulating levels of acute-phase mediators, C reactive protein in particular, and increased production of proinflammatory cytokines such as tumor necrosis factor (TNF)α, TNF-like weak inducer of apoptosis (TWEAK), TRAF6, interleukin (IL)-1, IL-6, and interferon (IFN)γ have been reported in cancer patients (5). In addition, mice hosting the C26 tumor show increased expression of messenger RNAs (mRNAs) coding for acute-phase proteins in the skeletal muscle (16). The other way round, there are studies showing that cachexia is associated with reduced expression of anti-inflammatory cytokines such as IL-10 (28). Finally, the association between cachexia and polymorphisms on genes coding for IL-8, IL-10, and TNFα has been reported in cancer patients (15, 34, 103).
Several lines of evidence demonstrate that proinflammatory cytokines contribute to cachexia. In healthy animals they induce loss of both muscle and adipose tissue mass, an effect dependent on the ability of these mediators to negatively affect protein and lipid anabolism, stimulating catabolism (134). Indeed, enhanced protein turnover rates associated with lipolysis, reduced activity of the lipoprotein lipase, and induction of the hormone-sensitive lipase have been reported upon cytokine administration to experimental animals (5). Of interest, tumor hosts frequently show comparable alterations that, in experimental settings at least, can be prevented by treating the animals with specific anti-cytokine antibodies (5).
Proinflammatory cytokines significantly contribute to the pathogenesis of cancer cachexia. They mainly act through signaling pathways dependent on NF-κB and p38 MAPK. Both of them contribute to increase the expression of muscle-specific ubiquitin ligases such as atrogin-1 and MuRF1, eventually resulting in muscle protein hypercatabolism (5). Consistently, inhibition of NF-κB transcriptional activity has been shown to protect against cancer-induced muscle wasting (79). In addition, proinflammatory cytokines can result in peripheral insulin resistance, impacting on the signal transduction pathways downstream the insulin receptor. Similarly, TNFα negatively impinges on the bioactivity of the insulin-like growth factor (IGF)-1, impairing tissue anabolism and stimulating catabolism (134). In some experimental models, cachexia is associated with increased expression of TWEAK that results in enhanced MuRF-1 levels and autophagy (67).
Particularly intriguing is the observation that cytokines produced by different tissues for different reasons may eventually target the skeletal muscle, eliciting a wasting response. This applies, for example, to transforming growth factor (TGF)β released from bone metastasis, proposed as a significant contributor to muscle wasting (147). In this regard, TGFβ can negatively regulate skeletal muscle mass by impinging on the Smad2/3 transcription factor complex, resulting in muscle wasting due to enhanced myostatin/activin signaling (23). Another mechanism by which both TGFβ and TNFα may mediate cachexia is the induction of the zinc transporter ZIP14, resulting in loss of myosin heavy chain in the adult muscle (146).
Together with mitochondrial alterations, supraphysiological levels of proinflammatory cytokines are among the main contributors to the disrupted redox homeostasis in cachexia. In this regard, the first evidence that ROS/RNS and inflammation are strictly interconnected is a rather old one (96). Subsequently, the discovery that at least some of the canonical proinflammatory cytokine-driven transcription factors such as NF-κB, AP-1, and Nrf2 can be regulated by the redox balance has clearly defined the relationship occurring between inflammation and oxidative stress. In this regard, ROS/RNS can activate redox-sensitive transcription factors, leading to the overexpression of both antioxidant molecules and proinflammatory cytokines. The latter further induces the production of oxidative species, feeding a vicious circle that amplifies the effects exerted by oxidative stress and inflammation on muscle protein and energy metabolism (Fig. 4). Along this line, TNFα has been shown to stimulate ROS production by mitochondria as well as by activating NADPH oxidase and xanthine oxidase, releasing high O2 − levels that are rapidly dismutated to H2O2 (1, 139). Accumulation of the latter in the muscle results in stress of the sarcoplasmic reticulum, altering the intracellular calcium homeostasis and likely impinging on the regulation of intracellular proteolysis by activating calpains (see also Role of Oxidative Stress in Muscle Wasting and Dysfunction section).
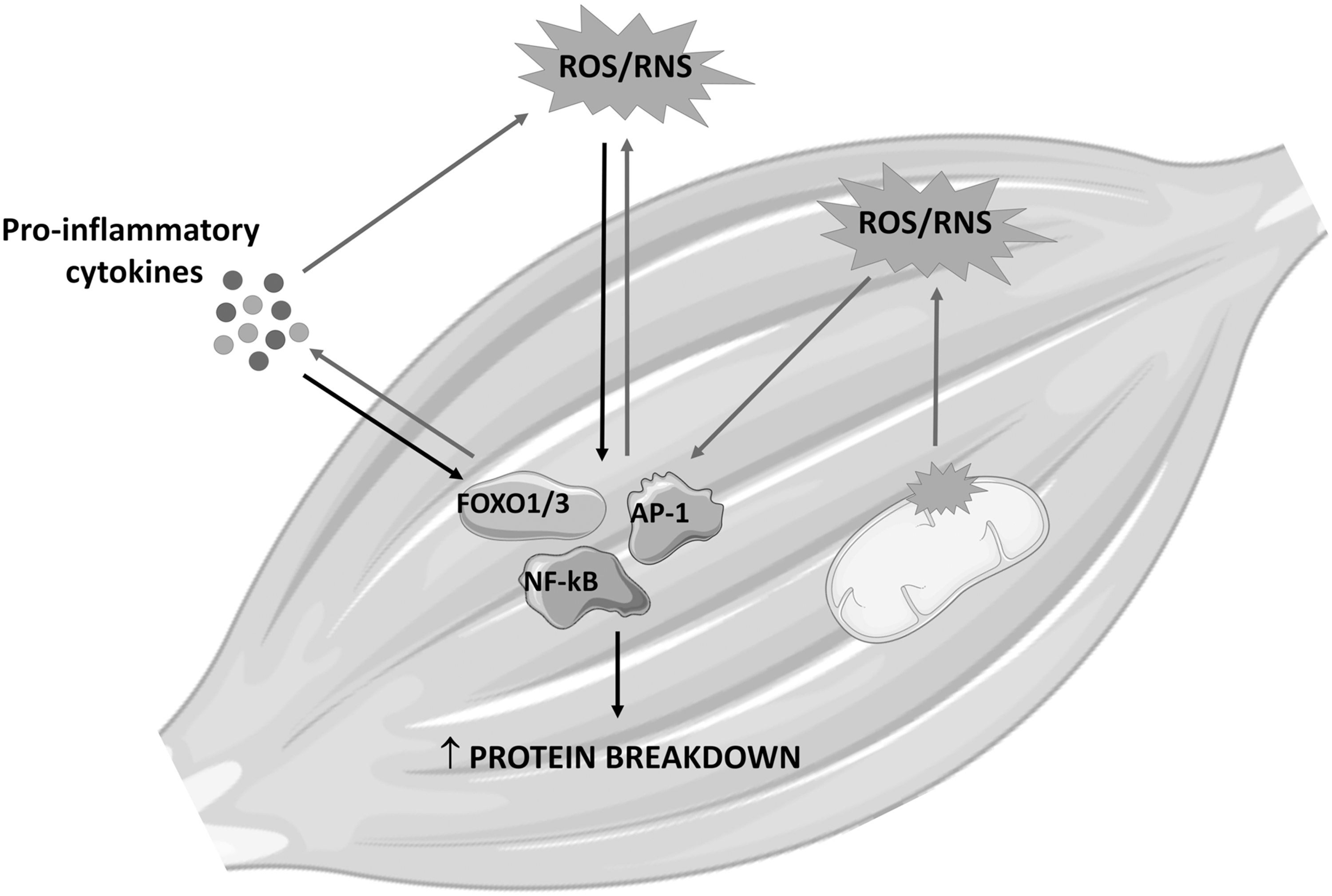
In addition, H2O2 can activate both NF-κB and Smad3, on one hand enhancing the inflammatory response, and on the other hand, further contributing to muscle protein hypercatabolism by inducing the transcription of genes coding for muscle-specific ubiquitin ligases and autophagy (see above) (7). Additional observations show increased O2 − levels in the muscle of tumor-bearing mice, associated with decreased expression of two NADPH oxidase subunits, and also of SOD1/2 and GPX, underlying the relevance of the antioxidant machinery to the maintenance of redox homeostasis (1).
Increased oxidative stress in the skeletal muscle has been shown to occur upon exposure to TGFβ derived from bone resorption induced by osteolytic metastasis. Indeed, by inducing the expression of NAPDH oxidase 4, TGFβ causes an increased ROS production, resulting in oxidation of RyR1 and in calcium homeostasis disruption, ultimately contributing to muscle wasting. Consistently, NADPH oxidase 4 expression is reduced by treatments able to interfere with TGFβ release or signaling (2, 147). Finally, the analysis of secretome from cancer cells has highlighted the release of a huge number of proinflammatory mediators that markedly enhance fatty acid oxidation in the skeletal muscle, leading to oxidative stress and tissue protein depletion (48).
Exercise Counteracts Cachexia Modulating the Redox Homeostasis
Exercise training is a nonpharmacological and cost-effective tool adopted for preventive and therapeutic purposes in different pathologic conditions and aging (143). The effects of exercise include increased muscle fitness and cardiovascular, metabolic, and psychological improvements (21, 41, 46). Notably, one of the mechanisms by which physical training affects muscle mass and function may rely on the regulation of the redox homeostasis (1). The beneficial effects of modulating the redox balance by exercise could again be explained by hormesis (see the Role of Oxidative Stress in Muscle Wasting and Dysfunction section) (59). Indeed, exercise is able to induce a transient increase of ROS production, resulting in the activation of different metabolic pathways according to the intensity and type of exercise (59).
Whereas regular and nonstrenuous exercise leads to low ROS production inducing metabolic adaptations poised to improve muscle function, strenuous exercise leads to excessive ROS levels, resulting in muscle damage (119). Among the adaptations induced by regular exercise, the reduction of systemic inflammation, the improvement of mitochondrial function, and the increase of the antioxidant defenses play a pivotal role in modulating the muscle redox homeostasis (84, 116), producing a potentially anticachexia response.
As described above, cachectic animals show impaired redox balance, likely affecting muscle metabolism (7). In this regard, physical exercise has proven effective in counteracting some of the redox alterations observed in tumor hosts. The improvement of muscle mass and function induced by moderate exercise in C26 tumor-bearing mice has been associated with improved oxidative stress, increased expression of antioxidant enzymes, and restoration of Nrf2 signaling (12). Since oxidative stress may also stimulate the proteolytic systems, the partial normalization of the redox homeostasis in the skeletal muscle of tumor-bearing mice might explain the reduction of autophagy, as suggested by the rescue of LC3B and p62 protein levels (12). Similarly, exercised rats bearing the Walker-256 tumor have shown a reduction of MDA levels coupled with the restoration of the GSH/GSSG ratio (89).
These findings have been associated with a reduction of systemic inflammation, as shown by reduced levels of proinflammatory cytokines such as IL-6 and TNFα and by increased concentrations of the anti-inflammatory IL-10 (89). Bearing in mind that inflammation activates a signaling cascade leading to ROS/RNS production and oxidative stress (see the Inflammation and Oxidative Stress Cross Talk in the Muscle and Beyond section), exercise could be considered an anti-inflammatory agent (50) even in cancer cachexia. In this regard, several studies concerning cancer cachexia have demonstrated that exercise may reduce the production of different proinflammatory factors (39, 90, 99), likely contributing to reduce oxidative stress and muscle wasting. Similar observations have been reported in cancer patients, in which physical training decreases markers of oxidative stress in the blood while increases the antioxidant capacity, resulting in reduced fatigue (108).
Beyond counteracting the detrimental effects of tumor growth, exercise is also effective in decreasing the oxidative stress induced by chemotherapeutic agents, the latter contributing to fatigue and reduced mobility in cancer patients (100). In this regard, several lines of evidence have shown that exercise is able to correct doxorubicin-induced cardiac alterations by modulating the redox balance. Notably, an acute bout of exercise decreases lipid peroxidation products, improving the cardiac function in rats treated with doxorubicin (149). Similarly, 8 weeks of aerobic exercise protect doxorubicin-treated mice from cardiac injury, decreasing 4-HNE-protein adduct in plasma and heart while increasing heart MnSOD levels (38).
The increased oxidative stress induced by doxorubicin is prevented by exercise also in murine skeletal muscle, reducing protein carbonylation and 4-HNE accumulation (125). Such pattern associates with reduced calpain/calpastatin ratio, calpain activity, and caspase-3 levels and activity (125). Consistently, another study has reported that aerobic exercise reduces the levels of markers of oxidative stress and increases MnSOD levels in heart mitochondria of doxorubicin-treated mice, resulting in increased mitochondrial content and oxidative function (74). However, exercise effectiveness in reverting oxidative stress is limited, in particular when tumor growth is combined with chemotherapy administration, resulting in severe muscle wasting. In this regard, moderate exercise, although being able to partially spare muscle mass and loss of function, does not correct the increase of ROS levels and protein carbonylation in chemotherapy-treated tumor-bearing mice (9).
Exercise may also affect the muscle redox balance modulating mitochondrial function (59). As described in the Mitochondria as Pivotal Elements in Muscle Oxidative Stress section, mitochondrial homeostasis is severely impaired in the skeletal muscle of cachectic subjects, leading to increased ROS production that in turn leads to mitochondrial damage in a vicious cycle (32). In this regard, exercise modulates mitochondrial homeostasis impinging on biogenesis, function, and disposal, likely contributing to improve mitochondrial fitness and reducing oxidative stress (84), see Figure 5.
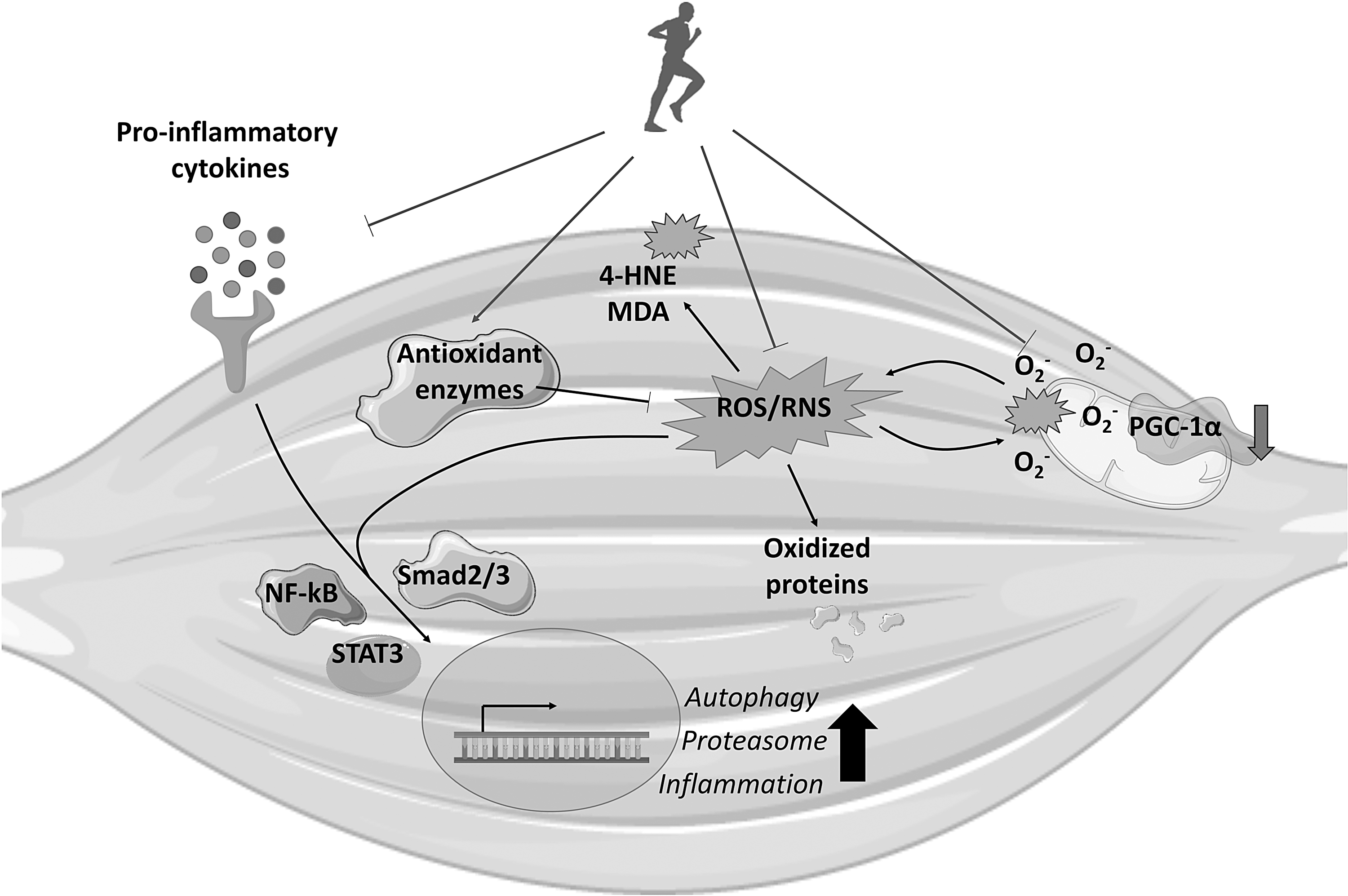
As previously discussed, one of the central regulators of the exercise-induced mitochondrial adaptations is the cotranscriptional factor PGC-1α. Beyond the regulation of the mitochondrial biogenesis, PGC-1α induces the activation of the antioxidant defenses (60). Consistently, the ablation of PGC-1α blunts some of the effects induced by exercise training in the skeletal muscle (11), and reduces the levels of different antioxidant enzymes (54). The other way round, PGC-1α overexpression in the tibialis anterior muscle decreases oxidative stress while increasing the antioxidant defenses in a model of immobilization-induced muscle atrophy (62). Consistently, some of the protective effects of exercise against muscle wasting may rely on the induction of PGC-1α. In this regard, several reports have demonstrated that exercise is able to induce the expression of this cotranscriptional factor in cancer cachexia. Notably, together with the reduction of markers of oxidative stress and increased antioxidant defenses, moderate exercise in C26-bearing mice increases PGC-1α levels and mitochondrial content (12). Similarly, PGC-1α expression increases in the C26 hosts upon combined exercise, including both aerobic and resistance training (107), and in chemotherapy-treated C26-bearing mice exposed to moderate endurance exercise (9). The increase of PGC-1α levels upon exercise training has been observed also in ApcMin/+ mice (148) and in C26 and LLC hosts, receiving also erythropoietin (EPO) or eicosapentaenoic acid (EPA), respectively (93, 99).
Together with the induction of mitochondrial biogenesis, exercise modulates mitochondrial fusion/fission events and mitophagy, thus regulating mitochondrial quality and function in the skeletal muscle (60). As reported above, the impairment of one of these events could lead to the accumulation of damaged mitochondria and oxidative stress. In aging, aerobic exercise increases the expression of mitochondrial protein related to both mitophagy and dynamics (64). Similarly, the effect of exercise training in tumor-bearing animals is poised toward the normalization of autophagy/mitophagy and mitochondrial fusion and fission. In this regard, the increase of Bnip3 levels is corrected by the combination of exercise and EPO administration to tumor hosts, coupled with reduced mitochondrial alterations and increased function (99). In chemotherapy-treated tumor-bearing mice, exercise increases Mfn2 gene expression, together with decreased Bnip3, PINK1, and Parkin levels (9). Similarly, a report by White et al. has shown that exercise increases Mfn2, while decreasing Fis1 protein expression in the skeletal muscle of ApcMin/+ upon IL-6 overexpression (148).
Potential Impact of Antioxidants and Exercise Mimetics in Cancer Cachexia
An intuitive way to reduce oxidative stress is the administration of exogenous antioxidants able to scavenge ROS/RNS, such as vitamins, carotenoids, and polyphenols. However, the studies concerning the use of this type of compounds in cancer growth and treatment, as well as in cancer cachexia, have produced controversial data. Although some antioxidants seem effective in reverting cancer-induced muscle wasting by reducing inflammation and oxidative stress (7, 17), other studies underline that antioxidants may promote tumor growth and reduce radio/chemotherapy efficacy (7). For instance, recent reports on preclinical models of cancer cachexia have shown that quercetin and an immunomodulatory diet (also enriched with antioxidant compounds such as vitamins A, C, E, and zinc and selenium) positively affect muscle mass in ApcMin/+ and C26-bearing mice, respectively (86, 142).
A beneficial action has been also achieved by resveratrol, although the effectiveness has been proven only in C26 hosts and not in other cachexia models, such as LLC or Yoshida AH-130-bearing animals (20, 121). Conversely, a report by Assi et al. has shown that the supplementation of a mixture of antioxidants increases tumor growth in C26 hosts, worsening muscle wasting and oxidative stress (6). On the same line, the maintenance of stable circulating antioxidant vitamins and β-carotene concentration seems to increase cancer patient mortality (7). In addition, the administration of antioxidants to cancer patients should be carefully evaluated when combined with exercise, since several lines of evidence suggest that antioxidants may hamper the beneficial adaptations of chronic exercise in the skeletal muscle (52).
Despite directly administering antioxidants, oxidative stress may also be targeted with compounds able to modulate the ROS/RNS-regulated pathways, to reduce inflammation or inhibit ROS-producing enzymes (Fig. 6). Using such an approach, different classes of drugs have been proven effective in reverting cancer-induced muscle wasting by mimicking some of the beneficial effects induced by exercise (11). Notably, trimetazidine (TMZ) increases muscle cross-sectional area and strength in C26-bearing mice, coupled with increased oxidative metabolism and mitochondrial mass (78). These improvements could also rely on the modulation of the redox balance.
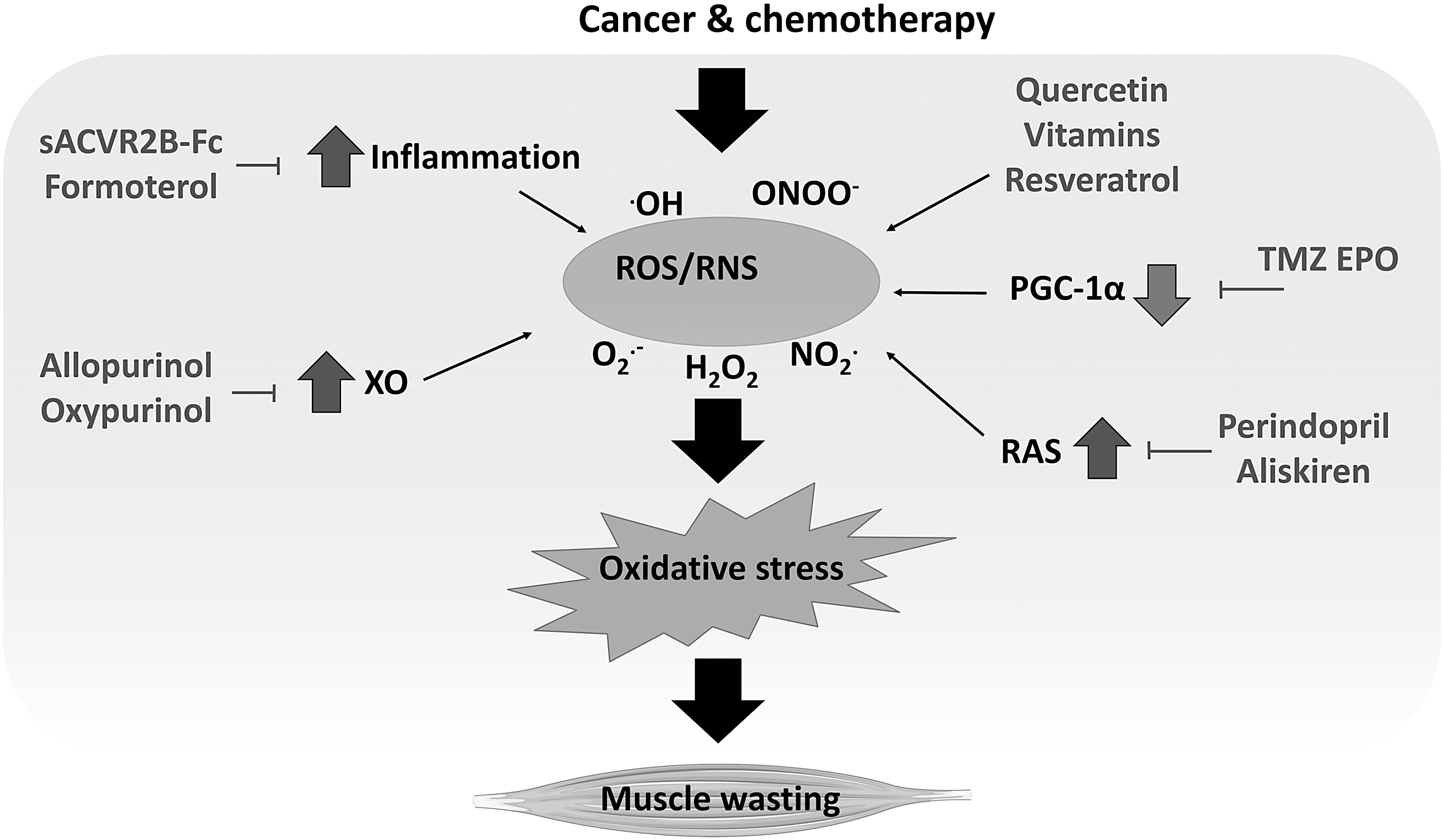
Notably, TMZ reduces the levels of markers of oxidative stress and increases SOD activity in human aortic smooth muscle cells (152) and decreases MDA levels in blood sample from young and aged rats (151). In this regard, one mechanism by which TMZ may modulate the redox balance is the inhibition of the β-oxidation (45), thus favoring glucose metabolism and reducing ROS production. Similarly to TMZ, EPO has been shown to improve mitochondrial function and oxidative stress. Indeed, in human cardiomyocytes, EPO contributes to correct doxorubicin-induced alterations by upregulating SIRT1, inducing the activation of PGC-1α and the transcription of genes involved in mitochondrial homeostasis and antioxidant defenses (31). Consistently, EPO-treated tumor-bearing mice show increased muscle expression of PGC-1α, mitochondrial function, and ATP intracellular levels (99).
Along with these drugs, molecules that are able to block or reduce the activation of inflammatory pathways could be useful to correct muscle oxidative stress. In a recent report by Hentilä et al., the blockade of activin receptor type 2 (ACVR2) ligands (e.g., myostatin, activins, and TGFβ) by the recombinant soluble activin receptor 2B (sACVR2B-Fc) has been proven to modulate the redox balance in healthy and tumor-bearing animals (57). In the latter, sACVR2B-Fc protects against muscle wasting, increasing survival and correcting GSH reduction and GSSG/GSH ratio increase (57, 87). Along with these effects, the soluble activin receptor increases the expression of heat shock protein 25 (HSP25) (57), which is involved in glutathione metabolism and resistance to ROS (43).
An adaptive response to oxidative stress is also induced by formoterol, a β2-adrenoceptor-selective agonist. Several studies report the beneficial effects of this compound in reverting cancer-induced muscle wasting in both animal models and cancer patients by triggering protein metabolism toward synthesis and reducing inflammation (53, 92, 114, 136, 137). Along with the anabolic/anticatabolic effects, formoterol also prevents the increase of oxidized proteins and MDA/HNE-protein adducts in the skeletal muscle of Yoshida AH-130-bearing rats (115). However, formoterol does not increase SOD1 and catalase protein levels in tumor hosts (115), suggesting that the mechanism by which this molecule acts on oxidative stress does not rely on the induction of the antioxidant defenses or, at least, the antioxidant induction is not detectable at the experimental time point considered by the authors. Conversely, formoterol could affect oxidative stress indirectly by reducing inflammation, as suggested by the decrease of inflammatory cell infiltration in the muscles of tumor hosts (115).
Another strategy to reduce oxidative stress is the direct targeting of enzymes responsible for ROS production. Among them, xanthine oxidoreductase (XOR) is an intracellular enzyme involved in purine catabolism that may exist in two different forms, xanthine dehydrogenase or xanthine oxidase (XO) (37). The latter is induced by stress conditions (e.g., inflammation) and uses molecular oxygen as an electron acceptor, producing superoxide anion and oxygen peroxide (37). Notably, XO activity increases in unloading-induced muscle atrophy and in the skeletal muscle of cachectic animals (35, 129). In this regard, the inhibition of XO by allopurinol prevents muscle and plasma XO activity and these effects associate with the reduction of the oxidative stress-induced protein p38 and atrogin-1, relieving unloading-induced muscle atrophy (35).
Similarly, in Yoshida AH-130-bearing animals, the inhibition of XO by allopurinol and oxypurinol partially prevents body and muscle wasting and increases survival, although the effects of oxypurinol vary depending on the dosage (129). The macroscopic improvements associated with the protection against several tumor-induced molecular alterations, such as increased inflammation, muscle protein carbonylation, markers of proteasome activation, and proteasome activity (129). Moreover, oxypurinol also improves cardiac mass and function in cachectic tumor hosts (128). However, particular attention should be given to tumor response, since oxypurinol administration increases the tumor mass in immunocompromised mice injected with MAC16 cells (140).
A final approach aimed at targeting the oxidative stress in experimental cancer cachexia is the use of inhibitors of the renin/angiotensin system (RAS). Beyond regulating blood pressure, this system is also locally active in different tissues, including the skeletal muscle. In this regard, the inhibition of the angiotensin converting enzyme (ACE) with perindopril prevents body wasting and muscle dysfunction, coupled with increased succinate dehydrogenase activity in C26 hosts with mild cachexia (82). Similarly, another RAS inhibitor specifically targeting renin, namely aliskiren, has proven effective in reverting muscle wasting in C26-bearing mice (145). In this regard, aliskiren increases gastrocnemius muscle mass and muscle function and decreases the levels of proinflammatory cytokines, of markers of autophagy, and of atrogin-1 and MuRF-1 (145). These effects associate with increased muscle SOD and GPX activity and with reduced MDA and hydroxyl radical levels (145). However, it is noteworthy that aliskiren also reduces tumor growth (145), so making it difficult to distinguish if the drug has a direct effect on the muscle or, by reducing tumor burden, indirectly improves cachexia. Similarly, the abovementioned drug perindopril is able to decrease tumor mass in severely cachectic C26 hosts (82).
Conclusions
Oxidative stress can be considered one of the most relevant triggers of muscle wasting in cancer cachexia, acting at the critical intersection between inflammation and mitochondrial dysfunction. Thus, it is likely that cancer patients experiencing cachexia might benefit from an intervention aimed at counteracting the excessive oxidative stress.
A desirable strategy to be set up should reinforce the muscle antioxidant defenses of cancer patients by means of exercise or exercise mimetic drugs rather than directly administering antioxidants. Such a strategy would maximize the beneficial aspects of oxidative stress control while minimizing the potentially detrimental action of antioxidants on host adaptive responses and tumor growth.
Footnotes
Funding Information
F.P. has received funding from Fondazione AIRC per la Ricerca sul Cancro under IG 2018—ID. 21963 project. F.P. and P.C. are supported by the University of Torino (ex-60% funds).