
Abstract
Significance:
According to the World Health Organization, noncommunicable diseases are the globally leading cause of mortality.
Recent Advances:
About 71% of 56 million deaths that occurred worldwide are due to noncommunicable cardiovascular risk factors, including tobacco smoking, unhealthy diets, lack of physical activity, overweight, arterial hypertension, diabetes, and hypercholesterolemia, which can be either avoided or substantially reduced.
Critical Issues:
Thus, it is estimated that 80% of premature heart disease, stroke, and diabetes can be prevented. More recent evidence indicates that environmental stressors such as noise and air pollution contribute significantly to the global burden of cardiovascular disease. In the present review, we focus primarily on important environmental stressors such as transportation noise and air pollution. We discuss the pathophysiology of vascular damage caused by these environmental stressors, with emphasis on early subclinical damage of the vasculature such as endothelial dysfunction and the role of oxidative stress.
Future Directions:
Lower legal thresholds and mitigation measures should be implemented and may help to prevent vascular damage.
Introduction
Environmental stressors and cardiovascular risk
The shift toward a modern society has brought with it a shift in the burden of disease toward noncommunicable diseases (NCDs). NCDs include cardiovascular diseases (CVDs), cancers, chronic respiratory diseases, diabetes, and mental health conditions, as well as other neurologic, endocrine, gastrointestinal, renal, allergic, and autoimmune disorders. According to the World Health Organization (WHO), this wide spectrum of disease is now the leading cause of mortality, with an estimated 38 million deaths—totaling 70% of global yearly deaths (23). Fourteen million of these deaths are those who “die too young”—that is, between the ages of 30 and 70 (130). In addition to accounting for the vast majority of global yearly deaths, NCDs also account for 80.6% (95% confidence interval 78.2–82.5) of years lived with disability, according to the Global Burden of Disease study (33).
Cardiovascular risk factors impact a huge proportion of the deaths that occur worldwide, about 71% of 56 million deaths. These risk factors include smoking, unhealthy diet, lack of physical activity, overweight, hypertension, high blood sugar, and cholesterol. Despite these risk factors being largely avoidable or alterable, it is estimated that 80% of premature heart disease, stroke, and diabetes stem from their presence and could be prevented (89). In addition to these classical risk factors, environmental risks pose additional implications to health and have thus far been underestimated.
To that end, epidemiological studies have established that exposure to traffic noise is correlated with increased cardiovascular morbidity and mortality [for review see Refs. (80, 84)]. Alongside these studies, the WHO recently estimated that 61,000 healthy life years are lost as a consequence of noise-induced CVD yearly in Western Europe alone. To add to that tally, the WHO also estimates that cognitive impairment in children due to noise costs 45,000 healthy years, noise-related sleep disorders cost 903,000, while 22,000 and 654,000 are lost to tinnitus and noise-related annoyance, respectively (1). In this context, genetic susceptibility is likely to play a role in response to environmental risk factors. For example, gene/environment interaction studies on the pathogenesis of hypertension in the general population (85) as well as in noise-exposed workers (139) found a significant association between ACE gene polymorphisms and occurrence of hypertension. These figures indicate that up to 1.6 million healthy life years are lost due to traffic-related diseases yearly in Western Europe.
Fossil fuels and biomass combustion, as well as industry, agriculture, and wind-blown dust, are all common and prevalent sources of fine particulate matter (PM) in the air (Fig. 1) (60). These sources are nearly ubiquitous and difficult to characterize, and so, air pollution is not often taken into account as a health risk. However, the Lancet Commission on pollution and health has made recommendations to enact air quality action plans to mitigate and prevent the growing impact of NCD (57). The commission further estimated that 9 million excess deaths were caused by poor environmental conditions and that about half of these deaths were attributable to ambient (outdoor) air pollution.

Although air pollution is often overlooked, there has been a general consensus that chronic exposure to fine particulates causes vascular inflammation, oxidative stress, and as a result, endothelial dysfunction and a subsequent rise in blood pressure. Over time, these physiological detriments can lead to myocardial infarction (MI), arterial hypertension, stroke, heart failure, and arrhythmia (82, 83, 99). In fact, mortality rates due to air pollution in 2017 as related to CVD were 2.4 million per year, with 269,000 in Europe (21). This number has been corrected by Lelieveld et al. (61) using a novel hazard ratio function to 790,000 excess deaths per year in Europe due to air pollution, demonstrating in particular high PM 2.5 μm levels for Germany (Fig. 2A, B and Table 1). This number means that because of a relatively dense population in combination with poor air quality, 133 deaths per year (and 129/year in the 28 countries of the European Union [EU]-28) per 100,000 occur, making the exposure in Europe among the highest in the world (61). When taken altogether, the data above indicate that in the EU-28, CVD alone is the cause for 264,000 deaths, a number that could potentially reach 513,000 (with substantial uncertainty) when other NCDs are included. There is, then, strong support for the special report of the European Court of Auditors that states that the health within the EU-28 is insufficiently protected (Table 1).

Premature Deaths Attributable to Air Pollution for Europe, European Union-28, or Specific Countries
Differentiation for different disease categories, deaths, and years of lost life or loss of life expectancy [with permission from Lelieveld et al. (61)].
CVDs are total cardiovascular diseases (CEV+IHD). The percentages refer to the fractional contributions of CVD and other NCDs to attributable mortality from all diseases.
Data for all EU countries, including 95% uncertainty intervals, are given in the supplement of reference (61).
CEV, cerebrovascular disease; EU, European Union; IHD, ischemic heart disease; LLE, loss of life expectancy; NCD, noncommunicable disease; YLL, years of life lost.
A healthy vascular endothelium is considered a central prerequisite for low cardiovascular risk (27, 81). Intact endothelial function relies on a delicate balance between the formation and degradation of nitric oxide (•NO), which it uses to modulate several critical functions of the vasculature: tone, inflammation, thrombus formation, and injury repair (27). As an oxygen-derived free radical, the half-life and biological activity of •NO are strongly impacted by the presence of oxidants such as the superoxide ion. Depletion of nitric oxide by superoxide disrupts the balance required by the vascular endothelium and leads to endothelial dysfunction; one of the first presentations of subclinical CVD and a consequence not only of classical risk factors (smoking, high cholesterol, diabetes mellitus, and hypertension) but also of novel risk factors such as exposure to noise and air pollution (27, 84). This redox biological concept also explains the central role of oxidative stress for environmentally triggered CVD.
Air pollution costs more lives than tobacco smoking
We recently used a new global estimate of exposure mortality model to calculate that 8.79 million premature deaths occurred in 2019 due to exposure to air pollution, in particular PM2.5. This estimate is in close correlation with the estimated 8.9 million global deaths attributable to air pollution by Burnett et al. (13). The WHO estimates for the yearly excess deaths attributed to tobacco smoking are at 7.2 million (131), implicating air pollution as the larger contributor to global mortality.
Data from the WHO suggest that 90% of people worldwide breathe polluted air, which lends itself to the simple explanation that there are fewer people who smoke than people who breathe polluted air. A corollary of this supposition is that since the air is polluted, humans are exposed to air pollution over a lifetime, rather than acutely as with tobacco exposure. Maternal PM2.5 exposure can limit fetal growth and contribute to preterm birth and since children are particularly sensitive to exposure, exposure to air pollution could cause lifelong health damage (57). Finally, exposure response differences between tobacco smoke and air pollution have recently come to light. Disease burdens at high PM2.5 concentrations (>35 mg/m3) were previously calculated based on hazard ratios from both active and passive smoking studies. In these calculations, the hazard ratios did not proportionately scale with the level of exposure (21). However, recent cohort studies have demonstrated that hazard ratios at high ambient PM2.5 concentrations (up to >90 μg/m3) are actually much higher than those expected when calculated on the basis on smoking studies (13).
In the next sections, the pathophysiology of the CVD burden of noise and air pollution is outlined, focusing on endothelial dysfunction and oxidative stress.
Pathophysiology of Noise-Induced CVD
The noise reaction model
Noise can exert physiological effects either directly or indirectly, as proposed by Babisch in his noise effect reaction scheme (78). The direct pathway involves the intuitive effects of noise wherein sleep disturbance can occur and at levels higher than 85 dB(A), can lead to cochlear damage and hearing loss. This path of the reaction scheme has been historically well-established, whereas the indirect pathway that occurs at subinjurious decibel levels is a rather novel field of investigation. In this pathway, perceiving sound causes cortical and sympathetic activation and subsequent emotional responses, such as annoyance, or physiological responses, such as disturbance of sleep, communication, or activities (Fig. 3) (78). Increases in corticosteroid and catecholamine release follow these disturbances (78), which is thereby responsible for a rise in blood pressure, an increase in blood viscosity, and activation of the coagulation cascade (64).

Stress responses are independent of cognitive involvement, meaning that the perception of noise is not necessarily a prerequisite for adverse cardiovascular effects. It is possible that noise exerts its effects directly through synaptic interactions, and also that there is cognitive and emotional involvement. Thus, both the decibel level of the noise and the inherent properties of the noise could affect the degree of stress response and subsequent neuroendocrine hemostasis. It is imaginable then that the persistence of this stress over long periods (years) could accelerate the development of CVD, including stable coronary artery disease, acute coronary syndromes, arrhythmia, heart failure, arterial hypertension, and stroke (80).
Translational noise studies in healthy subjects and patients with established coronary artery disease
We administered a field study involving both healthy subjects and subjects with existing coronary artery disease who would utilize an MP3 player overnight to deliver simulated aircraft noise. The simulation included either 30 or 60 noise events per night with a peak sound level of 60 dB(A) and mean sound level of 43 and 46 dB(A). Results from this field study indicate that following one night of noise exposure, both healthy subjects (115) and those with coronary artery disease (114) presented with endothelial dysfunction, as measured by flow-mediated dilation (FMD) (Fig. 4A), in addition to increased epinephrine levels and decreased sleep quality. The field study also yielded an important insight that administration of the antioxidant vitamin C (2 g p.o.) was successful in reversing the endothelial dysfunction in a subgroup of subjects, implicating oxidative stress as being at least partially responsible for the impairment of endothelial function following nighttime aircraft noise exposure (115).
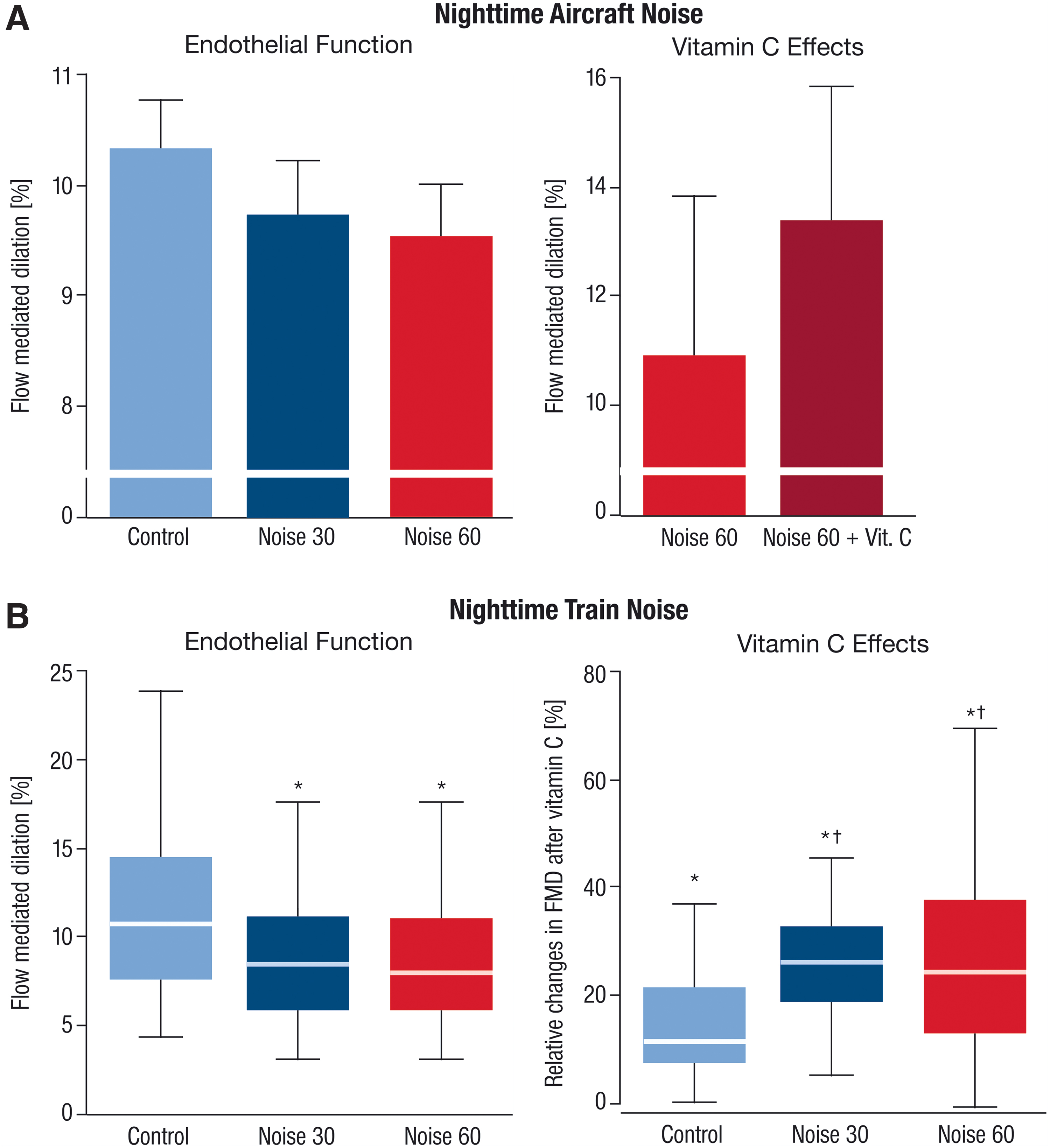
In a similar more recent study, 70 healthy volunteers were exposed to background noise, 30 train events (Noise30), or 60 train events (Noise60) at respective average sound pressure levels of 33, 52, and 54 dB(A) over the course of three nights in their home (Fig. 4B) (44). Following the exposures, the FMD of the brachial artery was reduced from 11.23% ± 4.68% for control to 8.71% ± 3.83% for Noise30 and 8.47% ± 3.73% for Noise60 (p < 0.001 vs. control). The quality of the participants' sleep was also impaired after both Noise30 and Noise60 nights (p < 0.001 vs. control). As in the previous study, a subset of randomly assigned participants were administered vitamin C, which was again found to improve endothelial function, with significantly stronger effects in subjects exposed to noise (Fig. 4B). Because the subjects who were administered vitamin C were randomly chosen, it is stronger evidence of the role of reactive oxygen species (ROS) in the mediation of noise-induced endothelial dysfunction. Further evidence for ROS involvement emerges with the targeted proteomic analysis in this study, which showed that after Noise60, plasma proteins within redox, prothrombotic, and proinflammatory pathways were significantly impacted versus controls (44). These data imply that exposure to nocturnal train noise both impairs endothelial function and also induces proteomic changes toward a proinflammatory and prothrombotic phenotype that could provide a molecular connection to the increased cardiovascular risk observed in epidemiological noise studies (Fig. 5).

Animal studies
In two recent animal studies, we performed further characterization of molecular mechanisms by which aircraft noise exposure affects vascular dysfunction (56, 76). The exposure protocol for mice consisted of 4 days of around-the-clock aircraft noise exposure to induce a significant increase in stress hormone levels (adrenaline, noradrenaline, and dopamine), angiotensin II, increased systolic and diastolic blood pressure, and impaired vascular function. The observed increases in oxidative stress were likely to be the cause for these biological implications (76).
With oxidative stress as a causative agent for the detrimental effects caused by noise exposure, two important enzymatic sources of ROS were characterized as playing a role in the vascular dysfunction and hemostatic consequences observed after noise exposure: the nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase; phagocytic form or NOX2) and an uncoupled and therefore dysfunctional nitric oxide synthase (NOS) (76). The dysfunction of these enzymes due to noise leads to an accumulation of superoxide, which as previously described, directly leads to a reduction in the bioavailability of NO. As NO is an important and powerful vasodilator, an increase of ROS blunts the dilatory ability of the vasculature and results in impairment of endothelial function and higher vascular tone. In addition to functional changes in the vasculature, comparative Illumina sequencing detected significant alterations in the aortic transcriptomes of noise-exposed animals, chiefly in genes involved in regulation of vascular function and remodeling and cell death (76).
To resolve the impact of a reduction in sleep quality on the noise-derived cardiovascular effects, both wild-type and Nox2 −/− (gp91phox −/−) mice were exposed to aircraft noise at a maximum sound level of 85 dB(A) for 1, 2, and 4 days around-the-clock, either during their sleeping phase or waking phase, and also investigated the protection by genetic Nox2 deletion (56). Of note, Nox2 −/− mice were protected from noise-induced endothelial dysfunction, which could be explained by a substantial decrease in vascular and cerebral superoxide formation as well as overall oxidative stress levels and inflammation (key data in Fig. 6A–E). Sleeping-phase noise exposure resulted in more severe adverse effects than waking phase and very similar effects to around-the-clock exposure, where noise caused higher blood pressure, impaired endothelial function, uncoupling of endothelial and neuronal nitric oxide synthase (e/nNOS), downregulation of nNOS mRNA and protein, as well as NOX2 activation, all resulting in cerebral oxidative stress and inflammation (key data in Fig. 7A–E). Next-generation sequencing revealed disruptions in the Foxo3/circadian clock genes in aortic and brain tissue, similar to the adverse gene regulation in around-the-clock exposed mice. As a transcriptional regulator of the circadian clock, induction of FOXO3 had a protective effect in noise-exposed mice and was able to mitigate endothelial dysfunction and prevent vascular oxidative stress. Patients with established coronary artery disease exposed to nighttime aircraft noise also displayed increased oxidative stress and inflammation biomarkers in serum (76). These data show that disruption of the circadian pathways by sleeping-phase noise exposure is more strongly correlated with endothelial dysfunction, increase in blood pressure, inflammation, and increases in oxidative stress-associated biomarkers than waking-phase noise exposure, implying that sleep disruption plays an important role in the maintenance of cardiovascular homeostasis (Fig. 7) (56).


A central role of oxidative stress on the circadian clock was previously reported and termed “redox control of cellular timekeeping” (97). The redox-regulatory mechanisms within the circadian clock were recently described in very close detail (62), for example, mammalian CRY1 contains redox-sensitive cysteine residues and a zinc/sulfur complex that controls binding of PER and FBXL3 to the CLOCK/BMAL1 complex leading to its inhibition (113). Also, redox-sensitive kinases such as AMPK or MAPK and stress-response proteins such as PARP-1, HO-1, transcription factors HIF-1α, PGC-1α, and histone deacetylase SIRT-1 contribute to the regulation of the circadian rhythm (62). Vice versa, the antioxidant and/or ROS-producing gene expression is under the control of the circadian clock as supported by daytime-dependent “redox oscillations” of peroxiredoxin redox state (107). Circadian rhythm dysregulation is a very severe trigger of pathophysiological pathways and is tightly associated with sleep deprivation and its known adverse biological changes such as severe inflammatory conditions (98), endothelial dysfunction, blood pressure increases, structural vascular changes, vascular senescence, and recruitment of immune cells in mice (16). This is the reason for nighttime noise being more detrimental than daytime noise (79). Besides noise exposure, a variety of other stressors, including restraint stress, social defeat stress, placement on an elevated platform, food or water deprivation, continuous lightning, cage tilts, and soiled cages, have been shown to cause circadian clock dysregulation (43, 62). Also, environmental exposures to heavy metals (91), pesticides (7, 46), or air pollution constituents such as fine particular matter (38) can dysregulate the circadian clock and initiate the onset/progression of CVD.
Besides regulation of coding RNA, noise and noise-induced mental stress are also known to cause changes in expression patterns of noncoding RNA, especially of health-relevant microRNAs (67). Noise-induced changes in microRNAs can be separated into the direct pathway (also leading to hearing loss) and the indirect pathway (mainly mediated by stress response pathways involving the sympathetic nervous system, the hypothalamic–pituitary–adrenal (HPA) axis, and stress hormones). The direct pathway (acoustic overstimulation) involves microRNA changes that influence ROS production via altered SIRT-1 and NRF-2 expression (miR-29/34/200), apoptotic pathways via altered EGR1, IRS1, P38/MAPK, and BCL11B signaling (miR-183/186/124/381), and protein misfolding via altered HSP70 expression (miR-90/451) as well as K+ homeostasis (miR-1/27), which is also reflected by upregulation of microRNA markers in plasma (miR-185-5p/451a) (67). For the indirect pathways, the upregulation of miR-134/183 in the central amygdala in response to acute stress seems to be important (66). Mental stress induced by restraint of the body or forced swimming in pregnant rats leads to altered hippocampal microRNA profiles in the offspring, for example, upregulation of miR-98/103/323 and downregulation of miR-145/151/425 (142). In addition, the miR-34 family is related to HPA-mediated stress responses (45) and anxiety-like behavior by regulation of corticotrophin releasing hormone receptor 1 (41). Finally, the activation of the glucocorticoid receptor leads to reduced expression of miR-132 (50). In conclusion, substantial epigenetic alterations by noise exposure seem feasible since a common denominator of noise-associated adverse health effects is oxidative stress and all epigenetic pathways are highly redox regulated (42, 52, 68) and have known impact on health and disease (25, 53).
Air Pollution
The air pollution constituents and cardiovascular events
Air pollution is a nonhomogenous mixture of gases and particulates that fluctuates in composition over time and by location [for review see Munzel et al. (77)]. Since the composition and content of air pollution display so much changeability, its effects are also extremely variable based on the chemistry of the constituent parts. As such, single-variable schemes and projections based on pollutant identity, size, or mass are often insufficient. Despite this, air pollution is currently broadly categorized by aerodynamic diameter: <10 μm (thoracic particles [PM10]), between 2.5 and 10 μm (coarse [PM2.5–10]), <2.5 μm (fine particles [PM2.5]), and <0.1 μm (ultrafine particles), all of which are quantified as mass per cubic meter (μg/m3). PM is not the only, or even primary, pollutant by mass in the air. In urban settings, 90% of the pollutant mass is from gases or vapors, also including volatile and semivolatile organic carbon compounds (77) and ozone, the most prevalent secondary pollutant. Despite these gaseous-phase compounds being the primary pollutants by mass, current data incriminate ultrafine particles and possibly coarse particles as the air pollutants posing the greatest threat to global public health (77). Many reviews have outlined the strong connection between cardiovascular mortality and events, including MI, stroke, and heart failure, and exposure to air pollution, particularly PM2.5 (10, 11, 82, 83, 87). For example, PM2.5, •NO2, SO2, and CO were all associated with increased risk of MI in a systematic review and meta-analysis of studies. Data of clinical/epidemiological studies on air pollution and cardiovascular events are presented in Table 2. The body of evidence supporting the connection between cardiovascular events and mortality is still growing, but there are several truths that are widely accepted. The first of these truths is that the chronic effects of exposure to air pollution are much more substantial than the acute effects. The second is that the elderly and other “at-risk” individuals, such as those with established CVD or obesity, are at higher risk. Third, there is no demonstrable “lower threshold” of exposure. There is a relationship between CVD and air pollution exposure at all levels of exposure, even those below current regulatory limits (31).
Air Pollution, Impaired Vascular Function, and Cardiovascular Events in Humans
MI, myocardial infarction; PM, particulate matter.
The epidemiological studies that have been conducted to date are not entirely consistent, but generally show that air pollution induces systemic oxidative stress in humans. The inconsistencies arise when factoring in the myriad of variables that must be considered, such as chemical composition, associated copollutants, and host susceptibility (103). The factors that influence the degree of susceptibility of individual patients are still poorly understood and underinvestigated. However, the limited evidence that exists suggests that certain polymorphisms in oxidative stress-related genes GSTM1, GSTP1, GSTT1, HFE C282Y, CAT, and heme oxygenase contribute to PM2.5 vulnerability (103). Based on data from the Normative Aging Study, Schwartz et al. have demonstrated that the negative association between PM2.5 and heart rate variability (HRV), a measure of cardiac autonomic dysfunction and independent risk factor for cardiovascular mortality, was modified by the presence of the allele for GSTM1 (116). Here, exposure to PM2.5 during the 48 h before measurement of HRV had no effect in subjects with GSTM1, whereas a significant decrease in HRV in subjects with deleted GSTM1 was observed. Likewise, Ren et al. found the gene polymorphisms of APOE, LPL, and vascular endothelial growth factor (VEGF) to modify the association between PM2.5 and HRV (106). Besides these genetic factors, the exposure concentration and duration, particle physical and chemical properties, also other parameters such as age, sex, lifestyle, and socioeconomic status determine the susceptibility of an individual for noise- and air pollution-induced health effects, which requires rigorous adjustment for these confounders. Strong support for this notion comes from data on differential impact of environmental pollution on mortality or loss of healthy life years during early and late life, indicating that the contribution of pollution to global deaths is highest in elderly people, whereas the impact on disability-adjusted life years (e.g., by severe illness) is most pronounced in infants (22, 26, 57).
Oxidative stress as a common denominator of the vascular effects of air pollution
The lungs have effective, but imperfect, endogenous protection in a layered mix of proteins and phospholipids that are regularly replenished. Alongside this surfactant protection, alveolar macrophages are tasked with preventing the penetration of particulates and reactive gases postinhalation from entering systemic circulation. Despite this protection, certain ultrafine particles have been reported to directly penetrate systemically (69). Larger sized particulates often have copollutants bound to their surface, which would allow for delivery of transition metals, endotoxins, and reactive quinones/aldehydes directly into lung tissue and possibly causing secondary toxicity (96). An additional complication of this multivariable equation is the possibility of chemical transformation over long-term exposure as well as with risk factors, which may be able to overwhelm the defenses of the lung and result in systemic exposure (103). The lung's native protection also does little to mitigate oxidative stress that is generally accepted to be the primary causative factor in the vascular complications following air pollution exposure. Oxidative stress has been reported in the lungs after immediate exposure (103) and is possibly potentiated in several ways. Important antioxidants (ascorbate, glutathione, and tocopherol) can be depleted and result in a reduction of necessary cofactors (NADPH, BH4) and aggravate production of ROS (96). Additional chemical transformations that can occur include alterations of thiols and fatty acids (•NO2 and •NO); lipid peroxidation, generation of reactive aldehyde, ketone, and endoperoxide fatty acid products; oxidation of thiols to disulfides or sulfoxides and DNA base oxidation. This concept is well supported; evidence is provided by all manner of studies, from in vitro cell culture and animal studies to human panel and interventional studies. These studies further suggest that both the solid and gaseous constituents of air pollution work in concert to increase oxidative stress, worsen endothelial function, and prime for or exacerbate acute and chronic cardiovascular sequelae. Whereas the majority of in vitro and in vivo studies suggest a central role of ROS formation for adverse health effects of air pollution (51), human studies of systemic oxidative stress (circulating biomarkers) do not regularly agree and appear to yield somewhat inconsistent results (29, 140). These inconsistencies could possibly be due to the utilization of only plasma and airway fluid, which limits the types of assays being used and could further skew the results. In addition, there is strong evidence that air pollution particles of different sizes and chemical compositions exert differential effects on circulating biomarkers of oxidative stress and inflammation (28, 141).
The atlas of knowledge regarding the role of oxidative stress in the PM-induced vascular dysfunction is growing, but there are still mechanistic questions that are yet to be answered. The role of endogenous immune and nonimmune cells and the extent to which they contribute to ROS formation and thereby vascular dysfunction are yet to be delineated. The manner in which the lung transduces the effects of air pollution is also not yet known, although central effects have been observed after activation of the HPA axis via the lung arc reflex, as well as by diffusion of reactive gases, ultrafine particles, and soluble, PM surface-bound compounds such as transition metals, endotoxins, and reactive (photoactivated) quinones/aldehydes through the lung and across the blood/brain barrier (103, 133). And last, the magnitude of the secondary damage-associated molecular patterns such as oxidatively modified proteins and lipids, and certain receptors such as Toll-like receptors (TLRs) and RAGE, has not yet been characterized or quantified (74, 103). Oxidative stress and inflammation have long been linked (129), but a conclusive characterization inflammatory phenotype or the role of systemic inflammation in the presentation of vascular dysfunction has not yet been presented. Environmental stresses can produce epigenetic or microRNA changes that lead to the alteration of signaling cascades, which can negatively impact ROS production and detoxification, as well as inflammation (67).
In vivo studies conducted using concentrated particles (PM2.5, ultrafines, diesel, gases, or mixtures) have demonstrated rapid (within hours) systemic effects following air pollution exposure (18, 128), which may also be related to increased incidence of MI shortly after exposure peak levels of fine PM (121). Human studies have complementarily utilized inhaled nanoparticles to demonstrate that these particles can cross the alveolar membrane and enter circulation, providing some insight into the possibility of air PM to do the same (69). Studies of acute inhalational exposure have demonstrated that vasoconstriction in both conduit vessels and microcirculation is hyper-responsive following acute exposure, which is further supported by findings of increased superoxide (O2 •−) and potentiation of vasoconstrictor responses in studies using subacute and chronic exposure protocols in mice and in rats, both in conjunction and without angiotensin II. Relatedly, exaggerated endothelin-1/ETA-receptor signaling has been reported following diesel exhaust and reactive gas exposure (Fig. 8). NADPH oxidase is known to activate the endothelin-1 promotor, and endothelin-1 is known to activate NADPH oxidase to produce superoxide, and so, these findings nicely imply a causative role for oxidative stress in the detrimental vascular responses seen after exposure to air pollution (27, 129). Abolishing NADPH oxidase-derived O2 •− production and uncoupled NOS-derived O2 •− production via apocynin and L-NAME had a protective effect in the vasculature, which suggests that the depletion of nitric oxide is an important mechanism for inducing adverse vascular effects (19, 123). Concentrated PM2.5 exposure has been shown to yield increased adhesion of inflammatory monocytes in the microcirculation of adipose tissue and increased deposition of mononuclear cells in perivascular tissue. Upon exposure to PM2.5, Nox2 and Tlr4 deficiencies were shown to improve vascular response (48, 122), while deficiency of the p47phox subunit of NADPH oxidase rendered the enzyme unable to produce O2 •− production in the visceral adipose tissue and improved insulin resistance (136). With respect to particle size, there is a dearth of studies comparing the effects of ultrafine particles relative to PM2.5, those that exist, however, generally show that ultrafine particles can exert equal if not greater effects than diesel exhaust on the vasculature. ApoE −/− mice exposed to ultrafine particles developed greater atherosclerosis when compared with those exposed to PM2.5. Ultrafine particle exposure may also inhibit the anti-inflammatory capabilities of high-density lipoprotein and thereby lead to greater systemic oxidative stress, supported by increased hepatic malondialdehyde, upregulation of Nrf2-regulated antioxidant genes, and lipid peroxidation products in the plasma and liver (4, 137).

According to two recent studies, the pulmonary antioxidant barrier could be a critical modulator of systemic responses (39, 40). The first demonstrated that the exposure to concentrated ambient PM2.5 (CAP) resulted in a reduction of insulin-stimulated Akt/eNOS activation in the aorta over the course of a 9-day exposure. The impairment of this pathway was successfully reversed by administration of antioxidant TEMPOL or through the overexpression of lung-specific extracellular superoxide dismutase (ecSOD) (40). The second study also exposed mice for 9 days to CAP, and thereafter reported a reduction in endothelial progenitor cells (EPCs), VEGF-stimulated aortic Akt phosphorylation, and plasma •NO levels in wild-type mice. These effects, however, were not seen in mice overexpressing ecSOD (ecSOD-Tg) in the lungs. The study further reports that EPCs from CAP-exposed wild-type mice had no effect when injected posthindlimb perfusion, but EPCs from CAP-exposed ecSOD-Tg were able to restore hindlimb ischemia (39). TIMP-1 and matrix metalloproteinases also participate in adverse remodeling and fibrotic/atherosclerotic processes that contribute to endothelial and vascular damage.
Ozone exposure studies consistently show an onset of endothelial dysfunction and enhanced vasoconstriction within hours (20, 90, 109), however, all exposures have been acute. The underlying mechanisms involve a rapid depletion of •NO and a decrease in aortic eNOS levels, effects of which are rescuable with O2 •− scavengers. Other studies suggest that the serum may contain (factors) that influence endothelial dysfunction and neuronal inflammation. An endothelial source for the factors likely to cause vascular and neuronal injury has been speculated, but is yet to be confirmed (109). For example, ozone exposure (1 ppm) for 4 h caused a dramatic degree of endothelial dysfunction (ACh-dependent relaxation) that was partially corrected by superoxide dismutase (SOD) and completely prevented by a mixture of SOD and catalase and the NADPH oxidase inhibitor apocynin in rats (90). Rodent studies are more numerous but tend to use concentrations of 0.5–1 ppm, which are far above those used in human studies. Furthermore, current regulatory standards for ozone exposure are 0.075 ppm averaged over 8 h in the United States and 0.057 in Europe. Overall, there is too little high-quality evidence to determine the impact of specific gases on vascular function [for reviews see Refs. (82, 83)].
Inflammation as a common denominator of the vascular effects of air pollution
Researchers using animal studies have utilized a variety of exposure parameters to study the effects of air pollution—differing in exposure route and duration, the strain and susceptibility of the animals, pollutant source, and particle characteristics (11, 103). Because of the inconsistency of exposure criterion, results can only be roughly generalized to state that exposure to air pollution has an important effect on ROS pathways, while reducing sources of ROS mitigates endothelial dysfunction, •NO availability, endothelial cell activation as measured by adhesion molecule expression, and inflammation to varying degrees. Especially the impact of air pollution on inflammatory pathways is well documented and the NLRP3 inflammasome was identified as a central player. Recently, PM2.5 exposure was demonstrated to reduce the viability of human endothelial cells and impair their activity through ROS-mediated NLRP3 inflammasome activation (117). In cultured RAW264.7 macrophages, exposure to ambient PM2.5 increased IL-1β downstream of TLR4/NF-κB signaling and NLRP3 inflammasome activation (135). Also, the gaseous air pollutant CO2 can cause high IL-1β and microparticle generation in neutrophils in an NLRP3-, mtROS-, protein kinase C-, and NADPH oxidase-dependent manner (125). According to animal experimental data, PM2.5 exposure also aggravated the development of atherosclerosis in ApoE −/− mice, involving CD36 signaling and the NLRP3 inflammasome (35). There is also human evidence for this concept as children of the Mexico City Metropolitan Area with high exposure to air pollution (e.g., ultrafine PM) exhibit increased myocardial inflammation that was associated with inflammasome activation (127).
Also underinvestigated to this point has been the effect of chronic air pollution exposure on the surfactant milieu in the lungs. As previously mentioned, the constituent phospholipids and surfactant proteins are an important defensive barrier in the lungs and distal airways and require continual repair to maintain integrity. However, it is possible that chronic exposure to air pollution could overcome the body's milieu defense (96) and thereby increase oxidatively modified derivatives of PAPC and the major oxidation product of cholesterol, 7-ketocholesterol, that represent important mediators of inflammation. The dysfunction of the endothelial barrier and inflammatory cell recruitment could be linked with these derivatives (Fig. 8), and enable air pollutants to prompt secondary mediator signals into circulation and to the bone marrow (48). Chemokine signals and other mediators may activate the TLR4 pathways in monocytes and macrophages to produce more oxidative stress (9, 63). Involvement of this mechanistic pathway is likely, due to the attenuation of ROS generation and reduction in inflammatory monocyte infiltration, in studies utilizing deficiencies of TLR4, NOX2, and p47phox , overall leading to an improvement in vascular response following inhalational exposure to concentrated PM2.5 (48, 136). Moreover, endothelial dysfunction is the first subclinical sign of atherosclerosis and PM2.5 exposure may have mechanistic ties to the onset of the disease. 7-ketocholesterol is formed in response to chronic PM2.5 exposure, which then accumulates within low-density lipoprotein and taken up by scavenger receptor CD36, which recognizes specific oxidized phospholipids and lipoproteins. 7-ketocholesterol accumulates within plaque macrophages, promoting endothelial dysfunction and the onset of atherosclerosis (104, 124). Impairment of endothelial regeneration owing to depletion of EPCs may also represent an important mechanism of sustained endothelial dysfunction.
PM2.5 exposure has been shown to exert its effects on blood pressure responses via activation of the sympathetic nervous system as well as by inflammation within the hypothalamus (138). Upon penetrating the central nervous system, air pollutants can cause inflammation in areas responsible for the regulation of blood pressure and metabolism (82, 101 –103). Airway sensory neurons have specific receptors such as the TRPA1 that are able to sense environmental toxins and oxidants, resulting in neurogenic inflammation (119). And last, inhibition of central IKKβ was able to prevent peripheral inflammation, insulin resistance, and abnormalities in whole-body metabolism (63, 138).
Changes in the epigenetic landscape as a novel denominator of the health effects of air pollution
Air pollution in the form of PM was shown to upregulate miR-128/302 with relevance for coronary artery disease, cardiac hypertrophy, and heart failure (8). Ambient PM (PM2.5) exposure of older subjects led to increased levels of multiple microRNAs (e.g., miR-126-3p and miR-150-5p) accounting for adverse CVD pathways by aggravation of inflammation, oxidative stress, and atherosclerosis (111). Another study found association of nine microRNAs with PM10 exposure and a prominent role of miR-101 for higher blood pressure in response to PM10 challenges (75). Other examples for air pollution-dependent changes in microRNA expression patterns related to phosphatase and tensin homologue regulation, control of apoptosis, inflammation, and cell cycle were summarized by Miguel et al. (67). Another epigenetic regulatory mechanism that is affected by air pollution is DNA methylation, which is typically lowered upon exposure with substantial impact on health (108). This is supported by data of the ENVIRONAGE cohort study indicating that transplacental in utero exposure to PM can induce changes to fetal and neonatal DNA repair capacity by altered placental promoter methylation of key DNA repair and tumor suppressor genes, leading to increased mutation rates and potentially carcinogenic insults later in life (86). Likewise, it was reported that airborne PM changes the acetylation and methylation of important histone marks via redox-regulated pathways that depend on mitochondrial ROS formation and also displays transgenerational epigenomic inheritance with transmission of epimutations from gametes to zygotes (118). These important findings are also reflected by the emerging concept that epigenetic changes during early-life environmental exposures can explain the late-life or long-term toxicity with a higher NCD burden (6), which may also explain different health status of monozygotic twins living in different geographic locations or under different life conditions (105). In conclusion, substantial epigenetic alterations by air pollution exposure seem feasible since a common denominator of air pollution-associated adverse health effects is oxidative stress and all epigenetic pathways are highly redox regulated (42, 52, 68) and have known impact on health and disease (25, 53).
Direct effects of air pollution exposure on vascular function and oxidative stress in humans
Vascular dysfunction in conduit vessels or the microvasculature arises rapidly but reversibly following acute exposure to PM2.5 and dilute diesel exhaust, as demonstrated in several controlled exposure studies. Some studies report a delay in the inhibition of endothelium-dependent vasodilatation, where effects are seen 24 h after exposure to concentrated PM2.5 (12). The degree of vascular dysfunction following exposure has been correlated with PM2.5 mass, and also with the level of TNF-α following exposure, suggesting that the degree and identity of the particle influence the level of systemic inflammation (12). Endothelial dysfunction has not been universally reported in response to concentrated PM2.5, which further underlines the importance of the particle's individual properties as well as the methods for determining vascular response (71). Particles of other sizes, ultrafine (including elemental carbon), and diesel particles have also induced endothelial dysfunction in the microcirculation (73, 92). In humans, impaired endothelium-dependent vasodilation as measured by acetylcholine response was found to persist for 24 h postexposure (126). Two hundred milligrams per cubic meter of diesel exhaust (fine particulates) rapidly increased blood pressure compared with filtered air in a randomized-controlled clinical trial (24). In addition, diesel exhaust exposure was found to further depress ST-segment and increase ischemic burden during exercise, also compared with filtered air (72). Filtered diesel exhaust did not retain the ability of its unfiltered counterpart to impair vasodilatory response to acetylcholine and bradykinin (70). Results from organ chamber experiments in isolated vessels show detrimental effects on endothelial dysfunction following exposure to diesel exhaust, but not carbon nanoparticles. Upon administration of SOD, the impairment was markedly improved, implying that the bioavailability of vascular •NO had been reduced by the diesel exhaust particles, likely due to increased O2 •− (70). However, it has also been reported that vascular •NO production by •NO-synthase is increased (59).
Ozone exposure studies have not yielded entirely consistent results—these generally show an impairment in conduit vessel endothelial function, but one study did not report endothelial dysfunction. In a similar vein, panel studies have forged a link between •NO2 levels and endothelial dysfunction, but upon acute exposure to •NO2 in humans, no abnormalities were detected (58). Studies on other gaseous pollutants, such as •NO2 and sulfur dioxide, are lacking but translational data suggest that these are most harmful and possibly toxic as an admixture and are less harmful independently (120).
There is an immense body of evidence that supports air pollution's role in causing endothelial dysfunction in both conduit and resistance vessels. Endothelial dysfunction is a hallmark of CVD, as it can negatively impact arterial stiffness and afterload, which upon chronic continuation can result in persistent hypertension. Air pollution is associated with hypertension at high and low levels (10, 15) and it is widely accepted that impairment in endothelial function and increases in blood pressure are important mechanisms for the initiation and propagation of atherosclerosis. Data from the MESA-Air cohort (n = 6795 across 6 U.S. regions) further support these mechanistic insights, reporting that each 5 μg/m3 increase in long-term PM2.5 exposure was associated with a greater progression of coronary artery disease (4.1 Agatston units/year) (49).
Air Pollution Generates Cardiovascular Risk Factors Such as Hypertension and Diabetes
Multiple systemic reviews and meta-analyses indicate that PM2.5 inhalation not only causes an acute “spike” in blood pressure over hours and days but can contribute to chronic hypertension development as well. One of these studies simulated lifetime exposure in an animal model over the course of >50 weeks. The animals displayed left ventricular hypertrophy, diastolic dysfunction, flow reserve abnormalities, and alterations in the myocardium's fetal gene expression and SERCA2a levels (134). Further support for these findings came from the MESA-Air and the Jackson Heart Study, which reported epidemiological evidence of traffic air pollutants and PM2.5 with left ventricular structural changes (10). Other studies have associated PM2.5 inhalation with insulin resistance, also supported by epidemiologic data (101). There is a well-established link between insulin resistance and endothelial dysfunction and general recognition that insulin is able to enhance its own vascular delivery. It is therefore logical that the insulin resistance seen after exposure to air pollution is a consequence of the endothelial dysfunction induced by such an exposure, although the mechanism is likely to be multifactorial (65, 100). Other consequential correlations for the vasculature following exposure to air pollution are obesity (103) and thrombosis (110), both of which are triggers for atherosclerosis and other cardiovascular events (Fig. 8).
Effects of Noise and Air Pollution Mitigation Measures
Noise
Transportation noise is a function of population density, making the densest populations the most vulnerable. In Europe, 30% of people are exposed to residential day-evening-night noise level (LDEN) levels above 55 dB(A) (87b) For those living in areas of high exposure to transportation noise, there is an increase in CVD incidence and mortality (87b), affirming the need to use mitigation strategies. The European Commission recently summarized a number of possible strategies for noise abatement (87a).
The most intuitive of strategies for a reduction in noise exposure is by improvement in noise insulation. However, while this strategy is effective in reducing exposure, it is less successful in implementation due to high costs and low cost-effectiveness. Technological methods such as development of quieter engines and production of low-noise tires for cars and brakes for trains are important for reducing noise levels from all transportation sources, but rely on industry participation. Governmental mitigation strategies can target the greatest contributor, road traffic noise, by reducing speed limits, building quiet road surfaces, and installing noise barriers along major roads. Because road traffic is ever-increasing, the need for traffic management and development of low-noise tires is imperative. Similarly, air transport has also been consistently increasing, which could be mitigated with the implementation of nighttime restrictions or curfews, as noise during sleeping hours has been shown to be particularly detrimental (32, 47, 114). Exposure during other hours are also detrimental and require separate strategies, for instance, changing descent procedure and limiting the running of engines on the ground (87a).
As the world modernizes, the demand for transportation of any mode only increases, exposing progressively more of the population to levels of noise detrimental to health and making new technological developments and legislation for noise abatement necessary for human health.
Air pollution
Significant improvements in air quality have been made in the most recent decades, to which improvements in life expectancy can be traced. In addition, these improvements in life expectancy have been made at no additional economic cost (57, 82). However, this success is not complete, as the population continues to increase, which increases energy and transportation demands and threatens to cause a backslide in these air quality gains. Geopolitical issues as well as population growth delay improvements in many parts of the world as well, putting millions of healthy and at-risk individuals “in the line of fire” of unsafe and polluted air. For these populations, personal protection in the form of masks and filtration devices is the only protection from air pollution-derived CVD. These personal-level mitigation strategies have been shown to reduce exposure and thereby blunt the adverse effects of air pollution exposure, including vascular dysfunction, rises in blood pressure and heart rate, and other cardiovascular outcomes (82).
Conclusions and Clinical Implications
The present article gives an overview of the data that demonstrates environmental pollutants as important and overlooked cardiovascular risk factors. These risk factors can initiate CVD and exacerbate it in at-risk and pre-established CVD groups. Herein we stress that these risk factors, because of their ubiquity, cannot be avoided or improved upon by lifestyle choices. Because of this, improvement in environmental pollution cannot be made by patients and doctors, but rather by politicians who introduce legislation to limit air and noise pollution to protect the population at large. As a future research perspective we would like to propose to investigate whether the cardiovascular side effects of noise and air pollution are additive. The additivity in risk between noise and air pollution has not been studied in animal models or in humans. Molecular mechanisms and pathways have been partially elucidated for each of these risk factors independently, which could provide direction for future studies. There is no shortage of foundational knowledge that needs to be established, including the magnitude and time course of response in coexposure, the extent of interactivity between the two factors on physiological parameters, the reversibility and recovery time following exposure, the effect of alterations in severity in the exposure, impacts on the circadian rhythm, and of course mitigation and prevention strategies. Finally, technological development using the interactivity data between the two exposures could be an extraordinary opportunity to map out interactions between environmental and nonenvironmental risk factors, and also to use this to provide personal measures of health (Fig. 8). In reality, however, the opportunity for technological growth and data collection is hindered by costs and subject burden. Environmental stressors are currently not, but should be, included as significant cardiovascular risk factors in the 2019 AHA/ACC guidelines for prevention and barely mentioned in the ESC guidelines for heart failure, arterial hypertension, arrhythmia, and acute coronary syndrome (5, 54).
Footnotes
Acknowledgment
We thank Margot Neuser for expert graphical assistance.
Funding Information
The present work was supported by continuous financial support by the Foundation Heart of Mainz and by a vascular biology research grant from the Boehringer Ingelheim Foundation for the collaborative research group “Novel and neglected cardiovascular risk factors: molecular mechanisms and therapeutic implications” to study the effects of environmental risk factors on vascular function and oxidative stress. T.M. is PI of the DZHK (German Center for Cardiovascular Research), Partner Site Rhine-Main, Mainz, Germany.