
Abstract
Significance:
Perivascular adipose tissue (PVAT), which is present surrounding most blood vessels, from the aorta to the microvasculature of the dermis, is mainly composed of fat cells, fibroblasts, stem cells, mast cells, and nerve cells. Although the PVAT is objectively present, its physiological and pathological significance has long been ignored.
Recent Advances:
PVAT was considered as a supporting component of blood vessels and a protective cushion to the vessel wall from the neighboring tissues during relaxation and contraction. Nonetheless, further extensive research found that PVAT actively regulates blood vessel tone through PVAT-derived vasoactive factors, including both relaxing and contracting factors. In addition, PVAT contributes to atherosclerosis through paracrine secretion of a large number of bioactive factors such as adipokines and cytokines. Thereby, PVAT regulates the functions of blood vessels through various mechanisms operating directly on PVAT or on the underlying vessel layers, including vascular smooth muscle cells (VSMCs) and endothelial cells (ECs).
Critical Issues:
PVAT is a unique adipose tissue that plays an essential role in maintaining the vascular structure and regulating vascular function and homeostasis. This review focuses on recent updates on the various PVAT roles in hypertension and atherosclerosis.
Future Directions:
Future studies should further investigate the actual contribution of alterations in PVAT metabolism to the overall systemic outcomes of cardiovascular disease, which remains largely unknown. In addition, the messengers and underlying mechanisms responsible for the crosstalk between PVAT and ECs and VSMCs in the vascular wall should be systematically addressed, as well as the contributions of PVAT aging to vascular dysfunction.
Introduction
There are two main types of adipose tissues in the body: white adipose tissue (WAT) and brown adipose tissue (BAT). WAT is widely distributed in the subcutaneous and the visceral cavity of the body, and is the main form of fat storage in the body. Its main function is to store excess energy in the form of fatty acids for the body to use when needed. However, visceral WAT contributes to atherosclerosis development by secreting proinflammatory adipokines, such as resistin, which lead to insulin resistance, dysfunction of vascular endothelial cells (ECs), and proliferation of vascular smooth muscle cells (VSMCs) (12, 58). Conversely, BAT promotes heat generation in the body by decomposing the lipids and burning them through uncoupling oxidation by the mitochondrial electron transport chain (77).
Currently, obesity is highly prevalent worldwide due to an imbalance between energy intake and energy expenditure. Due to the energy expenditure characteristics of BAT, activation of BAT has been explored as one of the strategies to treat obesity and associated diseases (125). However, the BAT amount is very limited in adult humans, and activation of BAT upon cold stimulation could only be found in 3% of males and 7.5% of females, respectively, by positron emission tomography-computed tomography (24, 71). Interestingly, cold stimulation can convert WAT into beige adipose tissue (BeAT) through a browning process (16). Similar to adipocytes in classic BAT, some clusters of adipocytes in BeAT express uncoupling protein 1 (UCP-1), which promotes energy expenditure (111). Thus, inducing BeAT instead of BAT is emerging as a strategy to treat obesity and associated diseases (23).
Of significance, and relevant to cardiovascular diseases (CVDs), the blood vessels such as the aorta, coronary, and carotid arteries are surrounded by adipose tissue, referred to as perivascular adipose tissue (PVAT). The fat attenuation index (FAI), which is the average reduction in the signal of adipose tissue within a volume of interest as measured from reconstructed computerized tomography scanning, in PVAT was inversely associated with the size of adipocytes and positively correlated with atherosclerotic plaque burden in the coronary artery (7). A recent large cohort study involving 3912 clinical patients further documented that high values of perivascular FAI were associated with a significantly higher adjusted risk of all-cause cardiac mortality (78).
However, PVAT displays heterogeneity according to its location. The PVAT from the thoracic aortic area is BAT-like, and the PVAT from the abdominal aortic area is BeAT-like in morphology and gene expression profile (103). Compared with visceral WAT, PVAT is irregular in shape and scattered (19). The expression levels of FABP4, FAS, GPDH, and LPSR are significantly lower in PVAT than in visceral WAT (19). Functionally, PVAT secretes a large number of angiogenic factors such as TSP-1, serpinE1, and IGFBP, which regulate vascular angiogenesis (87).
PVAT also secretes other vasoactive substances described in the sections below that contribute to vascular tone regulation and atherosclerosis development. In addition, adipocyte precursors and stromal cells in PVAT contribute to vascular remodeling (43, 81). Therefore, in addition to visceral WAT, PVAT should be included in the study of the roles of adipose tissues in the cardiovascular system and their contribution to cardiovascular health and disease.
PVAT and Hypertension
Hypertension is a major risk factor for stroke, aortic aneurysm, and coronary heart disease. Hypertension is a progressive chronic clinical syndrome characterized by elevated systemic arterial blood pressure. Apart from contributions to hypertension from the neurohemal fluid system, including the heart, kidney, and nervous system, this disease is prevalent in obese subjects, suggesting that changes in adipose tissue also contribute to hypertension development and progression.
PVAT produces many biologically active molecules including adipokines (e.g., leptin, adiponectin, and omentin), cytokines/chemokines (e.g., interleukin [IL]-6, tumor necrosis factor-α [TNF-α], monocyte chemoattractant protein-1 [MCP-1]), gaseous molecules (e.g., nitric oxide [NO] and hydrogen sulfide [H2S]), prostacyclin, angiotensin-1 to 7 (Ang 1–7), angiotensin II (Ang II), methyl palmitate, and reactive oxygen species (ROS) (16, 105). All these molecules contribute to vascular homeostasis. Adiponectin, NO, H2S, prostacyclin, Ang 1–7, and methyl palmitate induce vasodilation by targeting the underlying ECs and VSMCs in the blood vessel wall. Ang II, ROS, and other yet undetermined factors cause vasoconstriction. Thus, PVAT-derived factors synergistically and concurrently regulate vascular tone (105, 115).
PVAT mass is markedly increased in obesity. Compared with lean PVAT, obese PVAT secretes more inflammatory adipokines/cytokines, including TNF-α, leptin, IL-6, plasminogen activator, and resistin, which may further alter PVAT characteristics and secretomes, eventually affecting vascular homeostasis. Thus, the contribution of PVAT to hypertension has received increased attention in the last decade. Here, we provide an update on recent research progress regarding the roles of PVAT in blood pressure regulation.
Renin–angiotensin–aldosterone system in PVAT and hypertension
The mechanisms underlying hypertension development and progression are complex. Abnormal activation of the renin–angiotensin–aldosterone system (RAAS) is critical to the process of hypertension from the initial to the end stage. Interestingly, angiotensinogen (Agt) levels were found to be significantly elevated in adipose tissue of rats with essential hypertension and obesity. Also, the concentrations and activities of Agt, plasma rennin, and angiotensin-converting enzyme in adipose tissue were positively associated with the degree of obesity (29, 75, 120).
The adipose tissue might be the main source of RAAS in hypertensive patients with obesity (22). Locally, Ang II is derived from Agt, which is also found in PVAT. Angiotensin receptors including angiotensin type 1 receptor (AT1R), angiotensin type 2 receptor (AT2R), angiotensin type 3 receptor (AT3R), and angiotensin type 4 receptor (AT4R) are expressed throughout the vascular wall. It is known that AT1R activation can cause vasoconstriction, aldosterone synthesis, and secretion, increase vasopressin secretion and cardiac hypertrophy, and it regulates sympathetic activity.
Activation of AT1R in PVAT promotes vascular inflammation and endothelial dysfunction (48, 91). Aldosterone may directly act on PVAT to promote proinflammatory phenotypes. Aldosterone receptor antagonists have been shown to have beneficial effects on oxidative stress, endothelial function, blood pressure regulation, weight loss, and systemic insulin sensitivity (29, 50, 96). Angiotensin receptor blockers (ARBs) can reduce release of Ang II and aldosterone from PVAT, and prompt the release of perivascular-derived relaxing factors, which induce vasodilation by opening voltage-dependent K+ channels on VSMCs (11, 48, 60).
We recently reported that pretreatment of blood vessel rings with ARBs markedly blocked their contraction in response to PVAT extracts (24). Knockout of Agt in PVAT significantly reduced local Ang II production in PVAT from mice. The Agt-deficient mice showed hypotension during the circadian resting phase. In fact, Agt expression in PVAT showed a circadian pattern, suggesting that Ang II production in PVAT was controlled by the circadian clock. Indeed, knockout of Bmal1 in PVAT significantly blunted the circadian expression pattern of Agt and recapitulated the blood pressure phenotype of PVAT-Agt-deficient mice, showing hypotension during the resting phase. Circadian control of Agt expression in PVAT appears to involve direct transcriptional control of Agt by Bmal1 (Fig. 1) (18).
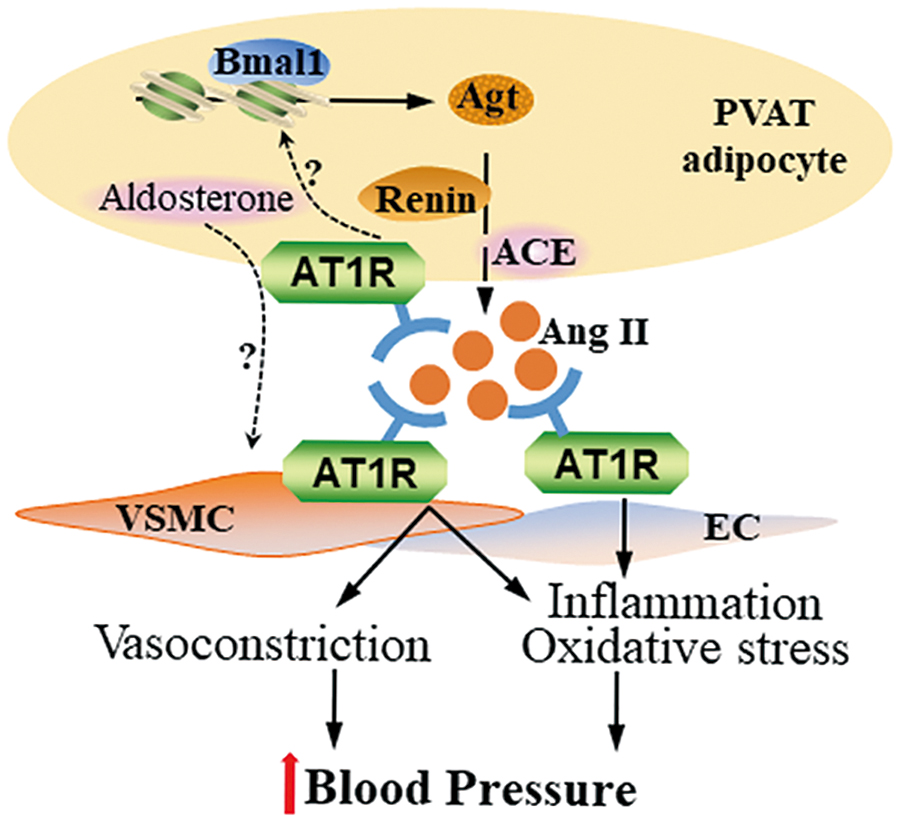
The relative contribution of other potential underlying mechanisms and the extent and role of the PVAT-dependent regulation of circadian rhythmicity of blood pressure and hypertension are still unknown, including their potential association with adverse outcomes in superdipper and nondipper phenotypes in humans, as observed in the clinic (69). Nevertheless, inhibition of RAAS by ARB and aldosterone inhibitors may reduce blood pressure and provide other cardiovascular benefits through direct effects on PVAT, beyond those on their well-established targets, such as kidney and heart (68).
Sympathetic nervous system in PVAT and hypertension
Electrophysiological studies confirmed the contribution of the sympathetic nervous system (SNS) in adipose tissues under conditions of glucose deficiency, cold stimulation, and free fatty acids (FFAs) release (95). SNS activation promotes lipolysis in adipose tissues. Local denervation of SNS resulted in increased adipose tissue expansion and inhibition of lipolysis (10), underscoring the contribution of adipose tissue SNS to hypertension. In the clinic, α/β-adrenergic receptor blockers, which reduce SNS activity, are used for the treatment of obesity-associated hypertension to reduce blood pressure (49).
It is unclear whether the activation of SNS in PVAT is associated with hypertension. Recent studies have shown that SNS activation is an important factor in PVAT-dependent vasoconstriction in vitro (9). Activation of the sympathetic nerves improves the contractility of the small mesenteric resistance arteries with PVAT, which can be eliminated by removing the sympathetic nerves in the adipose tissue. Stimulation of the sympathetic nerves has a diastolic effect on the superior mesenteric artery. However, in the aorta, there is a significant contraction-promoting effect by stimulation of the SNS (4). PVAT contains 5-hydroxytryptamine (5-HT). Fenfluramine, a 5-HT releaser, induced a greater contraction of aortic rings with PVAT than rings without PVAT. The α-1 adrenoreceptor antagonist prazosin and the norepinephrine transporter inhibitor nisoxetine significantly blocked fenfluramine-induced aortic ring contraction, indicating that PVAT-derived 5-HT or norepinephrine resulted in vessel ring contraction (56).
In addition, electrophysiological studies demonstrated that perivascular nerve activation by electrical field stimulation elicited a frequency-dependent contractile response in arterial rings, in which the amplitude of contraction was higher in arterial rings with PVAT than in those after PVAT removal. Electrical field stimulation of nerves in PVAT increased the release of O2 •−, Ang II, and chemerin, which induced vessel contraction (25, 38, 65, 108). Also, electrical field stimulation induced PVAT release of adiponectin, calcitonin gene-related peptide and methyl palmitate, which elicited endothelium-independent anticontractile effects on arterial rings (15, 94). These results suggest that PVAT releases both contractile and anticontractile factors in response to perivascular sympathetic nerve stimulation to maintain the physiological tone (Fig. 2).

Furthermore, renal sympathetic activity affects blood pressure in part by increasing renovascular resistance via release of norepinephrine from sympathetic nerves onto the renal arteries. PVAT surrounding the renal blood vessels contains a pool of norepinephrine (86). Sympathomimetic tyramine-induced vessel contraction was greater in the renal artery with PVAT versus that without PVAT, an effect that was reduced by nisoxetine. Renal denervation significantly reduced norepinephrine contents in renal PVAT but did not modify tyramine-induced contraction of renal arteries with PVAT (86).
Thus, these data support the hypothesis that activation of SNS in PVAT might coordinately regulate blood pressure through PVAT-derived factors. More studies are required to determine whether the activity of the SNS in PVAT of hypertensive subjects is altered, the roles in blood pressure regulation of the substances released specifically from PVAT upon SNS activation, and the underlying mechanisms.
Adipokines and immune cells in PVAT and hypertension
Hypertension is more prevalent in obese subjects than in lean subjects. The progression of hypertension is related to immune responses in adipose tissues (26). Under the obese condition, hypertrophy of adipose tissues causes alterations of the normal profiles of adipokines (36). Adiponectin is one of the most abundant adipokines that dilate the vascular wall. The plasma adiponectin level is reduced in hypertensive patients with obesity, and shows a significant upward trend after treatment and control of hypertension, indicating protective effects of adiponectin in hypertension (8, 121). The presence of PVAT reduced the contractile response to norepinephrine in blood vessel rings; however, PVATs anticontractile effect was significantly reduced in obese subjects and obese mice (44).
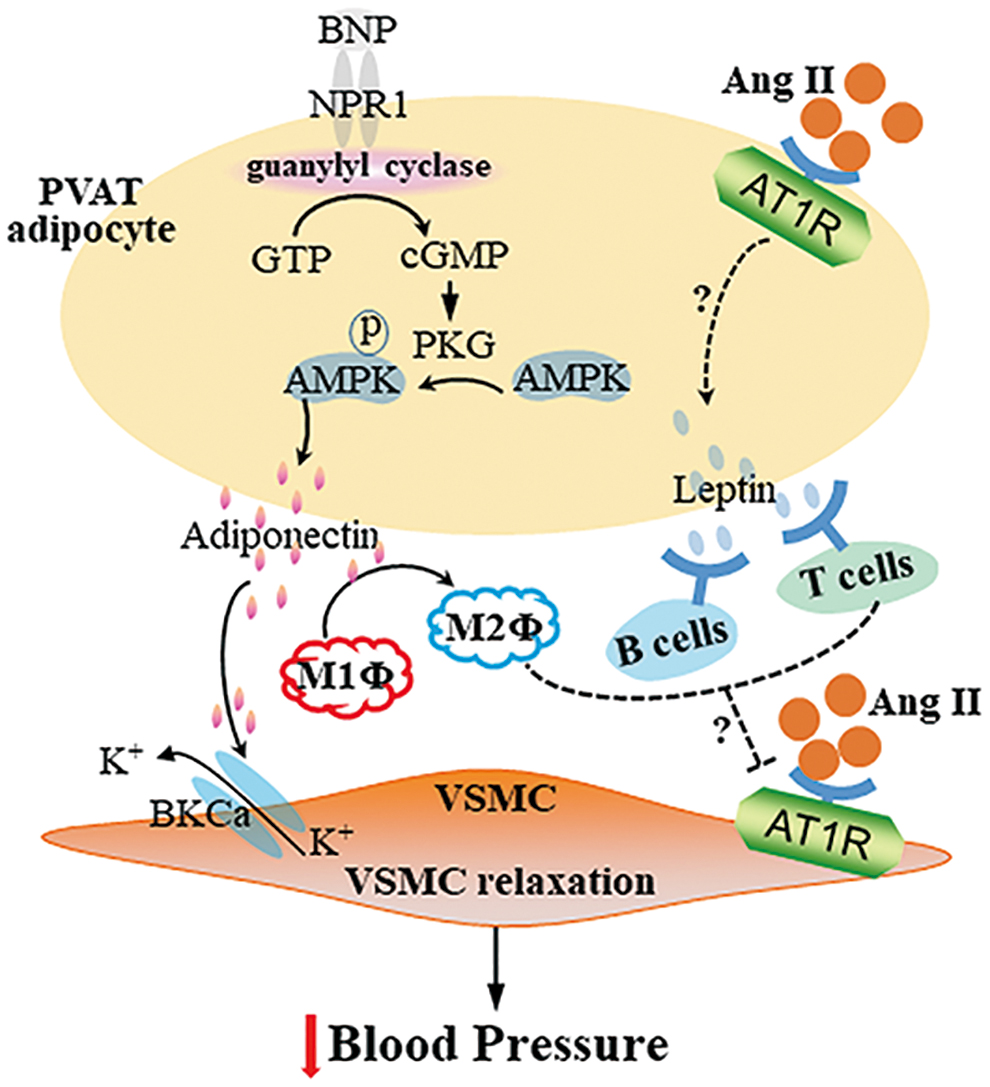
Multiple signaling pathways contribute to the anticontractile effects of PVAT-derived adiponectin. PVAT from knockout mice with loss of either adiponectin or mitochondrial Ca2+-activated large-conductance K+ (BKCa) channel did not show an anticontractile response in blood vessel rings, suggesting that BKCa channels are involved in adiponectin-induced vasorelaxation in arteries (66). cGMP-dependent protein kinase (PKG) was necessary for the anticontractile effects of PVAT on ECs and VSMCs, evidenced by the absence of anticontractile effects from PVAT of PKG−/− mice. The absence of PKG in PVAT was associated with reduced adipocyte adiponectin expression, suggesting that vasorelaxation induced by PVAT-derived adiponectin was associated with PKG (66).
AMP-activated protein kinase (AMPK) is a key mediator of cellular energy balance and may mediate the vascular effects of adiponectin (Fig. 3). High-fat-diet feeding reduced the anticontractile effect of PVAT as well as AMPK phosphorylation (6). PVAT from AMPKα1 knockout mice secreted significantly less adiponectin. PVAT from wild-type mice augmented relaxation of vessel rings without PVAT in response to cromakalim, a potassium channel-opening vasodilator, but not PVAT from AMPKα1 knockout mice. Addition of adiponectin to AMPKα1-deficient vessel rings without PVAT augmented relaxation in response to cromakalim (5).
In addition, the complement 3 (C3) activation in macrophages in PVAT was negatively associated with adiponectin expression in PVAT of mice with hypertension induced by DOCA salt. PVAT macrophage-derived C3 resulted in C5a generation, which promoted macrophage polarization from M2 to M1. C5a further induced TNF-α secretion and inhibited adiponectin expression in PVAT adipocytes of hypertensive mice. Consistently, macrophage infiltration and C5a expression were increased, which was associated with decreased adiponectin expression in adipose tissue from patients with aldosterone-producing adenoma (89, 90).
In the clinical setting, it was observed that the anticontractile activity of PVAT, lost in obese patients before bariatric surgery when compared with healthy volunteers, was restored 6 months after surgery. The improvement in anticontractile function after surgery was accompanied by improvements in inflammatory cytokines, adipokine profiles, and systolic blood pressure together with increased PVAT adiponectin and reduced macrophage infiltration and inflammation (1).
In mice, high-fat-diet feeding caused an increased infiltration of macrophages in PVAT with increased expression of the M1 macrophage markers Nos2 and IL-1β and the M2 marker Chil3 (6). Adiponectin secreted by adipose tissues stimulates M2-type polarization in macrophages, exerting anti-inflammatory effects (124). Sustained weight loss in rats led to an improvement in PVAT function associated with reduced inflammation and normalization of plasma adipokine levels, including leptin (13).
Leptin is another abundant adipokine released by adipose tissues, including PVAT. Leptin mRNA and protein expression were significantly lower in PVAT from spontaneously hypertensive rats (SHRs) (37). Apart from targeting the central nervous system, leptin receptors are expressed on CD4+ and CD8+ T cells, B cells, and monocytes/macrophages. In the absence of leptin, CD4+ T cells showed decreased proliferation and immune responses (82). While Ang II administration increased leukocyte and T cell content in PVAT (74), T regulatory cells (Tregs) adoptive transfer prevented Ang II-induced hypertension and microvascular injury (73). In T and B cell-deficient mice with knockout of recombination-activating gene 1 (Rag1), Ang II-induced increases in systolic blood pressure (but not in diastolic blood pressure) could be rescued by infusion of Tregs.
Ang II-induced endothelial dysfunction and oxidative stress in PVAT were exacerbated in the Rag1 knockout mice, while T cell transplantation from wild-type mice rescued the phenotype. Ang II increased monocyte/macrophage infiltration and proinflammatory polarization in PVAT in the Treg-deficient mice (73). Administration to mice of 1,2,3,4,6-penta-O-kgalloyl-β-
Apelin is widely found in the body, and is expressed in human and mouse adipocytes. Apelin levels are inversely associated with inflammation. Apelin-13 downregulates the expression of TNF-α and MCPs in macrophages (119, 123). Apelin-13 increased NO bioavailability, and reduced blood pressure in mice and rats (20, 63). Ang II also increased vasoconstriction in apelin-deficient mice (93). Acetylcholine-induced relaxation of arteries without PVAT was increased in the presence of exogenous apelin. Apelin mRNA level in PVAT was higher in obese SHRs than in control Wistar-Kyoto rats, perhaps reflecting a compensatory mechanism (47). Collectively, these findings suggest that apelin in PVAT might modulate blood pressure to promote vascular tone.
In addition, PVAT might mediate sex-dependent vascular function in hypertension. In the presence of PVAT, contraction in response to endothelin-1 and the thromboxane mimetic U46619 was significantly reduced in porcine coronary arteries from females, but not males. The adiponectin receptor agonist (adipoRon) produced greater relaxation in porcine coronary arteries from females compared with males (3). Vasorelaxation induced by cromakalim in rings with PVAT was significantly impaired in vessels from stroke-prone SHR males relative to females. A crossover study assessing the function of male PVAT on female vessels confirmed the reduced vasorelaxation response to cromakalim associated with male PVAT (99).
Further studies indicated that resistin expression was increased approximately twofold in PVAT from male stroke-prone SHR vessels. Preincubation with resistin significantly impaired the ability of female vessels with PVAT to relax in response to cromakalim (99). These findings suggest roles of PVAT-derived adipokines in mediating sex-dependent vascular function in hypertension and may underscore intrinsic differences in PVAT between the sexes, a possibility not yet addressed (110).
Overall, the findings summarized in this section highlight considerable progress in our understanding of the complexity behind the multifaceted roles of PVAT in the regulation of blood pressure and its contribution to hypertension, as well as define PVAT as a necessary target to be considered for clinical management of blood pressure.
PVAT and Atherosclerosis
Atherosclerosis is a chronic metabolic syndrome characterized by endothelial dysfunction, lipid deposition, and inflammatory infiltration (97). Endothelial dysfunction/injury caused by high shear stress promotes initiation of atherosclerosis, and subsequent adhesion of circulating inflammatory cells to the dysfunctional endothelium triggers accumulation of cholesterol in the arterial wall to further aggravate atherosclerosis (104). Recently, the roles of PVAT in atherosclerosis development have been extensively studied. Here, we review recent updates regarding the relationship between PVAT and atherosclerosis.
PVAT thermogenesis and atherosclerosis
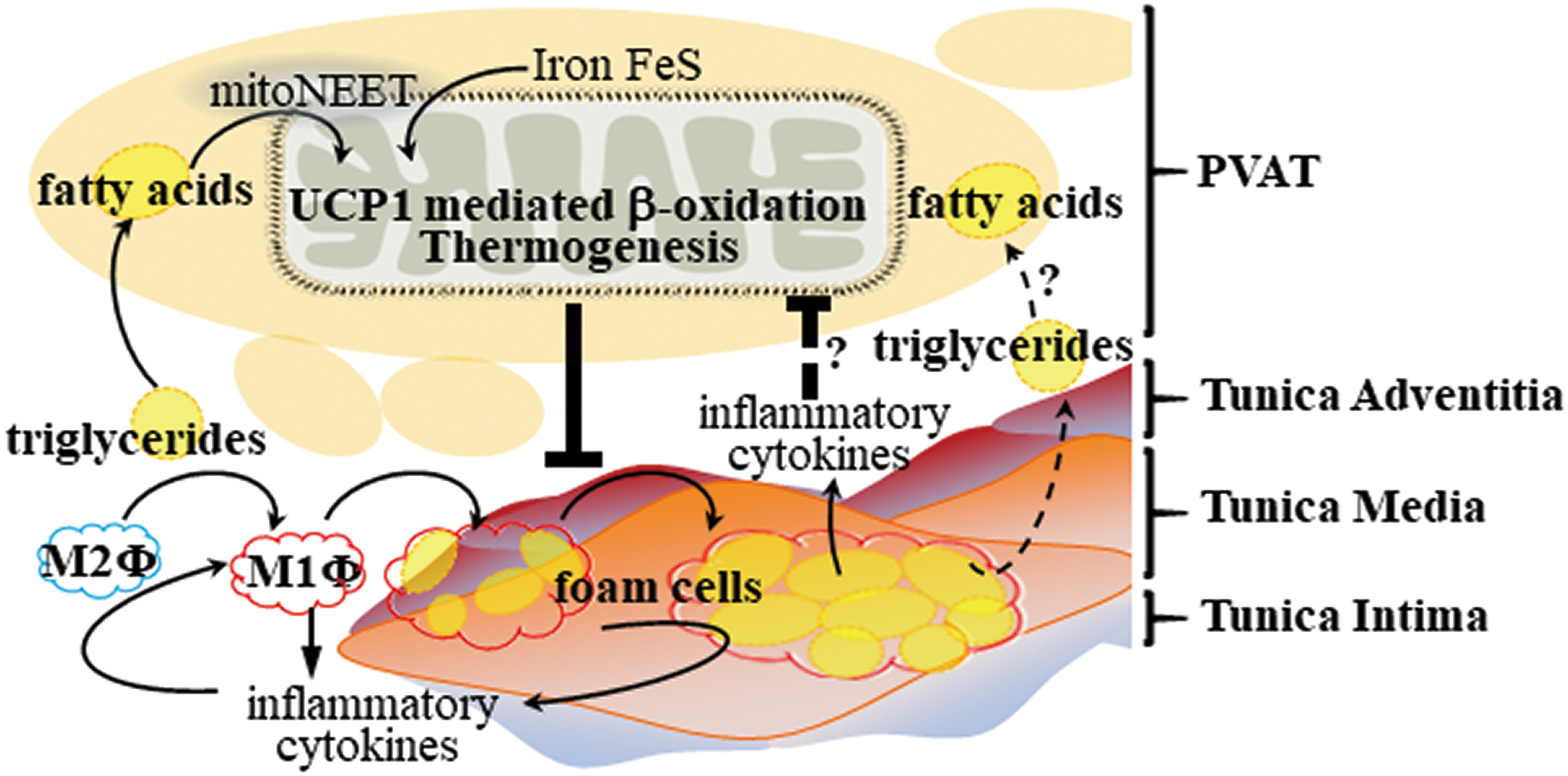
The PVATs in different locations resemble different types of adipose tissues. The thoracic aortic PVAT is BAT-like, while abdominal aortic PVAT is BeAT in rodents (79). The human aortic and coronary artery PVAT is BeAT as well (19, 31). As described above, both BAT and BeAT have thermogenic capability due to high UCP-1 protein levels in mitochondria. Unlike WAT, BAT and BeAT have anti-inflammatory characteristics.
We previously reported that thermogenic aortic PVAT was able to improve endothelial function, which was impaired in aging mice. Activation of PVAT in ApoE−/− mice with intact PVAT by mild cold exposure improved endothelial function and prevented atherosclerosis development. ApoE−/− mice lacking PVAT showed severe atherosclerotic lesions that could not be reduced by mild cold exposure (17). We further demonstrated that lack of PVAT augmented macrophage infiltration in the perivascular area of the aorta and increased production of inflammatory cytokines, which resulted in vascular inflammation and increased atherosclerotic lesions in the aortic wall (117).
Consistently, deletion of insulin receptors in brown adipocytes resulted in lipoatrophy in BAT, including PVAT, and induced visceral adiposity in the gonadal depot. Insulin receptor-deficient ApoE−/− mice showed severe atherosclerosis and increased expression of TNF-α and leptin, while there was decreased adiponectin in adipose tissues, including thoracic PVAT (42). These results suggest that promoting thermogenesis in thermogenic PVAT may inhibit atherosclerosis development. Even though cold exposure significantly induced thermogenesis in PVAT (17), cold exposure significantly increased blood pressure and heart rate (44), which could increase the incidence rates of stroke and heart attack (35), thus making it a nonfeasible treatment in humans.
We found that CDGSH iron-sulfur domain 1 protein (referred to as mitoNEET), a mitochondrial outer membrane protein that promotes adipose tissue browning and thermogenesis, was highly expressed and significantly increased in PVAT upon cold stimulation. ApoE−/− mice with mitoNEET overexpression in PVAT were cold resistant and showed increased expression of thermogenic genes. Furthermore, they showed significant downregulation of inflammatory genes and reduced atherosclerosis development upon high-fat-diet feeding (116). Therefore, targeted pharmacological approaches to induce thermogenesis in PVAT could inhibit PVAT inflammation and atherosclerosis development, and represent a more feasible treatment compared with cold exposure (Fig. 4).
PVAT and endothelial dysfunction in atherosclerosis development
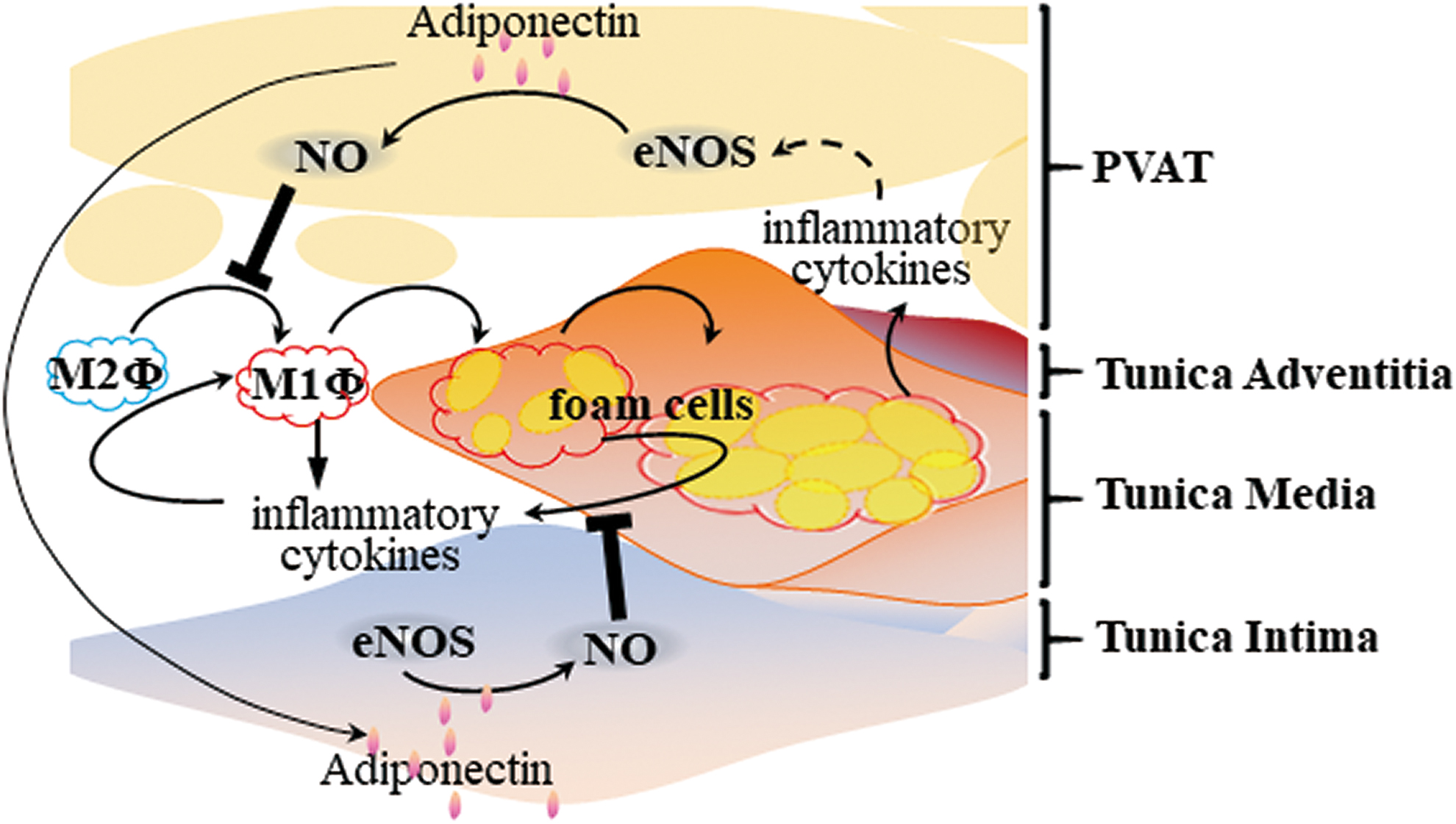
Inflammatory response and oxidative stress in PVAT in obesity ultimately lead to endothelial dysfunction (51). Endothelial dysfunction can also reduce endothelial nitric oxide synthase (eNOS) expression, reduce NO production and bioavailability, with ensuing adverse consequences on vasodilation, and significantly increase peripheral resistance of blood vessels. Recent studies have shown that PVAT expresses eNOS (114), and removal of PVAT reduces basal NO production in small arteries from healthy individuals, suggesting that PVAT contributes to vascular NO production (107).
It is known that adiponectin normalizes endothelial function through a mechanism involving increased eNOS phosphorylation and controls blood pressure through an endothelial-dependent mechanism (52). Furthermore, PVAT-derived adiponectin inhibits plaque formation via a reduced vascular inflammatory response, as indicated above (Fig. 5). PVAT secretes biologically active H2S, which improves endothelial function (32). However, the relationship between PVAT-derived H2S and atherosclerosis development is unknown. Further investigation is required regarding the antiatherosclerotic effects of PVAT-derived factors such as adiponectin, NO, and H2S operating through improved endothelial function.
PVAT immune cells and atherosclerosis development
PVAT-derived inflammatory mediators may have adverse effects on atherosclerotic plaque formation and stability. Obesity increases the number and activation levels of the multiple resident immune cells in PVAT, including macrophages, dendritic cells (DCs), T cells, and B cells, which locally regulate the inflammatory status of the vascular wall.
Macrophages can be divided into classically activated M1 macrophages and selectively activated M2 macrophages depending on their activation state, function, and secreted factors. IL-4 and IL-13 secreted by eosinophils stimulate M2-type polarization in macrophages, exerting anti-inflammatory effects (21, 62). In adipose tissue of obese animals, the polarization state of macrophage cells is transformed from M2 to M1. Metabolically activated macrophages express classic M1-type polarizing markers (such as CD38, CD319, and CD274) and secrete large amounts of inflammatory mediators, such as IL-6, IFN-γ, and TNF-α (55). M2 macrophages in PVAT increased the expression of brown adipocyte-specific markers, which resulted in reduced inflammation and lower oxidative stress (64, 76).
Mas receptors are expressed in PVAT. AVE0991, a nonpeptide Ang-(1 –7) receptor Mas agonist, has significant antiatherosclerotic properties in ApoE−/− mice. AVE0991 treatment reduced expression and production of the proinflammatory cytokines IL-1β, TNF-α, CCL2, and CXCL10, and differentiation to the M1 macrophage phenotype in PVAT (98). In addition, dysfunction of the low-density lipoprotein receptor-related protein-1 (LRP1) in liver and macrophages increased atherosclerotic risk (28, 72). Transplantation of adipose tissue from Lrp1−/− mice to the carotid artery regions of Ldlr−/− mice markedly increased atherosclerotic plaque in the carotid arteries, which was associated with increased expression of inflammatory genes such as IL6, TNF-α, and MCP-1 and infiltration of CD68+ macrophages in Lrp1−/− PVAT (53).
Macrophages also play an important role in regulating T cell activation through antigen presentation and costimulatory ligand expression, which lead to aggregation of other proinflammatory cells including CD4 T cells, CD8 T cells, B cells, and DCs (59). There are three types of T cells in PVAT; that is, CD4+ helper T cells (Th), CD8+ cytotoxic T cells (Tc), Treg (76). Th1, Tc1, and Th17 cells secrete proinflammatory cytokines such as IFN-γ, TNF-α, IL-17, and IL-6, which are related to atherosclerosis development (33, 112). In contrast, Treg have the ability to alter adipose tissue remodeling, improve insulin resistance, and reduce atherosclerosis and hypertension. They produce immunosuppressive factors, such as TGF-β and IL-10, regulating the immune response, reducing oxidative stress in blood vessels, endothelial dysfunction, infiltration of macrophages and T cells in the aorta, and proinflammatory cytokines in plasma (2, 41).
DCs are the most effective antigen-presenting cells found in PVAT (109). During PVAT inflammation, inflammatory mediators released from DCs stimulate T cells to produce proinflammatory cytokines by blocking the CD28/CD80/CD86 costimulatory axis between DCs and T cells (106). However, the contributions of DCs and T cells in PVAT to atherosclerotic lesion formation are still largely unclear.
B cells have emerged as important immune cells in the regulation of visceral adipose tissue inflammation and atherosclerosis. B cell-mediated effects on atherosclerosis are subset dependent, with B-1 cells attenuating and B-2 cells aggravating atherosclerosis (14, 30, 57, 70, 100, 101). PVAT from human coronary arteries near the diseased region harbors B cells, including B-1 cells. Moreover, mouse PVAT harbors high numbers of atheroprotective IgM-secreting B-1 cells. IgM from B-1 cells blocks oxidized LDL-induced foam cell formation and inflammatory cytokine production. ApoE−/− mice with B cell-specific knockout of the helix-loop-helix factor Id3 had increased numbers of IgM-secreting B-1b cells in PVAT (101), further suggesting that inhibition of local inflammation in PVAT may serve to provide atheroprotection (Fig. 6).

Anti-inflammatory response in PVAT prevents atherosclerosis development
Teneligliptin is a DPP-4 inhibitor, which is used for the treatment of type 2 diabetes mellitus. Oral administration of teneligliptin to ApoE−/− mice significantly inhibited atherosclerotic lesions in the aortic arch, which was associated with reduced expression of macrophage marker Mac3 and Nox-4 in the PVAT surrounding the aortic arch (92). Treatment of ApoE−/− mice with another antidiabetic drug, pioglitazone, also significantly downregulated oxidative stress and proinflammatory markers in PVAT (84).
CD40/CD40L signaling exerts a critical role in the development of atherosclerosis. Treatment of ApoE−/− mice with siRNA against CD40 significantly reduced the extent and severity of the atherosclerotic lesions, as well as the number of F4/80+, galectin3+, macrophages, and NF-κB+ cells in the intima, which was associated with downregulation of transcription factor Taf3 and macrophage differentiation regulator Xpr1 in PVAT (46).
PVAT-derived adiponectin might be one of the anti-inflammatory adipokines able to inhibit the development of atherosclerosis. Indeed, ApoE−/− mice that received PVAT transplantation from adiponectin-deficient mice exhibited advanced atherosclerotic lesions in the carotid artery, and this was associated with attenuated autophagy in macrophages (61). Transplantation of thoracic PVAT from wild-type mice to the carotid artery of ApoE−/− mice nearly abrogated plaque macrophage content without affecting plaque size, while transplantation of thoracic PVAT from ApoE−/− mice showed higher mRNA levels of inflammatory cytokines compared with wild-type PVAT (85).
However, transplantation itself may cause PVAT inflammation and underlying vascular remodeling. In fact, transplantation of thoracic aortic PVAT to carotid arteries of ApoE−/− mice markedly increased the intraplaque macrophage infiltration, lipid core, intimal and vasa vasorum neovascularization, and expression of matrix metalloproteinase-2/9 in plaques while decreased smooth muscle cells and collagen in the atherosclerotic plaques (122).
These data were obtained at the endpoint of the animal study. The roles of PVAT in the plaque progression during development of the atherosclerotic lesion remain still unaddressed. Recently, a novel imaging technique using microcomputed tomography (microCT) to characterize the temporal and spatial development of aortic PVAT and luminal plaque soft tissue has been developed (94). Using microCT, the vascular wall volume was found to be larger, while the aortic PVAT volume was reduced in ApoE−/− mice compared with those in the C57BL/6J mice. Plaque development was colocalized with luminal ostia and origins of branching arteries, which traveled through areas of greatest PVAT volume in ApoE−/− mice. This technique provides a new strategy to assess the relationship between PVAT and pathology in vascular walls in the same animal during lesion development (34).
Mechanisms underlying PVAT roles in atherosclerosis development
Obesity promotes expansion of adipose tissues, including PVAT, and increases the rate of basal fat breakdown, which in turn increase release of FFA and secretion of proinflammatory factors into circulation. Hypertrophic PVAT may release abundant FFA into the perivascular area and other layers of the blood vessel wall.
FFA activate nuclear factor-κB (NF-κB), protein kinase C (PKC) and Toll-like receptors (TLRs) which promote phosphorylation of insulin receptor substrate 1 (IRS-1) in PVAT and other vascular cells (64, 88). Phosphorylation of IRS-1 reduces its ability to activate downstream PI3K/Akt signaling, resulting in inhibition of eNOS synthesis and NO production, accompanied by a decrease in NO bioavailability, triggering changes in vascular function and accumulation of ROS, resulting in endothelial dysfunction and, ultimately, the inevitable development of atherosclerosis. In addition, FFA activate TLR2 and TLR4, which regulate the NF-κB signaling pathway in macrophages (88).
TLR4-deficient Ldlr−/− mice showed more mitochondria and capillaries in PVAT and were protected against atherosclerosis development associated with reduced TNF-α expression in PVAT (64). Intracellular oxidation of stored FFA in PVAT adipocytes may produce ROS, leading to local vascular inflammation and PKC activation (83). Vascular local ROS can activate PKC and NF-κB-mediated inflammatory responses, as well as induce mitochondrial dysfunction leading to more ROS production (27). PKC also triggers eNOS coupling, which increases the synthesis of endothelin-1, leukocyte adhesion, and cytokine activation, and leads to vasoconstriction, platelet aggregation, and endothelial dysfunction (40).
In addition, in rats with high-fat-diet-induced obesity, PVAT dysfunction can promote endothelial dysfunction by modulating the AMPK/mammalian target of rapamycin kinase pathway (67). The AMPK signaling pathway is involved in the regulation of PVAT inflammation and oxidative stress (118). AMPK is an anti-inflammatory and antioxidative-stress regulator that exerts anti-inflammatory effects by upregulating IL-10 and downregulating TNF-α and IL-6 (39, 67). AMPK agonists AICAR, metformin, and resveratrol inhibited expression of the inflammatory cytokines (TNF-α, IL-6, MCP-1) and promoted expression of the anti-inflammatory factors, such as adiponectin and peroxisome proliferator-activated receptor-γ, which are associated with decrease of endoplasmic reticulum (ER) stress in PVAT (102, 113).
The NACHT, LRR, and PYD domains-containing protein 3 (NLRP3), also known as cryopyrin, is predominantly expressed in macrophages and is a component of the inflammasome. NLRP3 activation occurs in response to ER stress-related inflammation (45). NLRP3-dependent inflammasome activation decreased adiponectin while increased IL-6 secretion in PVAT, suggesting that inflammation induces PVAT dysfunction associated with ER stress (126).
The findings discussed in this section underscore a causative role of PVAT dysfunction in atherosclerosis and point to potential targets for intervention aimed at restoring PVAT homeostasis in the treatment of atherosclerosis.
Conclusion and Perspective
PVAT is a unique adipose tissue that plays an essential role in maintaining vascular structure and regulating vascular functions. Under physiological conditions, PVAT functions as an anti-inflammatory tissue, depicts heat production, improved FFA metabolism, and can regulate vasodilation. However, under pathophysiological conditions, such as obesity, PVAT becomes dysfunctional and promotes infiltration of inflammatory immune cells and local oxidative stress, triggering an “outside-to-inside” pathological signaling within the vessel wall, which leads to dysfunction of the underlying VSMCs and ECs. Eventually, dysfunctional PVAT promotes hypertension and atherosclerosis progression, thus suggesting an “outside-to-inside” paradigm for vascular disease development. Whether and how these PVAT effects act concurrently and synergistically with “inside-to-outside” signaling from VSMC and EC to PVAT remain to be addressed.
Future studies should further investigate the messengers responsible for the crosstalk between PVAT and underlying VSMCs and ECs in the vascular wall. We also need to better understand the balance of anticontractile and contractile systems in PVAT in homeostasis and in pathology as it pertains to blood pressure regulation. CVDs display sex differences. It is yet unclear whether and which intrinsic features of PVAT exhibit sex differences and what are their consequences on the development of CVDs. In addition, the actual contribution of alterations in PVAT metabolism to the overall systemic outcomes of CVDs remains largely unknown. Finally, aging contributes to vascular dysfunction. Single-cell RNA sequencing and bioinformatics analyses revealed that the brown adipogenic differentiation capacity of the stromal cells in PVAT was decreased during aging, which contributed to vascular remodeling (81).
In addition, old mice were resistant to cold-induced browning of perivascular adipocytes (80), and the browning levels of thoracic PVAT of aging SHRs were reduced significantly more than those in age-matched Wistar-Kyoto control rats (54). Therefore, aging might result in PVAT dysfunction and contribute to hypertension development. It is unclear whether dysfunctional PVAT also contributes to atherosclerosis development during aging. The new imaging techniques may provide a unique advantage for research investigating the dynamic alterations in PVAT during aging as well as the progression of vascular diseases such as hypertension and atherosclerosis.
Funding Information
This work was partially supported by National Institutes of Health grant HL122664 (Lin Chang) and HL068878 and HL137214 (Yuqing Eugene Chen).