
Abstract
Significance:
Metabolic reprogramming is considered to be a critical adaptive biological event that fulfills the energy and biomass demands for cancer cells. One hallmark of metabolic reprogramming is reduced oxidative phosphorylation and enhanced aerobic glycolysis. Such metabolic abnormalities contribute to the accumulation of reactive oxygen species (ROS), the by-products of metabolic pathways. Emerging evidence suggests that ROS can in turn directly or indirectly affect the expression, activity, or subcellular localization of metabolic enzymes, contributing to the moonlighting functions outside of their primary roles. This review summarizes the multifunctions of metabolic enzymes and the involved redox modification patterns, which further reveal the inherent connection between metabolism and cellular redox state.
Recent Advances:
These noncanonical functions of metabolic enzymes involve the regulation of epigenetic modifications, gene transcription, post-translational modification, cellular antioxidant capacity, and many other fundamental cellular events. The multifunctional properties of metabolic enzymes further expand the metabolic dependencies of cancer cells, and confer cancer cells with a means of adapting to diverse environmental stimuli.
Critical Issues:
Deciphering the redox-manipulated mechanisms with specific emphasis on the moonlighting function of metabolic enzymes is important for clarifying the pertinence between metabolism and redox processes.
Future Directions:
Investigation of the redox-regulated moonlighting functions of metabolic enzymes will shed new lights into the mechanism by which metabolic enzymes gain noncanonical functions, and yield new insights into the development of novel therapeutic strategies for cancer treatment by targeting metabolic-redox abnormalities. Antioxid. Redox Signal. 34, 979–1003.
Metabolic Reprogramming and Redox Homeostasis
Cancer has historically been accepted to be an oncogene-induced disease with uncontrolled cell proliferation. However, since Dr. Otto Warburg observed the phenomenon that cancer cells are prone to employ glycolysis (an inefficient ATP-generating process) rather than oxidative phosphorylation (OXPHOS) even in the presence of oxygen, a growing body of studies have demonstrated the critical roles of metabolic rewiring in tumorigenesis (96). These metabolic alterations have been reported to be associated with the malignant transformation of cancer cells by promoting oncogenic mutations, leading to the concept that cancer is a metabolism-associated disease (37, 187). Enhanced glycolysis promotes the proliferation of cancer cells by converting metabolic flux to pentose phosphate pathway (PPP), which increases anabolic support (including intermetabolites, amino acids, nucleotides, lipids, etc.) via elevated glucose consumption and enhances antioxidant capacity (99). The alterations in metabolic profiles can be used to distinguish tumors from normal tissues in vivo by imaging the uptake of radio-labeled glucose analog ([18F] fluoro-2-deoxyglucose) using positron emission tomography (58). Given that metabolic reprogramming is manipulated by metabolic enzymes (many of them are druggable), which can be regulated by hypoxia-inducible factor 1 (HIF1), Myc, Ras, Akt, and p53 inactivation, targeting these metabolic abnormalities may provide a promising strategy for cancer therapy (35, 38, 82, 191).
Reactive oxygen species generation
During malignant transformation, cancer cells rewire metabolism to provide biomass, leading to excessive production of reactive oxygen species (ROS) (137). ROS are oxygen-derived chemical species including hydrogen peroxide (H2O2), hydroxyl free radicals (HO•), and superoxide (O2 •−), which are mainly generated in the mitochondrion, peroxisome, and endoplasmic reticulum (ER) (124, 169). In addition, some metabolic enzymes are reported to induce the production of ROS (Table 1) (63, 111, 137, 148). This evidence suggests that cellular metabolism directly or indirectly contributes to ROS generation in cancer cells.
The Sources of Intracellular Reactive Oxygen Species Production
ROS, reactive oxygen species.
ROS elimination
High levels of ROS could result in oxidative stress and excessive oxidation of proteins, DNA, and lipids, which are lethiferous to cancer cells (26). However, ROS at moderate levels can serve as second messengers, which are indispensable for physiological processes (168). To maintain the cellular redox homeostasis, cancer cells have developed robust antioxidant systems to antagonize elevated ROS levels (159). For instance, glutathione (GSH) is the most abundant and ubiquitous small antioxidant molecule in living cells (174). GSH is synthesized from cysteine, glutamate, and glycine through ATP-dependent reactions catalyzed by γ-glutamyl-cysteine ligase and glutathione synthetase (8). Under physiological conditions, reduced GSH is its major form and exists in millimolar concentrations in the bulk of cells (174).
GSH can maintain a reduced environment of subcellular compartments such as mitochondria, the nucleus, and cytosol (174). Approximately 10% of GSH is found in mitochondria, where GSH eliminates excessive ROS and protects cells from apoptosis (3, 43). Under oxidative stress, GSH can be converted to the oxidized form, GSH disulfide (GSSG), directly by ROS or indirectly via GSH-dependent reactions catalyzed by glutathione peroxidases (GPXs) (6). The ratio of reduced and oxidized forms of glutathione (GSH/GSSG) indicates the antioxidant potential and detoxifying capacity of cancer cells. Moreover, this ratio in different organelles reflects the metabolic burden in living cells (107). Sustained accumulation of GSSG is toxic for cancer cells (34). GSSG can be reduced to GSH by glutathione reductase at the expense of NADPH (34). NADPH possesses critical reductive power for many reactions and is fueled by multiple metabolic pathways such as PPP, folate cycle, pyruvate malate shuttle, and tricarboxylic acid (TCA) cycle (22, 109).
In addition to GSH, antioxidant enzymes, such as superoxide dismutases (SODs), catalase (CAT), peroxiredoxins (PRDXs), GPXs, and thioredoxins (TRXs), could prevent oxidative damage. SODs can convert O2 •− to H2O2, while CAT, GPXs, and PRDXs catalyze the conversion of H2O2 to H2O (67). By controlling these complex metabolic enzymatic systems, cancer cells will survive in response to oxidative stress, as well as maintain cancerous redox homeostasis. In addition, some dietary bioactive compounds including ascorbic acid, selenium, vitamin E, β-carotene, and N-acetylcysteine are natural antioxidants that can be used to regulate the intracellular redox status for clinical manipulation (30).
Moonlighting Metabolic Enzymes in Cancer Cells
Cellular metabolism maintains cell growth and proliferation by offering ATP and building blocks generated by metabolic enzyme-mediated catalysis of intracellular chemical reactions. Intriguingly, a study from Jong's group showed that duck lens є-crystallin protein is the same gene product as lactate dehydrogenase-B4, and its expression makes up to 10% of the total soluble proteins, which is far more than its metabolic need, suggesting that metabolic enzymes may have additional functions beyond their canonical metabolic activities (59). This is the first example of the moonlighting function of a metabolic enzyme. Afterward, increasing efforts have been made to investigate the noncanonical functions of metabolic enzymes in regulating cellular events. Specifically, some metabolic enzymes are thought to be regulated by redox modification, indicating that redox modification may be associated with the moonlighting function of these metabolic enzymes (2, 56, 83, 153). Here, we will discuss the recent findings of moonlighting functions for metabolic enzymes and molecules, with an emphasis on their regulation of epigenetics, gene transcription, and post-translational modifications (PTMs) (Table 2).
Summary of the Moonlighting Functions of Metabolic Enzymes in Cancer
α-HB, α-hydroxybutyrate; 3-PG, 3-phosphoglycerate; acetyl-CoA, acetyl-coenzyme A; ACSS2, acyl-CoA synthetase short-chain family member 2; ENO1, enolase 1; ET-1, endothelin-1; G6PD, glucose-6-phosphate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSSG, GSH disulfide; HK, hexokinase; LDHA, lactate dehydratase A; NMNAT-1, nicotinamide mononucleotide adenyltransferase-1; PFK1, phosphofructokinase 1; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PGAM5, phosphoglycerate mutase family member 5; PGK1, phosphoglycerate kinase 1; PKM2, pyruvate kinase M2; PPP, pentose phosphate pathway; PSAT1, phosphoserine aminotransferase; SRC-3, steroid receptor coactivator-3; TAZ, transcriptional coactivator with PDZ-binding motif; VDAC, voltage-dependent anion channel; YAP, Yes-associated protein.
Metabolic regulation of epigenetics in cancer
Epigenetics refers to the heritable changes of gene expression without altering the DNA sequence. Till now, various epigenetic events have been unraveled, including DNA methylation, histone methylation, histone acetylation, nucleosome remodeling, and noncoding RNA-mediated gene expression. (120). Epigenetic modifications play crucial roles in tumorigenesis and cancer progression through regulating gene expression and other DNA-related processes, such as DNA repair (81). The complicated modifications of DNA and histones are tightly regulated by a series of chromatin-modifying enzymes, such as DNA methyltransferases (DNMTs), ten-eleven translocation (TET), histone acetyltransferase (HAT), histone deacetylases (HDACs), and sirtuins (81). The functions of these epigenetic-associated enzymes require metabolism-derived cofactors, such as S-adenosylmethionine (SAM), α-ketoglutarate (α-KG), FAD, acetyl-coenzyme A (acetyl-CoA), and NAD+ (90). Therefore, metabolic network is linked with chromatin modifications, constituting a well-orchestrated axis to assist cancer cells with adaption to the environmental changes (Table 2). Recent studies indicate that the metabolic networks modulate chromatin modifications through changes in metabolite levels or noncanonical functions (e.g., DNA binding) (72). Importantly, several chromatin-associated metabolic molecules (including SAM, α-KG, FAD, acetyl-CoA, NAD+, and lactate) and enzymes are involved in redox regulation, providing an insight for the intertexture of redox and epigenetic alteration (83, 97, 128, 154).
S-adenosylmethionine
The methylation of DNA and proteins is catalyzed by DNMT and histone methyltransferases (HMTs), respectively, and both need SAM as the methyl donor (104) (Fig. 1A). SAM is generated from the addition reaction of ATP to methionine, which is catalyzed by methionine adenosyltransferase. SAM can be converted to S-adenosyl homocysteine (SAH), a potent inhibitor of SAM-dependent enzymes (such as DNMTs and HMTs) (115). Of note, cystathionine, the by-product of the SAM cycle, can be converted to cysteine, thereby contributing to GSH generation through the trans-sulfuration pathway (198). Therefore, the consumption of SAM during DNA or protein methylation may be coupled with the alteration of intracellular ROS levels. Notably, cystathionine β-synthase (CBS), which converts homocysteine to cysteine, can be oxidized to form a disulfide bond between Cys272 and Cys275. The activity of oxidized CBS is twofold to threefold less compared with reduced form (128). Besides, CBS can be S-glutathionylated at Cys346 under oxidative stress, resulting in ∼3-fold increase in CBS activity (129). These reports indicate that redox regulation may play an important role in the SAM cycle. The methyltransferase nicotinamide N-methyltransferase (NNMT) catalyzes the transfer of a methyl group from SAM to nicotinamide to form SAH (135).

It has been observed that NNMT is overexpressed in various cancers, including lung (146), liver (84), colon (142), and kidney cancer (184). The upregulation of NNMT is positively correlated with migration and invasion potential of colorectal cancer cells, indicating a prometastasis role of NNMT (142). Importantly, the overexpression of NNMT can inhibit the methylation of histones and other signaling proteins by elevating the SAH/SAM ratio (165). Notably, SAH alone is not sufficient to reduce the methylation of histones, implying that HMTs may be more sensitive to the decreased SAM concentration, rather than increased SAH alone (165). Therefore, targeting NNMT or replenishing SAM may hold potential for cancer treatment.
α-Ketoglutarate
The metabolic intermediate α-KG is a by-product of several reactions catalyzed by isocitrate dehydrogenase (IDH), aspartate aminotransferase, glutamate dehydrogenase (GLUD), the branched-chain aminotransferase (BCAT), or phosphoserine aminotransferase (PSAT1). DNA or lysine demethylation enzymes, such as TET proteins and JmjC domain-containing histone lysine demethylases (KDMs), mainly utilize α-KG and Fe2+ as cofactors to catalyze multistep oxidation or hydroxylation reactions on the alkyl group of their substrates (Fig. 1B) (180). Therefore, α-KG level is critical to maintain the activity of these α-KG-dependent enzymes. The point mutations of IDH are common in several types of tumors, including acute myeloid leukemia (AML) (36), low-grade gliomas (18), and secondary glioblastomas (9). IDH mutation abolishes its oxidative enzymatic activity and converts α-KG to 2-hydroxyglutarate (2-HG), resulting in the overproduction of 2-HG within tumors (which could accumulate to millimolar levels) (87). Due to the structural similarity, 2-HG can competitively inhibit the activity of α-KG-dependent enzymes, including KDM, TET, prolyl hydroxylases (PHDs), and collagen prolyl-4-hydroxylases, leading to the alterations of methylation levels of both DNA and histones (180). However, not all KDMs require α-KG as a cofactor. For example, lysine-specific demethylase (LSD, also known as KDM1) utilizes FAD to catalyze the demethylation of mono- and dimethylated H3K4 or H3K9 (Fig. 1C) (62).
Importantly, it has been reported that IDH can be glutathionylated at its Cys269 under oxidative stress, resulting in a decrease in its activity and α-KG production (83). In the presence of GSH, oxidation of IDH can be reduced and reactivated by glutaredoxin 2 (GRX2) (83). Whether the redox regulation of IDH is involved in tumorigenesis or tumor progression needs further investigation. In addition to 2-HG, fumarate and succinate are competitive inhibitors of α-KG-dependent enzymes, and their respective enzymes, fumarate hydratase (FH) and succinate dehydrogenase, are commonly mutated in human cancers (180). Fumarate can react with GSH to form a covalent adduct named succinicGSH, which will exaggerate intracellular oxidative stress (195). Interestingly, FH can be oxidized and inactivated under oxidative stress (199). It is worth noting that the cytosolic isoenzyme FH can be recruited to the nucleus in response to DNA damage, contributing to cancer cell survival (186). Both fumarate and succinate can promote histone methylation and increase the protein level of HIF1α by inhibiting the PHD activity (177). These important roles of TCA intermediates in controlling chromatin modifications, hypoxic response, and DNA methylation highlight the significance of TCA cycle and the involved key molecules in tumorigenesis and tumor development (113). Notably, the advent of “truncated” TCA cycle (featured by pyruvate and citrate shunting into lipid synthesis), which may result in decrease of α-KG and subsequent epigenetic alteration, is a hallmark of cancer (21). This may raise a novel insight to explain the interplay between tumorigenesis and metabolic reprogramming.
Acetyl-coenzyme A
Cellular acetyl-CoA is the sole acetyl group donor for histone acetylation, and is mainly produced from glucose and fatty acid metabolism (Fig. 1D) (13). In mammals, mitochondrial acetyl-CoA is predominantly generated from glucose-derived pyruvate via the pyruvate dehydrogenase complex (PDC). Alternatively, β-oxidation reaction can generate acetyl-CoA as an end product (105). In addition, branched-chain amino acids, including valine, leucine, and isoleucine, can be employed to generate acetyl-CoA, which is catalyzed by BCAT and the branched-chain α-ketoacid dehydrogenase complex (105). Cytosolic acetyl-CoA is mainly generated from the reductive carboxylation of glutamine, especially when glycolysis is inhibited. The conversion of glutamine to acetyl-CoA comprises multistep reactions catalyzed by glutaminase (GLS), GLUD2, and ATP-citrate lysase (ACLY), respectively.
Of note, PDC, ACLY, and acyl-CoA synthetase short-chain family member 2 (ACSS2) have been shown to translocate into the nucleus to promote the ectopic generation of acetyl-CoA, leading to the alteration of histone acetylation (160). The acetylation reaction is mainly catalyzed by HATs, including Gcn5 N-acetyltransferases (GNATs) and p300/CREB-binding protein (p300/CBP) HATs (155). GNATs family consists of Gcn5, PCAF, Elp3, Hat1, Hpa2, and Nut1 (155). The activity of HATs is influenced by the cellular nutrient availability, since the acetyl-CoA level is fluctuating with the alteration of metabolic conditions (112). Among these HATs, Gcn5 possesses similar binding constants for CoA and acetyl-CoA, indicating that the ratio of acetyl-CoA/CoA is important for the activity of Gcn5 (162). Different from histone methylation, which persists a half-life for hours, histone acetylation only has a half-life of few minutes, suggesting that histone acetylation is more sensitive to dynamic metabolic changes, while histone methylation may elicit the sustained epigenetic response (7).
NAD+
NAD+ is a product of nicotinamide mononucleotide adenyltransferase-1 (NMNAT-1) by catalyzing the conversion of nicotinamide mononucleotide (49). NAD+ is required for the activity of class III HDACs (so called sirtuins) (154). NAD+ can be depleted by poly(ADP-ribose) polymerase enzymes during DNA damage, which may lead to the impairment of the deacetylation activity of sirtuins (189). Intriguingly, the NAD+ synthase NMNAT-1 has been reported to localize exclusively in the nucleus and increase the deacetylase activity of SIRT1 (193). In general, increased acetylation of histones promotes transcription activation, while deacetylation of histones is associated with transcription repression. The overexpression of HDACs is found to be closely linked with several types of cancer (such as hematological malignancies) and predicts poor prognosis (Fig. 1E) (121, 132). Therefore, targeting the histone acetylation or deacetylation by modulating the cellular concentration or localization of acetyl-CoA and NAD+ may shed light on cancer treatment. HDAC inhibitors show promising anticancer efficacy in lymphomas (134) and breast cancer (167), and are now under extensive investigation in cancer therapy. However, the altered ROS levels induced by certain HDAC inhibitors should be taken into account to circumvent the possible side effects (66). In addition, the activation of sirtuins by NAD+ can improve mitochondrial homeostasis via the mitochondrial unfolded protein response (UPR) and expand the worm life span, whether NAD+ play a role in UPR of cancer cells remains to be further investigated (123).
Lactate
Lactate, which is converted from pyruvate and widely known as an energy source, is involved in the lactylation of histone lysine (190). The histone lactylation has been reported to stimulate gene transcription (97). Under hypoxic conditions, lactate production and subsequent histone lactylation level are enhanced in M1 macrophages, leading to the transcriptional activation of homeostatic genes such as Arg1 (190). This observation demonstrates a nonmetabolic role of lactate in regulating the epigenetic modification, suggesting that intervention of lactate level may be promising in the treatment of bacterial infection and cancers.
Metabolic enzymes in regulating cancerous gene transcription
In addition to epigenetic regulation, emerging evidence has demonstrated the roles of metabolic enzymes in regulating gene transcription. An important prerequisite is the nuclear translocation of metabolic enzymes. The first report of glycolytic enzymes translocating to the nucleus can be traced back to 1950s, since then bulk of the glycolytic enzymes were found in the nucleus (114), including pyruvate kinase M2 (PKM2) (173), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (152), lactate dehydratase A (LDHA) (102), phosphofructokinase (PFK) (40), ACSS2 (48, 93), and enolase 1 (42). Of note, the alteration in the subcellular localization of metabolic enzymes may lead to the reset of cellular metabolites, thereby regulating cellular signaling pathways and cell fates. Moreover, the translocation of metabolic enzymes may be elicited by hypoxia (91), glucose deprivation (89, 93), or oncogenic mutations (91), which are associated with ROS generation, suggesting a role of oxidative stress in regulating the subcellular translocation of these moonlighting enzymes.
Acyl-CoA synthetase short-chain family member 2
Acetyl-CoA is mainly produced in the mitochondrion, and is the major source of acetyl addition in protein acetylation (72). Moreover, acetyl-CoA is unstable, and its hydrolysis releases higher energy than the hydrolysis of ATP (14). Therefore, nuclear production of acetyl-CoA is vital to maintain the acetylation of histones (117). ACSS2 is a highly conserved metabolic enzyme catalyzing the conversion of acetate to acetyl-CoA (150). In response to glucose deprivation, ACSS2 can be phosphorylated at Ser659 by AMP-activated protein kinase (AMPK). The phosphorylated ACSS2 exposes its nuclear localization signal to bind to importin α5, leading to the nuclear translocation of ACSS2. Notably, glucose deprivation causes an increase of ROS levels, which may link with the nuclear translocation of ACSS2. Nuclear ACSS2 binds to the transcription factor EB, and then interacts with the promoter regions of autophagy- and lysosome-associated genes (93). Importantly, nuclear ACSS2 promotes the local generation of acetyl-CoA in the nucleus, contributing to the acetylation of H3 in the promoter region and subsequent transcription of autophagy- and lysosome-associated genes (48, 93). Furthermore, oxygen or serum limitation increases the nuclear translocation of ACSS2, leading to acetate recapture in the nucleus and maintenance of histone acetylation (15). Taken together, the distribution of metabolites in nucleus is essential for regulating the PTMs of nucleus proteins (especially histones), leading to genome-wide alteration of gene expression.
Phosphofructokinase 1
PFK1 phosphorylates the fructose-6-phosphate (F-6-P) to fructose-1,6-bisphosphate (F-1,6-BP), representing first committed step of glycolysis. PFK1 is encoded by PFKM (muscle), PFKL (liver), and PFKP (platelets), and each of them encodes a different isoform (119). Notably, PFK1 has been reported to translocate into the nucleus and bind with the transcriptional enhanced associate domain (TEAD), resulting in the stabilized interaction of TEAD with the Yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ). In this regard, PFK1 promotes the activation of YAP/TAZ and induces the transcription of its downstream target genes (40). Intriguingly, cancer cells with high activity of YAP/TAZ are accompanied with high levels of glycolytic gene signature (98, 145). These findings indicate that PFK1 can promote the suppression of the Hippo signaling pathway in an enzymatic activity-independent manner. Suppression of the Hippo signaling pathway could in turn regulate the transcription of glycolytic enzymes, suggesting a negative feedback loop between the aerobic glycolysis and Hippo signaling pathway (40).
Under hypoxic condition, PFK1 can be glycosylated at Ser529 to redirect the glucose flux from glycolysis to PPP, thereby protecting cancer cells from excessive ROS-induced cell death. Blocking the glycosylation of PFK1 inhibits cell proliferation and retards the tumorigenicity of lung cancer cells (185). However, whether the glycosylation of PFK1 results from hypoxia-induced oxidative stress needs further investigation. Therefore, targeting PFK1-mediated metabolism resetting may represent a promising approach for cancer treatment. In particular, PFK1 can be activated by fructose-2,6-bisphosphate (F-2,6-BP), which is a product catalyzed by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases (PFKFBs, also known as PFK2) (61, 88). PFKFBs are activated in many cancers (11, 71). Among these PFKFBs, PFKFB3 mRNA splice variant 5 has been found to localize in the nucleus and increase the expression of cyclin-dependent kinase 1 (CDK1), Cdc25C, and Cyclin D3 to promote cell proliferation (181). The enhanced cell proliferation rate can be crippled through mutating its active site or nuclear localization residues, indicating that the enzymatic activity of PFKFB3 may be required for the expression of CDK1, Cdc25C, and Cyclin D3 (181). PFKFB4 can phosphorylate steroid receptor coactivator-3 at Ser 857 and enhance its transcriptional activity, leading to increased transcription of PPP-associated genes to counteract oxidative stress (31). Taken together, these observations suggest that PFKFB is associated with gene transcription, and its product F-2,6-BP is essential for the expression of proliferation-related genes.
Metabolic regulation at post-translational level
In addition to metabolites such as α-KG and SAM, metabolic enzymes are involved in regulating PTMs in an enzymatic activity-dependent or -independent manner. For instance, the mitochondrial deoxyguanosine kinase (DGUOK), a rate-limiting enzyme in the mitochondrial deoxynucleoside salvage pathway, regulates the phosphorylation of YAP1 through modulating mitochondrial OXPHOS (100). Moreover, monocarboxylate transporter 1 (MCT1), a monocarboxylate transporter that promotes the passive transport of lactate, has been reported to induce the phosphorylation and subsequent activation of AMPK (76). Apart from DGUOK and MCT1, many metabolic enzymes, such as glucose-6-phosphate dehydrogenase (G6PD) (24), PSAT1 (73), and phosphoglycerate kinase (PGK) (138, 139), are involved in manipulating PTMs. Here, we highlight the moonlighting roles of several metabolic enzymes in modulating PTMs.
Glucose-6-phosphate dehydrogenase
G6PD, a limiting enzyme of the PPP, converts glucose-6-phosphate (G-6-P) to 6-phosphogluconate, with the consumption of NADP+ and the production of NADPH (116). NADPH is a critical reducing power that reduces GSSG to its reduced form GSH, which scavenges ROS to prevent cellular oxidative damage. Downregulation of G6PD decreases the level of NADPH and GSH, leading to increased ROS levels and enhanced chemotherapy-induced cell death (77, 130). Therefore, targeting G6PD impairs the cellular ROS-scavenging capacity and increases the susceptibility of cancer cells to oxidative stress (196). Recently, G6PD is found to be involved in the regulation of PTMs of several proteins. For example, inhibition of G6PD suppresses the phosphorylation of AKT, leading to reduced proliferation of bladder cancer cells (24). In addition, G6PD facilitates the phosphorylation of signal transducer and activator of transcription 3 (STAT3) at its Ser727 (192). The phosphorylation of STAT3 is responsible for G6PD-enhanced cell proliferation in clear cell renal cell carcinoma. Moreover, phosphorylated STAT3 can in turn enhance the gene expression of G6PD via binding to the G6PD promoter, constituting a positive feedback regulatory loop (192). These observations suggest that protein PTMs induced by G6PD are crucial in cancer proliferation. Targeting the moonlighting function of G6PD may hold great potential for cancer therapy.
Phosphoserine aminotransferase
Recently, our group found that PSAT1, an important metabolic enzyme involved in the serine biosynthesis, plays a critical role in regorafenib-induced autophagy in glioblastoma cells (73). Regorafenib can directly bind and stabilize PSAT1. Stabilized PSAT1 subsequently promotes the phosphorylation of AMPK and activates AMPK signaling pathway. Meanwhile, upregulation of PSAT1 leads to the inhibition of RAB11a transcription, leading to the impaired fusion of autophagosome and lysosome. The accumulated autophagosome is required for regorafenib-induced glioblastoma cell death, and inhibition of autophagosome formation could decrease the cytotoxicity of regorafenib (73). Intriguingly, PSAT1 catalyzes the conversion of 3-phosphohydroxypyruvate and glutamate to 3-phosphoserine and α-KG (70). α-KG is reported to activate CamKK2 (Ca2+/calmodulin-dependent protein kinase kinase 2), thus promoting the phosphorylation and activation of AMPK, which may account for PSAT1-induced AMPK activation (75). Whether α-KG is involved in PSAT1-mediated glioblastoma suppression needs further investigation.
Phosphoglycerate kinase
PGK is a crucial glycolytic enzyme, which converts 1,3-biphosphoglycerate (1,3-BPG) and ADP to 3-phosphoglycerate and ATP (65). PGK1 could translocate to mitochondrion in response to hypoxia, epidermal growth factor receptor (EGFR) activation, or oncogenic mutations (such as KRASG12V and BRAFV600E). Mitochondrial PGK1 phosphorylates pyruvate dehydrogenase kinase 1, leading to reduced mitochondrial pyruvate utilization and decreased ROS generation (91). It has been evidenced that Chlamydomonas reinhardtii PGK1 is a redox sensor and forms a disulfide bond between its Cys227 and Cys361 (122). However, the oxidative modification of PGK1 in human cells remains largely unknown, and whether the PGK1 oxidation regulates its mitochondrial translocation needs further investigation. In addition, nuclear PGK1 can be phosphorylated by CK2α at its Ser256 under EGFR activation. Phosphorylated PGK1 binds to cell division cycle 7-related protein kinase (CDC7) and alleviates ADP-mediated inhibition of CDC7 activity. The interaction between PGK1 and CDC7 is essential for EGF-induced DNA replication (92). Besides, it is evident that glutamine deprivation and hypoxia inhibit the mTOR-induced phosphorylation of acetyl-transferase ARD1, leading to the acetylation of PGK1 at Lys388. Acetylated PGK1 subsequently phosphorylates Beclin1 at Ser30 residue and enhances the activity of class III phosphatidylinositol 3-kinase VPS34. The PGK1 acetylation and Beclin1 phosphorylation are required for hypoxia- or glutamine deprivation-induced autophagy (138). Together, these observations highlight the importance of PGK1 in maintaining cell homeostasis by regulating DNA replication and autophagy. Since EGFR activation, hypoxia and oncogenic mutations are commonly associated with ROS production, whether ROS is involved in the moonlighting functions of PGK1 needs further investigation.
Redox Regulation of the Moonlighting Functions of Metabolic Enzymes in Cancer Cells
ROS can modulate cellular signaling pathways through altering the activities or expression of critical molecules involved in cellular events, including epigenetic alteration, gene transcription, and apoptosis (95). Notably, ROS have been reported to directly modify the active cysteine residues of proteins, resulting in the alteration of protein function. Redox regulation of protein function has been extensively studied and is becoming an important PTM pattern (16, 32, 79). The reduced form of thiol groups (-SH) in cysteine residues can undergo oxidative modifications to generate S-hydroxylated derivatives (-SOH), which may further be oxidized to disulfides (-S-S-), S-glutathionylation (RS-SG), sulfinic acids (RSO2H), or sulfonic acids (RSO3H). These modifications can cause changes in protein conformation or protein–protein interactions, leading to functional alterations of proteins (149). On the contrary, some of these oxidized forms can also be reduced by cellular antioxidant systems such as TRX, GRXs, and PRDXs. Therefore, the redox regulation of reactive cysteine residues can serve as molecular switches that control the function of redox-sensitive proteins. Importantly, several metabolic enzymes were found to undergo redox modifications, leading to the alteration of the subcellular localization and activity of these metabolic enzymes. These interconnections between oxidative stress and metabolism shed new light on the understanding of how metabolism senses varying cellular environment and gains moonlighting functions.
Recently, Gottlieb's laboratory reported a workflow based on stable isotope cysteine labeling with iodoacetamide (SICyLIA) to provide detailed insights into proteome-scale cysteine oxidation of metabolic proteins in diverse cellular models and primary tissues (166). FH-deficient mouse kidney epithelial cells as well as kidney tissues were used as chronic oxidative stress models. FH is an important TCA cycle enzyme, which converts fumarate to malate. Previous studies showed that fumarate can react with GSH to form succinicGSH, resulting in depletion of GSH and NAPDH, and subsequent chronic oxidative stress (136, 195). Wild-type cells were treated with physiologically recoverable concentrations of H2O2 as an acute oxidative stress model. This study indicated that metabolic and mitochondrial proteins were prone to be oxidized under acute oxidative stress. Furthermore, it has been suggested that cysteine residues in the active sites of proteins are more sensitive to oxidative stress, suggesting that the activities of metabolic enzymes may be tightly regulated by oxidative modifications, and are important for metabolic adaption to acute oxidative stress. However, the cysteine residues that underwent oxidative modifications are not predominantly engaged in the active sites of metabolic proteins in response to chronic oxidative stress, implying that the potential moonlighting functions of metabolic enzymes may be involved in the metabolic adaption to chronic oxidative stress (166). Together, the oxidative modifications of metabolic enzymes highlight a metabolic adaption pattern to the microenvironment alterations. Whether the moonlighting functions of metabolic enzymes are engaged in adapting varying environmental stimuli needs further elucidation.
Lactate Dehydratase A
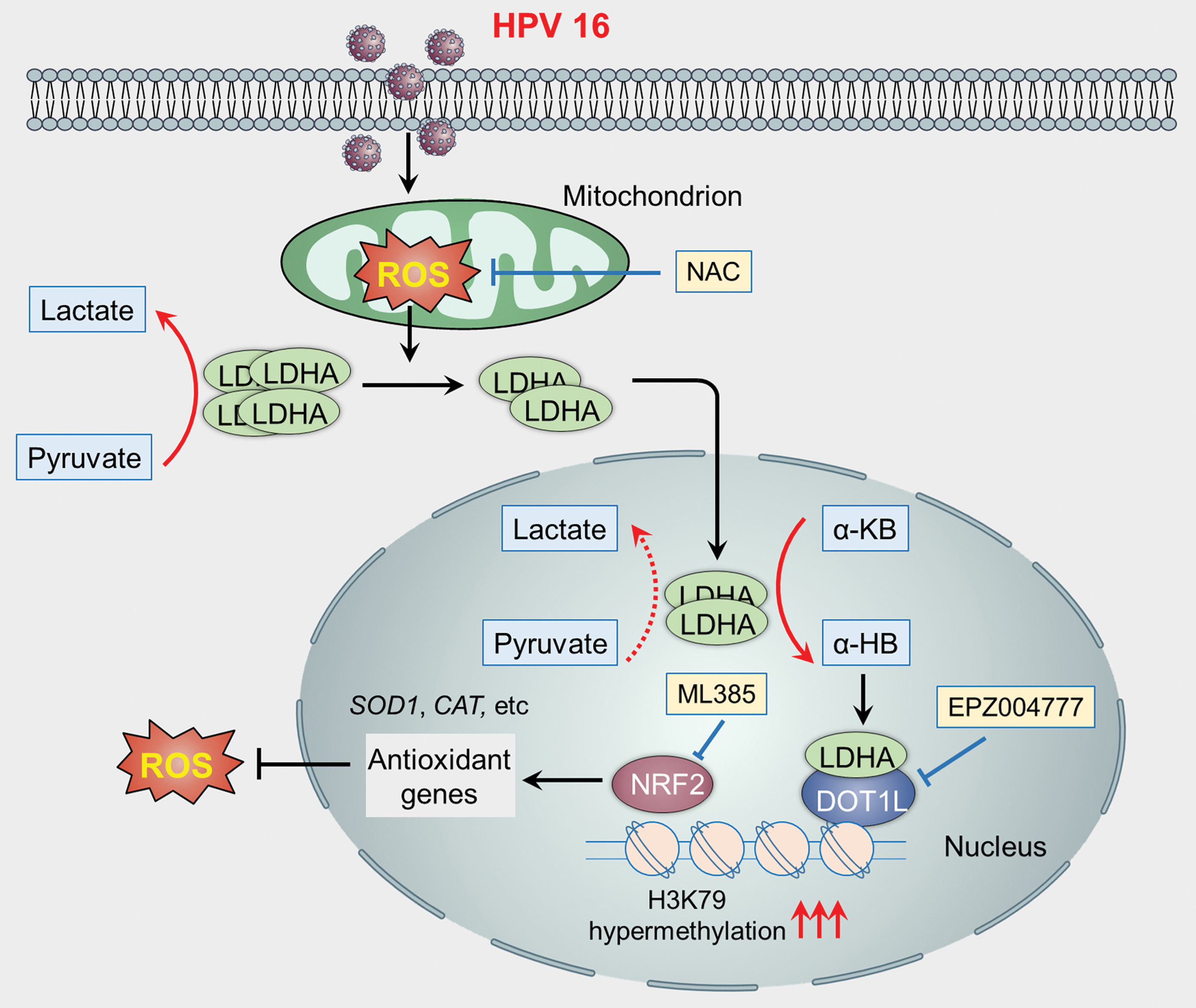
LDHA is an important glycolytic enzyme catalyzing the conversion of pyruvate to lactate. Numerous studies have reported that LDHA is upregulated in several cancer types, indicating an important role of LDHA in tumorigenesis and cancer progression (74). Recently, we found that LDHA can translocate into the nucleus and gain a noncanonical enzyme activity in response to human papilloma virus (HPV)-mediated oxidative stress (102). Mechanistically, the accumulation of ROS disrupts the tetramer of LDHA, leading to its translocation to the nucleus. Nuclear LDHA gains noncanonical activity to generate α-hydroxybutyrate (α-HB). Subsequently, accumulated α-HB induces the hypermethylation of H3K79 through enhancing the interaction between LDHA and disruptor of telomeric silencing 1-like. Next, the hypermethylation of H3K79 promotes the transcription of antioxidant genes to counteract HPV-induced ROS accumulation, leading to the survival and proliferation of cervical cancer cells (Fig. 2). This finding suggests that manipulating the intracellular redox status by antioxidants may disturb the nuclear translocation of LDHA and inhibit the proliferation of cervical cancer cells.
Pyruvate Kinase M2
PKM2 is a glycolytic enzyme that converts phosphoenolpyruvate to pyruvate along with ATP production (1). It is worth noting that PKM2 is directly involved in the metabolic rewiring. Regulation of PKM2 alone is capable of diverting metabolic flux from OXPHOS to aerobic glycolysis (27). Therefore, increased expression of PKM2 is essential for maintaining the Warburg effect and provides a selective growth advantage for cancer cells (64). Recently, nonenzymatic functions of PKM2 have been investigated, with an emphasis on its prosurvival and epigenetic regulating functions. Under oxidative stress, PKM2 can be oxidized at Cys358, leading to enhanced PPP and NADPH supply, allowing cancer cells to survive acute oxidative stress (2). Moreover, the oxidation of tetrameric PKM2 at its Cys423 promotes its binding with p53, leading to the repression of transcriptional activity of p53 and inhibition of apoptosis (143). Besides, it has been reported that PKM2 can translocate to mitochondrion under oxidative stress to phosphorylate Bcl2 through directly interacting with Bcl2. The phosphorylation of Bcl2 inhibits its binding with Cul3-based E3 ligase to prevent Bcl2 degradation, leading to apoptosis resistance of glioblastoma cells (Fig. 3) (94). In addition, PKM2 can translocate into the nucleus and bind to histone H3 upon EGFR activation, leading to H3 phosphorylation at Thr11. This phosphorylation is required for the dissociation of HDAC3 from the promoter of CCND1 and MYC, resulting in H3K9 acetylation and subsequent enhanced transcription of Cyclin D1 and c-Myc. Ultimately, EGFR activation-mediated PKM2 nuclear translocation facilitates the cell proliferation and malignant transformation of cancer cells (183). As EGFR activation is reported to increase cellular ROS levels, whether EGFR-mediated ROS accumulation is involved in the regulation of PKM2 nuclear translocation needs further investigation.

Hexokinase
Hexokinases (HKs) catalyze the first step reaction of glycolysis by which glucose is phosphorylated to generate the membrane-impermeable G-6-P (60). The HK isoform, HK I, inhibits the production of ROS via binding with the outer membrane of mitochondrion, thereby protecting cells from oxidative stress-induced cell death (170). HK II, another HK isoform, protects cells from redundant oxidative stress-induced apoptosis via binding with voltage-dependent anion channel (VDAC). The interaction of VDAC with HK II interferes with the translocation of Bax to mitochondrion, and thus inhibits cytochrome c-induced apoptosis (133). Moreover, HK II is important for maintaining the cellular NADP+/NADPH ratio, which is important for the regeneration of GSH (44). This antioxidant capacity of HK II is independent of its glycolytic enzyme function. HK III has also been reported to decrease cellular ROS for cell survival, which may be due to the preservation of mitochondrial membrane potential (175). However, differing from HK I and HK II, HK III cannot bind to mitochondrion (175). Intriguingly, it has been reported that several cysteine residues of HK I are susceptible to redox agents, such as 2-bromoacetamido-4-nitrophenol (69). This oxidative modification can affect the activity of HK I by causing conformational changes. Whether the oxidative modification of HK influences its moonlighting function remains largely unknown, which requires further investigation.
Glyceraldehyde-3-Phosphate Dehydrogenase
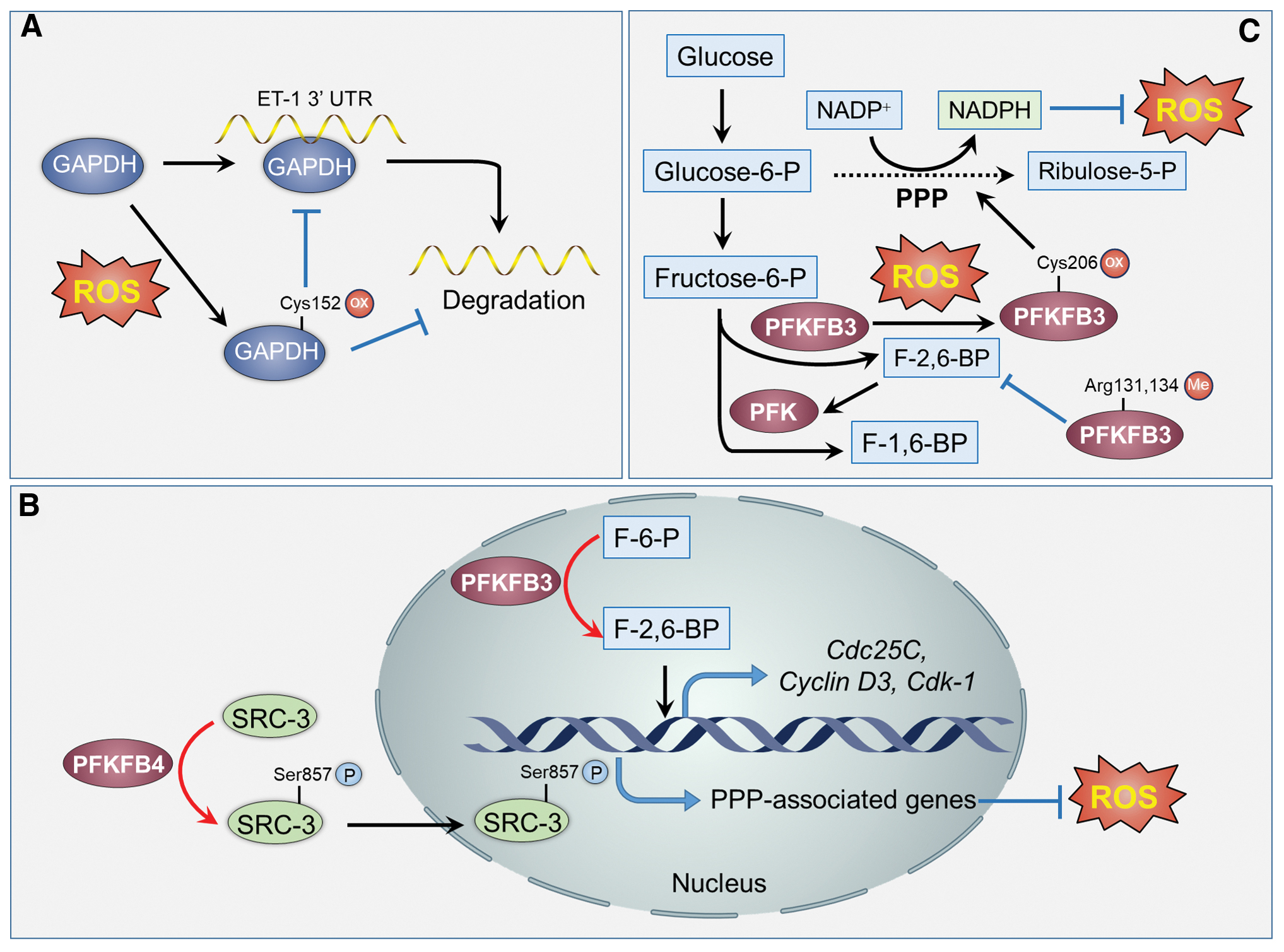
GAPDH catalyzes the conversion of glyceraldehyde-3-phosphate (G-3-P) to 1,3-BPG, which is accompanied with NADH generation. Besides its housekeeping function during glycolysis, GAPDH has several noncanonical functions, such as regulation of apoptosis, gene transcription, and DNA repair (17, 28, 56). Under oxidative stress, rat GAPDH can be oxidized at its Cys150 residue (corresponding to Cys152 for human GAPDH) and subsequently binds to Siah1. This interaction promotes the stabilization of Siah1 and subsequent nuclear translocation of GAPDH/Siah1 complex. Stabilized Siah1 in the nucleus can induce apoptosis by augmenting the degradation of nuclear proteins (56). Moreover, nuclear GAPDH could promote autophagy and glycolysis to protect cancer cells from caspase-independent cell death (17, 28). In addition, nuclear GAPDH can be acetylated at its Lys160 by the acetyltransferase CBP. GAPDH acetylation in turn stimulates the proacetylation activity of p300/CBP and increases the transcription of p300/CBP downstream targets (such as p53), leading to cell death (152). It has been reported that the active site Cys152 of GAPDH was also involved in the formation of GAPDH aggregate, which is responsible for the cell death induced by high concentrations of dopamine in brain (125). Alternatively, GAPDH can undergo S-glutathionylation at its catalytically active Cys152, leading to the inhibition of the interaction of GAPDH with the mRNA of endothelin-1 (ET-1) (Fig. 4A). The interaction of GAPDH with ET-1 mRNA contributes to the destabilization of ET-1 mRNA, resulting in decreased expression of ET-1 (141). Apart from the proapoptosis function, nuclear GAPDH can bind directly with the telomeric DNA to protect it from chemotherapy-induced degradation in human lung cancer cells. As the cysteine residue is sensitive to ROS, oxidative stress may be involved in regulating the binding of GAPDH to the telomere DNA, which awaits further characterization.
6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase 3
PFKFB3 is widely overexpressed in several cancer types and emerging as an important therapeutic target for cancer treatment (158). PFKFB3 has been reported to traffic to the nucleus via an MRN-ATM-γh2AX-MDCI-dependent manner. PFKFB3 mainly locates at the nuclear foci where ionizing radiation-induced DNA double-strand breaks occur. The relocalization of PFKFB3 is required for the recruitment of homologous recombination (HR) proteins to facilitate HR repair and cell survival (55). The hem oxygenase-1 (HO-1)/carbon monoxide (CO) pathway protects cancer cells from oxidative stress-induced cell death. It has been reported that PFKFB3 could be methylated at Arg131 and 134 by PRMT1 in response to HO-1 induction or CO treatment (Fig. 4B). The methylation of PFKFB3 in cancer cells inhibits the production of F-2,6-BP, leading to the shift of glycolysis toward PPP to generate NADPH for ROS scavenging (Fig. 4B) (182). Moreover, PFKFB3 can be S-glutathionylated at Cys206 in response to high levels of ROS or GSSG. The oxidized modification of PFKFB3 is coupled with decreased enzymatic activity, decreased glycolytic flux, and increased metabolic flux into PPP (Fig. 4C). Enhanced PPP flux could in turn induce the synthesis of GSH for ROS detoxification in the cancer cells. These observations suggest a new role of PFKFB3 as a switch that controls the cellular redox homeostasis by rewiring metabolism under oxidative stress (153).
Targeting Redox–Metabolism Abnormalities for Cancer Treatment
Given the moonlighting functions of metabolic enzymes have multifaceted roles in cancer cells and redox regulation is tightly associated with the moonlighting functions of metabolic enzymes, targeting redox or metabolism abnormalities may exhibit favorable therapeutic effects in cancer therapy. Preclinical and clinical studies of several compounds have presented encouraging outcomes (Table 3).
Promising Agents Targeting Redox–Metabolism Abnormalities for Cancer Treatment
γ-GCS, glutamylcysteine synthetase; 2-DG, 2-deoxyglucose; ACPEs, acetyl-CoA-producing enzymes; AIF, apoptosis-inducing factor; BPTES, bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide; DHFR, dihydrofolate reductase; GLS, glutaminase; GLUTs, glucose transporters; GSH, glutathione; H2O2, hydrogen peroxide; HR, homologous recombination; IR, ionizing radiation; mETC, mitochondrial electron transport chain; PDK, pyruvate dehydrogenase kinase; PX-12, 1-methylpropyl 2-imidazolyl disulfide; SAM, S-adenosylmethionine; SOD, superoxide dismutase; TRX, thioredoxin; TXNRD, thioredoxin reductase.
Modulating ROS for cancer treatment
Cancer cells exhibit persistently high levels of ROS and high dependence on antioxidant system for their survival, rendering them more susceptible to pharmacological ROS insult (Table 3). As low to moderate levels of ROS act as second messengers to support cancer cell survival, it is conceivable that the use of antioxidants may be a promising therapeutic approach to prevent tumorigenesis at the very early stage (52). For example, vitamin C has recently emerged as a flourishing anticancer agent, which delays cancer development at physiologic levels while exhibiting anticancer effects at pharmacological concentrations (157). Also, a nutritional intervention trial demonstrated that a combination of β-carotene, vitamin E, and selenium may decrease stomach cancer risk, and the beneficial effects still remain 10 years after the cessation of supplementation (10, 140). Although positive outcomes of several antioxidants have been obtained in preclinical and clinical studies, few of them are widely used in clinical practice. To address challenges lying ahead, more targeted drug delivery system and highly sensitive measurement of ROS levels in clinic may need to be further investigated.
Antineoplastic drugs such as cisplatin, doxorubicin, 5-fluorouracil (5-FU), paclitaxel, and tamoxifen have long been reported to induce ROS accumulation (19). These compounds exert their ROS-producing actions largely through interfering with the electron transport chain (ETC) (80, 118) or inducing ER stress (50, 78). In addition, agents impairing the ETC such as arsenic trioxide (127) may boost the overproduction of superoxide to kill cancer cells.
Apart from compounds elevating de novo production of ROS levels, targeting antioxidant capacity driving ROS detoxification can be an alternative strategy. The three major cellular antioxidant pathways include GSH, TRX, and CAT (52). The xCT inhibitor sulfasalazine limits the uptake of the substrate of cystine (a substrate of GSH synthesis), leading to reduced GSH level (103). Targeting GSH synthesis through glutamylcysteine synthetase inhibitor buthionine sulfoximine has been proven to be clinically beneficial (53). Further, benzyl isothiocyanate, phenethyl isothiocyanate, and sulforaphane could deplete cellular GSH through electrophile–nucleophile interactions (164, 179, 194), while the GSSG mimetic NOV-002 alters the intracellular GSSG/GSH ratio (163). As for the TRX system, TRX1 inhibitors such as 1-methylpropyl 2-imidazolyl disulfide (PX-12) are under investigation for cancer treatment (171). Notably, PX-12 are now in phase 2 clinical trial. Remarkably, dual inhibition of GSH and TRX systems can achieve synergistic antitumor effects (8) and reverse chemoresistance (147) in a wide spectrum of cancers.
Although the ROS-producing agents have shown promising therapeutic effects in several studies, cancer cells in advanced stages may adapt well to this extent of oxidative stress, thereby conferring drug resistance. Disequilibrating redox balance by concurrent administration of ROS-generating compounds and agents preventing antioxidant responses may have additive or synergistic cytotoxic effects on cancer cells. For example, treatment of breast cancer patients has been challenging due, in part, to the plasticity of breast cancer stem cells (BCSCs). Increasing ROS levels by 2-deoxyglucose (2-DG), H2O2, or other agents could accelerate the transition of quiescent mesenchymal-like (M) BCSCs to a proliferative epithelial-like (E) state, which are equipped with robust intracellular antioxidant systems. Taking advantage of their liability on various antioxidant pathways, coinhibition of NRF2, TRX, and GSH pathways could eliminate both types of BCSCs, leading to the suppression of tumor growth (106). A combination of the ROS-generating agents (arsenic trioxide and vitamin C, both of which are now in clinical trial) could provide a favorable clinical effect on refractory or relapsed multiple myeloma through depleting intracellular GSH (5). Furthermore, arsenic trioxide in combination with the SOD inhibitor 2-methoxyestradiol (2-ME) significantly enhances the antitumor activity in chronic lymphocytic leukemia (197).
Despite the promising perspective of exploiting unique ROS property of cancer cells for therapeutic benefits, there is still a great challenge lying ahead for the clinical use: when to use antioxidants for ROS scavenging and when to increase ROS levels to evoke cancer cell death, or what if the ROS levels are not adequately elevated to cause damage but further promote cancer cell growth (149)? Considering the double-edged functions of ROS in cancer cells as well as various redox statuses in different types of cancers, the ROS-generating agents or antioxidants may need to be carefully applied. In addition, studies conducted to determine the detrimental threshold in different cancer cells will probably enhance the therapeutic efficacy. Together, it may be extremely important to approach different ROS-modulating therapies in specific scenarios.
Targeting the metabolic susceptibility for cancer treatment
Since the folate analog aminopterin achieved clinical success in treating children with AML in 1947 (41), antimetabolites, which resemble nucleotide metabolites to target metabolism (in particular nucleotide biosynthesis), have been developed as a class of chemotherapeutic drugs in cancer cells. Metabolic hallmarks of cancer driven by oncogenes appear to be pleiotropic, including elevated glucose consumption, highly active glycolysis, rapacious consumption of glutamine, as well as other emerging metabolic alterations, thus paving the way for therapeutic strategies targeting these dependencies (Table 3).
Targeting the elevated glucose consumption in cancer cells, such as inhibiting glucose transporters (GLUTs), may significantly benefit clinical outcomes. Several compounds targeting GLUTs have been under investigation, including WZB117 (101), STF-31 (20), and silybin (silybin is now in phase 2 clinical trial) (188). GAPDH, HKs, and pyruvate dehydrogenase kinases (PDKs) are thought to be currently the most favorable therapeutic targets for cancer treatment among glycolytic enzymes. The fungal metabolite koningic acid is reported to specifically inhibit GAPDH (39) and selectively kill highly glycolytic cancer cells (86). Other GAPDH inhibitors have been documented to exhibit favorable antitumor activity in preclinical studies, including 3-bromopyruvate (3-BrPA) (47), nitroxyl (HNO) (131), and iodoacetate (144).
Although the primary intracellular target of 3-BrPA was GAPDH (46), it was originally reported that 3-BrPA was an inhibitor of HK (85). 3-BrPA caused the dissociation of HK II from mitochondrion, resulting in the release of apoptosis-inducing factor into cytosol and subsequent induction of cell death (25). The first anticancer agent demonstrated to directly bind with HK is methyl jasmonate, which detaches HK from mitochondrion and VDAC (51). Furthermore, the commonly used glycolysis blocker 2-DG can competitively inhibit HK through inducing the accumulation of 2-deoxyglucose-6-phosphate (DG6P), which cannot be further metabolized, and now 2-DG is in phase 2 clinical trial (172).
Apart from GAPDH and HKs, PDKs are promising therapeutic targets in cancer treatment. The well-characterized PDK1 inhibitor dichloroacetate could attenuate the proliferation of A549 lung cancer cells (12), and sensitize gastric cancer cells with high levels of PDK1 to 5-FU treatment (68). In addition, DAC could enhance the anticancer activity of sorafenib through activating OXPHOS in hepatocellular carcinoma cells (156). Another metabolic hallmark of cancer cells is that they consume more glutamine than other amino acids (33), making it a targetable metabolic demand. The selective ASCT2 inhibitor, V-9302, is the first identified pharmacological agent to block glutamine transport, which increases oxidative stress and inhibits cancer cell growth (151). GLS-specific inhibitors bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl) ethyl sulfide (BPTES) (176) and CB-839 (54) also show promising anticancer efficacy in a wide range of cancers harboring elevated GLS activity, CB-839 is now in phase 2 clinical trial.
Strategies targeting the moonlight functions of metabolic enzymes
As described above, the moonlight functions of metabolic enzymes are exploited by cancer cells to regulate many pivotal cellular events, thus these multifunction properties may also be explored therapeutically (Table 3). The mechanisms underlying proteins involved in moonlighting metabolism can be classified into two aspects: the moonlighting of metabolites and the noncanonical functions of metabolic enzymes.
Metabolism-derived cofactors play critical roles in regulating epigenetics in cancer, building the foundation for the metabolism–epigenome axis. Therefore, altering the levels of some metabolites may be clinically effective. Directly targeting the metabolic enzymes may be a potential strategy to regulate the metabolite levels. For example, all four acetyl-CoA-producing enzymes (ACPEs, including ACSS2, ACLY, carnitine O-acetyltransferase, and PDC) can translocate to the nucleus to produce acetyl-CoA and regulate gene transcription through histone acetylation (14). A high-throughput screening study identified the quinoxaline compound 1-(2,3-di(thiophen-2-yl)quinoxalin-6-yl)-3-(2-methoxyethyl)urea as a potent specific inhibitor of ACSS2, qualifying this enzyme as a druggable target for a wide range of tumors (29). Some ACLY inhibitors are also under preclinical or clinical development, including the weight loss agent hydroxycitrate (110) and the chemical inhibitor SB-204990 (57). In addition to the ACPEs, dihydrofolate reductase (DHFR) catalyzes the reduction of dihydrofolic acid to tetrahydrofolic acid (THF), thereby maintaining cellular THF pool and ensuring the supply of SAM, a substrate for DNMTs (4). Antifolates (such as methotrexate and pemetrexed) and other chemotherapeutics (taxanes, hydroxyurea) can inhibit DHFR and have been utilized as radiosensitizers in head and neck cancer, probably due to their effect on epigenetics (23). However, such nonspecific inhibition of metabolic enzymes may cause global changes in the metabolite levels or even uncontrolled cell signaling alterations due to unknown moonlighting functions of these enzymes.
Deleting or replenishing epigenetic-associated cofactors or their mimics including SAM, α-KG, FAD, acetyl-CoA, and NAD+ may be a more reliable alternative for cancer therapy. For example, the major methyl donor SAM was found to be able to inhibit cancer cell growth through methylating the promoter of the oncogenes such as c-Myc and H-Ras. SAH hydrolase (SAHH) catalyzes the hydrolysis of SAH, thus promoting the recycling of SAM. Paradoxically, DZNep (3-deazaneplanocin A), which inhibits the activity of SAHH and disrupts the recycling of SAM, has been found to influence the methylation of DNA and histones, and induce apoptosis in AML cells and breast cancer cells (45, 161). Another study highlighted a novel strategy to exploit the oncometabolite for glioblastoma treatment (108). IDH mutations occur frequently (>70%) in the early stage of lower grade malignant gliomas. The accumulated oncometabolite 2-HG competes with α-KG to inhibit several dioxygenases, leading to epigenetic alteration. IDH mutations in glioblastoma surprisingly confer superior therapeutic responses to alkylating agents due to the transcriptional repression of DNA repair protein MGMT, which mediates drug resistance. To exploit the mechanistic aspects of the oncometabolite 2-HG, researchers synthesized an α-KG mimic DMG to disturb epigenomic dioxygenase reactions and reverse alkylating agent resistance. They found that this compound was cytotoxic for glioblastoma cells, and could work synergistically with the first-line drug temozolomide (108). Thus, treatment with metabolite analogs may alter the composition of cellular metabolism and epigenome, paving the way for cancer therapies based on metabolism–epigenome axis. Notably, the distribution of epigenetic-associated metabolites in different subcellular compartments could lead to specific alteration of chromatin modifications (115).
Since the chemotherapeutic interventions mentioned above may lead to global cellular changes and potential adverse effects, there is an urgent demand for more specific and targeted strategies modulating the moonlight functions of metabolic enzymes. The development of novel specific compounds may alter the subcellular translocation, PTMs, or interaction of metabolic enzymes, thus modulating the moonlight functions. For example, the natural product saframycin A and several other members of the saframycin classes exhibit powerful antiproliferative activity in leukemia cells. Mechanistically, saframycins induce the translocation of GAPDH to nucleus where it forms a ternary complex with saframycin–DNA adducts to trigger cytotoxic effects (178). It was also found that GAPDH could induce breast cancer cell senescence through interacting with telomerase RNA component, inhibiting telomerase activity and shortening telomere. The Lys259 residue within its catalytic domain is imperative for the senescence-inducing function of GAPDH. Therefore, it is convincing that exploiting compounds targeting Lys259 of GAPDH may be a novel cancer therapeutic strategy (126). In addition, a potent PFKFB3 inhibitor, KAN0438757, has been developed to block PFKFB3 relocation to ionizing radiation-induced nuclear foci, leading to the impairment of PFKFB3-mediated HR repair. Pharmacological inhibition of PFKFB3 by KAN0438757 enhances the sensitivity of cancer cells to radiotherapy, indicating the potential of this selectively PFKFB3 inhibitor in cancer therapy (55).
In addition, the redox regulation of PKM2 is pleiotropic, involving its interaction with other proteins, subcellular translocation, and oligomerization. For example, anthracyclines such as doxorubicin can cause chemotherapy-induced cardiotoxicity due to nonspecific activation of p53 (a proapoptotic transcription factor) in the heart. Tetrameric PKM2 directly binds with p53 and differentially regulates the transcriptional activity of p53 in a redox state-dependent manner. Treatment with TEPP-46, a compound stabilizing tetrameric PKM2, can suppress p53-induced apoptosis in high-oxidative cardiomyocytes, while enhance the activity of p53 in relative low-oxidative tumor cells, thus enhancing the anticancer effect of anthracycline (143). Moreover, a recent study demonstrated that PKM2 389–405 peptide could effectively inhibit gliomagenesis and brain tumor development through disrupting oxidative stress-induced PKM2-Bcl2 interaction and promoting Bcl2 degradation, implying the promising perspective of using competitive peptide as a novel antitumor strategy (94).
Concluding Remarks
Cancer cells exploit metabolic enzymes and metabolites for sustained proliferation, leading to the general agreement that targeting metabolism abnormalities may benefit cancer treatment. However, given the moonlighting functions of metabolic enzymes beyond their metabolic roles, drugs targeting metabolism abnormalities may have some unexpected effects, which might affect therapeutic outcomes. Therefore, in-depth understanding the moonlighting functions of metabolic enzymes or metabolites is of great importance for improving the anticancer effect of metabolism-targeting strategies. Since cellular redox status and the moonlighting roles of metabolic enzymes are closely relevant, metabolic studies should be considered in parallel to redox status under certain situations. The fact that some moonlighting metabolic enzymes are regulated by cellular redox status highlights the promise of exploiting ROS modulators for cancer therapy. Specifically, ROS in different subcellular compartments and diverse species of ROS have distinct influence on the moonlighting function of metabolic enzymes. Thus, the redox–metabolism–epigenetics axis, redox–metabolism–apoptosis axis, redox–metabolism–transcription axis, and redox–metabolism–PTM axis may alter the traditional way we identify metabolic biomarkers in cancers. Moreover, understanding the connection of oxidative modifications and moonlighting functions of metabolic enzymes may improve the specificity of targeted strategy and decrease side effects.
Footnotes
Acknowledgments
The authors thank Prof. Lih-Wen Deng in National University of Singapore for her critical comments and suggestions.
Funding Information
This work was supported by grants from the Chinese NSFC (81821002, 81790251, 81430071, 81672381, 81872277), Guangdong Major Project of Basic and Applied Basic Research (2019B030302012), and the National 973 Basic Research Program of China (2013CB911300).