
Abstract
Significance:
Excessive and prolonged proinflammatory responses are associated with oxidative stress, which is commonly observed during chronic tuberculosis (TB). Such condition favors tissue destruction and consequently bacterial spread. A tissue remodeling program is also triggered in chronically inflamed sites, facilitating a wide spectrum of clinical manifestations.
Recent Advances:
Since persistent and exacerbated oxidative stress responses have been associated with severe pathology, a number of studies have suggested that the inhibition of this augmented stress response by improving host antioxidant status may represent a reasonable strategy to ameliorate tissue damage in TB.
Critical Issues:
This review summarizes the interplay between oxidative stress, systemic inflammation and tissue remodeling, and its consequences in promoting TB disease. We emphasize the most important mechanisms associated with stress responses that contribute to the progression of TB. We also point out important host immune components that may influence the exacerbation of cellular stress and the subsequent tissue injury.
Future Directions:
Further research should reveal valuable targets for host-directed therapy of TB, preventing development of severe immunopathology and disease progression. Antioxid. Redox Signal. 34, 471–485.
Introduction
M
Over time, Mtb infection triggers formation of immune cell structures, granulomas, in which different myeloid cells and lymphocytes are spatially organized to restrict further mycobacterial growth and dissemination to other tissues (106). A consequence of the interplay between the host and invading Mtb is a potent antimicrobial response that generates release of free radicals as well as production of cytokines. This host response helps the eradication of Mtb, although it may lead to bystander injury by promoting an unfettered necroimmunopathology (100). The latter scenario can be aggravated depending on pathogen virulence, which is most commonly associated with failure of the host immune response to prevent ongoing Mtb replication (Fig. 1).
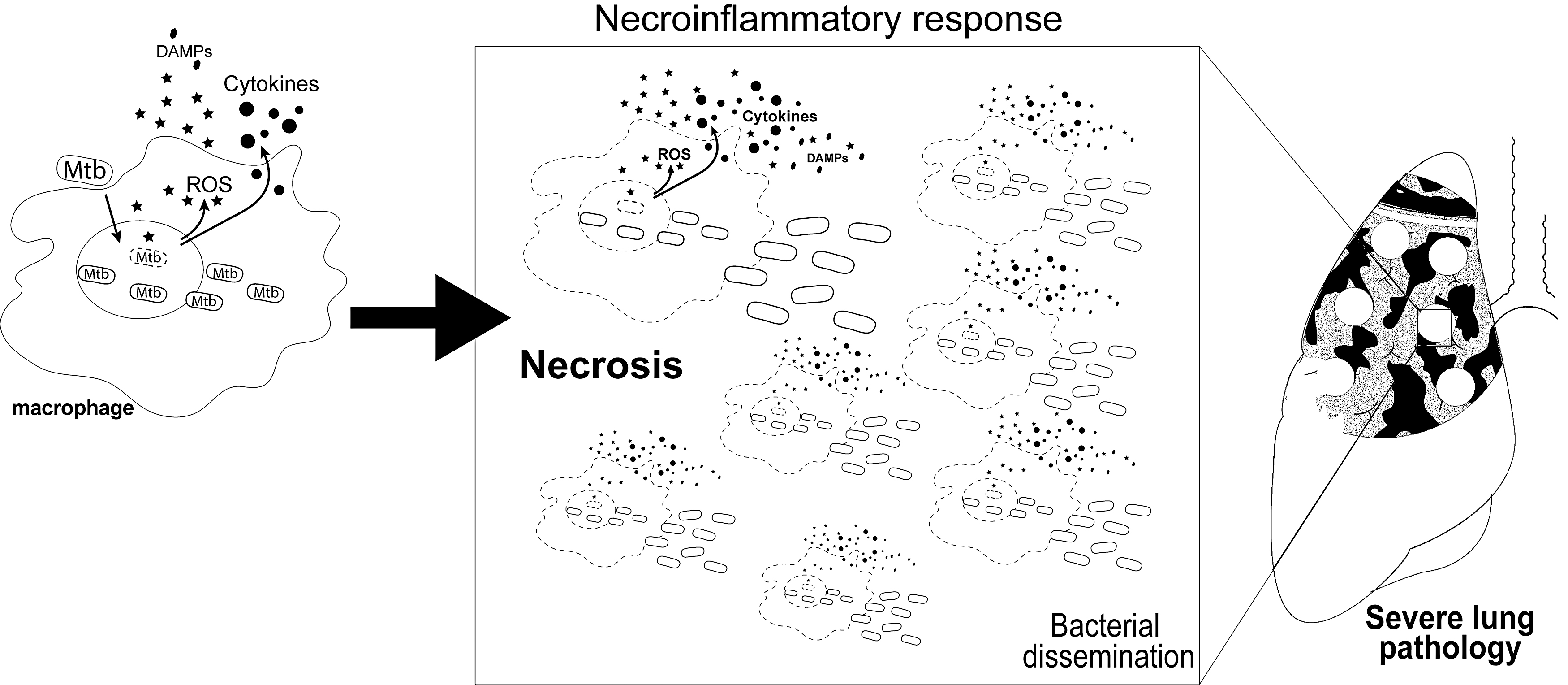
Furthermore, chronic inflammation leads to an imbalance between antioxidants as well as molecules with antioxidant properties such as glutathione (GSH), ferritin, coenzyme Q10, glutathione peroxidases (GPx) and free radical production, including superoxide (O2•−), hydroxyl radical (•OH), lipid hydroperoxide, and alkoxyl radicals, with subsequent tissue destruction and Mtb proliferation (118). This enables airway dissemination of Mtb through the lung and facilitates transmission to other susceptible hosts via cough. Persistent inflammation in response to different Mtb virulence factors occurs in parallel with resultant tissue remodeling, thereby leading to a wide spectrum of clinical manifestations.
Understanding how Mtb modulates the host immune response to evade host antimycobacterial effector functions is of critical importance to facilitate development of new therapies to treat TB. At present, antimycobacterial drugs are the mainstay of therapy focused on direct mycobactericidal activity or inhibition of bacterial replication. With the worldwide emergence of drug-resistant mycobacterial strains, alternative approaches are urgently needed. Innovative therapies modulating host immune responses may offer a promising approach to mitigate the necroinflammatory response, oxidative stress, and tissue remodeling. Novel therapies targeting unfettered immunopathology may avert the development of clinically severe TB and subsequent airborne transmission.
In this review, we discuss how the interplay between systemic inflammation, oxidative stress, and tissue remodeling influences Mtb infection and development of active TB. We also examine the current literature to highlight biochemical pathways and host immune mechanisms that may be useful as new targets for host-directed therapy of TB.
Mtb Infection Drives Systemic Inflammation
Perturbations in host tissue homeostasis triggered by infectious microorganisms result in activation of immune surveillance mechanisms that promote inflammation. In TB, the proinflammatory response is thought to play a crucial role in controlling Mtb infection. However, this antimicrobial host response may be detrimental when exacerbated, leading to unfettered inflammation and subsequent severe tissue injury (118). Systemic inflammation occurs in both pulmonary and extrapulmonary TB diseases, and is marked by increased concentrations of inflammatory markers in peripheral blood, such as acute phase proteins, lipid mediators (e.g., prostaglandin E2 [PGE2]), and a number of proinflammatory cytokines as well as chemokines (132).
This significant production of cytokines/chemokines induces migration of a large number of activated leukocytes, such as neutrophils, monocytes, dendritic cells (DCs), and effector lymphocytes into the affected tissue, thereby favoring establishment of immunopathology (106). Most of these cells can also be detected in the blood vasculature, as evidenced by a strong inflammatory signature in the case of extrapulmonary TB.
When Mtb is inhaled, it lodges in the lower respiratory tract where it is phagocytosed by AMs and DCs. To evade the antimicrobial response from those mononuclear cells, Mtb blocks phagosome fusion with lysosomes by inducing host expression of coronin-1/TACO, and thus favoring proliferation inside phagosome and continuous secretion of mycobacterial products, such as 6 kDa early secretory antigenic target (ESAT-6) (26, 62, 122). Cytosolic ESAT-6 triggers host sensors recognition of an infection eliciting proinflammatory response marked by the inflammasome activation and mature interleukin (IL)-1β generation (6). Recently, Mtb-infected AMs were shown to migrate into the lung interstitium and facilitate Mtb dissemination to other immune cells in the tissue (33).
AM translocation from alveoli into lung parenchyma depends on IL-1 signaling, since IL1R−/− and MyD88−/− mice display substantial accumulation of AMs in the alveolar space. Furthermore, Mtb ESX-1 secretion system, which is required for ESAT-6 release, has been shown to play a role in AM migration into lung interstitium (33).
Interestingly, other studies have demonstrated that Mtb escapes from the phagosome into the cytosol through a mechanism dependent on ESX-1/ESAT-6, resulting in the subsequent induction of macrophage necrosis (59, 122). The exact mechanism by which Mtb escapes from the phagosome into the cytosol is not yet well understood. One possible mechanism for Mtb escape is through a pore formation on the phagosomal membrane mediated by ESAT-6, since this Mtb protein has been described to exhibit pore-forming capability (74).
The presence of Mtb as well as its ESAT-6 antigen triggers activation of several molecular pathways, including the NLRP3-inflammasome assembly required for cleavage of pro-IL-1β (6), an essential cytokine for host defense against Mtb (85, 86). These findings suggest a possible role of mycobacterial escape into cytosol and IL-1β production by the NLRP3-inflammasome, promoting AM translocation into lung parenchyma and further mycobacterial dissemination.
A wide spectrum of individual lesions is formed in the lungs that vary from a solid structure to necrotic, caseous tubercles, and ultimately cavitary granulomas. Different stages of granuloma progression are concurrently apparent during active TB. Granuloma formation begins with an aggregate and ill-defined mass of immune cells resulting from cell recruitment to the site of infection, becoming more organized over time. The granuloma is composed of a macrophage-enriched center, which later differentiates into specialized cell types including multinucleated giant cells and epithelioid macrophages, surrounded by B and T cells, creating an efficient bacterial containment barrier to prevent dissemination of infection (22, 100, 106).
Tumor necrosis factor alpha (TNF-α) and IL-12, produced early in Mtb infection by antigen-presenting cells, play a critical role in the establishment of a Mtb-specific adaptative immune response and granuloma formation. Interferon (IFN)-γ, which is initially produced by natural killer cells during the early stage of infection, is subsequently enhanced secondary to expansion and activation of a Mtb-specific host adaptative immune response. IFN-γ plays an essential role in the host defense against Mtb infection by potentiating macrophage ability to phagocytose and kill the pathogen (94).
Mice deficient in IFN-γ production are extremely susceptible to mycobacterial infection (103). Recently, the production of this protective cytokine has shown to be regulated by adenosine receptors (A1, A2a, A2b, and A3), as a result of extracellular ATP degradation by ectonucleotidases that are enhanced during massive lung destruction caused by hypervirulent mycobacterial strains (5). The optimal generation of IFN-γ will determine the course of TB pathogenesis, in which either low or inappropriately high production of this cytokine is detrimental for the host (13, 14).
Interestingly, a study combining human disease and experimental models with nonhuman primates reported that the heterogeneity of granulomas may impact the extent of immune response and efficiency in controlling Mtb infection (130). A recent study has demonstrated that the immune response against Mtb varies in anatomically distinct compartments within granulomas through sophisticated high-resolution imaging and mass spectrometry imaging.
Proinflammatory enzymes responsible for the generation of lipid-derived inflammatory factors, such as eicosanoids, are highly expressed both in necrotic centers and in cells bordering the caseum. Moreover, high concentrations of eicosanoid precursors were found within the granuloma when compared with normal lung tissue, with marked accumulation of these mediators at the border of the caseum. Enhanced expression of leukotriene A4 hydrolase and lipoxygenases has also been observed at the caseous granuloma when compared with solid granulomas, suggesting increased generation of inflammatory eicosanoids, such as leukotriene B4 and lipoxins (81). In the following section, we discuss the implications of inflammatory eicosanoids and their generation/modulation on TB pathogenesis.
Eicosanoids are lipid mediators derived from the enzymatic or nonenzymatic oxidation of arachidonic acid, including prostaglandins, resolvins, lipoxins, and leukotrienes, which play an important role in regulating the immune response (116).
Two groups of enzymes, cyclooxygenases (COXs) and lipoxygenases, compete with each other for arachidonic acid generating prostaglandins, lipoxins, and leukotrienes. The latter two are bioproducts exclusively resulting from lipoxygenase activity. Mice deficient in 5-lipoxygenase are more resistant to Mtb infection, whereas prostaglandin E synthase-deficient mice display high susceptibility to infection (12, 29, 40). It is possible that lipid mediators may differentially regulate host protection against Mtb by interfering directly or indirectly with the regulation of programmed cell death in infected macrophages, but this issue is still not fully understood (15, 85, 128).
In infected macrophages, lipoxin A4 (LXA4) has a deleterious effect on host Mtb containment, promoting macrophage necrosis and Mtb dissemination into adjacent cells. Conversely, PGE2 appears to prevent cell necrosis and simultaneously stimulate apoptosis in Mtb-infected macrophages, thereby containing mycobacteria and reducing bacillary burden through a process called efferocytosis, in which apoptotic bodies are engulfed and removed from the milieu by neighboring tissue cells. Efferocytosis of Mtb trapped within an apoptotic body delivers it to the lysosomal compartment, where the pathogen will be killed (12, 29, 64, 83, 87, 97, 132, 138).
Mycobacterial virulence factors were shown to significantly influence the generation of different lipid mediators, in which LXA4 is provoked by virulent Mtb strains, whereas PGE2 was revealed to be triggered by avirulent strains (40). Interestingly, Mtb-infected mice genetically lacking IL-1 receptor signaling have been shown to display a profound reduction of PGE2 generation along with increased levels of LXA4 and LTB4 (both products of 5-LO) in the bronchoalveolar lavage fluid (BALF) (85).
In humans, plasma levels of prostaglandins and lipoxins are both higher in patients with active TB than in uninfected individuals (97, 132). Besides the role of PGE2 in inducing apoptosis of Mtb-infected macrophages, this lipid mediator has been reported to trigger several tissue remodeling enzymes such as the extracellular matrix (ECM) metalloproteinase-1 (MMP-1), highlighting the role of PGE2 in tissue repair during TB (49).
Leukocytes generate reactive oxygen species (ROS) such as hydrogen peroxides and nitric oxide to control Mtb growth in the hyperinflammatory environment of the granuloma (81, 118). To prevent collateral cellular damage from excessive ROS, infected and activated cells concurrently induce production of antioxidants and transcription of antioxidant enzymes.
With persistence of infection, when excessive or sustained ROS production overwhelms the available antioxidant defense systems, key molecules are denatured, with subsequent dampening of immune cellular functions, leading to cell damage and death (Fig. 2) (11, 118, 133). This inefficient control of excessive ROS-mediated toxicity occurs due to reduced activity of endogenous antioxidative enzymes and reduced intake or absorption of antioxidants usually obtained from the diet (104). Mtb thrives in this proinflammatory environment and increases its metabolic activity leading to unrestrained proliferation and augmentation of mycobacterial burden.

We have recently shown that increased Mtb replication in macrophages is associated with intracellular iron overload and cell death that facilitates Mtb dissemination (Fig. 3) (4). Moreover, when ferrostatin-1, a drug known to prevent lipid peroxidation (41), is used to treat both macrophages in vitro and Mtb-infected mice in vivo it is possible to detect a drastic suppression in macrophage death, tissue necrosis, and reduced Mtb loads in lungs of infected mice (4). Interestingly, several reports have associated vitamin E deficiency, an antioxidant that dampens lipid ROS (41), with increased host susceptibility and disease severity, supporting the idea that the loss of antioxidant response and excessive ROS generation are both detrimental to the host (2, 38, 70, 75, 96, 131).

Thus, manipulation of oxidative stress may potentially serve as an adjunct therapy for TB, when added to conventional long-term treatment. In support of this approach, a body of clinical studies has shown that patients given vitamin E (a major fat-soluble antioxidant that scavenges peroxyl radicals and dismisses the oxidation of polyunsaturated fatty acids [PUFAs]), selenium [a micronutrient important for the function of some antioxidant enzymes, such as Gpx4 (61)], and/or N-acetylcysteine [a precursor of GSH, an important antioxidant (3)] as adjunct to Mtb antibiotics demonstrated improved treatment outcomes when compared with those receiving placebo, which was coincident with an improved immune response against Mtb (23, 25, 55, 60, 65, 76, 117).
Along with the detrimental effect of excessive ROS production, type I IFNs (IFN-α and IFN-β) have been shown to promote Mtb infection. In addition to well-characterized antiviral effects, type I IFN response generates proinflammatory immune signatures in the context of active TB infection (17). Type I IFN response promotes unrestrained inflammation-driven tissue damage in mice and patients with more severe forms of TB (97, 115, 132). Absence of type I IFN signaling in ifnar-deficient mice results in increased PGE2 and IL-1β production in BALF, suggesting that type I IFNs antagonize the protective effect of the IL-1 pathway during Mtb infection (84).
We have recently demonstrated that the overall disturbances in cytokine plasma levels are heavily influenced by IFN-α, the signal of which has been found elevated in blood from TB patients (97). These findings argue that type I IFNs act to downregulate protective host immune functions against Mtb.
IL-1 has an important role in host resistance against Mtb through an inflammatory cascade that stimulates PGE2 production via a pathway requiring COX-2, as described previously (85). Aberrant levels of this cytokine, however, may be detrimental to host defense control of Mtb infection by increasing tissue damage (90). Specifically, elevated production of IL-1β is associated with more extensive radiographic disease (27, 134) as well as with larger cavitary lesions (7, 32, 120, 129). Furthermore, IL-1 is involved in fibroblast activation (19) and recruitment of neutrophils, which has been in turn shown to augment tissue damage, promoting loss of pulmonary function and host death (16, 17, 51, 72, 89, 123).
As a major modulator of inflammation, IL-10 is a potent cytokine in modulating exaggerated antimycobacterial immune responses. The induction of IL-10 production during infection inhibits macrophage functions and suppresses proinflammatory cytokine production including TNF-α, another cytokine required for optimal granuloma formation (1, 94). Interestingly, TNF-α blockade has been shown to facilitate granuloma disintegration and thus promote Mtb dissemination (28, 44, 56, 121). IL-10 production occurs within the granuloma and may also facilitate mycobacterial persistence by preventing Mtb-phagosome maturation in macrophages (35). In contrast, production of IL-10 by B cells may aid counterbalancing of chronic inflammation in the lungs of those with more advanced stages of TB disease characterized by intense production of ROS (108).
The complex interplay between cytokines, chemokines, and lipid mediators promotes loss of optimal regulation of the inflammatory cascade. This chronic process leads to cell stress, inducing deregulation in the redox reactions (imbalance of oxidant and antioxidant products) affecting tissue remodeling response, thereby culminating in the destruction of pulmonary parenchyma.
Tissue Remodeling in Pulmonary TB
TB initially triggers an intense inflammatory response that in most cases is followed by a chronic process of inflammation mediated by numerous cell types, proinflammatory cytokines, and chemokines. This inflammatory response results in significant tissue remodeling of the ECM with destruction of pulmonary parenchyma leading to bronchiectasis, restrictive and obstructive lung disease (109). How Mtb promotes development of cavitary lesions is not completely understood. Mycobacterial virulence factors are directly responsible for significant tissue remodeling, reinforcing the perception that Mtb needs to promote ECM disruption to disseminate.
Tissue remodeling is a physiological process initiated after cellular damage that aims to restore tissue function. Intracellular components released into the extracellular milieu trigger immune responses promoting the recruitment of cell types such as neutrophils and macrophages into the tissue parenchyma. After the initial stage, a nonspecific type of collagen is deposited in the tissue being repaired, which is replaced later by tissue-specific collagen in a slow and gradual process, involving the organization of collagen fibers. This process is necessary for repairing damaged ECM in response to innumerable infectious and noninfectious pulmonary diseases, including pneumonia, chronic obstructive pulmonary disease, sarcoidosis, and acute respiratory distress syndrome (49).
Through immune evasion mechanisms, Mtb begins its struggle to survive in the midst of the host immune system resulting in dysregulated production of cytokines, chemokines, and lipid mediators. Chronic persistence of Mtb leads to a host response that increases bystander tissue destruction involving MMPs, cathepsins, and kallikreins, which outpaces collagen deposition, TIMPs (metalloproteinase inhibitors), and other protease inhibitors, resulting in tissue remodeling. Elucidation of this process is crucial to understanding TB immunopathology and may highlight novel adjunctive treatment of pulmonary TB (46).
In particular, cathepsins play an important role in bone, cartilage, and collagen processing, with cathepsin K being the most studied. Cathepsin K has the capacity to cleave collagen triple helix, and its activity differs from MMPs. Generally, pathological collagen degradation is associated with high levels of cathepsin K (66, 139). Other cathepsins such as cathepsins B, L, H, and S have been identified as potential targets for tissue remodeling based on in vitro studies (135), although there is no evidence of involvement of these cathepsins in tissue remodeling in animal models of TB.
In the lungs, cathepsin K is highly expressed by pulmonary epithelial cells as well as AMs, playing an important role in remodeling the ECM (20, 21). This finding is supported by studies that found cathepsin B and K upregulation in serum/plasma of pulmonary TB patients (6, 66). Employing an animal model of lung cavitation, Kubler et al. infected rabbits with a virulent Mtb strain and evaluated levels of cathepsin K as well as MMPs. Upregulation of several MMPs including MMP-1, MMP-13, MMP-14 as well as cathepsin K was found in the lungs of Mtb-infected animals (66). These findings suggest that inhibition of MMPs and cathepsin K may serve as a potential target for adjuvant therapies in TB as blockade of these enzymes may reduce pulmonary pathology.
MMPs are a family of proteases (which can be associated with either zinc or calcium) that are able to degrade all components of the ECM. Moreover, only certain MMPs are capable of cleaving lung fibrillar collagens at neutral pH, which facilitates tissue remodeling (45, 47). MMPs can be classified on the basis of their substrate specificity into different subfamilies, namely collagenases (MMP-1, MMP-8, and MMP-13), gelatinases (MMP-2 and MMP-9), stromelysins (MMP-3, MMP-10, and MMP-11), elastases (MMP-7 and MMP-12), and membrane-type MMP (MMP-14, MMP-15, MMP-16, and MMP-17) (111).
In the lungs, MMP-1 (known as interstitial collagenase), MMP-8 (referred to as neutrophil collagenase), MMP-13 (collagenase 3), and MMP-14 play important roles in cleaving the primary architectural collagen in the tissue parenchyma (43). Most MMPs are tightly regulated requiring gene transcription induction before their secretion, except MMP-8 and MMP-9 in neutrophils. The majority of MMPs are not expressed in healthy tissues, with their expression being observed only in inflamed tissues, or in those undergoing repair or remodeling (102). These enzymes are usually secreted by monocytes-derived cells, neutrophils, and stromal cells (98).
Epithelial cells in the lungs are a significant source of MMPs since they express MMP-1, MMP-2, MMP-7, and MMP-9. In addition, highly differentiated macrophages express a broader profile of MMPs than monocytes. Under all circumstances, cytokines, exogenous stimuli, and cell–cell contact are required for MMP expression (24). MMPs have several physiological functions in development, reproduction, maintenance of homeostasis as well as facilitation of cell migration, cleavage of cytokines and activation of defensins.
In TB, exaggerated generation of mainly MMP1 is associated with progression of consolidated regions to cavities with high bacterial loads as demonstrated in a rabbit model of pulmonary TB cavitation (67). In addition, reduced level of a metallopeptidase inhibitor TIMP-1 is found in consolidated areas, leading to a potent activity of metalloproteinases and promoting tissue destruction (48, 67). Several studies have reported significant increase in MMP-1 levels in blood of TB patients (8, 67), although the exact mechanism by which this happens is poorly understood.
Interestingly, MMP-1 has been shown to be regulated by mitogen-activated protein kinase (MAPK) signaling in human macrophages after Mtb infection (8, 49, 95, 107). In addition, a number of studies have shown that increased MMP-1 levels are critical for cavitation in TB pathogenesis (45). It has been shown that mice do not express MMP-1, impeding in vivo study on how this enzyme contributes to TB pathogenesis. Of note, murine granulomas rarely progress to necrosis as observed in humans, suggesting that expression of MMPs, such as MMP-1, might be important for tissue damage induction. This issue was addressed by infecting transgenic mice overexpressing human MMP-1 with virulent Mtb. The upregulation of this enzyme has been shown to be detrimental via significant destruction of alveolar walls and lung parenchyma, through enzymatic collagen degradation (45).
Furthermore, Mtb-infected mice deficient in MMP-9 display reduced macrophage migration into the lung and defective granuloma formation, while being counterintuitively more resistant to infection (126). Elevated migration of neutrophils is also observed during early stages of Mtb infection in mice and humans, with the corresponding high MMP-9 levels after infection in both host species (57). Mtb promotes MMP activity and collagen degradation by eliciting cytokines such as TNF-α, which was shown previously to augment levels of MMP-1 as well as MMP-10 and MMP-3, thus propagating collagenase activity and causing tissue damage. Interestingly, active TB patients display higher levels of both heme oxygenase-1 (HO-1) and MMP-1 in plasma compared with those with latent disease (8).
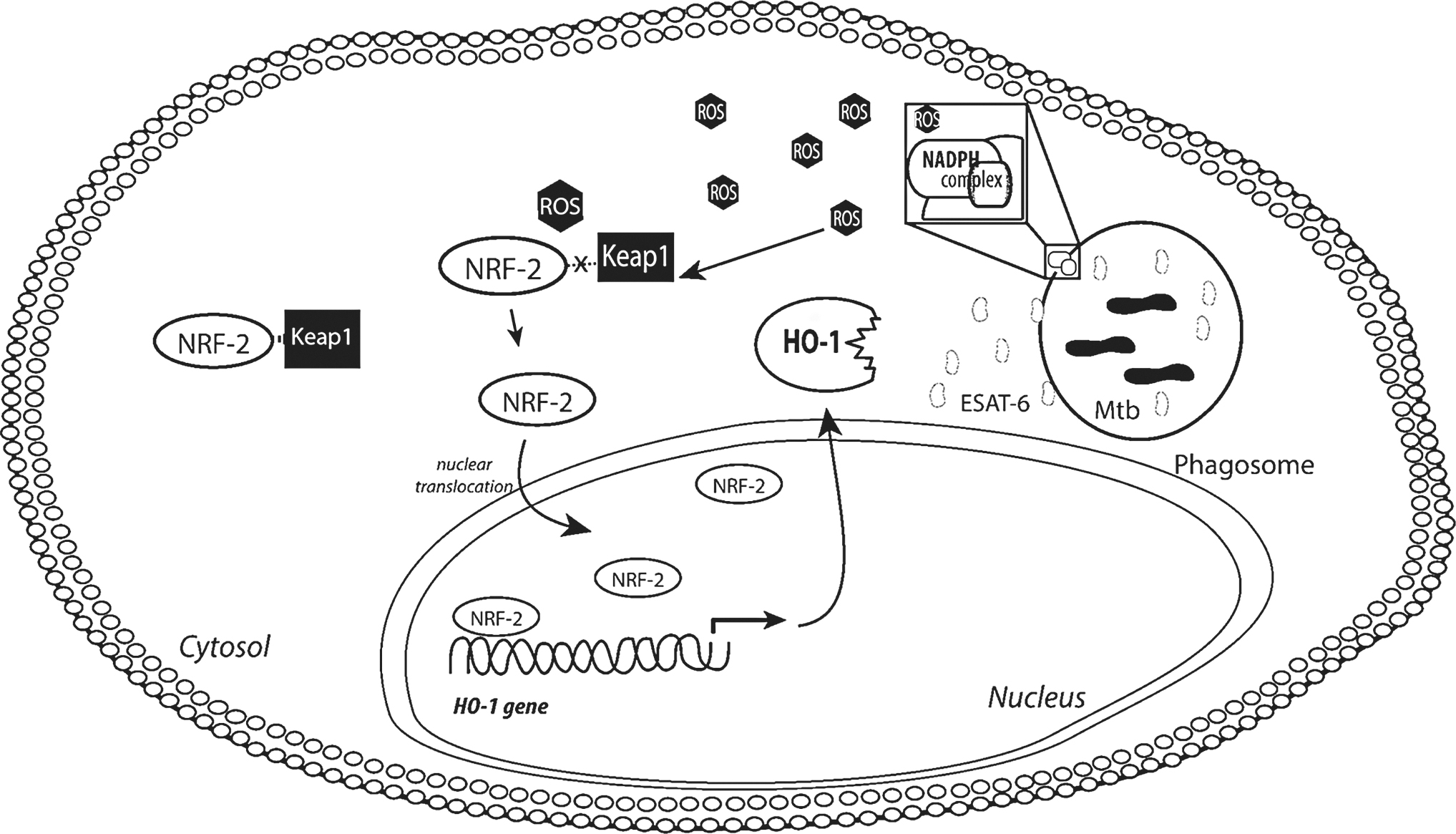
HO-1 cleaves heme, which is generally obtained from heme-containing proteins, and generates three by-products such as biliverdin, carbon monoxide (CO), and free iron. HO-1 is induced in Mtb-infected macrophages by a mechanism associated with ROS production and increased activity of NRF-2 (Fig. 4) (110). Human monocyte-derived macrophages, when exposed to Mtb or intracellular ESAT-6, were shown to upregulate HO-1 and simultaneously downregulate MMP-1. Inhibition of MMP-1 is associated with CO production as a result of HO-1 activity (8). However, free iron, another by-product of HO-1 activity, was shown to trigger high levels of MMP-9 through activation of AP-1/ERK/AKT pathway (63), but did not impact induction of MMP-1 (8, 63). Notably, increased cellular iron was associated with enhanced secretion of both MMP-9 and MMP-1 (79).
With recruitment of inflammatory cells and their subsequent death as a result of cell stress, caseous necrosis appears within days after Mtb infection, creating an optimal environment for bacterial proliferation. Granuloma formation is an effective way to contain Mtb proliferation and with an appropriate host immune response maintain an intact ECM, thereby preventing TB transmission by infected individuals. However, when an imbalance between MMP-1 and TIMPs occurs, the granuloma progresses to instability, facilitating bacterial dissemination through tissue as well as between individuals (98). MMP inhibitors have been shown to enhance both delivery and/or retention of TB drugs in the lungs resulting in improved drug efficacy (137). In contrast, the inhibition of MMP-7 in mice has been associated with more severe pathology (99), suggesting a different role for MMPs in regulating host resistance against Mtb.
Oxidative Stress Mechanisms in Pulmonary TB
Oxidative stress results from the imbalance of total oxidant over total antioxidant status. After Mtb infection, the first immune response is characterized by significant production of oxidants by AMs (18), a critical process for the initial destruction of Mtb. During the initial stages of infection, immune cells, primarily macrophages, produce ROS to promote the death of Mtb. Under physiological conditions, high levels of ROS occur concurrent with increased production of antioxidants to neutralize potentially harmful effects of ROS in tissues (Fig. 5A). The sensitive balance between antioxidant and oxidants is the key of tissue homeostasis and, when disturbed, results in irreversible cell damage with pathological consequences.

While initial infection with Mtb triggers intense production of ROS, in certain situations there is no commensurate and proportional increase in antioxidants, thereby leading to an imbalance between ROS and antioxidants in the tissue (Fig. 5B). Through its continuous interaction with the host immune system, Mtb triggers progressive increases in ROS production by macrophages that culminate in excessive oxidation and lipid peroxidation (Fig. 5C) (3, 4). Human monocyte-derived macrophages infected with Mtb in vitro exhibit increased lipid peroxidation, DNA oxidation, and more frequent cell death resulting from dramatic accumulation of intracellular ROS (3, 4, 110).
Via an unknown mechanism, macrophages lose their primary antioxidant enzymatic functions involved in the regulation of lipid peroxide accumulation, resulting in increased pathogen burden and proliferation. Excessive macrophage stress leads to DNA peroxidation, which, consequently, downregulates cellular metabolism resulting in a cascade of alterations that increases ROS generation along with reduction of antioxidants, culminating in significant tissue damage and pathogen dissemination.
Peroxidation of lipids is a natural process that occurs in steady state in small proportion in the body mainly by the effect of several ROS, including hydroxyl radical, hydrogen peroxide, and others. These ROS attack most often the PUFAs initiating a self-propagating chain reaction. In brief, lipid peroxidation is initiated by the hydrogen abstraction or addition of an oxygen radical causing a rearrangement of the double bonds in polyunsaturated lipids, which eventually culminates in destruction of membrane lipids. Due to the presence of methylene CH2-groups, which contain hydrogen that is particularly reactive to ROS, PUFAs are considered to be more sensitive than saturated lipids to this peroxidation process (34).
Several by-products such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4-HNE) are generated during homolytic decomposition of lipid hydroperoxides in this complex process. The accumulation of these by-products has severe consequences for cell function. Both MDA and 4-HNE have mutagenicity properties due to their high ability to form DNA-adducts (82). As mentioned above, failure of the natural biological membrane repair leads to destruction of these cellular membranes, favoring the accumulation of lipid peroxides (lipid ROS), which are dangerous for cellular and tissue integrity. Interestingly, high levels of lipid peroxidation by-products have been reported in several noninfectious maladies, including Alzheimer's disease, renal failure, hepatotoxicity, fibrosis as well as infectious diseases such as HIV/AIDS, malaria, and TB (3, 39, 91, 114).
HO-1 is an antioxidant enzyme highly expressed in lung tissue that functions as a key stress-response component involved in degradation of heme molecules resulting in generation of three by-products free iron, CO, and biliverdin, as mentioned previously (30, 36, 119, 127). HO-1 displays a dual role in the host stress response as it may work as an anti-inflammatory component that improves cell viability through generation of two important antioxidant molecules, CO and biliverdin, or may conversely augment oxidative stress, induce cell death, and stimulate pathogen growth by releasing free iron from heme (4, 53). HO-1 has emerged as an important mediator in several diseases, such as malaria (88), leishmaniasis (73), TB (8, 30, 36, 110, 119), and sepsis (113), being frequently associated with severity of disease (9).
As mentioned above, Mtb drives production and secretion of HO-1 and oxidant factors, such as MMPs (8). HO-1 and MMP-1 are differentially regulated in infected macrophages as a result of different inflammatory responses leading to distinct clinical presentations, and the balance between these molecules influences the extent of lung lesions and bacterial loads. Mtb infection is associated with substantial increase in total oxidation status in pulmonary TB patients when compared with those with latent infection and uninfected controls (3, 8). HO-1 has also been implicated in the promotion of lipid peroxidation by liberating free iron and thus regulating ferroptotic cell death (68, 80). Cells overexpressing HO-1 undergo ferroptosis, whereas cells deficient in HO-1 display increased resistance to this form of necrotic cell death in several in vitro models (68). How HO-1 is involved in the induction of lipid ROS as well as cell death triggered by Mtb remains unclear.
Lungs are continually exposed to high levels of free radicals under stress conditions. Excessive production of ROS has been highly associated with the oxidation of proteins, DNA, and lipids, causing direct lung injury. Biological membrane lipids are highly susceptible to free radical damage, which is detrimental to cell functions and viability. Lung functions are significantly impaired by accumulation of free radicals. Initial oxidation of few lipid molecules is sufficient to result in significant tissue damage, as lipid peroxidation is a self-propagating chain reaction. Interestingly, lipid ROS are also involved in the induction of MMP-1 expression (105).
The depletion of GSH, an important antioxidant, triggers the accumulation of ROS along with increased levels of MMP-1. Inhibition of lipid peroxidation through treatment of cells with Trolox (a potent antioxidant) and by free iron chelation reduced the expression of MMP-1 in vitro (105). Despite extensive efforts in the field of lipid peroxidation, it is not clear whether lipid peroxide accumulation is the cause or consequence of pathological conditions. It seems that tissue damage is initiated by excessive ROS production, and thus contributes to the generation of several MMPs culminating in ECM degradation, which in turn amplifies lipid ROS accumulation as well as widespread necrosis. Since MMPs, HO-1, and lipid peroxidation are all associated with severe TB disease (10, 30, 36), it is reasonable to consider the prevention of excessive ROS production through augmentation of host antioxidant response as a target for host-directed therapy in TB.
Interplay Between Oxidative Stress and Tissue Remodeling: Therapeutic Perspectives
Despite the fact that Mtb infects humans over millennia, and that intense research toward identification of new therapies has been done, TB treatment remains ineffective, with multidrug therapy required for long periods focused on halting replication and driving Mtb death. This treatment strategy has limitations with ongoing significant mortality resulting from TB disease and emergence of resistant strains. Alternative and adjunct therapies are thus needed against Mtb infection. A thorough understanding of the three main drivers of pathology in TB, as shown in this review, highlights areas for new therapeutic strategies that target aberrant host responses to Mtb infection, an approach that will complement pathogen death by reducing pulmonary injury.
Several studies have associated oxidative stress with various lung diseases, such as chronic obstructive pulmonary diseases, asthma, acute pulmonary distress syndrome, and TB (93). Prior clinical trials have evaluated the efficacy of therapies aiming to dampen inflammation in TB through decreased ROS secretion and tissue remodeling in pulmonary TB, focused primarily on steroids that have been used as adjunctive treatment for several decades (37, 42, 123) to reduce IFN-γ, TNF-α, and IL-1β (77). Dampening inflammation through reduction of proinflammatory cytokines may impact the ability of immune cells to control Mtb infection.
Although ROS production is important for pathogen death, it can also trigger a series of oxidative reactions of PUFAs on host cells inducing cell death by accumulating lipid peroxides on biological membranes (4, 50). High levels of lipid peroxidation have been reported in patients with pulmonary TB compared with healthy individuals (3, 52). As mentioned previously, lipid peroxides can also induce MMPs, which in turn promote ECM degradation. Thus, it seems that targeting excess generation of ROS is a promising therapeutic strategy for TB, since overproduction of these molecules initiates a series of chemical reactions and causes damage to cellular components and excessive immune response activation by interfering transcription factors, kinases activation, and expression of proinflammatory mediators (31, 58, 92).
Convergence of these pathological pathways during TB disease promotes oxidative stress and tissue remodeling through mediation of the immune response. MMP activity is balanced by TIMP function, while other mechanisms including the prostaglandin signaling cascade and extracellular signal-regulated kinase (ERK) MAPKs also regulate MMP-1. In this context, p-aminosalicylic acid (PAS) has been shown to be effective as one of the first treatments for TB. Despite the absence of a well-understood mechanism of action, PAS is now known to immunomodulate PGE2 production, suppressing the release of MMP1 (107). These findings point PAS as a promising novel approach to adjunctly treat TB.
Similarly, doxycycline has been shown to modulate MMP secretion, primarily MMP-1 and MMP-3, in Mtb-infected primary human macrophages at 72 h in a dose-dependent manner as well as in bronchial epithelial cells (134). Moreover, doxycycline suppresses TNF-α secretion by macrophages, downregulating host inflammation in response to Mtb infection (134). Concurrent with this anti-inflammatory function, doxycycline has a direct bacteriostatic action against Mtb. By acting on multiple pathways involved in TB pathogenesis, doxycycline may serve as a potential adjunctive drug to combat TB and reduce tissue complications related to excessive inflammation. Interestingly, several studies have shown that broad spectrum inhibition of MMPs improves TB drug treatment and also reduces tissue damage in different animal models (78, 125). To date, there is one clinical trial investigating the effect of doxycycline in the modulation of tissue destruction in pulmonary TB patients (retrieved from
Statins are widely used as inhibitors of cholesterol biosynthesis. It is known that these drugs have broad anti-inflammatory effects, such as preventing excessive ROS generation and also inducing autophagy in vitro, a host strategy for eliminating the pathogen without triggering a necroinflammatory response (54, 101). Of note, there is evidence of an association between statin use and reduced risk of developing active pulmonary TB (69, 124). In murine models of pulmonary disease, statin treatment reduces lung pathology. Nevertheless, there is still no direct evidence that this drug prevents tissue damage in TB patients. Thus far, there are two clinical trials of statin use as adjunctive therapy for TB and TB-IRIS (retrieved from
Conclusions
TB infection has persisted as a significant cause of morbidity and mortality despite the availability of antimicrobials that aim to inhibit replication or cause mycobacterial death. While much research to date has focused on novel antimycobacterial therapies to address emerging drug resistance, it is now clear that a shift in focus is needed to modulate aberrant host responses. Emerging pathways involved in tissue remodeling, excess lipid ROS generation, and systemic inflammation offer novel targets for adjunctive therapies to temper host immune responses. Methods to dampen systemic inflammation must be balanced against the need for a robust initial immune response driven by infected macrophages to promote granuloma formation and Mtb containment (Fig. 6).
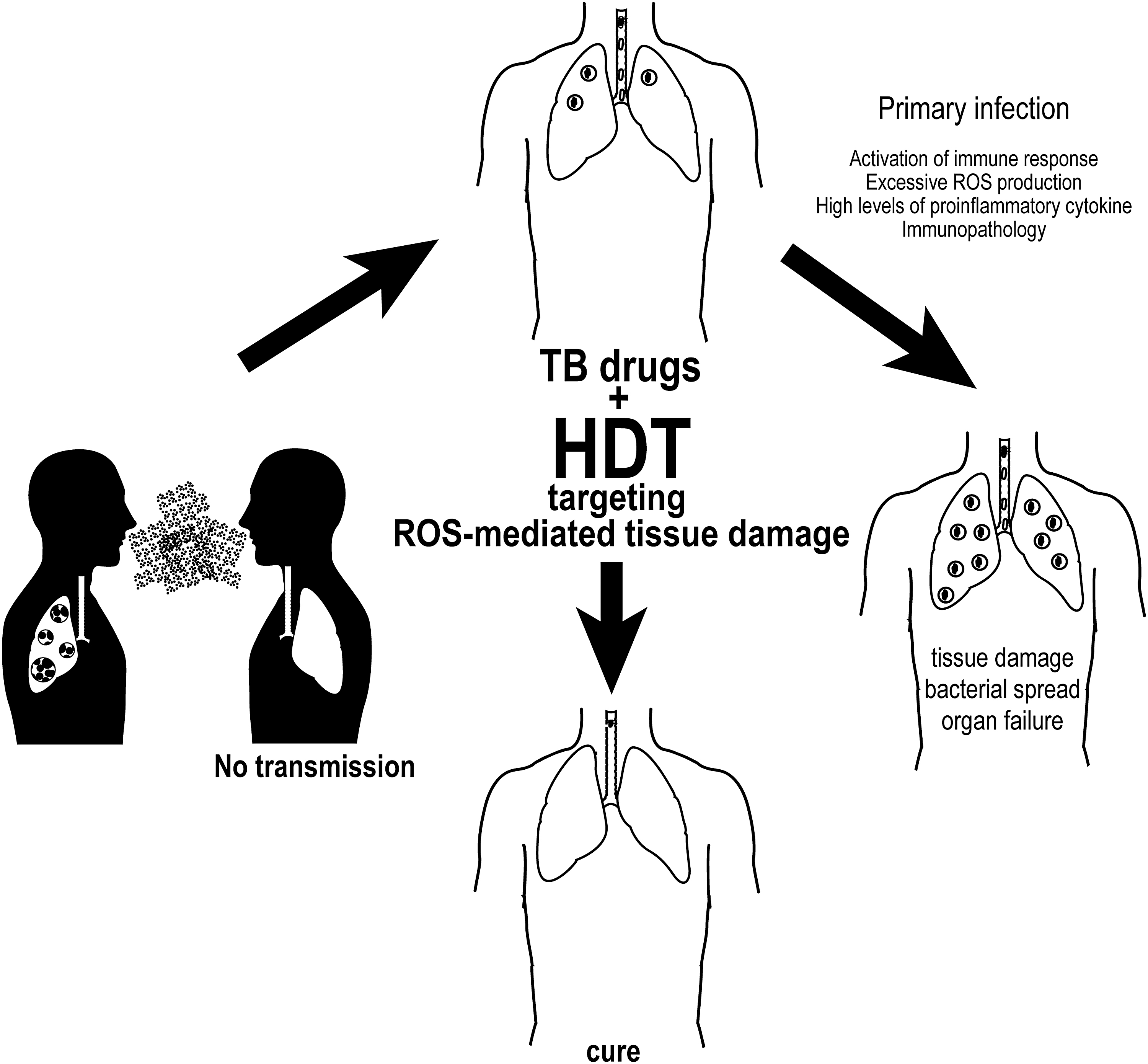
Prolonged macrophage activation in the course of chronic infection leads to imbalance of oxidants and antioxidants. Existing therapies including PAS and doxycycline, in addition to systemic steroids, are currently available options that may modulate the host immune response via inhibition of MMP-1 activity in chronic Mtb infection. Moreover, novel therapies that target inflammatory pathways early in infection may represent an effective strategy to avoid disease progression and tissue damage.
Footnotes
Funding Information
No funding was received for this article.