
Abstract
Significance:
Levels of platelet noncoding RNAs (ncRNAs) are altered by disease, and ncRNAs may exert functions inside and outside of platelets. Their role in physiologic hemostasis and pathologic thrombosis remains to be explored.
Recent Advances:
The number of RNA classes identified in platelets has been growing since the past decade. Apart from coding messenger RNAs, the RNA landscape in platelets comprises ncRNAs such as microRNAs, circular RNAs, long ncRNAs, YRNAs, and potentially environmentally derived exogenous ncRNAs. Recent research has focused on the function of platelet RNAs beyond platelets, mediated through protective RNA shuttles or even cellular uptake of entire platelets. Multiple studies have also explored the potential of platelet RNAs as novel biomarkers.
Critical Issues:
Platelet preparations can contain contaminating leukocytes. Even few leukocytes may contribute a substantial amount of RNA. As biomarkers, platelet RNAs have shown associations with platelet activation, but it remains to be seen whether their measurements could improve diagnostics. It also needs to be clarified whether platelet RNAs influence processes beyond platelets.
Future Directions:
Technological advances such as single-cell RNA-sequencing might help to identify hyperreactive platelet subpopulations on a single-platelet level, avoid the common problem of leukocyte contamination in platelet preparations, and allow simultaneous profiling of native megakaryocytes and their platelet progeny to clarify to what extent the platelet RNA content reflects their megakaryocyte precursors or changes in the circulation. Antioxid. Redox Signal. 34, 1200–1216.
Introduction
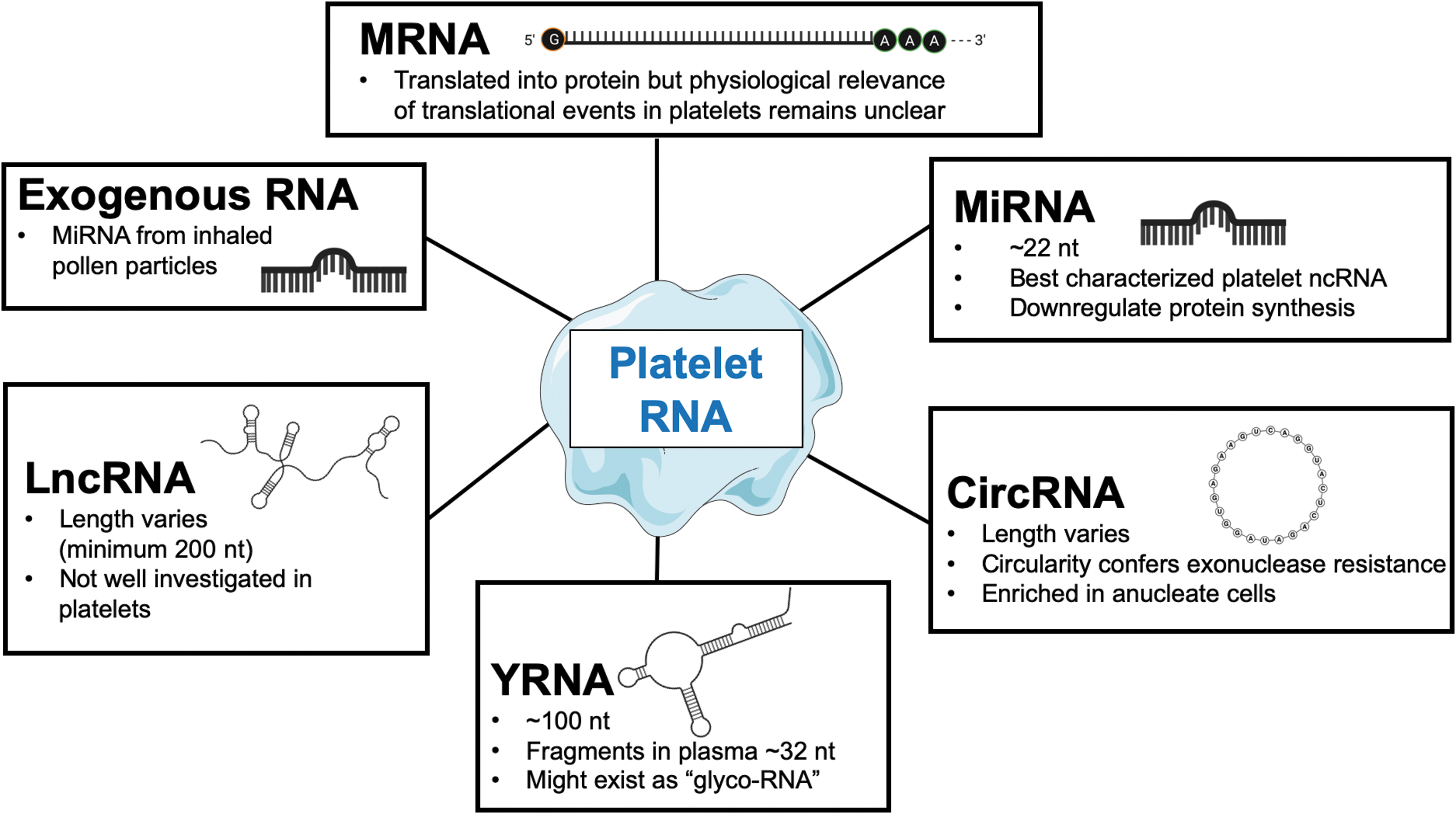
Platelets are the main effectors of physiologic haemostasis and pathologic thrombosis. Despite being anuclear, they contain coding and noncoding RNAs (ncRNAs). Notably, platelets are particularly rich in ncRNAs. These include microRNAs (miRNAs), circular RNAs (circRNAs), long noncoding RNAs (lncRNAs), and YRNAs (Fig. 1). Further, platelets may be the primary source of ncRNAs in the circulation and may also take up exogenous RNAs. Thus far, platelet RNAs were investigated in platelet function, as potential biomarkers and mediators of cross-talk with other cell types. More recently, studies have focused on how ncRNAs are released from platelets and whether they are compartmentalized in extracellular vesicles or complexed to RNA-binding proteins. This review will provide an overview with a particular focus on implications for cardiovascular disease.
The Coding Platelet Transcriptome
Platelets are anuclear and devoid of DNA but retain both coding and ncRNAs from megakaryocytes. Certain genomic mutations in the genome will cause alterations of the platelet RNA profile, for which several datasets are publicly available (21, 79, 94, 120). Genome-wide association studies have identified numerous genes linked to platelet count, volume, and function (61). Gray platelet syndrome is one example: Mutations in the Neurobeachin Like 2 gene cause more than hundreds of transcripts to be differentially expressed in platelets. The Neurobeachin Like 2 gene encodes a protein that is predicted to be involved in vesicular trafficking and may be critical for the development of platelet α-granules. Patients with Gray platelet syndrome lack these organelles (72). Development of megakaryocytes is under control of transcription factors such as transcription factor NF-E2 (135) and erythroid transcription factor (151), highlighting the importance of megakaryocyte transcriptional events for thrombopoiesis. Abnormalities in the medullary and/or extramedullary megakaryocyte niche could be reflected in the transcriptional profile of primary platelets. Megakaryocytes and their precursors can otherwise only be studied with imaging and most recently with single-cell RNA-sequencing (scRNA-seq) of bone marrow material (42, 43).
Differences in the transcriptomic profile of platelets are currently assumed to result from changes in megakaryocytes. There are emerging concepts that challenge this assumption (Fig. 2): First, it has been proposed that platelets contain a functional spliceosome along with pre-messenger RNAs (pre-mRNAs). Formation of messenger RNAs (mRNAs) through pre-mRNA-splicing and subsequent mRNA translation to proteins has been shown to be induced by thrombin-mediated platelet activation (44) as well as by endotoxin (133). Translation has been reported for procoagulant proteins such as tissue factor (132), as well as cytokines such as interleukin-1β (133). Second, it has been observed that platelets can take up RNA from other vascular cells (36) or tumor cells (106), providing a potential explanation that smaller (a possible surrogate for older) platelets contain a more diverse set of transcripts than larger (supposedly younger) platelets (36). Finally, the emerging concept of “tumor-educated platelets” relies on the detection of cancer-specific RNA in platelets from patients with neoplasms, when sampled remotely. These “liquid-biopsy” approaches to genotyping tumors based on perturbations in the platelet transcriptome might have potential to enter clinical practice (69) and have been replicated by independent groups. However, their pathophysiological relevance and the contribution of transcriptional changes in circulating platelets to platelet protein content remain unclear. Although platelet protein synthesis has been reported (155), it may be rather limited (60, 121). “Inheritance” from megakaryocytes or uptake from plasma (11) is likely to play a more important role in platelet protein composition and function than endogenous translation in platelets.
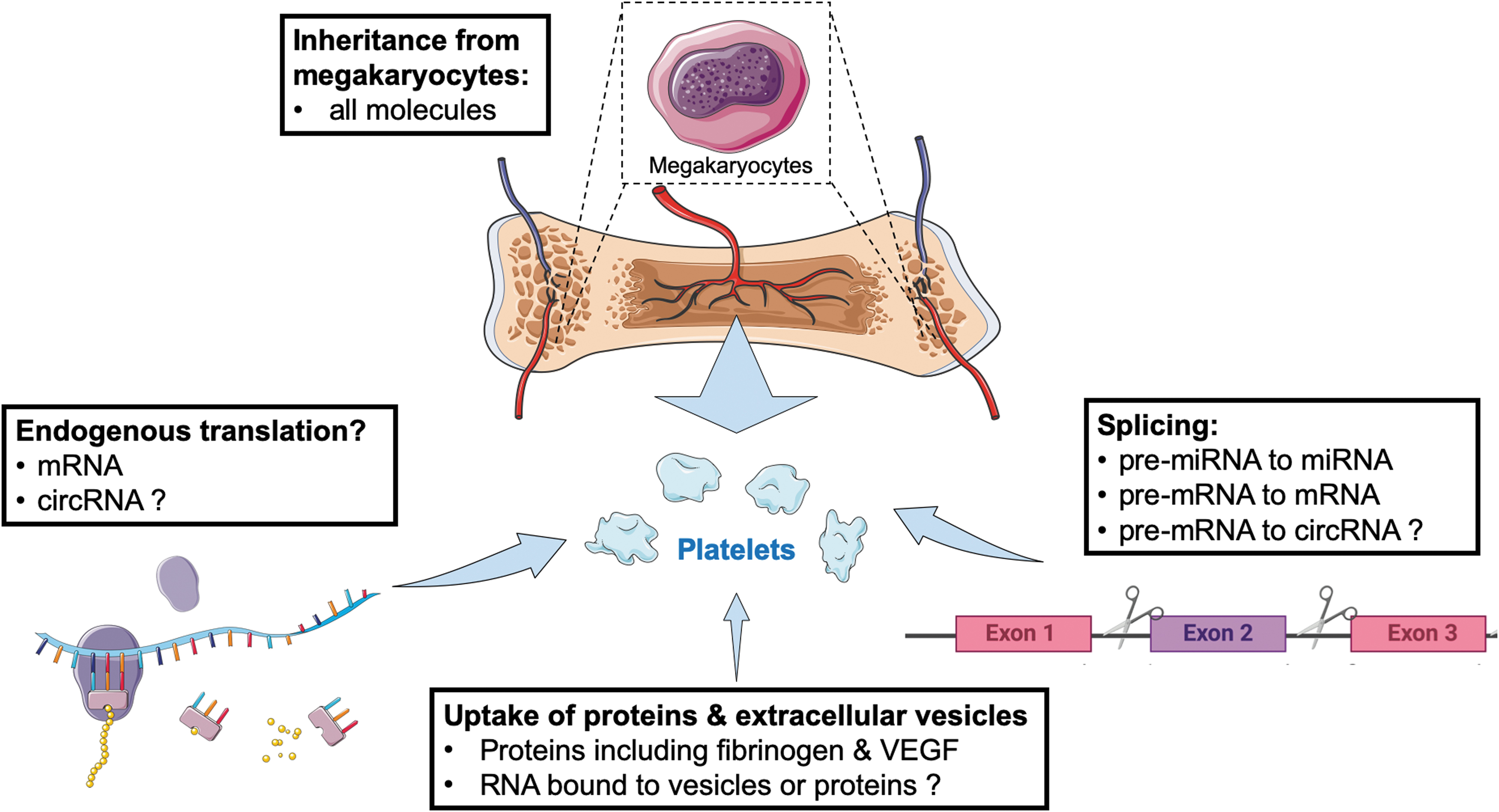
Thus far, the megakaryocyte transcriptome has been studied in immortalized cell lines or megakaryocyte cultures differentiated from CD34+ progenitors. Although transcriptomes of different megakaryocyte cultures showed agreement for the majority of constituents (17), caution has to be applied when individual transcripts are compared between megakaryocyte-like cells and freshly isolated platelets. The mRNA profile of primary megakaryocytes will differ from immortalized cell lines and in vitro differentiated CD34+ cells. Cecchetti et al. (29), for instance, studied how megakaryocytes differentially sort mRNAs into platelets. RNA-sequencing (RNA-seq) and subsequent quantitative polymerase chain reaction (qPCR)-validation was applied to isolated platelets as well as megakaryocytes that were differentiated in vitro from CD34+ progenitor cells. Based on the discordant presence of specific mRNAs for matrix metalloproteinases and tissue inhibitors of metalloproteinases in these two cell types, the authors concluded that this is likely the result of selective mRNA transfer from megakaryocytes to platelets. Intracellular mRNA sorting pathways exist in asymmetric cells such as neurons, in which certain mRNA sequences facilitate their movement along microtubules (29, 96). Microtubules are also essential for proplatelet formation (117). Similar RNA sorting mechanisms may, therefore, occur during platelet biogenesis. The authors experimentally ruled out differential degradation of the studied transcripts as an alternative explanation.
The development of single-cell transcriptome analysis based on a next-generation sequencing platform (scRNA-seq) by Tang et al. (141) in 2009 opened exciting new avenues for research. Megakaryocyte transcriptomics is one of them, as insufficient sample numbers of this rare cell type make it nearly impossible to perform transcriptomic analyses by conventional methods. In 2018, Davizon-Castillo et al. (42) applied this technique for the first time to native murine megakaryocytes. They could identify seven transcriptionally different clusters of cells, with three of them appearing to represent late states of maturation and possibly, the proplatelet-forming groups of cells due to the elevated expression of megakaryocyte transcripts. A year later, the same group identified tumor necrosis factor-α as the key aging-associated proinflammatory cytokine responsible for platelet hyperreactivity (43). Interestingly, this hyperreactive platelet phenotype was reflected in significant reprogramming of inflammatory, metabolic, and mitochondrial gene pathways in bone marrow megakaryocytes, as determined by scRNA-seq and subsequent bioinformatic pathway analysis.
The platelet transcriptome is known to be differentially regulated in several cardiovascular conditions such as coronary heart disease (CHD) (38, 49, 65), coronary artery bypass graft (CABG) surgery (116), and atrial fibrillation (158). In the case of CHD, studies have shown platelet transcriptomic differences between ST-segment elevation myocardial infarction (STEMI) and stable CHD (65); between non-ST-segment elevation myocardial infarction (NSTEMI) and stable CHD (38); and between STEMI and NSTEMI (49). It is not clear whether these differences are causal; they could also be a consequence of the disease or medication. Answering this question might offer diagnostic or therapeutic opportunities. During and shortly after CABG surgery, there is an increase in thromboembolic events. The strong inflammatory stimulus of the extracorporeal circulation and the surgical procedure itself induce a hypercoagulable state and augmented thrombopoiesis. Platelets isolated from CABG patients have elevated mRNA and protein levels of key molecules involved in platelet aggregation (116). Sepsis is another acute condition with excessive activation of the immune system. A recent study has shown upregulation of many platelet mRNAs in sepsis patients (100), consistent with previous studies in mice (56).
The Noncoding Platelet Transcriptome
Similar to coding transcripts, it is believed that the megakaryocyte precursors pass on ncRNAs, including miRNAs, YRNAs, circRNAs, and lncRNAs, to the budding platelets. Of these, miRNAs were the first to be discovered in platelets (85) and have since then been investigated in detail with regards to their importance for platelet biology or their potential as biomarkers.
MicroRNAs
In 1993, Lee et al. (88) described the first miRNA by characterizing a mutant of the nematode Caenorhabditis elegans. Till then, it was thought to be implausible that transcripts as short as 22 nucleotides would have regulatory functions. Thus, RNA fragments of this size were overlooked (87). In the next few decades, miRNAs became the most widely studied class of ncRNAs. It became clear that miRNAs are remarkably conserved between species, highlighting their biological importance for regulatory processes. Through sequence complementarity of the miRNA seed region to the 3′-untranslated region in their respective target mRNA, protein synthesis can be attenuated by mRNA degradation or translational repression. Computational prediction indicates that single miRNAs regulate multiple mRNAs. Estimates suggest that around one third to half of human genes are targeted by miRNAs (16, 57, 110). The number of biologically relevant interactions, however, is likely to be lower for the following reasons (111): (i) Copy numbers of miRNAs need to reach at least several hundred per cell to exert biological functions (103). This dose sensitivity appears to be a critical factor (9, 19, 45, 127) and is not taken into account by computational target predictions. Single-cell protein measurements in the presence or absence of a respective miRNA revealed that miRNAs can generate thresholds levels for their target mRNAs, below which protein production is highly repressed (miRNA acting as a switch), whereas protein synthesis responds to target mRNA input when close to the threshold (miRNA acting as a fine-tuner) (103). Recent single-cell miRNA-mRNA co-sequencing experiments confirmed that the negative correlation of miRNAs with their targets is lost for low-abundant transcripts (154). (ii) Studies with miRNA mutants have furthermore suggested that in many cases a main target is responsible for most of the phenotypic changes in vivo [with a few exceptions such as miR-965 in Drosophila (148)], thereby questioning whether targeting of multiple mRNAs really is a widespread and biologically relevant feature of miRNAs (111).
miRNAs are essential for regulatory functions in complex organisms. Indeed, recent technological breakthroughs have revealed that cell-to-cell heterogeneity can be the product of miRNA expression variability alone, independent of genotype. This was recently shown by Wang et al. (154), who simultaneously sequenced miRNAs and mRNAs in the same single cells. For this purpose, they used a “half-cell genomics approach,” splitting single-cell lysates into two equal parts. By correlating miRNA-miRNA (r = 0.93) and mRNA-mRNA (also r = 0.93) sequencing data from each half against each other, they demonstrated that their approach was robust. When they next correlated levels of miRNA-mRNA interaction pairs against each other, predicted mRNA targets were negatively correlated with the variation of abundant, but not of low abundant miRNAs. As their study made use of clonal cells sharing the same genotype, they could show that cell-to-cell heterogeneity can result from variability in miRNA expression alone (mediated by different cellular molecular states).
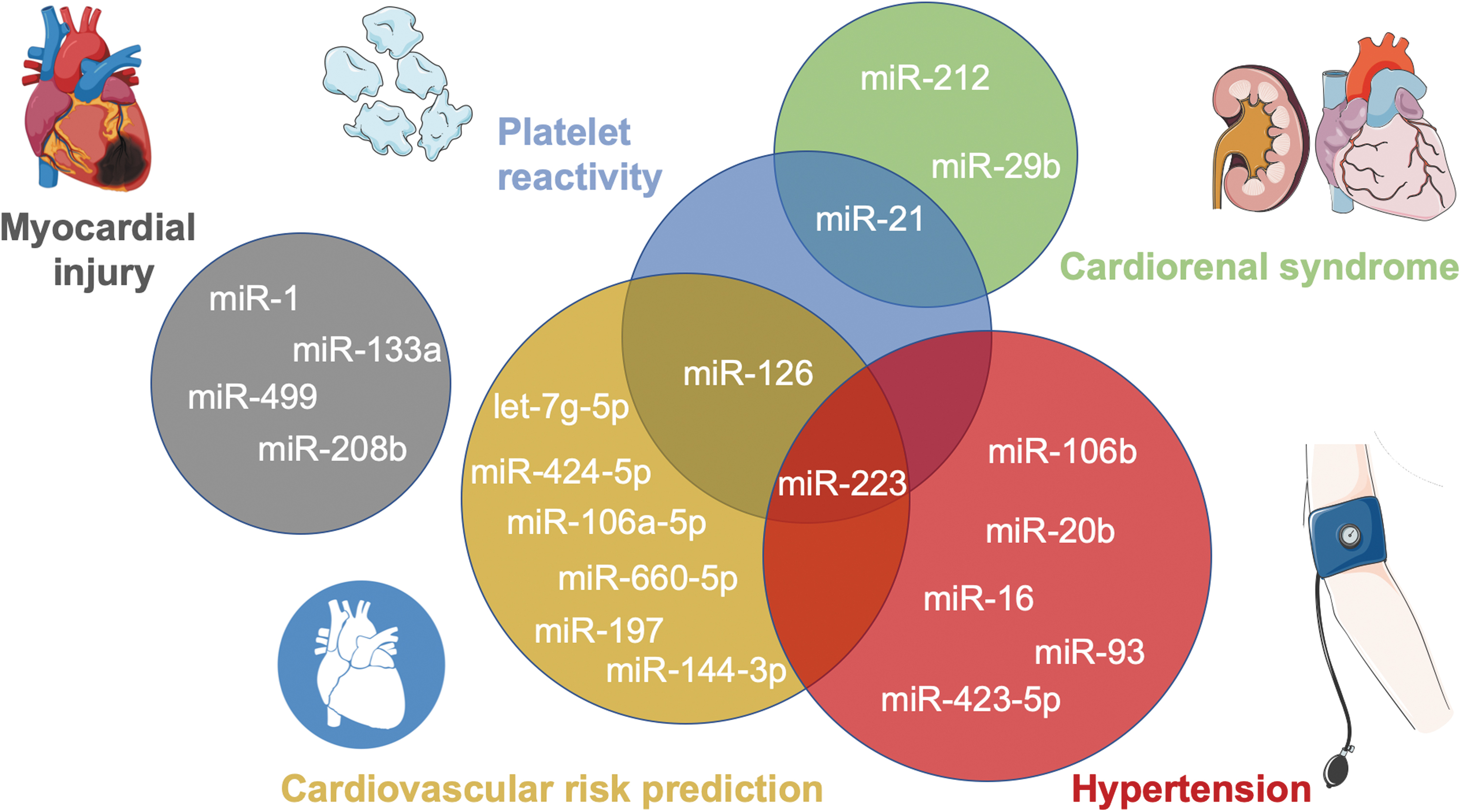
Given their importance, most miRNAs are ubiquitously expressed, whereas a small subset of miRNAs are known to be tissue- or cell specific and are dysregulated in disease (99). In 2008, miRNAs were first discovered to not just be restricted to the intracellular space but to also be stably detectable in the circulation (101). Later, their detectability was confirmed in other biofluids (e.g., tear fluid, urine, breast milk, seminal fluid, saliva, amniotic fluid, bronchial lavage, cerebrospinal fluid, pleural fluid, peritoneal fluid, colostrum) (156). Their remarkable stability in RNase-rich extracellular environments was surprising and was later shown to be due to various protective mechanisms. Extracellular miRNAs may originate from different cells and reflect a (patho-)physiological state, which led to their extensive investigation as biomarkers including for cardiovascular diseases (Fig. 3), such as myocardial injury (130), cardiovascular risk prediction (24, 161), hypertension (48), cardiorenal syndrome (34, 115, 126), and platelet reactivity (140).
Levels of many circulating miRNAs are highly correlated, indicating a common origin (97). The existence of miRNAs as well as their processing proteins Dicer and Argonaute 2 (AGO2) in platelets was reported in 2009 (85). Platelets contain ∼750 different miRNA transcripts (21). Based on later findings that have partly emanated from our lab, it is now clear that the majority of circulating miRNAs derive from platelets and other circulating cells (157).
MicroRNAs in platelet formation
In megakaryocyte precursors, miRNAs regulate several steps that are important for platelet formation (Fig. 4). Early studies on megakaryocyte differentiation in culture have revealed a concerted regulation of several miRNAs targeting genes involved in megakaryocyte differentiation (59). Later functional studies using knockdown and/or overexpression approaches conducted in vitro and in vivo have revealed that most established factors involved in megakaryocyte proliferation, maturation, and platelet formation are regulated by a miRNA network.
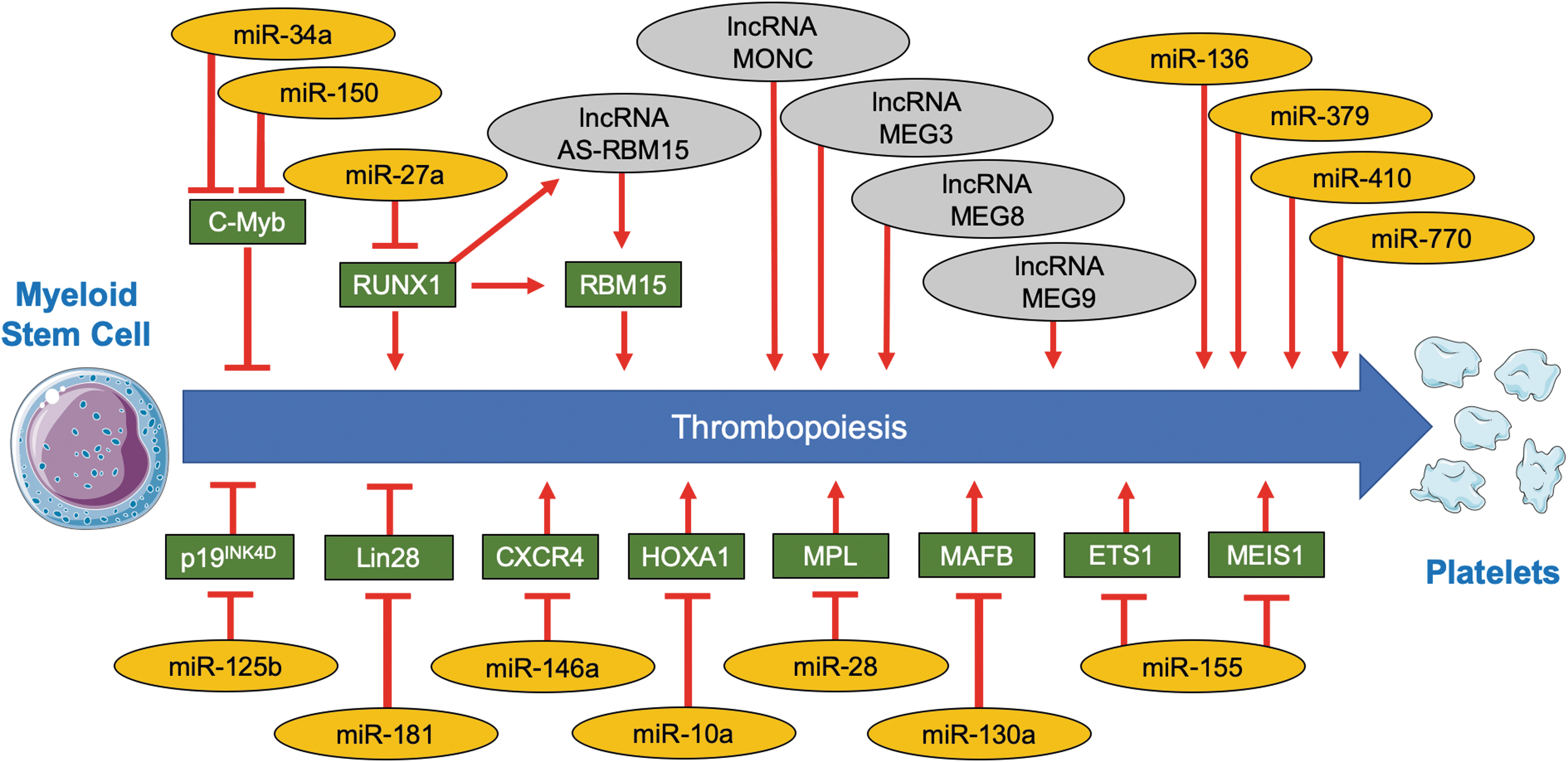
For instance, the liver- and kidney-derived hormone thrombopoietin increases expression of miR-150 and miR-34a in megakaryocytes, which, in turn, downregulates the transcription factor transcriptional activator Myb and enhances thrombopoiesis (13, 14, 104). This finding illustrates how systemic signals are able to change the noncoding transcriptome of megakaryocytes and as a consequence alter gene expression in megakaryocytes. In case of miR-150, these findings are based on in vitro studies, whereas miR-150 knockout mice exhibit normal platelet counts. Differences, however, are seen on recovery from acute bone-marrow depletion, suggesting a role for miR-150 in acute injury rather than constitutive platelet turnover (3). Other established transcription factors with miRNA-dependent regulation in the context of megakaryopoiesis (Fig. 4) are transcription factor MafB (miR-130a) (59), homeobox protein Hox-A1 (miR-10a) (59), zinc finger and BTB domain-containing protein 16 (miR-146a) (83), runt-related transcription factor 1 (miR-27a) (15), protein C-ets-1 (miR-155) (119), and homeobox protein Meis1 (miR-155) (119). At the receptor level, the thrombopoietin receptor (miR-28) (62) and the C-X-C chemokine receptor type 4 receptor (miR-146a) (83) are controlled by miRNAs (Fig. 4). Similarly, the cell-cycle inhibitor cyclin-dependent kinase 4 inhibitor D, a negative regulator of megakaryocyte maturation, was shown to be under control of miR-125b (114). The RNA-binding protein lin-28 homolog A (LIN28) is both modulator and target of miRNAs: It represses the biogenesis of miRNA let-7 (LIN28/let-7 feedback circuit) and is itself downregulated by miR-181. Disruption of the LIN28/let-7 feedback circuit by miR-181 has been shown to promote megakaryocytic differentiation (91). Other recently identified miRNAs, for which overexpression and knockdown experiments revealed a causal role in megakaryocytic differentiation, include miR-136, miR-379, miR-410, and miR-770 (131).
The main limitations of these studies are that due to the rarity of bone marrow megakaryocytes, immortalized cell lines or megakaryocytes differentiated from isolated CD34+ progenitor cells were used. Depending on the culture conditions, the reported findings might therefore not replicate the in vivo situation. Although an attempt has been made to isolate megakaryocytes from formalin-fixed paraffin-embedded tissue sections by laser-microdissection and subsequently profile their miRNA content (68), no comprehensive quantitative dataset of the noncoding transcriptome of isolated primary bone marrow megakaryocytes is available to date. Given that platelet protein synthesis is rather uncertain (23, 60, 94), post-transcriptional regulation by ncRNAs may not occur in platelets. Instead, functional effects in platelets may be a consequence of miRNA-mediated repression of protein translation in megakaryocyte precursors.
Circulating platelet miRNAs in cardiovascular disease
Increased platelet reactivity predisposes to a wide range of diseases, including stroke and myocardial infarction (MI), whereas impairment of platelet function results in bleeding disorders (140). Since the first randomized controlled trial on aspirin in the secondary prevention of mortality from MI in 1974 (51), inhibiting platelet function has reduced cardiovascular morbidity and mortality. However, conditions such as high on-treatment platelet reactivity are seen in patients with diabetes (76) or inflammatory diseases (129), and they may contribute to thrombotic events despite antiplatelet therapy. A number of assays for determining platelet reactivity are available, but they do not inform clinical decision making for choice or dosage of antiplatelet therapy. In fact, the European Society of Cardiology has issued a position paper, which discourages their use (6). Over the past decade, platelet miRNAs have been correlated to platelet function and thereby generated interest as markers of platelet reactivity.
Kaudewitz et al. (75) demonstrated in patients with acute coronary syndrome on different antiplatelet therapies that circulating levels of platelet miRNAs showed significant positive correlations with the vasodilator-stimulated phosphoprotein (VASP) phosphorylation assay (e.g., miR-223, r = 0.28; miR-24, r = 0.25; miR-191, r = 0.24; miR-126, r = 0.22), but not with optical aggregometry. Kaudewitz et al. (75) also revealed that miR-126, a miRNA that was considered to be endothelial specific, is also abundantly expressed in megakaryocytes. When miR-126 was inhibited in mice using antagomirs, platelet aggregation was reduced. Interestingly, a single-nucleotide polymorphism in the primary transcript region of miR-126 that facilitates processing to mature miR-126 was associated with a rise in circulating protein markers of platelet activation in the Bruneck study. In vitro experiments in megakaryocyte-like MEG-01 cells further showed that miR-126 inhibits the protein disintegrin and metalloproteinase domain-containing protein 9 (ADAM9, which inhibits platelet adhesion to collagen), whereas P2Y purinoreceptor 12 (P2Y12) receptor expression (which mediates the adenosine diphosphate platelet activation pathway) is downregulated when miR-126 is inhibited by antagomir treatment. Two independent studies have since confirmed this effect of miR-126 on ADAM9 (58) and P2Y12 (128) regulation. One recent study showed that transfection of CD34+-derived megakaryocytes with miR-126 leads to 156% higher reactivity of its descendent platelet-like structures when assessed in a fibrinogen-coated flow chamber (58). As expected for a platelet-associated miRNA, the rise of circulating miR-126 levels after platelet activation can be inhibited by treatment with aspirin (18).
Similar to miR-126, a previous study has shown that the P2Y12 receptor is also a target of miR-223 in platelets. P2Y12 mRNA was found to be present in AGO2 immunoprecipitates (85). Deficiency for miR-223 leads to larger thrombi and delayed clot retraction in mice (50). Diabetic patients were found to have reduced intraplatelet levels of miR-223, leading to elevated expression of the P2Y12 receptor and P-selectin, thereby increasing platelet reactivity (54). The P2Y12 receptor is of particular clinical relevance, because it is the target of several antiplatelet drugs such as clopidogrel, ticagrelor, and prasugrel and further insight into its regulation might be relevant to assess drug responses. A study in patients with NSTEMI has shown that patients with a poor treatment response to clopidogrel had lower levels of miR-223 (134). Other studies also found that high platelet miR-223 levels correlated with the degree of platelet inhibition (35), whereas low plasma miR-223 levels were associated with high on-treatment platelet reactivity in patients with NSTEMI (166). Results from the prospective, community-based Bruneck study revealed that miR-223 levels (in particular when adjusted for miR-126) negatively correlated with risk of MI (161). Data from two other prospective cohorts have since shown that low levels of plasma miR-223 are associated with increased mortality (77). In a recent study, Braza-Boïls et al. have demonstrated that miR-223 along with miR-223* as well as miR-197 can indicate platelet activation in endotoxemia despite P2Y12 inhibition (22).
Zeng et al. (162) proposed platelet-derived miR-223 as a key regulator of vascular smooth muscle cells (VSMCs) contributing to intimal hyperplasia under hyperglycemic conditions. Using lineage tracing reporter mice, they visualized internalization of entire platelets (expressing platelet factor 4-cre driving membrane-bound Tomato/membrane-bound green fluorescent protein recombination) by VSMCs on arterial injury. Using fluorescence in situ hybridization, platelet-derived miRNAs were detected within VSMCs. Platelet-derived miR-223 was shown to downregulate platelet-derived growth factor receptor β (PDGFRβ) levels. Target engagement could be demonstrated both in vitro (through downregulation of a 3′-untranslated region-reporter construct) and in vivo (using platelet-miR-223 knockout mice and rescue with local agomir-223 delivery) in VSMCs. On endothelial denudation, activated platelets exert a pro-proliferative effect on VSMCs through the release of growth factors such as PDGFβ. PDGFβ promotes VSMC dedifferentiation from a quiescent contractile phenotype to a synthetic and proliferating VSMC phenotype. Subsequently, activated platelets are taken up by VSMCs, releasing miR-223 and applying a brake on VSMC dedifferentiation. This negative regulatory feedback appears to be compromised by hyperglycemia. In diabetes, platelet miR-223 levels are significantly decreased. It remains to be seen whether this platelet miRNA-dependent mechanism on neointima formation is also relevant in patients with diabetes who often suffer from severe intimal hyperplasia.
The protein vesicle-associated membrane protein 8 (VAMP8 or endobrevin) is responsible for platelet α-granule secretion and was previously suggested as being negatively regulated by miR-96, based on correlation analysis in hyperreactive human platelets and subsequent computational target prediction and experimental mRNA/protein measurements in megakaryocyte-like cells (80). Studies in patients on antiplatelet therapy found that platelet miR-96 levels are unaffected by treatment with the P2Y12 inhibitor clopidogrel (134), whereas another study found that a therapeutic switch from clopidogrel to ticagrelor leads to an increase of miR-96 levels (27).
Circular RNAs
circRNAs are a subgroup of lncRNAs (105). They predominantly arise from pre-mRNA back-splicing of exons, leading to the generation of a circRNA molecule with a 3′,5′-phosphodiester bond, termed “backsplice junction.” A subset of circRNAs may also derive from intronic or intergenic regions via alternative splicing (30). Their generation seems to be controlled by the cis-regulatory elements and trans-acting factors that regulate splicing (30). The RNA-binding proteins Muscleblind (10) and Quaking (39) were shown to promote circRNA expression, whereas Adenosine deaminase 1 acting on RNA (124) and DExH-Box Helicase 9 (4) downregulate circRNA production. circRNAs were discovered early in 1976 by Sanger et al. (125) in plants. Shortly afterward, electron microscopic evidence was presented for circRNAs in the cytoplasm of eukaryotic cells (67). A circRNA that is antisense to the sex-determining region Y mRNA (responsible for initiation of male sex determination) was reported in 1993 (25). Apart from this discovery, the class of circRNAs was largely forgotten and remained understudied until 2012, when Danan et al. (41) developed a combined experimental/computational approach called circRNA-seq, which allows profiling of circRNAs in a whole-genome, unbiased manner and revealed that circRNAs are highly prevalent. Methods used for circRNA detection target the mentioned backsplice junction: primers in reverse transcription qPCR (RT-qPCR), probes in microarrays, and sequencing reads in RNA-seq. This approach can yield false-positives due to reverse transcriptase template switching, trans-splicing, and genomic duplication (12). Validation experiments take advantage of the relative resistance of circRNAs to the exonuclease RNase R or include combined Northern blot analyses after polyacrylamide gel electrophoresis with RNase H cleavage (138).
First, functional studies have shown that certain circRNAs can act as so-called miRNA-sponges, which sequester and downregulate miRNAs by containing multiple miRNA target sites (64). These sponging functions were shown to have physiologically relevant effects: (i) CRISPR-Cas9-mediated knockout of the locus encoding circRNA Cdr1 led to miRNA deregulation, affecting brain function (112); (ii) circRNA forkhead box P1 was shown to sustain mesenchymal stem cell identity via miRNA inhibition (31); and (iii) circRNA LDL receptor related protein 6, a circRNA enriched in VSMCs, arising from alternative splicing of the lipoprotein receptor 6 gene (LRP6), was able to sponge miR-145 and thereby hinder miR-145 mediated VSMC migration, proliferation, and differentiation (63). Other circRNAs can act as transcriptional activators (92, 165) or be translated into proteins, for example, circRNA mannose-binding lectin family member 3, circRNA reticulon 4, and circRNA zinc finger protein 609 (89, 102, 108, 160). Due to their highly variable sequence length ranging from hundreds to thousands of nucleotides (105), the functional characteristics of the few circular transcripts that have been characterized so far cannot be extrapolated to the entire class of circRNAs. Recent methodological advances such as the development of an easy-to-use circRNA overexpression system (pCircRNA-DMo) (102) will facilitate the functional characterization of circRNAs.
The circRNA profiling in 20 human tissues revealed that this RNA class is highly abundant in platelets (95). Enrichment of circRNAs in platelets relative to nucleate cells (17- to 188-fold, depending on transcript) was also confirmed by Alhasan et al. (5) and proposed to be a consequence of differential degradation: Linear transcripts are more susceptible to degradation by exonucleases, and platelets lose >90% of their megakaryocyte mRNAs over time (Fig. 5). In contrast, circRNA enrichment of platelets relative to megakaryocytes could be due to circRNAs being produced in platelets, rather than being inherited from megakaryocytes. Nicolet et al. (105) demonstrated that expression of circRNAs in human hematopoietic cells is cell-type specific, and it increases on maturation. A recent study has shown that circRNAs can also be of functional importance in the hematopoietic system, as circRNA cyclic GMP-AMP synthase was shown to regulate differentiation of hematopoietic stem cells (159).

Preußer et al. (113) used RNA-seq and subsequent semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) validation to analyze circRNAs in human platelets and exosomes isolated from in vitro activated platelets. After replicating previous reports showing high abundance of circRNAs in platelets, their RNA-seq data revealed a novel circRNA (Plt-circR4). Experimental validation of circularity for this transcript included both RNase R treatment and combined Northern blot analysis after polyacrylamide gel electrophoresis with RNase H cleavage. Based on expression analysis by semi-quantitative RT-PCR in 11 different cell types, this transcript was identified as the first platelet-specific circRNA. However, the study did not include erythrocytes in the expression analysis, which are also known to be highly enriched in circRNAs (113). Further, Preußer et al. (113) could show that circRNAs are organized in large circRNA-protein complexes inside platelets. Finally, their data suggested selective release of circRNAs into extracellular vesicles, with smaller circRNAs being preferentially released.
Long ncRNAs
lncRNAs were arbitrarily defined to be longer than 200 nucleotides. The first discovery of a lncRNA (long noncoding RNA H19 imprinted maternally expressed transcript) dates back to the 1980s (107), although the transcript was wrongly classified as mRNA until its function in the form of an RNA molecule was finally acknowledged in 1990 (20). As one might expect from a class of RNA molecules that is so heterogenous in sequence length and structure, lncRNAs can function in numerous ways. lncRNAs can act as signals, decoys, scaffolds, or guides by interactions with proteins, DNAs, mRNAs, or miRNAs (40). miRNAs, circRNAs, and YRNAs are predominantly found in the cytosol, whereas lncRNAs are believed to be mainly confined to the nucleus (46). Thus, it was believed that they are less likely to be passed on to platelets from their megakaryocyte precursors (140). Recently, it has become apparent that lncRNAs show a much more diverse subcellular localization (28).
A preprint communication by Sun et al. (139) reports an abundance of lncRNAs in platelets. Based on large-scale deep sequencing of human platelets, they showed that lncRNAs in platelets show differential expression depending on platelet reactivity. Bioinformatic analysis further revealed that this subset of differentially expressed lncRNAs is related to platelet function. In a small cohort of patients with acute MI (n = 12) and matched controls, the lncRNA long intergenic nonprotein coding RNA 1269 was substantially downregulated in hyperreactive platelets, an effect reversed by aspirin treatment.
Another recent study has shown that the lncRNA metallothionein 1 pseudogene 3 (MT1P3) (167) is upregulated in megakaryocytes of patients with type 2 diabetes and is positively correlated with P2Y12 mRNA levels. Knockdown of MT1P3 by small interfering RNA reduced P2Y12 expression, and it inhibited platelet activation and aggregation in an animal model of diabetes. In contrast, overexpression of MT1P3 had the opposite effect on P2Y12 levels. Luciferase reporter assays suggested that MT1P3 sponges miR-126, known to be involved in P2Y12 receptor regulation (75).
lncRNAs can also be important for processes that lead to the formation of platelets (Fig. 4). Several lncRNAs control hematopoietic stem cell differentiation. Paralkar et al. (109) revealed hundreds of lineage-specific lncRNAs that are differentially expressed during mouse and human erythro-megakaryocytic development, many of which are regulated by transcription factors that are involved in hematopoietic lineage differentiation, such as T cell acute lymphocytic leukemia protein 1 and erythroid transcription factor. Knockdown of 21 highly expressed erythroid-specific mouse lncRNAs revealed that 7 abrogated terminal erythroid differentiation. The long intergenic ncRNA HOTAIRM1 regulates cell cycle progression during myeloid maturation (164), and lncRNA UCA1 was shown to regulate heme biosynthesis and erythrocyte development (93). lncRNA RNA-binding protein 15 (AS-RBM15) was found to fine-tune the relative proportion of erythroid and megakaryocytic differentiation in the hematopoietic lineage (144). AS-RBM15 can upregulate translation of the protein RBM15 (involved in megakaryocyte differentiation). Transcription of AS-RBM15 and RBM15 is under control of runt-related transcription factor 1, which is known to repress erythroid gene expression and promote megakaryocyte differentiation.
Other recently identified lncRNAs involved in megakaryopoiesis are lncRNA maternally expressed 3, 8, 9 (131) as well as lncRNA miR-99a-Let-7c cluster host gene (52). As highlighted by Paralkar et al. (109), inter-species sequence conservation should be considered for the interpretation of lncRNA data, since more than 80% of mouse erythroid lncRNAs are not detected in the human erythroid lineage (109). Thus, it is still questionable whether all lncRNAs, especially the ones that are present in very low copy numbers, are functional.
YRNAs
YRNAs are around 80–120 nucleotides in length and were discovered in 1981 in the cytoplasm of mammalian cells (90). In humans, four noncoding YRNAs have been described (hY1, hY3, hY4, hY5), being encoded by the same chromosomal locus (82). They are required for human chromosomal DNA replication (33) and when bound to Ro60 proteins they form Ro small ribonucleoprotein complexes, which are involved in the regulation of RNA stability and cellular stress responses (82). A recent preprint communication by Flynn et al. (55) has shown that YRNAs undergo glycosylation—a way to mediate inter- and intramolecular interaction that was only known for proteins and lipids. Although it is still unknown as to how this “glycoRNA” is generated and regulated, the concept represents an interesting new aspect of RNA biology and could be of relevance for platelet research, given the high abundance of YRNAs in platelets (75).
YRNA fragments were first detected by Dhahbi et al. (47) in human serum and plasma. Although single YRNA fragments have a predicted mass of only ∼10 kDa, Dhahbi et al. showed that YRNA fragments circulate as complexes of 100–300 kDa in blood. In experiments performed by our group, a platelet origin of circulating YRNA fragments (32–33 nucleotides) was confirmed by platelet spike-in experiments, a positive correlation to the VASP phosphorylation assay in patients with acute coronary syndrome and to circulating levels of platelet-derived proteins in the community-based Bruneck study (75). Fragmentation of YRNAs was initially discovered in apoptotic cells and associated with cellular stress responses (123). In the case of platelets, it is believed that YRNA fragments are formed during the budding process from the plasma membrane of megakaryocytes (75).
Exogenous plant RNAs in platelets
The transfer of RNA molecules between animal and plant species is controversial. It has been proposed that plant miRNAs can be found in the circulation and tissues. MiR-168 found in rice could be detected in serum samples of a Chinese cohort (163). An obvious concern is a food-related contamination of the RNA samples, as other studies concluded that food-related RNA uptake does not seem to be a widespread phenomenon (137). A recent study by Koupenova et al. (81) has shown that RNA derived from pine pollen can be found in human platelets, mediated through inhalation and subsequent platelet-pulmonary vascular cell interactions. In this study, plasma-derived RNA of participants of the Framingham Heart Study Offspring cohort was first analyzed by RNA-seq to identify the most abundant extracellular transcripts, which were then validated in 2779 individuals with RT-qPCR. In this dataset, the authors could identify three sequences belonging to pine pollen miRNAs, one of which was present in 20% of the participants. Consistent with the presumed pollen origin, a modest association with hay fever was found. When the authors delivered pollen intranasally to mice, pine miRNAs could be detected in platelets of mice exposed to pollen but not in the control group. Interestingly, platelets of the pollen-treated group were less reactive after thrombin activation. Previous studies have shown that proteolytic enzymes in pollen can degrade tight junctions between epithelial cells (122), and in vitro experiments in the study by Koupenova et al. (81) have shown that endothelial and epithelial cells can transfer pollen miRNA to platelets, providing a potential mechanistic explanation. Although computational target prediction revealed that the identified pollen miRNAs have several possible human targets, the functional consequences of these potential interactions remain unknown. The clinical characteristics of the study participants further revealed no association with disease.
Methodological Approaches in Studying the Noncoding Platelet Transcriptome
Substantial improvements in research methods for studying ncRNAs have been made. Northern blots have been used to detect ncRNAs for decades but are limited in their throughput. The sensitivity of this method was greatly improved by the introduction of locked nucleic acid-modified probes (78). Microarrays enable large-scale expression profiling of ncRNAs but have the drawback of requiring predesigned probes and therefore rely on a priori knowledge. RNA-seq represents a less biased approach that enables the discovery of novel transcripts. Bias in RNA-seq experiments can still be introduced by selective amplification, differences in library preparations, as well as the subsequent data interpretation. TaqMan- and SYBR Green-based RT-qPCRs offer highly sensitive and specific measurements of predefined targets and are commonly used to validate profiling data obtained from microarrays or RNA-seq experiments. Although RT-qPCR is suitable for research purposes, the workflow is labor intensive, less amenable to automation, and confounded, that is, by the presence of heparin (74). The latter limitation can be overcome by heparinase treatment of the RNA, which further increases processing time (37). Recently, miRNA immunoassays have been developed, which might enable absolute quantification of predefined miRNAs at scale, without the need for reverse transcription. The method uses a biotinylated DNA probe to directly capture the miRNA and subsequently a streptavidin, horseradish peroxidase conjugate to quantify the resulting enzymatic color reaction, similar to enzyme-linked immunosorbent assays (73). However, the method is limited in sensitivity and at present most suited for abundant miRNAs.
Preanalytical Considerations
Accurate measurements rely not only on the capability of the measurement platform but also on the control of preanalytical variation, in particular sample processing. For example, measurements of platelet-derived transcripts in serum and plasma may not be directly comparable (140): Unlike plasma, the preparation of serum is the result of the activation of the coagulation cascade. This inevitably leads to platelet activation and the release of more platelet ncRNAs than in plasma preparations. Since the coagulation cascade involves several proteolytic reactions, ncRNAs might lose their protein carriers and be more vulnerable to preanalytical degradation. Plasma samples are, therefore, deemed most suitable for analyses of circulating ncRNAs. However, differential platelet activation (and ncRNA release) can occur due to differences in centrifugation speed and sample processing time. Conventional plasma preparations usually contain residual platelets and leukocytes. Especially contaminations with leukocytes, which are known to interact with activated platelets (26), can confound RNA analysis, because platelets contain around 1000 times less RNA than leukocytes (143). A protocol for the preparation of platelet-poor plasma, which includes two consecutive centrifugation steps at low and high speed (140), may thus be preferable.
scRNA-seq of small RNAs was first shown in 2016 by Faridani et al. (53) and represents a promising technological advancement. With this method, the ncRNA content of single cells, including miRNAs, transfer RNAs, and small nucleolar RNAs can be measured in a less biased way. Similarly, ncRNAs with longer sequences such as lncRNAs or circRNAs can be measured by conventional scRNA-seq. The feasibility of scRNA-seq in platelets remains to be demonstrated. Their diverse repertoire of RNAs would offer an interesting substrate for scRNA-seq: 9500 mRNAs as determined by bulk RNA-seq (21) as well as ncRNAs (140) with ∼750 different miRNAs based on RNA-seq (21) and ∼21,000 circRNAs identified by circRNA-seq of RNase R-treated platelet RNA (5). The relatively small size of platelets (2–3 μm diameter) is also not a limiting factor for commercial scRNA-seq platforms (1). However, the RNA amount in platelets is low, and platelets might get activated during the single-cell isolation process. A comprehensive, quantitative dataset of the noncoding platelet and megakaryocyte transcriptome at different maturation states could define to what extent the noncoding platelet transcriptome is determined by their megakaryocyte precursors rather than processes in mature platelets. The mechanisms by which changes in the noncoding platelet transcriptome arise are likely to differ under conditions that preferentially affect megakaryocytes or conditions that predominantly have an impact on circulating platelets, such as altered shear stress by stenosed or artificial heart valves (146). Changes to the noncoding transcriptome in platelets could involve the following mechanisms: (i) Similar to splicing of some pre-mRNAs to mRNAs in platelets (44), miRNAs can be processed from residual precursor miRNAs (85); (ii) horizontal transfer of miRNAs has been reported to other cell types such as endothelial cells (84) or monocytic cells (118); and (iii) it is also plausible that platelets might alter their miRNA content by shedding or selective degradation, although no evidence has been presented to date.
Extracellular RNA Shuttles and Horizontal RNA Transfer
The extracellular environment is characterized by high RNase activity (145). However, ncRNAs are detectable in the circulation due to their association with small and large extracellular vesicles (70, 71, 113, 147) and/or protein complexes (Fig. 6) (8, 71, 149). Microvesicles are 150–1000 nm in size. These large extracellular vesicles are generated through shedding of the surface membrane. In contrast, exosomes are only 40–150 nm in size and derived from endosomes. The lack of abundant cytosolic proteins in exosomes suggests that their content is selectively controlled (71). In contrast to microvesicles, however, their miRNA content appears to be rather low (32). A recent study has shown that caveolin-1 selectively regulates sorting of miRNAs into microvesicles, following noxious stimuli such as oxidative stress (86). In platelets, the release of exosomes and microvesicles on platelet activation was first shown in 1999 (66). Later studies revealed that the quantity and quality of extracellular vesicles, which are also formed constitutively (2), are in part dependent on the agonist that induced platelet activation. Estimates suggest that 25% of all plasma vesicles are platelet derived (7).
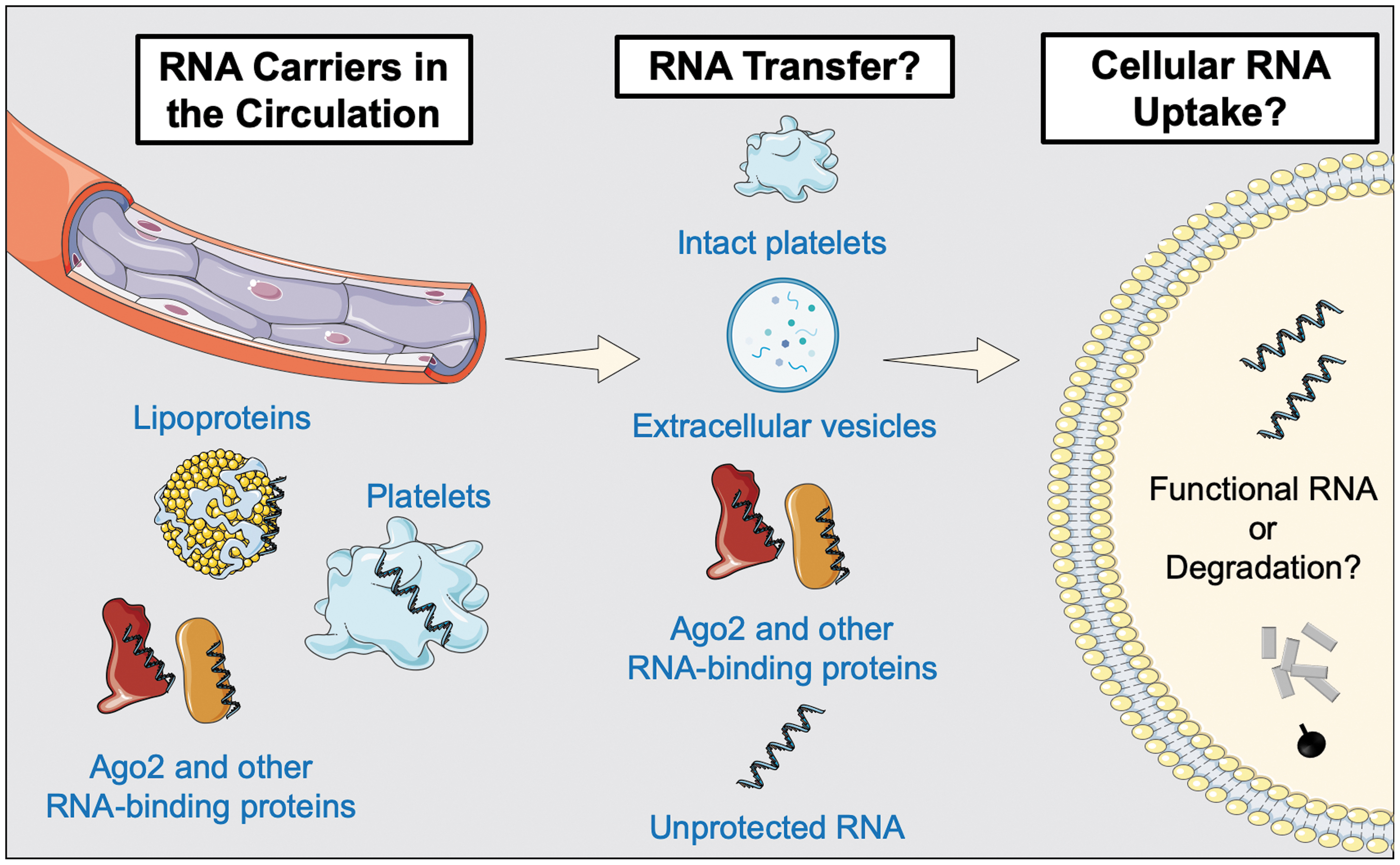
Jeppesen et al. (71) have recently explored how purification strategies may have led to an underestimation of the heterogenous landscape of extracellular vesicles. According to their findings, there are three classes of small-, three classes of large-, and two classes of small to large extracellular vesicles. Particularly relevant for the ncRNA field was the finding that many of the most abundant miRNAs were associated with the nonvesicular fractions. However, an RNA enrichment could be confirmed in nonexosomal small vesicles. Moreover, many RNA-binding proteins previously linked to loading of RNAs in exosomes were not found in classical exosomes when alternative purification strategies were applied. This includes the RNA-binding proteins Y-box-protein 1 (136), heterogeneous nuclear ribonucleoproteins A2/B1 (150), and major vault protein (142). Similarly, the miRNA-processing proteins AGO1–4, previously implicated in exosomal miRNA biogenesis (98), were not detected. Instead, AGO1–4 were found to be secreted independently of exosomes. Vesicle-free Ago2 protein has been shown to carry a substantial proportion of the extracellular miRNAs in plasma (8). Similarly, the protein nucleophosmin 1 was found to protect miRNAs from degradation (153).
Vickers et al. (149) implicated high-density lipoprotein (HDL) as a carrier of plasma miRNAs. Their study suggested that cellular export of miRNAs to HDL is regulated by neutral sphingomyelinase, whereas HDL-mediated delivery of miRNAs to recipient cells depends on the scavenger receptor class B type I. The authors speculated that transfer through this receptor would likely prevent the miRNAs from entering the lysosomal pathway and instead divert them to the cytoplasm, explaining the downregulation of cellular mRNA targets after coincubation of HDL-miRNAs with cultured cells. Although this represents a tempting concept of intercellular communication, it has to be noted that these effects were observed in vitro with miRNA copy numbers exceeding physiological concentrations (152).
Indeed, this limitation can be generalized to most previous studies related to horizontal miRNA transfer. These studies have relied on cellular overexpression of certain miRNAs in vitro, transfer of the cellular releasate to target cells, and subsequent analysis for miRNA uptake and function. As pointed out previously (140), the physiological relevance of these processes remains unclear and there has been little progress to clarify whether the abundance of circulating miRNAs is sufficient to induce relevant biological effects (Fig. 6). The copy numbers of circulating miRNAs are several orders of magnitude below the cellular miRNA content. Unless there is a signaling cascade that amplifies the initial miRNA signal, it is difficult to envisage how a few copies of miRNAs that are transferred to recipient cells can mediate biological function.
Also, staining for miRNAs by in situ hybridization tends to be confined to their expected cells of origin (128), thus arguing against a widespread exchange of miRNAs between different cell types. Another mechanism as to how platelet-derived RNA species could exert biological effects on other cells has been demonstrated in the study by Zeng et al. (162). As described earlier, the authors suggest that on vascular injury entire platelets are incorporated into VSMCs in vivo. Abundant platelet miRNAs such as miR-223 then contribute to the regulation of the phenotypic switch of VSMCs.
Conclusions and Perspectives
Recent studies revealed important functions of different RNA classes that have been identified in platelets. Certain platelet-derived transcripts may exert functions outside of platelets, that is, by transfer via extracellular vesicles, proteins, or uptake of whole platelets into recipient cells—assuming that the findings seen in specific transcripts and conditions represent a more widespread in vivo phenomenon. Although different platelet RNAs have been shown to be involved in important pathophysiological processes such as thrombopoiesis and platelet reactivity, potential platelet RNA-based clinical applications are still unclear, but one of the more tangible applications is the use of platelet RNAs as biomarkers. Platelets are a main source of circulating RNAs. Potential applications might include the identification of hyperreactive platelet subtypes by their transcriptomic profile. Platelet-derived ncRNAs and circRNAs, in particular, are remarkably stable compared with mRNAs. In addition to previous studies linking RNAs with platelet reactivity, a direct comparative analysis of RNAs and proteins released from platelets on treatment with various agonists with conventional platelet reactivity measurements could improve our understanding of the clinical utility of these measurements. Also, the integration into “biomarker signatures” combining protein and RNA measurements, as recently shown for biomarker combinations of myocardial injury (130), is a logical next step. Nonetheless, several technical challenges make the comparability of platelet-RNA studies difficult (140). scRNA-seq could overcome limitations that have hampered platelet research for decades, such as leukocyte contamination in platelet preparations, and identify potential hyperreactive platelet subpopulations with a greater propensity to trigger thrombotic events. Development of treatment strategies directed at these subpopulations might help to decrease morbidity and mortality in cardiovascular disease.
Footnotes
Acknowledgments
Author Disclosure Statement
M.M. has filed and licensed patent applications on miRNAs as platelet biomarkers.
Funding Information
C.G. is funded by a British Heart Foundation (BHF) PhD studentship (FS/18/60/34181). A.J. was a British Heart Foundation Clinical Research Training Fellow (FS/16/32/32184). M.M. is a BHF Chair Holder (CH/16/3/32406) with BHF program grant support (RG/16/14/32397) and member of a network on “MicroRNA-based Therapeutic Strategies in Vascular Disease” funded by the Foundation Leducq. This study is also supported by VASCage—Research Centre on Vascular Ageing and Stroke. As a COMET centre, VASCage is funded within the COMET program—Competence Centers for Excellent Technologies by the Austrian Ministry for Climate Action, Environment, Energy, Mobility, Innovation and Technology; the Austrian Ministry for Digital and Economic Affairs; and the federal states Tyrol, Salzburg, and Vienna.