
Abstract
Significance:
Osteonecrosis (ON) is characterized by bone tissue death due to disturbance of the nutrient artery. The detailed process leading to the necrotic changes has not been fully elucidated. Clinically, high-dose corticosteroid therapy is one of the main culprits behind osteonecrosis of the femoral head (ONFH).
Recent Advances:
Numerous studies have proposed that such ischemia concerns various intravascular mechanisms. Of all reported risk factors, the involvement of oxidative stress in the irreversible damage suffered by bone-related and vascular endothelial cells during ischemia simply cannot be overlooked. Several articles also have sought to elucidate oxidative stress in relation to ON using animal models or in vitro cell cultures.
Critical Issues:
However, as far as we know, antioxidant monotherapy has still not succeeded in preventing ONFH in humans. To provide this desideratum, we herein summarize the current knowledge about the influence of oxidative stress on ON, together with data about the preventive effects of administering antioxidants in corticosteroid-induced ON animal models. Moreover, oxidative stress is counteracted by nuclear factor erythroid 2-related factor 2 (Nrf2)-dependent cytoprotective network through regulating antioxidant expressions. Therefore, we also describe Nrf2 regulation and highlight its role in the pathology of ON.
Future Directions:
This is a review of all available literature to date aimed at developing a deeper understanding of the pathological mechanism behind ON from the perspective of oxidative stress. It may be hoped that this synthesis will spark the development of a prophylactic strategy to benefit corticosteroid-associated ONFH patients. Antioxid. Redox Signal. 35, 357–376.
Introduction
Osteonecrosis (ON) is reported in the literatures as being characterized by the death of bone tissues due to a disturbance of the nutrient artery (121). Clinically, osteonecrosis of the femoral head (ONFH) is mainly seen in patients who require high-dose corticosteroid therapy for underlying diseases, or in those who smoke or drink excessive amounts of alcohol (68). As for the associated ischemic process, researchers have proposed various intravascular mechanisms including oxidative stress, blood thrombosis or coagulation, lipid metabolism abnormality, vasospasm, and cell death such as apoptosis, necrosis, or necroptosis (53, 65 –67, 74, 117, 122, 178). However, the micropathways leading to ischemia has not been fully elucidated.
Among all the reported risk factors, it has become increasingly clear that oxidative stress is closely involved in the pathogenesis of a large variety of diseases, through numerous pathological conditions such as degeneration of organs, autoimmune diseases, metabolic disorders, cancer, and aging as well as ONFH (50). It is thought that oxidative stress plays a role in ON largely by causing bone and vascular damage. The possible relationship between oxidative stress and ONFH has been shown by using samples from the patients (27, 82, 106, 132, 157, 158, 181, 198). Excessive corticosteroid use has been reported to cause elevated oxidative stress in an animal model of corticosteroid-induced ON, and this model has in fact confirmed bone and vascular endothelial cell dysfunction (64, 185). Vascular endothelial cell dysfunction is implicated in blood thrombosis or coagulation, resulting in ischemia or vascular occlusion (41, 92, 171). Besides, oxidative stress can have a significant impact on the pathology of ischemia–reperfusion injury and vasospasm (19, 189). These angiopathies are also known as possible factors associated with ischemic events in ON (58, 67).
Interestingly, although a multifactorial event is considered essential to the occurrence of ONFH, one previous study has reported that findings characteristic of ON were induced in a rat model by a single-agent administration of a pro-oxidant (65). Using established corticosteroid-induced ON rabbit models (185), various kinds of antioxidants have also been reported to be useful in preventing ON (64, 87, 94, 96, 108, 114, 153, 194, 196). Taken together, these results indicate that there may be a relationship between oxidative stress and ON development. But there has been little discussion of the antioxidant response mechanism against oxidative stress in ON, and considering that antioxidant monotherapy has still not succeeded in preventing ONFH in humans, understanding this mechanism would not only further the clinical prevention of ONFH and the contractionary effect of the necrotic lesion but would also elucidate the mechanisms behind ON. Achieving this goal requires a more thoroughgoing interpretation of the pathological connection between ON and the molecular response to oxidative stress.
The organisms adapt to oxidative stress by inducing redox regulatory mechanisms to maintain homeostasis. Upon sensing oxidative stress, a complex cellular defense mechanism protects cells against exposure to toxic radicals by setting off an antioxidant response reaction (160). There are several essential transcriptional factors for the redox regulation, including nuclear factor erythroid 2-related factor 2 (Nrf2), Forkhead box-O 3 (FOXO3), and nuclear factor-kappa B (NF-κB) (107, 159). Among them, transcription factor Nrf2 is considered to be a master regulator of antioxidant defenses through the activation of various vitagenes and other protective molecules and plays a central role in the maintenance of cellular and tissue redox homeostasis. Nrf2 has been reported to play a crucial role in preventing several degenerative diseases in multiple organs (4, 180). Considering that Nrf2 is closely involved in bone-related as well as vascular injuries such as delayed bone healing or cardiovascular diseases (57, 84, 101), it may be closely tied to the pathogenesis of ON through the regulation of antioxidants.
To the best of our knowledge, there have been few articles that compile the evidence linking oxidative stress with ON focused on the efficacy of various antioxidants or address a possible role of Nrf2 on ON. Based on data collected in the past, we decided to conduct an up-to-date review summarizing the current state of knowledge about the role of oxidative stress in ON and gathering data on the effects of antioxidant administration in preventing ON. In discussing the regulation of Nrf2, we highlight its potential role in the pathology of ON. It is hoped that this review helps clarify the pathological mechanism of ON from the perspective of oxidative stress and will ultimately lead to the development of a prophylactic strategy for ONFH patients.
Role of Oxidative Stress in ON
Overview of oxidative stress
Oxidative stress generally arises due to an imbalance between the generation of oxidants (specifically here, free radicals and reactive oxygen species [ROS]) and their elimination by protective mechanisms such as antioxidants. In a biological context, ROS form as the natural by-product of normal metabolism of oxygen and are important in cell signaling and homeostasis (33). But when other oxidative events occur, an oxidation reaction takes place whereby triplet (3O2) and molecular (O2) oxygen combine to produce dramatic levels of free radicals or ROS, such as superoxide (O2 −), hydroxyl radical (•OH), hydrogen peroxide (H2O2), and singlet oxygen (1O2). The dose–response phenomenon of ROS, characterized by low-dose stimulation and high-dose inhibition, is based on the concept of hormesis (21, 22, 35). It is considered that low concentrations of ROS trigger the expression of antioxidant enzymes and other defense mechanisms, whereas high levels of ROS in the cell make the situation detrimental through inflammation or mitochondrial dysfunction (21, 22, 30).
Mitochondria, intracellular organelles with double membrane, are considered to be the main source of ROS in most eukaryotes. Mitochondria play a central role in the energy metabolism by generating adenosine triphosphate (ATP) in the complexes of respiratory chain (electron transport chain) in the inner mitochondrial membrane. During ATP synthesis, ROS such as O2 − and H2O2 are produced. Despite the continuous intracellular ROS production in mitochondria, redox homeostasis is normally maintained through an adaptive defense system that eliminates ROS by the function of antioxidant enzymes. However, excessive amounts of ROS can disrupt this redox balance and subsequently lead to mitochondrial DNA mutation (116). Mitochondrial DNA is an important target for such exogenous and endogenous ROS as it is more vulnerable to damage than the nuclear DNA due to their limited repair capacity by the lack of the chromatin complex structure consisting of protective histones (80, 90). The mitochondrial DNA mutation caused by excessive ROS leads to consecutive respiratory chain dysfunction, resulting in further ROS and mutation in mitochondrial DNA (a vicious cycle of ROS). This can lead to irreversible damage to the cell and contribute to various diseases such as cancer, cardiovascular disease, diabetes mellitus, ischemia–reperfusion injury, liver disease, and neurodegenerative disease (33, 45, 174).
Oxidative stress and cell death
ROS are known to be involved in cell death, such as apoptosis, necrosis, and necroptosis. Apoptosis is one of the programmed cell deaths characterized by chromatin condensation, nuclear fragmentation, cell shrinkage, and membrane blebbing. This is tightly regulated by caspase-3, Bcl-2, and Fas proteins and is not accompanied by inflammation. ROS are generally associated with the regulation of major pathways of apoptosis evoked by mitochondria or death receptor (133, 142). In mitochondria, exogenously or endogenously generated ROS activate proapoptotic proteins, and caspase-9 and -3, leading to DNA fragmentation by nucleases located in the intermembrane space of mitochondria. These processes lead to apoptosis. ROS also induce apoptosis by activating death receptors including Fas receptor, the tumor necrosis factor-α (TNF-α) receptor, and TNF-related apoptosis-inducing ligand receptor, and ROS directly provoke nucleus or mitochondrial DNA fragmentation (14, 142).
However, necrosis is nonprogrammed cell death that occurs when cell is severely damaged by external forces to nonphysiological states, such as complement-mediated cytolysis, heat stress, hypoxia, infection, or injury. Necrosis is specified by swelling of cells and organelles, disruption of membrane, and release of damage-associated molecular patterns, which can cause damage to surrounding tissues through inflammation. Moderate ROS can only induce cell apoptosis, whereas excessive stress can cause necrosis (91). ROS generated from mitochondrial or nonmitochondrial sources also play an important role in TNF-α-induced necrosis in a caspase-independent manner (170).
In recent years, the concept of necroptosis has been proposed as a regulated cell death with morphological features of necrosis that has important roles in the pathology of various diseases through inflammation. Necroptosis shares a characteristic of necrosis in that it is TNF-α-mediated cell death by excessive ROS (146, 182). In addition, ROS is potentially involved in the two critical kinases, receptor-interacting protein-1 (Rip1) and Rip3, or mixed lineage kinase domain-like protein, which is essential for the induction of necroptosis (100, 146, 182). In addition to necrosis, it has been considered that both apoptosis and necroptosis are involved in the pathogenesis of ON (66, 75). In particular, these kinds of cell death in bone and vasculature caused by oxidative stress are considered to be a significant risk factor for ONFH (64, 185).
Influence of oxidative stress on bone metabolism
ROS-induced oxidative stress adversely affects bone homeostasis by altering the functions of bone metabolism-related cells, such as mesenchymal stem cells (MSCs), osteoblasts, osteoclasts, and osteocytes (24, 162, 175). Bone marrow MSCs are pluripotent cells found in bone marrow tissue that proliferates and differentiates into different cell types, including osteoblasts or adipocytes (139). Oxidative stress is thought to be a major cause of decreased osteogenesis, but it also causes increased adipogenesis by impairing MSC function (172). Intraosseous fat embolism is one of the possible pathomechanisms of ONFH (74), and bone marrow fat cell enlargement is a cardinal histological feature of corticosteroid-associated ON in rabbits (118). Therefore, redox balance may be an inseparable link of ON in MSC differentiation.
Osteoblasts are required for the synthesis and mineralization of extracellular matrix. During osteogenesis, proliferous osteoblasts lay down osteoid and subsequently evolve into osteocytes embedded in mineralized bone matrix. ROS cause osteoblast dysfunction and restrain the expression of multiple osteoblastic markers including alkaline phosphatase, Runx2, type I collagen, bone morphogenetic protein-2 (BMP-2), osteonectin, and osteocalcin (7, 120). Bai et al. (7) reported that H2O2 stimulates extracellular signal-regulated kinases 1 and 2 (ERK1/2) phosphorylation, as well as phospholipase C-γ1 and ERK-dependent NF-κB activation in primary osteoblasts and bone marrow MSCs, resulting in reduced osteoblastic differentiation. Besides the osteoblast differentiation, Arai et al. (5) reported that H2O2 suppressed mineralization in osteoblast cell line. The canonical Wnt pathway enhances an accumulation of β-catenin in the cytoplasm, which promotes osteoblast differentiation after the translocation of β-catenin into the nucleus through the T-cell factor lymphoid-enhancer binding factor family of transcription factors (110). In high ROS environment, p66shc protein is phosphorylated and β-catenin is lost to ROS-activated FOXO transcription factors, which causes increased osteoblast and osteocyte apoptosis by inhibiting Wnt-induced osteoblast gene expression (110). ROS also play roles in the osteoblast differentiation and survival in association with BMP-2/Smad or Ca+-mediated mitochondrial pathways (34, 127). The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is a well known enzyme system that actively produces ROS. In bone, NADPH oxidases (Nox) family is a major source of ROS, such as H2O2 and O2 −. Especially, Nox4-derived ROS are involved in the mitochondrial dysfunction and apoptosis in osteoblast or osteocyte, potentially resulting in weakening bone tissue (179).
Conversely, ROS can have a positive effect on osteoclasts in bone metabolism (186). Osteoclasts are multinuclear cells derived from hematopoietic precursor cells and uniquely have bone-resorbing function. The receptor activator of NF-κB ligand (RANKL) is known as a type II transmembrane protein expressed on the surface of osteoblasts and stimulates RANK expressed in osteoclast progenitor cells to control osteoclast differentiation and maturation. Basically, ROS work on osteoclast differentiation as a potent multifunctional signal molecule in intercellular pathways, including RANKL, mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinases (PI3Ks), and NF-κB pathways (54, 175). The MAPK family such as ERKs, c-Jun N-terminal kinase (JNK), and p38 MAPK has a responsibility for the translocation of c-Fos and nuclear factor of activated T cells c1 (NFATc1) to drive osteoclast lineage (162). Bai et al. (8) reported that evoked intracellular ROS activated RANKL mRNA and protein expression in osteoblast-like cell line and primary bone marrow MSCs and osteoblasts and that ROS stimulated phosphorylation of ERKs, whereas ERKs inhibitor reduced ROS-induced RANKL expression in these cells. These findings reveal that ROS have a potential to induce osteoclastogenesis through the activation of RANKL expression via ERKs and 5′-cyclic adenosine monophosphate (cAMP)/protein kinase A/cAMP response element binding protein pathway in osteoblasts. Lee et al. (89) described the involvement of JNK and p38 in osteoclastogenesis through ROS generation. Plasma membrane-associated Nox protein induces endogenous ROS production through RANKL pathway, and then, ROS activate MAPK, PI3K, and NF-κB signaling, subsequently driving osteoclast maturation by expressing genes, such as cathepsin K, matrix metalloproteinase-9, and NFATc1 (162, 163).
Oxidative stress-related vascular events
Vascular endothelial cells are squamous cells that constitute the inner cellular lining of arteries, veins, and capillaries. The vascular endothelium contributes the maintenance of vasculature homeostasis by producing and releasing vasorelaxant factors, such as nitric oxide (NO), prostaglandin I2 (PGI2, prostacyclin), and endothelium-derived hyperpolarizing factor (EDHF) (44, 161). Under physiological conditions, H2O2 acts as an EDHF to protect the vascular tissues (112, 161). However, excessive ROS production can lead to the endothelial dysfunction that underlies the development of cardiovascular diseases, such as atherosclerosis, complications from diabetes, or ischemia–reperfusion injury.
Intracellular ROS generation in vascular endothelial cells comes primarily from four major enzyme systems, including Nox, xanthine oxidoreductase, uncoupled endothelial nitric oxide synthase (eNOS), and mitochondrial respiration complexes (16, 20). Although Nox-derived ROS are crucial for cell signaling within the vascular compartment, endothelium-specific overexpression of Nox clearly promotes vascular dysfunction and oxidative stress, leading to disease progression (18, 95). Xanthine oxidoreductase, a urate-producing enzyme, is converted into xanthine oxidase by processes including thiol oxidation and/or proteolysis and is thought to increase the cardiovascular diseases via oxidative radical production (16). In vascular system, ROS reduce eNOS expression and NO production. Moreover, ROS also increase O2
− production through Nox activation. The peroxynitrite (ONOO−) forms by combining generated O2
− with the free radical NO, thereby leading to a decrease in NO bioavailability in vascular endothelial cells (18, 149). Then, ONOO− causes eNOS uncoupling by a depletion of eNOS cofactor tetrahydrobiopterin (BH4), an
Oxidative stress in cerebral or coronary artery vasospasm causes decreased blood flow to distal tissues resulting in ischemia (49, 190). Cerebral vasospasm is generally induced by the presence of a blood clot in the subarachnoid space after subarachnoid hemorrhage, and oxyhemoglobin is identified as the main mediator of cerebral vasospasm. There are several sources of excessive free radical generation associated with cerebral vasospasm, including mitochondrial oxidative stress, oxyhemoglobin free radical generation, enzymatic sources of free radicals, and disrupted antioxidant protection (189). In these vasospasms, elevated oxidative stress and endothelial dysfunction lead to vasoconstriction through NO degradation or increased coronary smooth muscle Ca2+ sensitivity by member A Rho kinase/Rho-associated kinase pathway activation (190). Ikemura et al. (67) reported that an anti-vasospasm agent had a preventive effect on the development of corticosteroid-induced ON in rabbits via the regulation of the Rho-kinase pathway.
Environmental factors such as hypoxia or ischemia produce a large amount of ROS. Sato et al. (148) induced complete ischemia of the femoral head in rats by transverse diaphysis osteotomy and hip dislocation and reported that two stress proteins (oxygen-regulated protein 150 and heme oxygenase 1 [HO-1]) were elevated in the acute phase of ischemia and that apoptosis of osteoblasts or osteocytes was subsequently observed in the proximal fragment of femur. This result suggests that necrotic changes can be caused by ischemia as a result of oxidative stress. Ischemia–reperfusion injury is defined as the (paradoxical) exacerbation of cellular dysfunction and death following restoration of blood flow to previously ischemic tissues. Antioxidants can play an important role in cardioprotection and have therefore emphasized that ROS-mediated stress injury is involved in the pathology of myocardial ischemia–reperfusion injury (86). Previous studies suggest that ischemia–reperfusion injury also plays a major role in the development of ON using animal models or angiography for ONFH patients (6, 58).
Oxidative Stress-Related Markers in ONFH Patients
Nontraumatic ONFH occurs mainly in young people, and 10,000–20,000 new ONFH cases are diagnosed every year in the United States (168). Some patients suffer from severe hip pain due to femoral head collapse and subsequent hip joint dysfunction, and most of them require surgical treatment (121). ONFH is observed largely in patients who receive high doses of corticosteroid, drink excessive amounts of alcohol, or smoke (68, 78). Previous studies have shown that these substances induce oxidative stress and result in harm to several organs, including bone tissues and blood vessels (13, 25, 26, 70, 125, 144). Indeed, samples taken from ONFH patients (such as blood samples, extracted femoral head, or cells) show evidence that emphasizes the presence of oxidative stress in ONFH.
Oxidative stress-related studies (genetics, epigenetics, metabolomics, etc.) for ONFH patients
Several studies have found abnormal oxidative stress and/or antioxidant markers in ONFH patients (27, 82, 106, 132, 157, 158, 181, 198). Okura et al. (132) analyzed serum levels of antioxidant nutrients in 39 ONFH patients and 78 age- and gender-matched healthy people as control and found significantly lower total carotenoid [a class of lipid-soluble natural pigments known as antioxidant nutrients (115)] levels in ONFH patients. Liu et al. (106) examined plasma samples from 30 ONFH patients and 30 normal subjects and reported abnormal antioxidant-related metabolites in ONFH patients through the active nucleotide and reduced glutathione (GSH) pathways. GSH inhibits an increase in lipid peroxide [LPO, a biochemical indicator of tissue injured by active oxygen (167)] and maintains a cellular oxidation balance that favors reduction (17). Wu et al. (181) investigated serum proteome profiles of 11 adult ONFH patients compared with 11 healthy volunteers and found ONFH patients to have lower serum levels of apolipoprotein A-IV, which attenuates lipid oxidation (154). Zheng et al. (198) reported eNOS gene polymorphisms in 125 nontraumatic ONFH patients compared with 126 healthy controls by genotyping, suggesting that eNOS gene polymorphisms may be a risk factor for ONFH. Kim et al. (82) conducted an association analysis of genotyped single nucleotide polymorphisms and haplotypes in 443 ONFH patients and 273 control subjects and reported a genetic effect of catalase on the risk of ONFH. Catalase is an endogenous antioxidant enzyme that protects cells against ROS damage by converting H2O2 into oxygen and water (143). Using extracted bone tissue from corticosteroid-associated ONFH patients, Chen et al. (27) reported that genes encoding for antioxidant enzymes, including those for catalase, NAD(P)H quinone dehydrogenase 1 (NQO1) and HO-1, were notably downregulated and that the protein levels of antioxidant enzymes such as catalase, γ-glutamylcysteine synthetase (γ-GCS) and superoxide dismutase 1 (SOD1) were lower in the necrotic bone. Sun et al. (157, 158) isolated MSCs from the proximal femur during hip surgery, reporting that MSCs from corticosteroid-associated ONFH patients showed elevated ROS level and depressed oxidative stress-related genes such as SOD and catalase compared with patients with femoral neck fracture. Bosco et al. (15) reported that hyperbaric oxygen therapy reduced plasma levels of TNF-α and interleukin 6 by temporary ROS production in ONFH patients, suggesting the benefit of moderate ROS by modulating inflammation (15). These results show that antioxidant function has already been depleted or become deficient in ONFH patients and thus raise the possibility that insufficient antioxidant function due to excessive ROS might be one of the critical factors behind irreversible events during the developmental process of ONFH (Table 1).
Oxidative Stress-Related Markers in Osteonecrosis of the Femoral Head Patients
γ-GCS, γ-glutamylcysteine synthetase; CAT, catalase; eNOS, endothelial nitric oxide synthase; HO-1, heme oxygenase 1; MSC, mesenchymal stem cell; NQO1, NAD(P)H quinone dehydrogenase 1; ONFH, osteonecrosis of the femoral head; ROS, reactive oxygen species; SOD1, superoxide dismutase 1.
An Oxidative Stress-Induced ON Animal Model
Experimental animal models suggest that oxidative stress can closely related to the pathogenesis of ONFH (64, 65, 148). In a notable example, Ichiseki et al. (65) reported on the development of oxidative stress-induced ON in a rat model. For this study, male Wistar rats were subcutaneously injected with 500 mg/kg of buthionine sulfoximine (BSO, an inducer of oxidative stress) for 14 consecutive days. The authors reported significantly decreased production of the antioxidant GSH in rats treated with BSO compared with the nontreatment group, and they detected ON in 71.4% treated with BSO based on empty lacunae and adjacent fatty bone marrow cells in histopathology. They also found up to 40% ON with only one-time injection of 500 mg/kg BSO (63). These authors, moreover, suggest that BSO may impair redox function by blocking synthesis of GSH in the organism, thereby inducing oxidative stress. It is interesting to note that total blood cholesterol and triglyceride levels did not change significantly despite a marked decrease in blood GSH levels. This suggests that necrotic changes in bone can be induced by oxidative stress alone and that oxidative stress induced by decreased GSH may suffice to cause ON even without lipid abnormalities. Although the employed model does not fully simulate clinical ON in humans (in light of the multiple factors associated with ONFH), these results nonetheless suggest that oxidative stress by itself can induce ON (at least in rats) and may play a major role in the development of ON (at least among other factors).
Oxidative Stress and Corticosteroids
Corticosteroid administration and ONFH
Corticosteroids are irreplaceable medications used broadly to treat chronic autoimmune diseases such as systemic lupus erythematosus, or for immunosuppression subsequent to organ transplants (51). Frequent occurrence of corticosteroid-associated ONFH has been reported in patients who receive high-dose corticosteroid therapy for these underlying diseases. Epidemiology studies from East Asia showed that 47.4% of all cases diagnosed as nontraumatic ONFH were directly associated with corticosteroids (68). Despite a clinical correlation between corticosteroid use and ONFH, the molecular mechanisms behind the development of ONFH have not been completely understood. With corticosteroid-associated ONFH appearing mainly in young patients, and with relatively many requiring surgery to alleviate severe hip pain after femoral head collapse or secondary arthritis of the hip (98), it would be highly desirable to clarify the pathophysiology of corticosteroid-associated ONFH and to develop and implement a preventive strategy for such patients.
Effects of corticosteroid on bone and vascular tissues
Presumably, corticosteroid-associated ONFH is attributed to multiple effects of the corticosteroid on cells (78). Several studies have demonstrated corticosteroid-induced osteoblast or osteocyte dysfunction under in vitro conditions (166, 177, 184, 200). Most hematopoietic bone cells, osteoblasts, and osteocytes in the femoral cortex undergo apoptosis after long-term corticosteroid medication in an animal model (12, 75). Widespread apoptosis of these cells, with accompanying necrotic lesion, is also observed in corticosteroid-associated ONFH patients (23, 178). But other studies have shown that the early bone loss associated with excess corticosteroid use is caused by a direct effect of corticosteroids on osteoclasts: to extend their life span (176). One report has shown significant bone loss with elevated tartrate-resistant acid phosphatase (TRAP)-positive osteoclast counts in the femoral head in a corticosteroid-induced ON animal model (27). The same study also reported a robustly upregulated expression of osteoclast-related proteins such as TRAP or cathepsin K in necrotic lesions of extracted femoral head in corticosteroid-associated ONFH patients. Therefore, excess corticosteroids are capable of directly affecting bone metabolism (at least through suppression of osteoblast and osteoclast precursor production in bone marrow) and of increasing apoptosis and necrosis of osteoblasts and osteocytes as well as of activating osteoclasts (27, 78).
Previous research has also investigated the effects of corticosteroids on vascular endothelial cells (3, 131). High-dose corticosteroids have been reported to induce cell cycle arrest and apoptosis of vascular endothelial cells, and to cause severe effects in combination with transfection of hypoxia-inducible factor-1α (131). Direct injury from corticosteroids and an indirect effect from corticosteroid-induced ROS overproduction on vascular endothelial cells may combine to affect the synthesis of NO; this would aggravate blood coagulation and thrombosis formation (78). High-dose corticosteroid use also promotes a procoagulant effect in the blood through decreased tissue plasminogen activator and increased plasma plasminogen activator inhibitor-1 antigen levels (78, 173).
As for the effects of direct corticosteroids on the femoral head-feeding vessels in animal models, Drescher et al. (37 –39) reported that corticosteroid treatment is capable of affecting vasoactive mediators, such as endothelin-1, bradykinin, and noradrenalin, and can regulate local blood flow by modulating vascular responsiveness to vasoactive substances. Corticosteroids were reported to cause bone marrow fat cell enlargement, sometimes with serious intramedullary infiltration and raised intraosseous pressure, in a corticosteroid-induced ON model (118). Zhou et al. (199) reported a fatty tamponade in the medullary cavity of the femoral head and narrow intramedullary vascular sinusoids compressed by an excess of lipocytes in a corticosteroid-induced ON rabbit model. The authors also report sparse intramedullary vessels and a decreased vascular area in the femoral head after corticosteroid treatment. Yin et al. (191) reported a high number of adipocytes in corticosteroid-induced marrow stromal cell cultures as well as increased adipose-specific gene expression, but decreased osteogenic gene expression, compared with the control. This indicates that corticosteroids can directly induce differentiation of marrow stromal cells into a large number of adipocytes and inhibit their osteogenic differentiation. These studies suggest that corticosteroids may cause ischemic ON by elevating intraosseous pressure and subsequently decreasing blood flow to the femoral head through adipogenesis and fat hypertrophy in the bone marrow.
Corticosteroid-induced oxidative stress
Corticosteroids are usually adaptable, mobilizing energy to tissues in an emergency and restraining unnecessary anabolism, whereas overdose or long-term administration of corticosteroids can have various deleterious effects on multiorgan systems. Generally, corticosteroids medication is closely related to ROS generation. Indeed, corticosteroid treatment can directly inhibit intracellular ROS production, but large amount of corticosteroid activity can elevate oxidative stress to protein through mitochondrial dysfunction (113, 147).
However, the mechanism of corticosteroid causing redox imbalance has not been completely understood due to the extraordinarily diverse cellular responses. Du et al. (40) have shown that glucocorticoid receptors (GRs) interact with the antiapoptotic protein Bcl-2 and translocate into mitochondria in response to corticosteroid, subsequently regulating mitochondrial functions. They also reported that long-term treatment with high-dose corticosterone decreased GR and Bcl-2 levels in mitochondria. Considering the inhibitory effect of Bcl-2 for mitochondrial ROS production, it is speculated that corticosteroids can be involved in ROS generation in mitochondria. Interestingly, a recent article (29) reported that long-term incubation of exogenous corticosteroid increased intracellular ROS production and reduced mitochondrial functions such as ATP production or basal respiration rate in osteoblasts, regardless of no significant change of ROS production or elevated mitochondrial functions in short-time corticosteroid exposure. Conversely, this treatment did not clearly induce intracellular ROS production and mitochondrial dysfunctions in osteoclasts. Iuchi et al. (70) reported an increase in H2O2 production and a decrease in NO availability by high-dose corticosteroid treatment in human umbilical vein endothelial cells. They also showed that the major production sources of ROS by high-dose corticosteroid treatment were mitochondrial electron transport chain, Nox, and xanthine oxidase by using inhibitors against metabolic pathways for ROS generation. These findings reveal that excess corticosteroid exposure can cause ROS overproduction and thereby perturb NO availability in the vascular endothelium, leading to various complications in vasculature.
The presence of oxidative stress acceleration has been confirmed soon after corticosteroid administration in an established corticosteroid-induced ON model (62, 64, 114). Ichiseki et al. (64) examined vascular permeability using a corticosteroid-induced ON rabbit model and reported that blood circulatory dysfunction (such as elevated vascular permeability and impaired hemostasis) occurred as soon as 5 days after corticosteroid administration when oxidative stress was shown to occur. The presence of oxidation injury in bone has been confirmed in a corticosteroid-induced ON model by the detection of strong immunohistochemical staining for oxidative stress in bone marrow vessels, myelocytes and adipocytes (62, 87, 119). These suggest that the early occurrence of these two events created conditions in which vascular and tissue injuries were also likely to occur quite soon after corticosteroid administration. It is noteworthy that such effects of corticosteroids largely overlap with those of ROS on bone and blood vessels. In particular, the unfortunate fact appears to be that oxidative stress is indirectly involved in NO inactivation (through vascular endothelial cell injury) in the pathology of corticosteroid-associated ONFH (78), which causes ischemia.
In brief, from these previous reports, overadministration of corticosteroid can not only directly exert oxidative stress on bone and vascular cells but also cause ROS-induced tissue damage in corticosteroid-induced ON animal models, which strongly supports the hypothesis that ROS may be an essential (although themselves an insufficient) part of the mechanism behind corticosteroid-associated ONFH (64).
Preventive Effects of Antioxidants in a Corticosteroid-Induced ON Animal Model
The establishment of a preventive therapy for ONFH is a long-standing challenge, especially since some patients cannot avoid receiving high-dose administrations of corticosteroids. Many researchers have attempted to establish methods of preventing corticosteroid-associated ONFH in humans by examining corticosteroid-induced ON models in rabbits (185). Many types of drugs, including lipid-lowering drugs (71, 122, 128, 137), anticoagulants (11, 122), and vasodilation drugs (36), have been reported to effectively suppress the development of corticosteroid-induced ON in this model.
Various antioxidants have also been reported on the rabbit model as useful in preventing ON through inhibition of oxidative stress (59, 64, 87, 94, 96, 108, 114, 153, 194, 196) (Table 2). Li et al. (96) examined the protective effects of molecular hydrogen [reported as a novel selective antioxidant in preventive and therapeutic applications (130)] on corticosteroid-induced ON and reported that the incidence of ON was significantly lower in the hydrogen group (29%) than in the model group (68%). The authors also demonstrated that molecular hydrogen increased blood levels of GSH but decreased malondialdehyde (MDA, a biochemical indicator of oxidative stress-related injury). Zhai et al. (194) demonstrated that the sequential injection of resveratrol, a natural compound found in grapes, decreased ON from 75% to 44%. Huang et al. (59) also reported that hydrogen-rich saline [an alternative form of molecular hydrogen known as a safe and easily available antioxidant for medical purposes (130)] significantly decreased LPO even as it increased GSH plasma levels compared with the nontreatment group; and they observed a significant increase in microvessel density. Song et al. (153) reported that grape seed proanthocyanidin extract [widely used compounds with free radical scavenging and antioxidant properties (46)] reduced the incidence of ON to 18% as opposed to 88% in the corticosteroid-only group. They also found that this medication supported a decrease in tissue SOD and GSH peroxidase activities. These antioxidant enzymes reportedly counteracted the accumulated peroxides produced by the corticosteroid. Oppositely, it suppressed the increased tissue MDA levels and 8-hydroxy-2′-deoxyguanosine (8-OHdG), positive cells in immunohistochemistry in bone tissue. 8-OHdG is produced when DNA sustains oxidative injury from ROS (169). Li et al. (94) reported a protective effect of edaravone against ON. Edaravone is a strong, novel free radical scavenger with promising antioxidant functions, including enhancement of prostacyclin production, inhibition of •OH-dependent and -independent LPO, and quenching of active oxygen (56). The authors found an incidence of ON in the treatment group (20%) significantly lower than in that without treatment (73%) and observed higher blood GSH and lower LPO levels in the treatment group. Histopathologically, the incidence of thrombosis and numbers of intraosseous vessels and hematopoietic cells damaged by oxidative injury were significantly lower in the treatment group. Lu and Li (108) investigated the effect of lipoic acid [a both water- and fat-soluble natural antioxidant capable of penetrating tissues to protect them from free radical damage (152)] on preventing corticosteroid-induced ON and observed a significantly lower incidence of ON in the treatment group (21%) versus nontreatment (73%), and higher GSH and lower MDA plasma levels in the treatment group. Mikami et al. (114) reported preventing corticosteroid-induced ON with intravenous administration of vitamin E, a well-known fat-soluble antioxidant that can protect polyunsaturated fatty acids from oxidation and regulate ROS production (88); they thereby demonstrated complete prevention of corticosteroid-induced ON (as opposed to a 93% rate of developing ON absent treatment) as well as suppression of the GSH blood level decline associated with treatment. Kuribayashi et al. (87), moreover, reported an effect of α-tocopherol [known as the most potent form of vitamin E (99)] on the prevention of corticosteroid-induced ON. The authors showed that a diet supplemented with α-tocopherol reduced ON incidence from 70% without treatment to 24% in the treatment group. Zhang et al. (196) described a dose-dependent effect of icaritin [a component of Epimedium flavonoid isolated from Chinese Herba Epimedii that reportedly has an antioxidant effect (104)] in reducing the incidence of corticosteroid-induced ON. These authors showed a significantly lower incidence of ON in high-dose treatment (6%) as opposed to nontreatment (94%) as well as a higher GSH/LPO ratio in plasma. Ichiseki et al. (64) reported the rate at which ON developed when treated with the antioxidant GSH as 0% as opposed to a nontreatment rate of 70% as well as higher GSH and lower LPO blood concentrations in the treatment group. Antioxidants such as sesamin, a porous control release of Selenium (Se@SiO2), Coenzyme Q10 aliphatic acid D-003, or resveratrol, have already proven therapeutic in corticosteroid-induced ON in rats (31, 32, 83, 123, 129).
Experiments Describing Preventive Effect of Antioxidants for Osteonecrosis Onset in Corticosteroid-Induced Rabbit Model
8-OHdG, 8-hydroxy-2′-deoxyguanosine; GSH, glutathione; GSPE, grape seed proanthocyanidin extract; GSH-Px, glutathione peroxidase; IHC, immunohistochemistry; LPO, lipid peroxide; LPS, lipopolysaccharide; MDA, malondialdehyde; MPSL, methylprednisolone; ON, osteonecrosis.
Several reports have furthermore described cytoprotective effects of antioxidants on bone cells under corticosteroid induction (Table 3). Yamamoto et al. (184) reported that the administration of hemin (an inducer of HO-1) significantly reduced osteocyte cell death due to corticosteroids and hypoxia. Jia et al. (72) indicated an inhibitory effect of vitamin E on osteocyte apoptosis and DNA oxidative damage in bone marrow hematopoietic cells in early-stage corticosteroid-induced ON. Using quantitative TUNEL and caspase 3 assay, Song et al. (153) demonstrated lower apoptosis of osteocytes due to corticosteroid administration where grape seed proanthocyanidin extract had been given. Sun et al. (155) reported that edaravone had a protective effect against osteoblastic cell death from dexamethasone exposure as it inhibited oxidative stress and mitochondrial permeability transition pore opening. Yang et al. (187) described the effect of hydrogen sulfide [a substance that functions as an antioxidant by scavenging ROS directly (28)] on corticosteroid-induced osteoblast cell damage and reported that the treatment group exhibited dramatically inhibited dexamethasone-induced viability reduction, apoptosis, and lactate dehydrogenase release in osteoblastic MC3T3-E1 cells. Inkielewicz-Stepniak et al. (69) demonstrated that fisetin [a dietary flavonoid whose antioxidant effect has been touted as promoting health (79)] prevented dexamethasone-induced oxidative damage in osteoblastic MC3T3-E1 cells. Liu et al. (103) have reported that fullerol [a water-soluble fullerene derivative demonstrated to act as a potent antioxidant (52)] works against dexamethasone-induced oxidative stress and adipogenesis while simultaneously enhancing osteogenesis through increased RUNX2 and osteocalcin expression in a cloned bone marrow MSC.
Cytoprotective Effect of Antioxidants for Bone Cells Under Corticosteroid Induction
Such results suggest that various types of antioxidants might greatly reduce the incidence of ON (by a range of 31% −93%) in a corticosteroid-induced ON rabbit model compared with the nontreatment group (64, 87, 94, 96, 108, 114, 153, 194, 196). Briskly put, antioxidant substances may help prevent corticosteroid-associated ONFH by alleviating oxidative injury. And this, moreover, indirectly supports the contention that oxidative stress is associated with the onset of corticosteroid-induced ON. Most of the cited antioxidants are nontoxic, safe, and widely used to treat patients with other diseases in clinic. Using these antioxidants thus would seem highly practical. Mikami et al. (114) stand out in having completely prevented corticosteroid-induced ON by administering vitamin E intravenously regardless of an extremely high dose (40 mg/kg) of methylprednisolone. Lu and Li (108) also reported a possible effect of combining lipoic acid and vitamin E because of its inhibitory effect on both water and fat soluble. In light of the few side effects of antioxidants, clinical use of vitamin E in combination with other antioxidants may be useful in preventing ON.
But some rabbits did eventually develop ON despite antioxidant treatment in most of these studies. This may be because ON is caused by multifactorial events including lipid metabolism disorders and abnormalities in the coagulation and fibrinolytic systems. Indeed, so far no preventive effect with respect to ONFH has been reported for a single antioxidant administration in humans. Agent sensitivity and the anatomical mechanism behind ON in humans and rabbits may differ. Additionally, in many of these reports, increased antioxidant activity was confirmed by blood levels of antioxidant or oxidant markers (64, 94, 96, 108, 114, 196). At the same time, several reports have demonstrated a decrease in oxidant markers upon antioxidant administration by using immunohistochemistry (87, 94, 96, 153). Given that antioxidants can reduce local corticosteroid-induced oxidative injury in bone and vessel tissues, parallel coverage with antioxidants during high-dose corticosteroid administration may well reduce necrotic events by suppressing local oxidant injury—even in humans.
In addition, the role of metabolic enzymes for corticosteroid is essential in considering corticosteroid-induced ON. Hepatic cytochrome P4503A (CYP3A) enzymes play a central role in corticosteroid metabolism. Kaneshiro et al. (76) reported lower hepatic CYP3A activity in patients with corticosteroid-induced ONFH compared with non-ONFH control patients, and they indicate that low hepatic CYP3A activity can significantly contribute to the risk of corticosteroid-induced ONFH. In the corticosteroid rabbit ON model, Masada et al. (111) reported that the CYP3A inducer phenobarbital suppressed the occurrence of ON from 83% to 33%, whereas Iwakiri et al. (71) reported that simvastatin, a lipid-lowering drug, not only reduced the incidence of ON but also increased hepatic CYP3A activity. However, the effect of antioxidants on hepatic CYP3A activity is unknown but intriguing.
Role of Nrf2 on Bone and Vascular Tissues Under Oxidative Stress
Nrf2 and antioxidant system
A complex cellular defense mechanism protects cells against exposure to toxic radicals by activating an antioxidant response reaction upon sensing oxidative stress (160). In this system, Nrf2 plays a central role in redox homeostasis as a major stress–response transcription factor. Nrf2 enhances resistance to many types of harmful stress by strongly inducing expression of genes that encode for detoxification enzymes and antioxidant proteins. Stabilized Nrf2 migrates into the nucleus, where it binds to antioxidant response elements (AREs) in the promoter regions of target genes and, together with small Maf proteins, activates nearly all the antioxidant enzymes (160, 180). Nrf2 is considered a master regulator of the ARE-driven cellular defense against oxidative stress. In the absence of oxidative stress, however, Nrf2 is ubiquitinated by the Kelch-like erythroid cell-derived protein with cap “n” collar homology-associated protein 1 (Keap1) dimer and the cullin-3 (Cul3-) ring box (Rbx) complex, which subsequently leads to Nrf2 degradation by proteasome (126, 195). Nrf2 has been reported to serve a crucial function in preventing several degenerative diseases in multiple organs (4, 180).
Recently, there have been emerging researches focused on biological relevance for cell or tissue protection in various diseases of the redox homeostasis elicited by the activation of vitagene signaling pathway (164). Several redox-sensitive genes, such as 70 kDa heat shock protein, HO-1, thioredoxin/thioredoxin reductase, and sirtuins system, are upregulated as part of a cytoprotective response that protects against various electrophiles and oxidants (93, 138). All these cytoprotective genes can be transcriptionally modulated by Nrf2 in case of electrophile counterattack coordinately with a battery of cytoprotective proteins, including NQO1, uridine 5′-diphospho-glucuronosyltransferase, and glutathione S-transferases (21, 93). Given the relationship between redox status and the Nrf-2-dependent vitagene network and its possible biological relevance in cellular redox homeostasis (21, 35), Nrf2 is thought to be essential for regaining its homeostasis in the development process of ONFH that causes a disturbed redox state due to ROS excess. We discuss below the role of Nrf2 in ONFH-related bone and vascular cells.
Role of Nrf2 on bone
By counteracting ROS through various antioxidant genes expression, Nrf2 can help avoid ROS-induced events, including cell damage by nuclear and mitochondrial DNA mutation, various cell deaths (apoptosis, necrosis, and necroptosis), reduced osteoblast differentiation, and elevated osteoclast activation (30, 91, 116, 146, 172, 182, 186). Nrf2-target antioxidant gene HO-1 is important in determining whether bone marrow-derived MSCs differentiate into osteoblasts or adipocytes through specific signaling pathway (9). HO-1 induction has been reported to decrease the expression of peroxisome proliferator-activated receptor γ through activation of Wnt/β-catenin signaling and to suppress adipocyte differentiation (172).
There have been previous articles describing the protective effects of noncoding RNA in osteoblasts from oxidative stress through Nrf2 activation (43, 183). Fan et al. (43) tested the role of the long noncoding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) in human primary osteoblasts and osteoblastic cell lines, reporting that dexamethasone-induced ROS production was significantly attenuated by lentiviral vector-MALAT1 transfection in osteoblasts through AMP-activated protein kinase (AMPK) and Nrf2 signaling. Xu et al. (183) investigated the effect of a putative Cul3, an indicator of Nrf2 degradation, targeting microRNA “miR-455” on human primary osteoblasts and osteoblastic cell lines under high H2O2 condition, showing that forced expression of miR-455 prevented cell apoptosis by Cul3 degradation and Nrf2 protein stabilization and led to subsequent transcription of ARE-dependent genes NQO1 or HO-1. Liu et al. (105) also demonstrated the potential effect of icariside II on dexamethasone-induced murine osteoblast cell damages by activating epidermal growth factor receptor/Akt/Nrf2 signaling. Regarding osteoclastogenesis, Kanzaki et al. (77) showed that RANKL stimulation upregulated Keap1 while downregulated Nrf2, resulting in a decrease in the Nrf2/Keap1 ratio and downregulation of cytoprotective enzymes such as HO-1 and γ-GCS in murine osteoclast progenitor cells. Conversely, Nrf2 overexpression upregulated cytoprotective enzymes and reduced ROS levels, leading to decreased number of TRAP-positive multinuclear cells, and attenuated osteoclast differentiation and bone resorption in both in vitro and in vivo models (77). Li et al. (102) revealed that glycyrrhizin inhibited RANKL-induced osteoclastogenesis and reduced ROS production by inhibiting the MAPK and NF-κB pathways and activating the AMPK/Nrf2 signaling.
Nrf2 deficiency causes increased intracellular ROS levels and defects in the production of numerous antioxidant enzymes in both osteoblasts and osteoclast precursors (60, 141). Rana et al. (141) reported that calvarial osteoblasts isolated from Nrf2-knockout and wild-type (WT) mice showed an approximately 50% decrease in matrix formation in the absence of Nrf2. Hyeon et al. (60) reported that RANKL-induced osteoclast differentiation, actin ring formation, and osteoclastic bone resorption were all substantially promoted in Nrf2-deficient osteoclast precursor cells compared with WT cells. Currently, the influence of chronic Nrf2 deficiency on bone acquisition is quite controversial (61, 81, 135, 136, 156). Micro-computed tomography analysis of several animal studies has shown that the deletion of Nrf2 in bone tissue leads to lower bone mass, as demonstrated by bone parameters such as bone mineral density, bone volume fraction, or trabecular thickness (61, 81, 156). Conversely, Park et al. (135) reported that bone parameters such as bone volume fraction or trabecular thickness were significantly higher in Nrf2-knockout mice compared with age-matched WT mice. Pellegrini et al. (136) have reported that Nrf2 can regulate mass accrual in bone differently depending on age and gender.
Some reports have shown bone mass formation in Nrf2-activated bone tissue in a shortage of Keap1 (192, 193). Yoshida et al. (193) have shown lower bone mineral density in Keap1-knockout mice, indicating that Nrf2 hyperactivation can lead to hypoplasia in bone. Yin et al. (192) reported that male Keap1-heterozygotic mice had a significantly higher bone volume fraction compared with WT mice despite no difference appearing in female mice; this would suggest that moderate Nrf2 activation with disruption of Keap1 may improve bone mass by regulating bone remodeling in male mice. One prior study showed reduced callus formation and osteoblast activity in Nrf2-knockout mice, indicating that Nrf2 deficiency impairs fracture healing (84, 101). It is therefore clear from the foregoing that Nrf2 plays an important role not only in long-term bone acquisition but also in acute bone repair and regeneration. These previous studies indicate that Nrf2 not only can strongly influence bone metabolism through osteoblastogenesis and osteoclastogenesis, but the experiments with Nrf2 or Keap1 knockout mice have shown that Nrf2 is also essential in bone acquisition and bone regeneration.
Role of Nrf2 on vascular diseases
Against these ROS-induced vascular endothelium dysfunctions, the following possible role of Nrf2-target gene HO-1 to prevent eNOS and subsequent NO reductions has been reported (134). Batzlsperger et al. (10) reported that continuous HO-1 overexpression by oncoretroviral vectors reduced eNOS expression in human endothelial cells, suggesting that continuously elevated HO-1-activity can protects vascular endothelium from oxidative stress through a decrease in eNOS expression and activity (10, 134). Under the redox-stressed endothelium condition, avoiding the risk of eNOS uncoupling may be necessary to maintain vascular homeostasis. Heiss et al. (55) have reported that active Nrf2 contributes to keep eNOS in the coupled state through elevated HO-1. HO-1 prevents the accumulation of O2 − through inhibiting Nox activity and/or BH4 oxidation and increasing SOD and catalase (73, 165). Also, Pae et al. (134) have suggested the possibility that the HO-1/carbon monoxide (CO) pathway can compensate for the loss of NO bioavailability by progressing soluble guanylate cyclase/cyclic guanosine monophosphate levels, anti-inflammatory, antiproliferative, and antiapoptotic effects.
Nrf2 contributes to the angiogenic potential of both vascular endothelial cells as well as bone marrow-derived proangiogenic cells (47). It is implicated in the regulation of vascular events including thrombosis (188), vasospasm (197), and ischemia–reperfusion injury (150). Nrf2-HO-1 signaling is reported to ameliorate oxidative stress injury in vascular endothelial cells and in the subsequent thrombotic response through inhibition of prothrombotic von Willebrand Factor and enhancement of NO and PGI2, implying a promising strategy for thromboembolism (188). Previous studies have also reported on the role of the Nrf2-ARE pathway in cerebral vasospasm, where it suppresses the release of proinflammatory cytokines (97, 197). The Nrf2-ARE pathway is one of the essential signaling pathways to attenuate myocardial infarct size and to preserve cardiac function after myocardial hypoxia and ischemia–reperfusion injury (42, 150). As the clinical relevance of HO-1 expression in vascular disease has been highlighted previously (1), the Nrf2/HO-1 pathway may offer a promising therapeutic strategy for vascular diseases associated with reduced NO bioavailability, and it may be no exception for ONFH.
Conclusions and Future Directions
This review summarizes the current state of knowledge about the pathogenesis of ON with a special focus on oxidative stress. Oxidative stress damages nuclear and mitochondrial DNA in cells and induces various cell deaths such as apoptosis, necrosis, and necroptosis via the mitochondrial or death receptor (91, 116, 133, 142, 146, 182). In bone cells, oxidative stress directly affects bone metabolism by activating osteoclasts and reducing osteoblastogenesis in MSCs, osteoblasts, and osteocytes (24, 162, 175). The abnormalities in bone metabolism caused by ROS overproduction may contribute considerably to the irreversible necrotic changes of bone tissue in ONFH through a decrease in capacity for bone repair and regeneration. Oxidative stress leads to vascular endothelial cell dysfunction by reducing NO bioavailability (16, 18, 85), resulting in a variety of vascular events including blood coagulation, platelet aggregation, thrombosis formation, vasospasm, and ischemia–reperfusion injury (20, 86, 92, 109, 151, 190). This may ultimately lead to ischemia, hypoxia, and occlusion of the nutrient arteries in the femoral head, given the link between these vascular mechanisms and ONFH reported in previous publications (53, 58, 67, 74, 122) (Fig. 1).
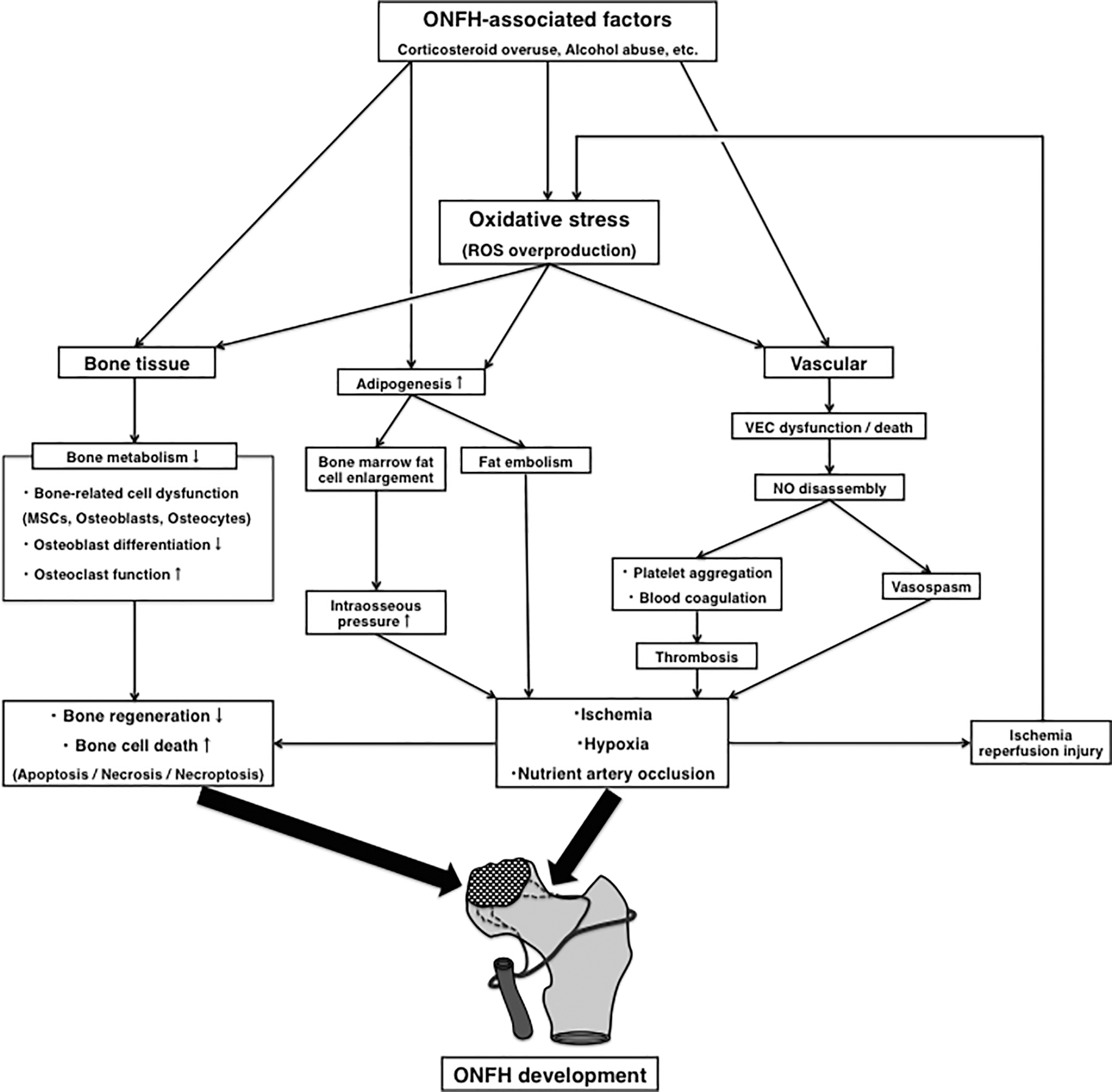
Previous studies have detected ON in rats treated with a single oxidant-inducing agent by empty lacunae and adjacent bone marrow fatty cells in histopathology (63, 65). This strongly supports the possibility that oxidative stress may be directly involved in ON development. Additionally, corticosteroids, one of the main causes of ON, can lead to ROS overproduction via mitochondria only in case of overdose beyond the physiological range or long-term administration (113, 147). Indeed, various oxidative stress markers have been detected in etiological studies of corticosteroid-associated ONFH patients (27, 82, 106, 132, 157, 158, 181, 198). Other reports have shown that by inhibiting oxidative stress, various antioxidants are useful in preventing corticosteroid-induced ON in rabbits (64, 87, 94, 96, 108, 114, 153, 194, 196), suggesting that antioxidants may have a preventive effect on ON. However, no previous studies have reported that single-agent antioxidants effectively prevent ONFH in human, and the pharmacological prevention for ONFH by using other types of drugs is still under debate (2, 124, 140, 145). Drugs that target the activation of cellular defense systems in a comprehensive manner, as well as multidrug therapy including antioxidants, may offer a way to prevent the development of ONFH in the future. The Nrf2-dependent cytoprotective network helps to maintain redox homeostasis with vitagene expressions and further antioxidant enzymes (21, 35, 93). This review has shown that induction of the Nrf2/HO-1 pathway has the potential to protect bone and vascular cells from toxicity, preserve their capacity for bone repair or regeneration (9, 43, 77, 102, 105, 172, 183), and prevent ONFH-related vascular events (1, 10, 42, 97, 134, 150, 188, 197)
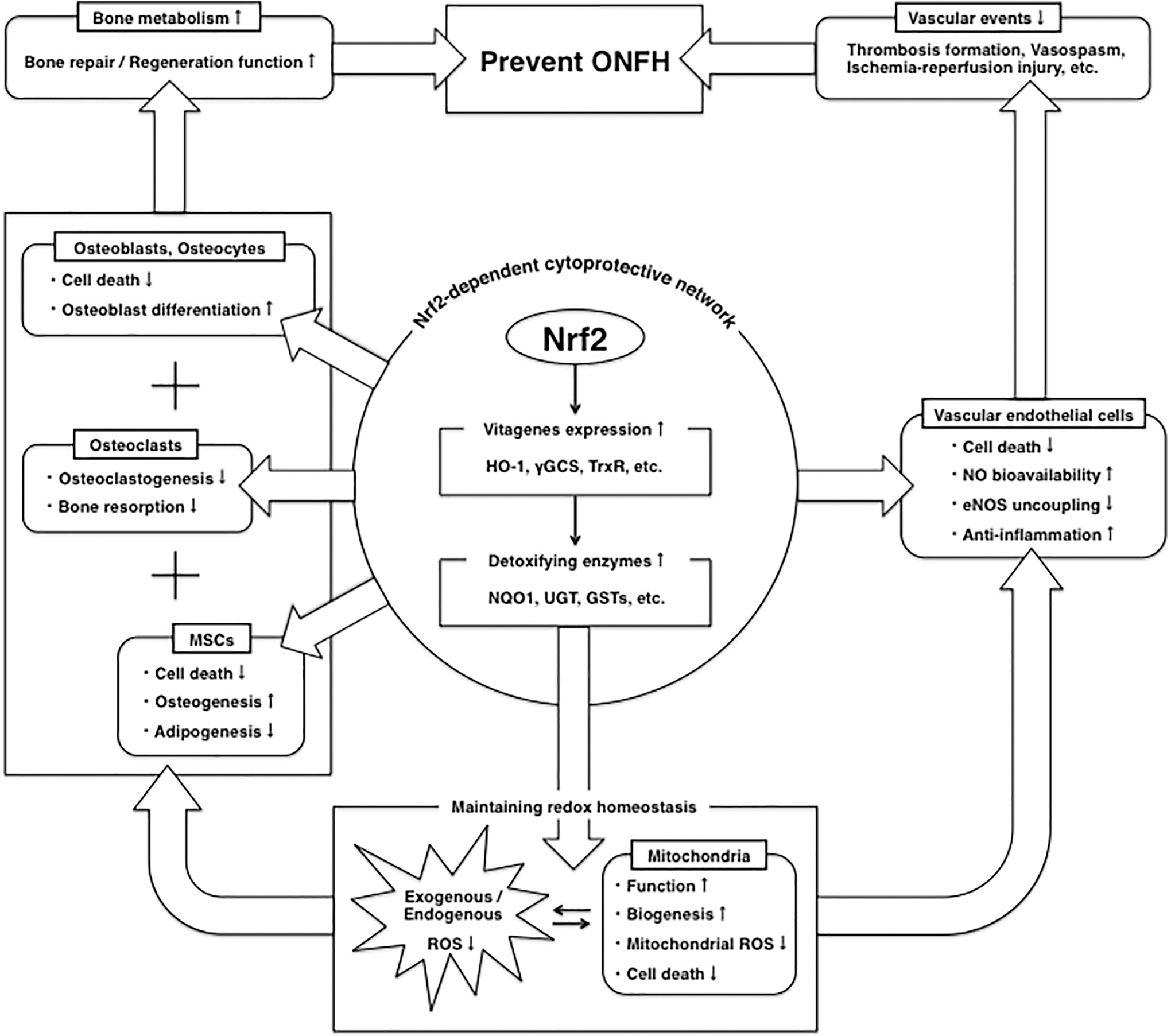
Footnotes
Acknowledgment
The authors thank Mr. John Foulks for his contribution to edit English in the article.
Authors' Contributions
Y.K. designed and wrote the first draft of the article. C.J.W. and T.P. did the literature search and recommended the articles to be evaluated. All authors contributed in reading and writing sections of the review and approved the final version.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by a grant from the Interdisciplinary Center for Clinical Research within the faculty of Medicine at the RWTH Aachen (OC1-1), the