
Abstract
Aims:
Thioredoxin (TRX)-fold proteins are ubiquitous in nature. This redox scaffold has evolved to enable a variety of functions, including redox regulation, protein folding, and oxidative stress defense. In bacteria, the TRX-like disulfide bond (Dsb) family mediates the oxidative folding of multiple proteins required for fitness and pathogenic potential. Conventionally, Dsb proteins have specific redox functions with monomeric and dimeric Dsbs exclusively catalyzing thiol oxidation and disulfide isomerization, respectively. This contrasts with the eukaryotic disulfide forming machinery where the modular TRX protein disulfide isomerase (PDI) mediates thiol oxidation and disulfide reshuffling. In this study, we identified and structurally and biochemically characterized a novel Dsb-like protein from Salmonella enterica termed bovine colonization factor protein H (BcfH) and defined its role in virulence.
Results:
In the conserved bovine colonization factor (bcf) fimbrial operon, the Dsb-like enzyme BcfH forms a trimeric structure, exceptionally uncommon among the large and evolutionary conserved TRX superfamily. This protein also displays very unusual catalytic redox centers, including an unwound α-helix holding the redox active site and a trans-proline instead of the conserved cis-proline active site loop. Remarkably, BcfH displays both thiol oxidase and disulfide isomerase activities contributing to Salmonella fimbrial biogenesis.
Innovation and Conclusion:
Typically, oligomerization of bacterial Dsb proteins modulates their redox function, with monomeric and dimeric Dsbs mediating thiol oxidation and disulfide isomerization, respectively. This study demonstrates a further structural and functional malleability in the TRX-fold protein family. BcfH trimeric architecture and unconventional catalytic sites permit multiple redox functions emulating in bacteria the eukaryotic PDI dual oxidoreductase activity. Antioxid. Redox Signal. 35, 21–39.
Introduction
A central step in the folding of many secretory and membrane proteins is the introduction of disulfide bonds (Dsbs) between cysteine residues, a process called oxidative protein folding. In Gram-negative bacteria, the thiol oxidation process occurs in the periplasmic space and is facilitated by the action of the Dsb family of proteins (38). In the classic bacterial Dsb pathway, disulfide bonds are introduced into proteins through the action of two enzymes, DsbA, a monomeric thioredoxin (TRX)-like enzyme containing a catalytic CxxC motif, and its associated oxidase DsbB (3). Two additional Dsb proteins, dimeric DsbC and its partner reductase DsbD, form a supplementary Dsb system that reshuffles non-native disulfide bonds in misfolded proteins (54).
Bacterial pathogens often carry an extended repertoire of disulfide catalysts (26, 38, 67), which contribute to their pathogenic potential by catalyzing the folding of many virulence factors, including surface adhesins and toxins, secretion and motility systems (26, 38). This is indeed the case for Salmonella where the periplasm harbors the classical DsbA–DsbB thiol oxidation and DsbC–DsbD disulfide isomerization pathways (27, 34), an additional DsbL/I redox pair involved in the oxidative folding of the periplasmic aryl-sulfate sulfotransferase (27, 41) and a set of Dsb-like proteins encoded by the suppressor of copper sensitivity (scs) locus, which are involved in Dsb reduction and copper detoxification (20, 57, 61), and SrgA, which is a virulence plasmid-encoded DsbA homolog essential for the biogenesis of plasmid-encoded fimbriae (Pef) (5, 27).
Innovation
The present work represents a substantial leap forward in our understanding of the diversity of thioredoxin (TRX)-fold proteins, which are ubiquitous in nature where they regulate essential functions for life. Through evolution, this ancestral redox scaffold has been modified with insertions, extensions, and oligomerization events to enable a variety of functions, including thiol oxidation, along with disulfide reduction and isomerization, all vital for redox regulation and signaling, protein folding and oxidative stress defense (2). Our findings on bovine colonization factor protein H (BcfH) emphasize the structural and functional malleability of TRX proteins, whereby trimerization enables bacterial proteins to acquire multiple redox functionalities similar to eukaryotic PDI redox systems. Importantly, given the central role of disulfide bond (Dsb) proteins in promoting bacterial virulence, our research may also be the basis for future antivirulence approaches targeting this family of medically important proteins.
The diversity of Dsb formation machinery in Salmonella is further highlighted by bovine colonization factor protein H (BcfH), a putative Dsb-like protein encoded by the bovine colonization factor (bcf) fimbrial operon (75). Bcf are the only chaperone-usher (CU) fimbriae that are conserved across all Salmonella enterica serovars (75) and have been shown to contribute to Salmonella intestinal persistence in mice (40, 73). In addition to BcfH, the bcf gene cluster encodes for the typical components of CU fimbriae, including a putative major fimbrial subunit (BcfA), the usher protein (BcfC), tip adhesin (BcfD), two putative chaperones (BcfB and BcfG) and two accessory fimbrial subunits (BcfE and BcfF) (75).
With the discovery of new Dsb proteins and systems, it is becoming increasingly apparent that the conventional monomeric DsbA and dimeric DsbC systems are not applicable to all Dsb-like oxidoreductases. One of these new groups includes the trimeric disulfide isomerases such as suppressor of copper sensitivity protein C from Proteus mirabilis (PmScsC) (18) and α-DsbA2 from Wolbachia pipientis (72). In this study, we show that this is also the case for the putative disulfide oxidoreductase BcfH, associated with Salmonella Bcf fimbriae. Our crystal structure of the BcfH trimer and detailed functional investigation reveal an oligomeric Dsb enzyme with both thiol oxidase and disulfide isomerase activities. Further, this study provides insights into the conformational changes that may allow this protein to function with dual activities. Finally, using in vitro redox and in vivo assays we show that BcfH mediates oxidation of a specific bcf fimbrial component, and that this oxidoreductase is required for bcf-mediated biofilm formation in Salmonella.
Results
Bcf fimbrial proteins are cysteine rich and BcfH is a conserved Dsb-like oxidoreductase
The S. enterica bcf CU fimbrial operon consists of eight genes, mainly encoding putative fimbrial components (bcfA-G) and the bcfH gene at the 3′ end, which encodes a Dsb-like putative disulfide oxidoreductase (75). A discontiguous MegaBlast search revealed conserved bcf-like operons among 10 distantly related Enterobacteriaceae family members from Salmonella spp., Klebsiella spp., Enterobacter spp., and Kluyvera intermedia, with nucleotide sequence identities ranging between 56% and 100% (Supplementary Fig. S1A). Subsequent phylogenetic analysis of the bcf operon showed that sequences fell into two clades with those from Enterobacter spp. mostly forming their own clade (Supplementary Fig. S1B). Found in all but the Enterobacter spp., the bcfH genes are highly conserved; sharing >76% sequence identity. While the Enterobacter spp. lacked bcfH, they contained an additional bcfA-like homolog at the 5′ end.
Analysis of the predicted fimbrial components of the bcf operon revealed that all of the encoded BcfA-G proteins were likely substrates for a disulfide oxidoreductase such as BcfH, as they each contained at least two cysteines (Supplementary Fig. S2A). Indeed, sequence comparison with other structurally characterized fimbrial proteins predicts that all of the Bcf components could include disulfide bonds in their three-dimensional structures (Supplementary Table S1). The reliance of the Bcf proteins on a disulfide oxidoreductase is consistent with previous findings that have shown the key role of the Dsb oxidative folding system in the assembly of different components of fimbrial adhesins (26, 38). It is also not unprecedented for bacteria to acquire additional oxidoreductases for the efficient folding of specific substrates (27).
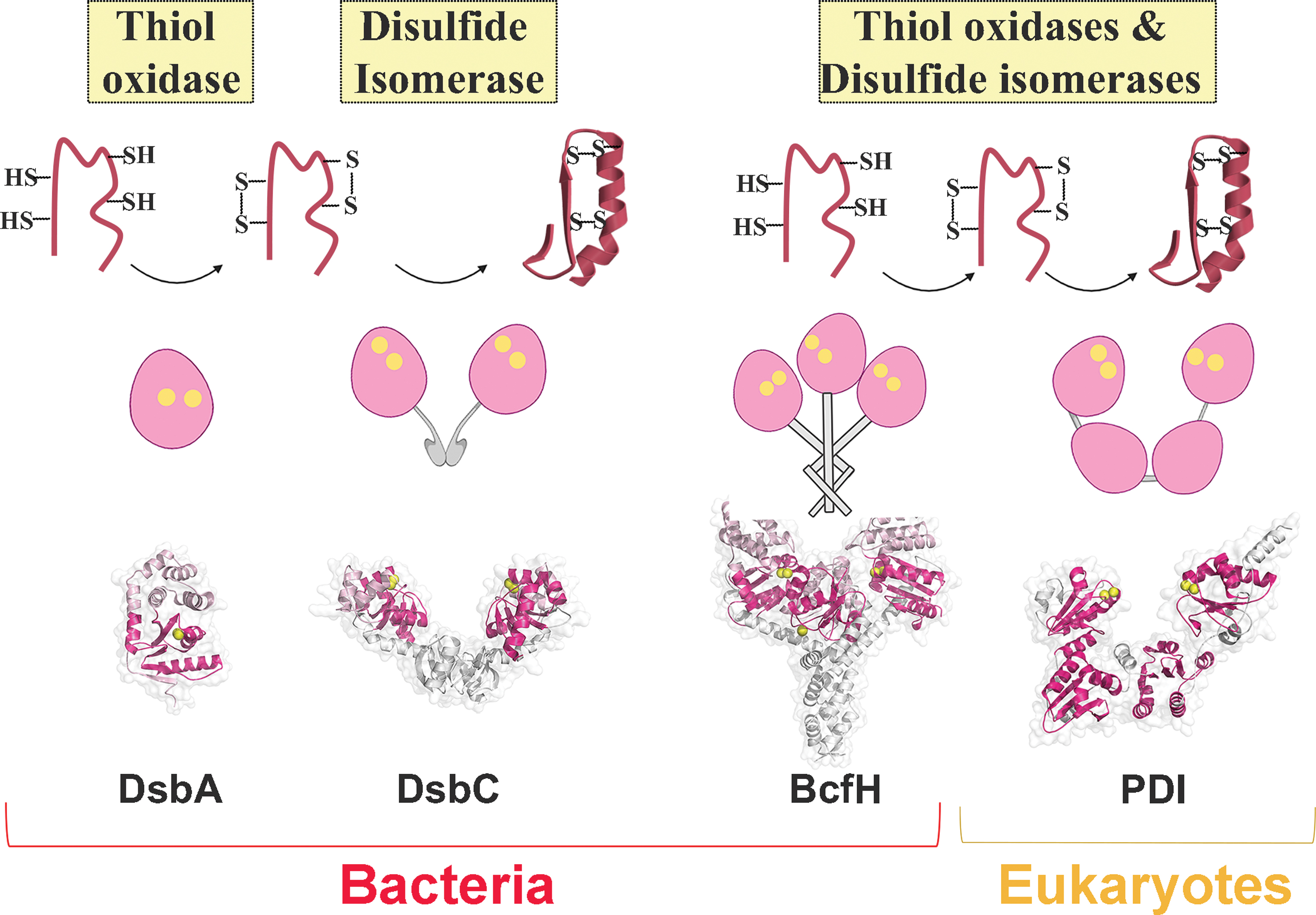
To begin investigating the potential Dsb-like properties of BcfH, a sequence alignment with other previously characterized Salmonella Dsb proteins showed that although sequence identities varied between 10% and 18%, sequence similarities ranged between 60% and 65% (Supplementary Fig. S2B). As with other Dsb-like proteins, BcfH is predicted to incorporate a TRX fold with the characteristic CxxC motif (CSWC) and a putative cis-proline loop (Supplementary Fig. S2B). An additional feature of BcfH is the presence of an N-terminal extension (Supplementary Fig. S2B), consistent with the dimerization domain of disulfide isomerases such as DsbC and DsbG (24, 74). Although the long N-terminus resembles disulfide isomerases, the inserted helical domain in the TRX-fold mimics that of DsbA-like thiol oxidases, which typically consist of a long insertion with four consecutive α-helices compared with the short two α-helical insertions of DsbC-like isomerases (19, 58).
Structural characterization of BcfH
The mixed sequence properties of BcfH led us to determine the three-dimensional structure of BcfH by X-ray crystallography. The crystal structure of BcfH was solved using the multiwavelength anomalous diffraction (MAD) method with selenomethionine (SeMet)-labeled protein. BcfH crystals belong to the orthorhombic space group P212121 with six molecules in the asymmetric unit. After iterative cycles of model building and refinement, the structure of BcfH was refined to a resolution of 2.31 Å with R-factor and R-free values of 16.7% and 20.9%, respectively (Fig. 1A and Table 1).

BcfH Data Collection and Refinement Statistics
Statistics for the highest resolution shell are shown in parentheses.
Analysis of the BcfH structure revealed structural features akin to Dsb-like proteins but more interestingly a trimeric association, making this only the second reported example of an atomic resolution trimeric Dsb protein structure, with the others being monomers or dimers (18). The six molecules in the asymmetric unit were divided into two trimers (chains ABC and chains DEF). Comparison of both trimers gives an overall root mean square deviation (r.m.s.d.) value of 1.59 Å over 715 Cα atoms aligned, indicating that both trimers are structurally similar (Supplementary Fig. S3A). In each BcfH trimer, the monomers adopt a ladle-like shape where the N-terminal extension and C-terminal catalytic domain form the handle and the bowl of the ladle, respectively (Fig. 1B).
The N-terminal region (residues 1–49) has an α-helical conformation encompassing three short α helices (α1–α3) (Fig. 1B). BcfH trimerization occurs via these N-terminal α-helical extensions, which wrap around each other forming a right-handed coiled-coil structure (Fig. 1C). Analysis of the buried residues within the trimerization stalk showed that each monomer contributes a ladder of hydrophobic amino acids (Ile9, Ala13, Ile17, Val24, Val28, Leu32, Phe38, and Leu39) to form the central hydrophobic core. Although these homotrimeric contacts are largely hydrophobic, three salt bridges further stabilize the complex, residue Arg49 in the α3 helix of each protomer forms an electrostatic interaction with Glu34 or Glu37 on the α3 helix of the adjacent protomer (Fig. 1C). The trimer is further stabilized by an antiparallel β-sheet between β strands (β1) at the N-terminus of two of the monomers in the trimer (Fig. 1C).
BcfH protomers adopt two different conformations
In each protomer, the N-terminal trimerization domain is followed by a flexible linker region. This linker region starts at residue 50 as a continuation of the α3 helix and concludes at residue 84 on a loop preceding the catalytic domain (Fig. 1B, D).
This flexible linker is responsible for two distinct conformations found in the BcfH trimer. Two of the protomers assume an extended configuration (r.m.s.d. value of 1.82 Å, 239 Cα aligned), whereas the third protomer is in a compact conformation (superposition of the compact and extended monomers yielded an r.m.s.d. value of 0.9 Å with only 185 Cα of the catalytic domains aligned) (Fig. 1B and Supplementary Fig. S3). A ∼90° bend in the flexible linker, most notably at a glutamine-rich motif in the α3 helix (residues 50 and 56), moves the catalytic domain into the interior of the trimer resulting in a compact orientation (Fig. 1B and Supplementary Fig. S3). By comparison, this flexible region forms a continuation of the α3 helix in the other two protomers, holding the catalytic domains outward in an extended conformation (Fig. 1B and Supplementary Fig. S3).
The compact orientation of one catalytic domain in the trimer allows interactions to form between all the catalytic domains. A total of 13 hydrogen bonds and three salt bridges form between these domains (Supplementary Table S2), over an interacting surface of 1200 Å2. Although somewhat stabilizing, these interactions are sparse over a large area, so it could be assumed that under certain conditions the compact catalytic domain can move out and also form an extended orientation (Supplementary Fig. S4 and Supplementary Movie S1). The position of the compact catalytic domain in the crystal structure also completely covers the CxxC active sites of the other two extended catalytic domains, which would preclude substrate binding. Consequently, the protein would require a different conformation to function as a thiol-disulfide oxidoreductase, which supports the potential for all three domains to adopt other conformations.
BcfH C-terminal domain features the canonical DsbA architecture
The C-terminal domain of BcfH (residues 85–254) preserves the structural characteristics of DsbA thiol oxidases, including a TRX fold, with the classic βαβ (residues 85–123) and ββα (residues 204–254) motifs along with an inserted five α-helical domain (residues 124–203) (Fig. 2A–C). As with other TRX-like proteins, the CxxC motif adopts a right-hand hook conformation at the N-terminus of the active site α-helix (30) (Fig. 2A, inset b). This BcfH domain also retains the hydrophobic surface properties adjacent to the catalytic motif of DsbA-like proteins (Fig. 2D).

Unlike most Dsb proteins, the BcfH catalytic motif (Cys94-Ser95-Trp96-Cys97) contains a Trp residue. In two monomers (one extended and one compact) of the BcfH trimer, the indole ring of the Trp side chain shields the catalytic cysteines (Fig. 2A, inset a). This is an infrequent catalytic motif in DsbA-like proteins where serine and tryptophan are only present with a frequency of 6% and 0.5% at the N- and C-terminal positions, respectively, in the CxxC motif of DsbA homologs (52).
Another defining characteristic of TRX-like proteins is the presence of a conserved cis-Pro loop adjacent to the CxxC motif (53). The equivalent loop of BcfH is located in the α9-β4 connecting loop and consists of Pro217 preceded by Thr216. Surprisingly, in one of the extended protomers from each trimer, this proline is found in a trans-configuration mapping away from the active site (Fig. 2A, inset a), a feature only previously reported for the protein disulfide isomerase (PDI)-like protein Eps1p from Naumovozyma dairenensis (4). The trans-proline loop is distant from the CxxC motif, and disrupts the hydrogen bond interactions between the residue preceding the proline (Thr215 in BcfH) and the catalytic cysteines, interactions that modulate the redox properties in TRX-like proteins (53). Furthermore, the noncanonical characteristics of this protomer in BcfH may suggest a different substrate binding mode from other TRX-like proteins, which rely on the cis-proline loop adjacent to the catalytic cysteines for binding incoming substrates (36).
Even though the protein was oxidized before crystallization experiments, the active site CxxC of all the extended catalytic domains in the BcfH structure was found in a reduced state, whereas the CxxC was oxidized in the catalytic domain of the compact protomers. This could be a result of incomplete oxidation by oxidized glutathione (GSSG) and/or high sensitivity to radiation-induced disulfide reduction during X-ray data collection. Despite this, pairwise comparison of the six symmetrically independent catalytic motifs showed that this domain overall shows limited structural variation (r.m.s.d. between 0.2 and 1.7 Å for all Cα atoms; Supplementary Fig. S5A, B). However, in the monomers with the oxidized CxxC motif there is also a partial unwinding of the active site helix α4 (Fig. 2A, inset b). This changes the configuration of both the active site and associated Trp96, Ser98, and Lys99, which protrude and mediate intra- and interdomain interactions that stabilize the compact conformation.
BcfH exists as a trimer in solution
To confirm the trimeric structure of BcfH in solution, we analyzed recombinantly expressed and purified BcfH by small-angle X-ray scattering (SAXS) followed by analytical ultracentrifugation (AUC). SAXS experiments were performed on BcfH at concentrations ranging from 0.3 to 5 mg/mL. The data are essentially independent of concentration, and consistent with BcfH existing as a trimer in solution with a maximum dimension of 115 Å (Fig. 3A, B and Supplementary Table S3). The p(r) for BcfH shows a single peak that differs only slightly from the p(r) predicted from the crystal structure. This difference is due to the crystal structure being slightly more compact with a maximum dimension of 109 Å (Fig. 3B and Supplementary Table S3), most likely due to the protomers sampling multiple conformations caused by the flexible linkers as seen for the other trimeric TRX-like protein PmScsC (18). It was notable that the nature of the BcfH trimer is different from that observed for PmScsC, where the p(r) for the latter displays a bimodal curve characteristic of a symmetric trimeric rotor. Both rigid-body and dummy-atom modeling against the BcfH scattering data yielded models in excellent agreement with the scattering data, and with shapes similar to the crystal structure (Fig. 3C).

AUC sedimentation velocity experiments were conducted on BcfH at 0.2 to 3.0 mg/mL concentrations. The effect of redox state on BcfH oligomers was investigated using oxidized and reduced BcfH. Sedimentation coefficient distributions (c(s)) for oxidized BcfH showed a single sedimenting 4.7 S 20,w species (Fig. 3D) consistent with the molecular weight of a 83.7 kDa trimer (Supplementary Fig. S6). Upon reduction, there was a reproducible shift in the c(s) to a 4.45 S 20,w, species (Fig. 3D) with an associated change in the frictional ratios from 1.35 to 1.40. Such a shift may be indicative of a conformational change of the trimer upon reduction of the CxxC motif, to a more elongated shape. This result is consistent with the compact oxidized catalytic domains and elongated reduced catalytic domains observed in the BcfH crystal structure.
BcfH and PmScsC trimers share structural similarities
Comparison of BcfH against the Protein Data Bank (PDB) using the DALI tool revealed that this protein shows the highest structural similarity with the PmScsC crystal structures (18) that include transitional (PDB: 5IDR), compact (PDB: 4XVW), and extended (PDB: 5ID4) conformations (Fig. 4B, C), with 24% sequence identities, Z-scores of 17.5–23.0, and r.m.s.d. values of 1.7 to 5.5 Å. PmScsC is the only other trimeric TRX-like protein where the entire crystal structure has been reported. Similar to BcfH, PmScsC encompasses an N-terminal trimerization domain, a flexible linker followed by a C-terminal catalytic domain (Fig. 4A, B) (18).

Overall, the BcfH structure with its mixed conformations of extended/compact protomers most resembles the conformation of the PmScsC transitional form (PDB: 5IDR), which is also in a similar mixed state (Fig. 4C). The PmScsC transitional state is thought to be the intermediate between the 5ID4 form with three extended protomers and the 4XVW conformation with three compact protomers. Accordingly, the extended BcfH protomers do most resemble those from the extended PmScsC with an r.m.s.d. of 3.0 Å over 202 residues, and the compact BcfH protomer with the compact PmScsC with an r.m.s.d. of 2.2 Å over 152 residues (Supplementary Fig. S7A, B). The large conformational flexibility of PmScsC is a consequence of a glutamine-rich shape-shifting region linking the trimerization and catalytic domains (18). Structure-based sequence alignment of BcfH and PmScsC showed that these proteins share 65% sequence identity in this linker region, further suggesting an equivalent conformational plasticity in BcfH (Fig. 4A).
BcfH is a multifunctional thiol-disulfide oxidoreductase
Given that the structural properties of BcfH resemble thiol-disulfide oxidoreductases, we determined the redox properties of BcfH. We measured the disulfide exchange reaction of BcfH with glutathione, by monitoring the relative amounts of oxidized and reduced protein at different reduced glutathione (GSH)/GSSG ratios. The resulting sigmoidal curve contained two plateaus, at ∼0.65 and 1 fraction of reduced protein. Curve fitting to a biphasic sigmoid model yielded two equilibrium constants, Keq of 2.04 ± 0.6 × 10−5 M and 1.01 ± 0.2 × 10−3 M, which convert into intrinsic redox potentials of −101 and −151 mV, respectively. The former value is comparable with the redox potential of PmScsC (−108 mV) (18) and more oxidizing than Escherichia coli DsbA (−126 mV) (27). The second BcfH redox potential is more reducing than that of DsbC (−130 mV) (Fig. 5A) (76).
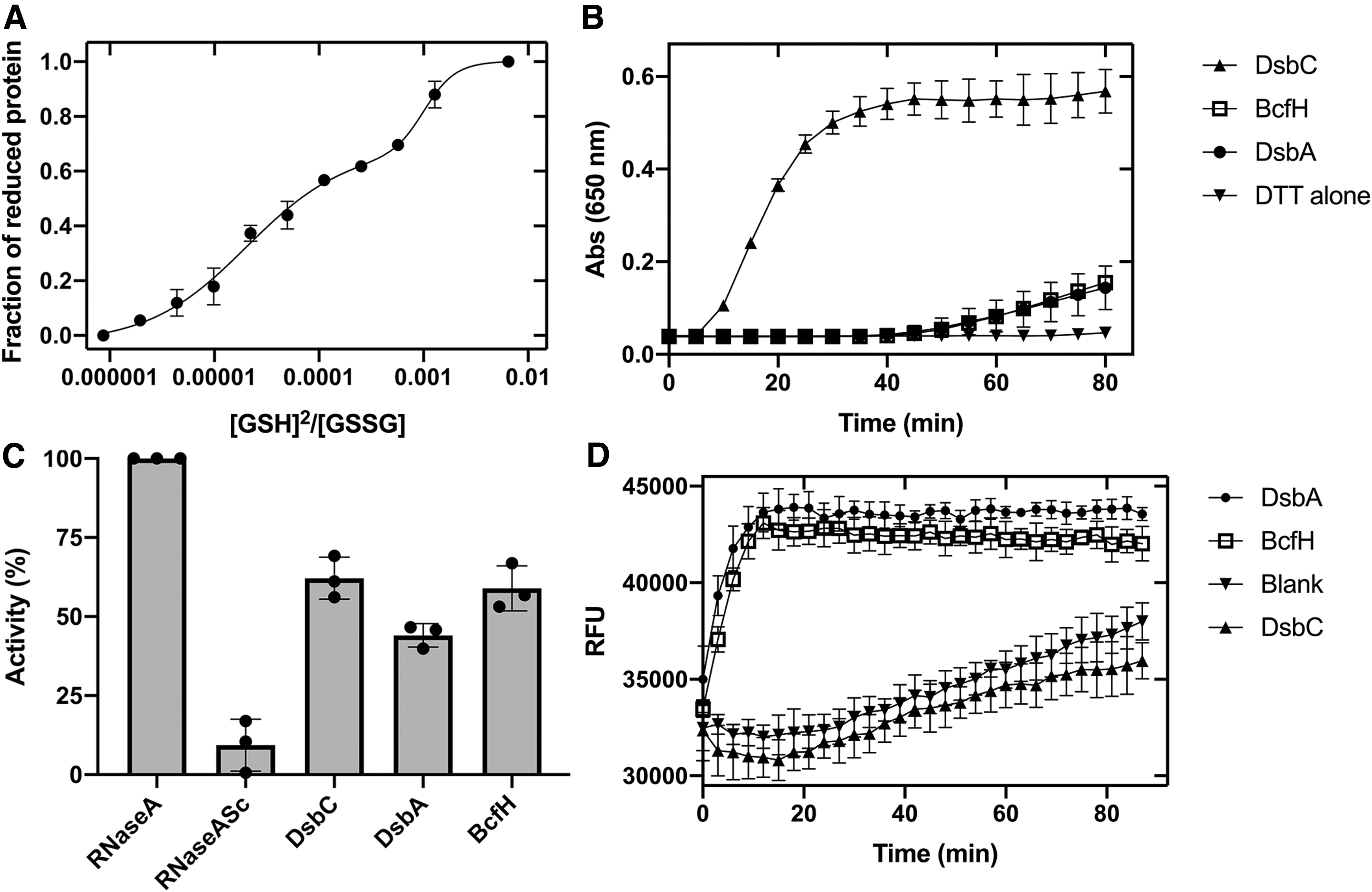
The two reduction potentials indicate different relative stabilities of the reduced and oxidized states in the BcfH active sites. To gain understanding of the local environments surrounding each redox center, we analyzed the residues within 4 Å distance of the catalytic cysteines, which have the potential to form hydrophobic or polar interactions and therefore modulate the stability of the dithiol/disulfide states (Supplementary Fig. S8). One of the extended monomers found in the reduced form exhibits short contacts between the Cys94 Sγ and the main-chain nitrogen atoms of Ser95, Trp96, and Cys97 in the CSWC motif as well as the Thr216 hydroxyl group in the cis-proline loop. These types of hydrogen bond interactions have been previously shown to stabilize the reduced state and increase the redox potential of TRX-like proteins (53). The higher network of interactions may account for the −101 mV redox potential. The second monomer in extended conformation, also found in a reduced state, shows fewer thiol-stabilizing interactions, with contacts just between the Cys94 Sγ and the main-chain nitrogen atoms of Ser95, Trp96, and Cys97. This feature is due to the unusual trans-conformation of the Pro217, causing the α9-β4 loop to be more distant from the Cys94, thereby removing stabilizing interactions with the Thr216 and Pro217. This would lower the stability of the reduced form relative to the oxidized state and may reduce the reduction potential. Finally, the active site of the BcfH monomer in a compact conformation was found in the oxidized state with only two close contacts between Cys94 Sγ with Cys97 main-chain nitrogen and Trp96 NE1. This catalytic site is unusual because it localizes in an unwound helix (Fig. 2A, Supplementary Fig. S8), which lacks most of the thiol-stabilizing interactions between Cys Sγ and the main-chain nitrogen atoms in residues forming the conventional CxxC right-hand hook, which would increase the redox potential. However, this effect may be offset by the unfolded less constrained helix, favoring the reduced dithiol over the disulfide state lowering the redox potential.
We then evaluated the redox activity of BcfH using standard disulfide reductase, isomerase, and thiol oxidase assays. The reductase activity of BcfH was determined spectrophotometrically in the classic insulin-reduction assay (28). In this assay, the positive control S. enterica DsbC fully reduced the disulfide bonds of insulin within 30 min. Conversely, the oxidant S. enterica DsbA and also BcfH showed lower activity, reducing only ∼25% of insulin after 80 min of incubation (Fig. 5B).
We next determined the disulfide isomerase activity of BcfH by measuring its ability to refold scrambled RNase A (scRNase A). After a 5 h incubation, the control disulfide isomerase DsbC recovered ∼70% of the activity of RNase A while the activity of DsbA was significantly different, only refolding ∼45% of scRNase A (Fig. 5C and Supplementary Table S4). By comparison, DsbC and BcfH scRNAse A refolding activities were not significantly different (Supplementary Table S4) with BcfH restoring ∼60% of the RNase A activity, thus showing capacity to function as a disulfide isomerase.
To function as an isomerase, the active site cysteines in BcfH must be maintained in the reduced dithiol form. In Salmonella, both DsbD and its functional homolog ScsB are known to maintain Dsb-like proteins reduced in the periplasm. We investigated the ability of these proteins to reduce BcfH in vitro by adding stoichiometric amounts of reduced N-ScsB (N-ScsBred) or reduced N-DsbD (N-DsbDred) to oxidized BcfH (BcfHox) with incubation for 30 and 120 s. However, BcfH was found not to be reduced by either protein when its redox state was assessed by 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) thiol alkylation (Supplementary Fig. S9A, B). As such BcfH must rely on a separate system for redox state maintenance.
We then assessed the dithiol oxidase activity of BcfH using an in vitro oxidation assay with a fluorescently labeled DsbA model peptide (69). The oxidation activity was monitored by the increase in europium fluorescence resulting from peptide cyclization via disulfide bond formation between the terminal cysteines. BcfH catalyzed peptide oxidation at a similar rate to that of the disulfide oxidase DsbA (Fig. 5D), suggesting that BcfH can also function as an oxidase. By comparison, the isomerase DsbC showed no oxidase activity under similar experimental conditions (Fig. 5D).
BcfH acts as an oxidase/isomerase in fimbrial biogenesis
Given that BcfA-G proteins contain at least two cysteines in their primary sequence, with BcfC and BcfD having four cysteines, and all predicted to form disulfide bonds (Supplementary Fig. 2A and Supplementary Table S1), we hypothesized that BcfH serves as a dedicated oxidase and isomerase for Bcf fimbriae in S. enterica. Overexpression of Bcf fimbriae in S. enterica was previously shown to increase biofilm formation (12). To investigate the role of BcfH in Bcf fimbrial biogenesis, we used a strain of S. enterica serovar Typhimurium (S. Typhimurium) devoid of DsbA homologs dsbA, srgA, and dsbLI (SL1344Δ3KO) (27), which is attenuated for biofilm formation compared with wild type (WT) (Fig. 6A). Overexpression of Bcf fimbriae lacking bcfH in this SL1344Δ3KO strain was found to result in a significant lack of biofilm formation, compared with the wild-type S. Typhimurium with ∼2-log reduction in biofilm cell numbers (colony forming unit [CFU]/mL), which is similar to the empty vector control (Fig. 6A). Notably, upon coexpressing Bcf fimbriae with BcfH in this strain background, biofilm production was restored to WT levels, identifying the important role of BcfH in Bcf fimbrial formation. Further, we selected the putative chaperone BcfB, which based on sequence analysis we predicted would contain a disulfide bond (Supplementary Table S1). We were able to express and purify BcfB, and subsequently show by AMS alkylation and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis that BcfH could partially oxidize this Bcf chaperone over the time course investigated (Fig. 6B).
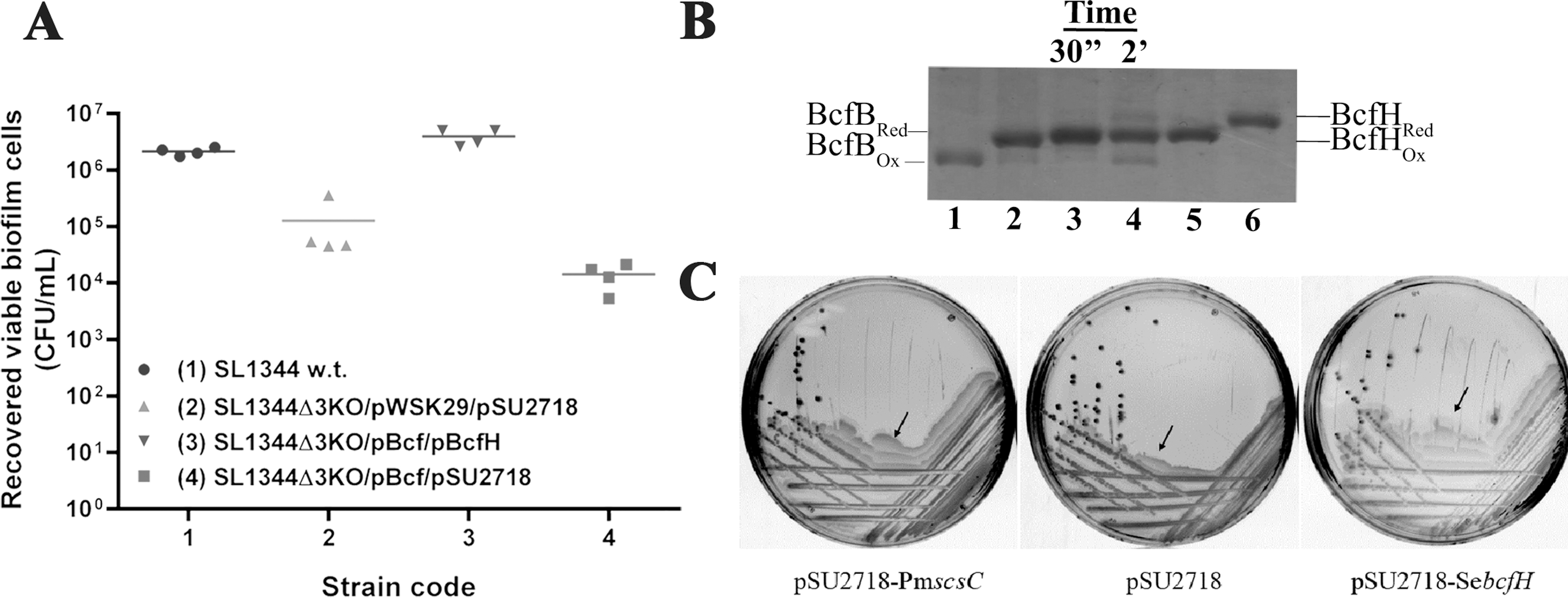
To further define the role of BcfH as an isomerase, we turned to the well-characterized and structurally similar trimeric isomerase PmScsC. PmScsC was recently shown to mediate P. mirabilis swarming, as a ΔscsC mutant had reduced motility (18). Here, we showed that the swarming defect of a P. mirabilis ΔscsC mutant could be functionally restored by complementation with Salmonella bcfH, similar to complementation with the native PmscsC (Fig. 6C).
Following this result, we investigated the target specificity of BcfH using the well-defined oxidase/isomerase model systems of E. coli motility, mucoid colony formation, and cellular alkaline phosphatase activity that depend on the oxidase DsbA or isomerase DsbC. In all cases, BcfH was unable to complement these E. coli Dsb enzymes, despite being expressed in the periplasm (Supplementary Fig. S10). Together, these findings indicate that BcfH can catalyze formation/isomerization of disulfide bonds but not in specific DsbA or DsbC substrates within E. coli (Supplementary Fig. S10A–C). This substrate specificity of BcfH could ensure that functional Bcf fimbrial production is preserved during bcf gene cluster mobilization via horizontal gene transfer between species.
Discussion
Nontyphoidal Salmonella serovars such as S. Typhimurium are significant pathogens that account for the majority of food-borne infections in humans and animals worldwide (22). Globally, nontyphoidal Salmonella strains are associated with >100 million food-borne infections, with >150,000 deaths every year (16). These pathogens use an arsenal of virulence factors to cause infection, including multiple adhesins, toxins, secretion machinery such as type III secretion systems and flagella for motility and cellular spread (11). Among these, a large catalog of CU fimbrial adhesins, bcf, fim, sti, sth, lpf, saf, stc, stj std, stf, pef, and stb, have been described in S. Typhimurium, which mediate attachment to and colonization of a wide range of host cells (73, 75).
The bcf fimbrial operon is conserved in all S. enterica serotypes (75), and has been shown to be important for Salmonella attachment and persistence in different animal models of infection (73, 75). The bcfA-E gene cluster contains fimbrial structural subunits along with chaperone and usher proteins required for fimbrial assembly (45, 75). This operon also has an additional bcfH gene (45), encompassing the hallmarks of Dsb oxidoreductases, including a TRX fold with a CxxC motif.
This study aimed to fully characterize BcfH, and investigate its function in fimbrial biogenesis and Salmonella virulence. The trimeric architecture present in the crystal structure of BcfH is reminiscent of that reported for the P. mirabilis Dsb-like protein PmScsC but with significant differences. Each BcfH monomer consists of an N-terminal α-helical trimerization domain and a C-terminal DsbA-like catalytic domain joined by a flexible linker. In BcfH, this linker motif adopts a helical configuration in two of the monomers, which in turn assume an extended conformation, and loop conformation in one monomer, which assumes a compact conformation and folds toward the extended monomers (Fig. 1). The protomer in compact configuration, which is found in the oxidized state, displays an unwinding of the first turn of the α4 helix, which contains the redox CxxC motif. This partial uncoiling of the catalytic α4 helix is a clear departure from the canonical TRX-like structure and has only been reported in a limited number of examples including DsbA3 from Neisseria meningitidis (70) and the PDI-like protein N. dairenensis Eps1p (4), but this phenomenon was attributed to a crystal packing artifact. The finding that this conformation is present in more than one crystal structure of very different TRX-like proteins suggests a possible role in the mechanism of action of these proteins, whereby unwinding of the α-helix harboring the catalytic CxxC motif may allow efficient thiol–disulfide exchange reactions with substrate proteins.
One of the extended protomers displays a trans-proline loop in the active site (Fig. 2A), a feature extremely uncommon in this family of proteins. The canonical cis-proline loop configuration has been previously shown to regulate the activity of TRX- like Dsb proteins by facilitating interactions with the proximal CxxC motif and regulating its redox properties (53). In BcfH, the trans-proline loop may also be altering its redox properties by affecting the stability of the reduced state. Furthermore, the cis-proline has been found to enable the binding and correct positioning of substrates for efficient thiol–disulfide interexchange, whereby mutants lacking this cis-proline accumulate in Dsb-substrate intermediates (36). As such, the cis- to trans-switching of the proline loop could also affect substrate binding. The significance and functional implications of the trans-proline loop in BcfH protomer could therefore be multifactorial; this configuration could change the substrate repertoire interacting with this particular BcfH protomer, affect the resolution time of the catalyst-substrate mixed disulfide, and contribute to the ability of BcfH being able to function as both a thiol oxidase and isomerase.
A notable observation in the crystallized BcfH trimer is that the protomer in the compact configuration occludes two of the three catalytic sites of BcfH, which would indicate that they are not solvent accessible and functional. However, our biochemical data have shown that this protein can be fully oxidized and reduced in the presence of GSSG and GSH (Fig. 6B), indicating the solvent accessibility of all catalytic cysteines in solution. Furthermore, analysis of BcfH by AUC demonstrated a redox-state-dependent conformational flexibility in BcfH in solution. Previous studies have shown that the linker motif in PmScsC allows a large dynamic flexibility to this disulfide isomerase (17, 18). Based on the amino acid sequence conservation of the glutamine-rich linker motifs in BcfH and PmScsC, we hypothesize a dynamic flexibility for BcfH, which is supported by our solution studies. It would appear that our structure represents a BcfH transitional form, between a compact and a fully extended form.
Functionally, PmScsC is a strong disulfide isomerase, an activity that is dependent on its trimeric architecture (18). Biochemical characterization of BcfH showed that the active sites in this protein have distinct redox potentials, one more oxidizing than EcDsbA and a second more reducing than EcDsbC. This differs from PmScsC where all three active sites have the same redox potential. The distinct redox properties of BcfH may be due to the ability of the Pro217 to display a trans-configuration, thereby conferring a different stabilizing bond network to the reduced state. In comparison, the corresponding proline loop in all three PmScsC structures was only found in a cis-conformation. Remarkably, BcfH also displayed both strong thiol oxidizing activity similar to DsbA and isomerase activity comparable with those of DsbC. The latter activity is supported by the observed functional redundancy between BcfH and PmScsC in P. mirabilis, along with significant restoration of RNase A activity by BcfH. Likewise, the oxidase activity of BcfH was evident in its ability to oxidize a peptide substrate. Cysteine residues are present in all the fimbrial subunits of the bcf operon. The role of thiol oxidases in the folding of different fimbrial components is well documented (8, 32, 51). Moreover, Salmonella has a dedicated thiol oxidase SrgA essential for the biogenesis of Pef fimbriae (5). Interestingly, SrgA is coexpressed with Pef fimbrial genes under the same environmental conditions (acidic pH), ensuring that this Pef-specific foldase can catalyze functional Pef production as required (5). Most other Salmonella fimbrial operons, including bcf, are also under tight regulation control with gene expression only detected under certain growth conditions (29). It was therefore tempting to propose that the multifunctional oxidoreductase BcfH is encoded as part of the bcf operon to promote the oxidative folding of all disulfide-bonded components required for Bcf fimbrial biogenesis. Indeed, we have shown that BcfH can oxidize the fimbrial chaperone BcfB in vitro and catalyze Bcf-mediated fimbrial formation in vivo.
BcfH dual activity is intriguing given that these redox functions in bacterial Dsb proteins are conventionally carried out by different proteins in separate redox systems, whereby monomeric DsbA proteins catalyze thiol oxidation and dimeric DsbC proteins mediate disulfide isomerization (Fig. 7) (10, 25). The dual redox functionality, although uncommon among bacterial proteins, is well recognized for eukaryotic oxidoreductases such as PDI (21). This U-shaped modular protein contains two catalytically active TRX domains flanking two structural TRX domains (19, 66), which can mediate efficient thiol oxidation or disulfide reduction, and reshuffling depending on the redox state of the catalytic CxxC motifs (19, 66).
Materials and Methods
Sequence selection
The Discontiguous MegaBlast program was used to compare diverged nucleotides at interspecies levels (43). We omitted bcfH from our query since dsbA-like genes are not always found within fimbrial operons. Thus, only bcfA-G is blasted against the nr/nt database, with a query coverage from 40% and the E-value cutoff at 1E-140. The bcf-like gene cluster is then manually extracted from each species. These gene clusters were aligned with Clustal Omega (59). This alignment was then used for neighbor-joining phylogenetic tree constructions by Geneious Tree Builder with a bootstrap value of 10,000 (Geneious Prime 2020.04.28). BcfH (CBW16129.1) and BcfH-like proteins, including CBW16129.1, QGF74828.1, AVE97242.1, and AWF33847.1, were compared using MUSCLE alignment (13). All BcfA-like sequences were validated by Smart Blast to confirm the identity of dual adhesins in the gene clusters analyzed.
Bacterial strains and plasmids
Bacterial strains and plasmids are included in Supplementary Table S5. LB-Lennox medium was used for bacterial growth at 37°C unless otherwise indicated. 1.5% (w/v) bacteriological agar was added for solid growth media.
Cloning and protein production
S. Typhimurium SL1344 genomic DNA was used as the template to clone bcf genes (GenBank accession no: 1251546). Cloning of bcfABCDEFG (bcf operon minus bcfH) in pWSK29 occurred over two steps by the introduction of a synonymous mutation in bcfC to add a PstI restriction site. The 5′ end of bcf operon was first cloned into SacI-PstI-digested pWSK29, followed by insertion of the 3′ end of the bcf operon up to and including bcfG but excluding bcfH gene by PstI-HindIII sticky ends. The bcfH gene was cloned separately into EcoRI-HindIII-digested pSU2718 (for cloning primers, see Supplementary Table S5).
The coding sequence for the mature BcfH protein (residues 28–281, lacking the first 27 residues corresponding to the signal sequence) was amplified by PCR from S. Typhimurium SL1344 genomic DNA. The amplified product was then cloned into a modified version of the expression vector pMCSG7 (pMCSG7*), which encodes the targeted gene as a fusion protein containing an N-terminal 6 × His-TRX tag, followed by the Tobacco Etch Virus (TEV) protease cleavage site (for cloning primers, see Supplementary Table S5) (15). The recombinant protein was expressed in E. coli C43 (DE3) cells using the autoinduction method (60). The SeMet-labeled BcfH was expressed in minimal medium containing 50 μg/mL SeMet as previously described (23). The recombinant protein was purified by nickel affinity chromatography, followed by TEV protease cleavage and a reverse nickel affinity chromatography purification step. BcfH was oxidized or reduced with a 50-fold molar excess of GSSG and GSH (Sigma-Aldrich, Australia), respectively, and then subjected to a final purification step by size exclusion chromatography.
The mature form of BcfB, the putative chaperone component of the Bcf fimbriae (residues 27–228), was synthesized by Epoch Life Science (Epoch Life Science) and inserted into a modified version of pMCSG7 (pMCSG7**, Supplementary Table S5). BcfB was expressed in E. coli BL21(DE3) cells using the autoinduction medium and purified using similar methods, as described for BcfH.
S. Typhimurium DsbA, DsbC, N-ScsB, and N-DsbD proteins were expressed and purified as previously described (33, 61).
Crystallization and diffraction data collection
Before setting up crystallization experiments, the purified protein was oxidized with 30 molar excess of GSSG for 1 h at 4°C and buffer exchanged into 25 mM HEPES, 50 mM NaCl, pH 7.0. The sitting and hanging-drop vapor diffusion methods were used for the crystallization of native and SeMet-labeled BcfH. High-throughput crystallization trials were carried out in-house, using a Crystal Gryphon Protein Crystallography System (Art Robbins Instruments) and commercial screens, including Crystal and PEG/Ion Screens (Hampton Research, San Diego, CA), JCSG+, and PACT Suites (QIAGEN, Netherlands). Each drop consisted of 200 nl protein solution (30 mg/mL) and 200 nl well solution, and crystallization plates were incubated at 20°C. Small crystals of native BcfH were obtained within a few days in a JCSG+ Suite condition comprising 0.2 M magnesium chloride (MgCl2), 100 mM Tris 8.5, and 20% (w/v) PEG 8000 and in a PACTscreen condition containing 0.2 M sodium nitrate, 0.1 M Bis-tris propane pH 6.5, 20% (w/v) PEG 3350. Focused screens were then set around those initial hits as well as combination of those conditions. Larger (approximate dimensions 0.1 × 0.05 × 0.4 mm) diffracting crystals were obtained in 0.2 M MgCl2, 0.1 mM Bis-tris propane, pH 6.0, 20% (w/v) PEG 3350 over 3–4 days. SeMet BcfH crystals were grown using the same protein concentration and identical crystallization conditions as for the native protein. Native and SeMet BcfH crystals were cryoprotected in a solution comprising the crystallization condition supplemented with 20% (v/v) ethylene glycol, and crystals were then flash cooled and stored under liquid nitrogen.
X-ray data for BcfH were collected at the protein crystallography beamlines (MX1 and MX2 beamlines) at the Australian Synchrotron (Melbourne, Australia). For native BcfH, 180° (oscillation 0.1° with 0.1 s exposure) were collected at 100 K using the MX2 beamline housing an EIGER × 16M detector. The native data were processed and scaled using HKL2000 (46). Two wavelength MAD (2W-MAD) data (0.97909 Å [12.663 keV, peak], 0.97938 Å [12.65 keV, inflection]) were collected at 100 K using the MX1 beamline. A total of 720 images were collected from one crystal using an ADSC detector, with 360 images for each wavelength and oscillation of 1° with 1 s exposure. The X-ray data from each wavelength were integrated using XDS and scaled using XSCALE (35). The data-collection statistics are given in Table 1.
BcfH structure determination
The structure of BcfH was solved using the 2W-MAD phasing protocol of the experimental AutoRickshaw automated crystal structure determination platform (47) at the synchrotron facility. The marker atom structure factors (FA values) were calculated using the program SHELXC, and initial selenium positions were found with the program SHELXD (55) and the substructure was completed using PHASER (42). The occupancy of all substructure atoms was refined using the program BP3 (49). The sixfold noncrystallographic symmetry (NCS) operator was found using the program RESOLVE (65). Density modification and NCS averaging were performed using the program PARROT in the CCP4 software suite to resolution 3.1 Å (7). A partial model was produced using the program BUCCANEER (6) with SAD refinement and refined to R/Rfree of 37.3%/43.0%. The partial model was further improved using MRSAD phasing protocol of AutoRickshaw (48) against the peak dataset, which underwent six rounds of phase improvement, model building, and refinement cycles. In the resulting model, of a total of 1524 residues in six chains, 1452 residues were correctly built, and refinement of the partial model converged to 26.1/34.32%, R/Rfree. The resulting model was refined against the native dataset to 2.34 Å resolution. The structure was iteratively improved by alternating refinement in REFMAC5 (44) and manual model building in COOT (14). Final refinement was performed using PHENIX (1), including TLS refinement (Table 1). The structural superposition of molecules was carried out using the SSM options from the COOT. Molecular figures were generated using PyMOL (9), and figures of the electrostatic potential were generated using APBS Electrostatics options from PyMOL.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed on both oxidized and reduced BcfH on multiple occasions at concentrations ranging from 0.2 to 3.0 mg/mL. In all cases, samples were obtained after size exclusion chromatography (Superdex™ S-75 16/60; GE Healthcare Life Sciences) in 25 mM HEPES, 150 mM NaCl pH 7.0 with 0.1 mM EDTA. AUC experiments were performed using a Beckman Optima XL-A analytical ultracentrifuge. Protein samples (380 μL) and reference solutions (400 μL) were loaded into double sector quartz cells using a Beckman 8-hole An50 Ti rotor. Initial scans were performed at 725 g to determine the optimal wavelength and radial positions. Absorbance readings were collected at 280 nm and 128,794 g at 20°C. Solvent density, solvent viscosity, and estimates of the partial specific volume of BcfH (0.7331 g/mL) at 20°C were calculated with SEDNTERP (39). Data were analyzed using c(s) and c(M) with SEDFIT (56).
SAXS studies
SAXS data for BcfH were collected on the SAXS-WAXS beamline at the Australian Synchrotron. Four serial dilutions of ∼5 mg/mL stocks were prepared in 25 mM HEPES, 150 mM NaCl pH 7.0 with 0.1 mM EDTA and loaded into a 96-well plates, yielding solutions at 0.3, 0.6, 1.3, 2.5, and 5.0 mg/mL. Data reduction was carried out using Scatterbrain software (v 2.71), and corrected for solvent scattering and sample transmission. The estimated molecular mass was calculated using contrast and partial specific volumes determined from the protein sequences. Data processing and Guinier analysis were performed using Primus (v 3.2) (37). The pair-distance distribution function (p(r)) was generated from the experimental data using GNOM (v 4.6) (63), from which I(0), R g, and D max were determined. The program DAMMIN (v 5.3) (64) was used to generate 16 dummy-atom models for BcfH (assuming a C 1 point-group symmetry), all of which were averaged using the program DAMAVER (v 2.8.0) (71). A scattering curve was calculated from the BcfH crystal structure (PDB ID: 7JVE, chains A, B, and C) using CRYSOL (62), and this was transformed into a p(r) using GNOM. Rigid-body modeling was performed using CORAL (v 05) (50) against the 0.3 mg/mL data. Chains A, B, and C in 7JVE were oriented, such that the z-axis passed through the approximate threefold axis of the dimerization domain, and the origin positioned at the geometric center of the three domains. During optimization, the three N-terminal domains (up to residue R49) were fixed in place, while the C-terminal domains (residues A57-G254) were free to move, and the glutamine-rich linker was represented by a flexible dummy-atom linker. The rigid-body modeling was repeated 16 times, and the model with the best fit was chosen as the representative model. SAXS data and models for BcfH have been deposited in the SASBDB with accession ID: SASDJL7). Details are given in Supplementary Table S3.
Measurement of redox potential
The redox potential of BcfH was measured using AMS gel shift analysis as described previously (31). Recombinant BcfH (22 μM) was incubated overnight at room temperature in degassed redox buffers containing 100 mM sodium phosphate, pH 7.0, 1 mM EDTA supplemented with 100 mM GSSG and varying concentrations (50 μM–15 mM) of GSH. After incubation, the reactions were quenched with 10% (w/v) trichloroacetic acid (TCA), free sulfhydryl groups were alkylated AMS, and samples were analyzed by 15% SDS-PAGE. The obtained biphasic sigmoid shaped curve was fitted to the biphasic sigmoid equation in OriginPro (OriginLab Corporation), and the resulting equilibrium constants were then used to calculate the corresponding redox potentials using the Nernst equation (Equation 1).
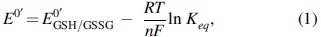
where E 0′GSH/GSSG = −240 mV, R = 8.314 J K−1 mol−1, T = 298 K, n = 2 (number of electrons transferred), and F = 9.649 × 104 C mol−1.
The data presented are the mean ± standard deviation error (SD) from three experimental replicates.
Insulin-reduction assay
The disulfide oxidoreductase activity of BcfH was measured by the ability of the protein to catalyze insulin reduction in the presence of DTT, as described previously (28). The reaction mixture (total volume 200 μL) was prepared directly in a 96-well microtiter plate by adding a final concentration of 5 μM of protein (DsbA, DsbC, or BcfH) in a buffer that consists of 100 mM sodium phosphate, pH 7.0, 2 mM EDTA and 0.33 mM DTT. Insulin (Sigma-Aldrich) at 0.13 mM concentration was added to the reaction mixture just before measurements commenced. The reduction of insulin was monitored by measuring the optical density at 650 nm for 80 min in a Spectramax M5 spectrophotometer (Molecular Devices). The uncatalyzed reduction of insulin by DTT without added enzyme served as the negative control. The data presented are the mean ± SD from the three independent replicates.
Disulfide isomerase activity
The in vitro disulfide isomerase activity of BcfH was evaluated using the scRNase A assay performed as previously described (18). In brief, reshuffling of scRNase A (40 μM) was carried out at room temperature in 100 mM sodium phosphate pH 7.0, 1 mM EDTA, 8.2 μM DTT, and 10 μM BcfH, DsbA or DsbC. At various time points, 50 μL of the reaction sample were mixed with 150 μL of 4 mM cytidine 2′,3′-cyclic monophosphate (cCMP) in 100 mM sodium phosphate, 1 mM EDTA, pH 7.0. The ability of RNase A to hydrolize cCMP was monitored at 296 nm in a Spectramax M5 spectrophotometer (Molecular Devices). Samples containing native RNase A and scRNase A without added enzymes served as positive and negative controls, respectively. The assays were performed in triplicate using three independent protein samples.
Electron transfer experiments
Protein samples (BcfH, N-ScsB, N-DsbD, and DsbA) were prepared at 50 μM concentration, and oxidized or reduced by incubating at 4°C for 1 h using 10 mM GSSG or GSH (Sigma-Aldrich), respectively. BcfB was oxidized or reduced by using 25 mM GSSG or 30 mM tris(2-carboxyethyl)phosphine (TCEP; Astral Scientific, Australia), respectively. Excess GSSG and TCEP in the reaction mixtures was removed by SEC (Superdex 75 10/300 GL; GE Healthcare Life Sciences) equilibrated in 100 mM sodium phosphate, pH 7.0, 50 mM NaCl, and 1 mM EDTA. The final redox state was confirmed by AMS gel shift analysis.
For electron transfer experiments, 100 μL of 20 μM reduced proteins (BcfB, N-ScsB, and N-DsbD) were mixed with 100 μL of 20 μM oxidized BcfH. Fifty-microliter aliquots were taken at 30 and 120 s time points and quenched with 10% (w/v) TCA. After centrifugation, the protein pellets were then washed with cold acetone and solubilized in 50 mM Tris, pH 7.0, 1% SDS containing 4 mM AMS (Life Technologies, Australia). The alkylation reaction was stopped by adding nonreducing loading dye, and the oxidized and reduced proteins were separated by 15% SDS-PAGE. Because N-ScsB and BcfH have similar sizes, the N-ScsBred–BcfHox electron transfer reaction was also analyzed by Western blotting using a polyclonal BcfH antibody.
Peptide oxidation assay
The thiol oxidation activity of BcfH was assessed as previously described using a synthetic model peptide (69). In brief, the lyophilized peptide substrate (CQQGFDGTQNSCK with a 1,4,7,10- tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelate at the N-terminus and a methylcoumarin amide group coupled to the lysine) was reconstituted at 2 mM in 100 mM imidazole pH 6.0. Europium trifluoromethanesulfonate (Sigma-Aldrich) was added to the peptide solution at a molar ratio of 1:2. Assays were run on an Envision Multilabel Plate Reader (PerkinElmer) using 96-well Optiplates, with excitation at 340 nm and emission at 615 nm. Total reaction volume in each well was 50 μL, containing 80 nM DsbA or BcfH and 2 mM GSSG in a buffer of 50 mM MES, pH 5.5, 50 mM NaCl, 2 mM EDTA. The reactions were initiated by the addition of 8 μM peptide substrate. Time-resolved fluorescence was used to measure disulfide bond formation, with a 100 ms delay before reading and a 400 ms reading time. Measurements were carried out in triplicate for each protein, and data were analyzed using GraphPad Prism version 5.0 (GraphPad Software, Inc.).
Biofilm culture using the Calgary Biofilm Device
Biofilms were grown and established in the Calgary Biofilm Device (CBD) (MBEC assay; Innovotech, Inc., Canada), which was used unmodified. The device consists of a two-part reaction vessel; the top component contains 96 identical pegs protruding down from the lid, which fits into a standard, flat-bottom, 96-well plate (the bottom component). Biofilm assays were performed as previously reported (68) with the following modifications: overnight bacterial cultures in lysogeny broth (LB-Lennox) were diluted to 107 CFU/mL in LB and used to inoculate the enclosed, flat-bottom, 96-well plate with ∼106 bacterial cells (170 μL) in each well. The peg lid was inserted into the inoculated wells, and the complete CBD was statically incubated for 48 h at 37°C and 95% relative humidity. The peg lid was removed from the growth plate and rinsed once for 5 s in phosphate buffered saline (PBS; 96-well plate, with 200 μL in each well). The rinsed CBD lid, with attached pegs and biofilms, was transferred to a new 96-well plate containing fresh PBS recovery medium. To facilitate the transfer of any remaining viable cells to the recovery medium, the device was sonicated for 30 min at <20°C. The peg lid was then discarded, the biofilm-recovered bacteria from each well of the recovery plate were serially diluted in PBS, and then triplicate 5-μL aliquots of each dilution were plated onto LB agar and incubated overnight at 37°C for viable CFU enumeration. Assays consisted of 4 biological replicates. Data were analyzed for group differences using the Kruskal–Wallis test in GraphPad Prism 8.
Swarming motility assay
P. mirabilis strain PM54 ΔscsC carrying pSU2718 (vector control), pSU2718-PmscsC (positive control), or pSU2718-SebcfH were streaked onto NaCl-free LB-Lennox containing 1.5% agar and 100 μM isopropyl b-D-1-thiogalactopyranoside (IPTG). The plasmids were maintained with chloramphenicol at 30 μg/mL. Plates were incubated at 37°C for 20 h before imaging.
Swimming motility assay for DsbA cellular activity
Statically grown 37°C stationary E. coli cultures were adjusted to OD600 ∼ 2.0, and spotted 2 μL onto LB-Lennox motility agar containing 1 mM IPTG and 0.25% bacteriological agar. Media were supplemented with 50 μg/mL ampicillin to maintain pWSK29-based plasmids. The plate was incubated at 37°C, and motility was monitored over time with images taken at 12 h. Plate images shown in figures are representative of four biological replicates.
Mucoidal phenotypic reporter assay for DsbC activities
Single bacterial colonies were patched onto M9 media containing 0.2% glucose, 1.5% bacteriological agar, and 1 mM IPTG. The plate is supplemented with 17 μg/mL chloramphenicol to maintain pSU2718-based plasmids. Plates were incubated at 21°C, and image was taken at 40-h time point. The figure is representative of three biological replicates.
Alkaline Phosphatase Assay for DsbC cellular activity
Stationary grown cultures diluted 1:50 in LB-Lennox with appropriate antibiotics for plasmid maintenance were grown at 37°C with aeration. At OD600, 10 mM L(+)-arabinose was added to the cultures to induce AppA phosphatase expression for an additional 50 min. A culture aliquot of 1 mL was transferred into a reaction tube containing 100 μL 1 M iodoacetamide (dissolved in ddH2O) and incubated on ice for 20 min. Samples were washed twice in wash buffer (200 mM Tris-HCl pH 7.3, 250 mM NaCl, 40 mM NH4Cl) and resuspended in 1 mL wash buffer. One hundred microliters of resuspended samples were transferred to reaction tubes containing 12 μL 0.1% SDS, 12 μL chloroform, and 900 μL reaction buffer (1 M Tris-HCl pH8.0, 1 mM MgCl2, 1 mM ZnCl2). Samples were shortly vortexed and incubated at 37°C for 5 min. For each sample, 1.53 μL 500 mM PNPP was added, mixed by inversion, and incubated at 28°C until a yellow color was developed in the positive control. The reaction was stopped with 120 μL stopping buffer (83.33 mM EDTA, 416.67 mM K2HPO4). Cell debris was removed by centrifugation at 20,000 g for 3 min, and 200 μL of supernatant were aliquoted to 96-well plates for OD420 and OD550 readings. The Units activity of phosphatase was calculated as a function of OD420, OD550, the time it took for color development and volume of cells used.
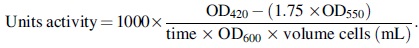
Periplasmic extraction method
Overnight stationary cultures were diluted 1 in 50 in fresh LB containing 20 μg/mL chloramphenicol and grown to OD600 0.5 at 37°C with aeration (25 mL). IPTG was added at 1 mM concentration, and cultures were further incubated for 2 h. Cells were harvested by centrifugation at 5°C for 3 min and washed once with PBS (1 × ). Pellets were resuspended in 900 μL 0.1 M Tris pH 8.0, 500 mM sucrose, 0.5 mM EDTA, and incubated for 5 min on ice. Cells were pelleted by centrifugation at 5°C for 3 min; supernatants were carefully removed and pellets were gently resuspended in 400 μL 1 mM MgCl2 hypotonic solution. Pellets were shortly incubated on ice for 15 s, then 20 μL MgSO4 (20 mM) was added, and cells were centrifuged at 5°C for 3 min. Periplasmic fractions were harvested from supernatants.
Ponceau S visualization and immunoblotting
Transferred nitrocellulose membranes were stained briefly with Ponceau S solution (0.1% Ponceau S in 5% acetic acid). Membranes were then rinsed with ddH2O to visualize the transferred protein contents before blotting. Immunoblot analyses were performed using a rabbit α-BcfH primary antibody (1:6000) and Rabbit α-GroEL primary antibody (1:100,000; Invitrogen), followed by goat α-rabbit IgG-alkaline phosphatase (1:20,000; Invitrogen) for colorimetric detection using BCIP/NBT substrate (Sigma-Aldrich). The Ponceau S stained membrane and developed blots were imaged in the BioRad GelDocXR+ system using the Image Lab software (v5.1; BioRad).
Movie generation
Supplementary Movie S1 was created in PyMOL (9) by generating the molecular morphs between the crystal structure of BcfH in an intermediate conformation (where one of the monomers adopts a compact conformation) and the modeled structure of BcfH in an extended conformation (all protomers adopting a fully extended configuration). The movie was generated using the following parameters: 3 refinement cycles, 30 output states, with the interpolation method RigidMOL.
Footnotes
Authors' Contributions
B.H. and P.S. conceptualized and designed the study and discussed the data. M.T. contributed to microbiological study design and data analysis. P.S., J.J.P., G.W., L.H., Y.H., A.D.V., A.E.W., and S.P. performed the experimental work and the analysis of the data. All authors contributed to writing of the article. All authors have approved the version to be published.
Acknowledgments
This research was undertaken on the MX1, MX2, and SAXS/WAXS beamlines at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. The authors also acknowledge the use of the La Trobe University Comprehensive Proteomics Platform.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Australian Research Council (ARC) project grants (DP 190101613, DP180102987, and DP150102287), an ARC Future Fellowship (FT130100580), ARC DECRA (DE130101169), the National Health and Medical Research Council (NHMRC) Project Grants (GRT1144046, GRT1143638), and a Vera and Clive Ramaciotti Foundations Health Investment Grant (2017HIG0119). M.T. was supported by a Vice-Chancellor's Research Fellowship from the Queensland University of Technology.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Movie S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.