
Abstract
Significance:
Reactive species have been classically considered causative of age-related degenerative processes, but the scenario appears considerably more complex and to some extent counterintuitive than originally anticipated. The impact of reactive species in precocious aging syndromes is revealing new clues to understand and perhaps challenge the resulting degenerative processes.
Recent Advances:
Our understanding of reactive species has considerably evolved, including their hormetic effect (beneficial at a certain level, harmful beyond this level), the occurrence of diverse hormetic peaks in different cell types and organisms, and the extended type of reactive species that are relevant in biological processes. Our understanding of the impact of reactive species has also expanded from the dichotomic damaging/signaling role to modulation of gene expression.
Critical Issues:
These new concepts are affecting the study of aging and diseases where aging is greatly accelerated. We discuss how notions arising from the study of the underlying mechanisms of a progeroid disease, Cockayne syndrome, represent a paradigm shift that may shed a new light in understanding the role of reactive species in age-related degenerative processes.
Future Issues:
Future investigations urge to explore established and emerging notions to elucidate the multiple contributions of reactive species in degenerative processes linked to pathophysiological aging and their possible amelioration. Antioxid. Redox Signal. 37, 208–228.
Introduction
From the mitochondrial free radical theory of aging (61) to present notions of reactive species in biological processes, new players have joined the stage, and the role of the original players has been to some extent overturned. Due to the very large number of investigations, reactive oxygen species (ROS) and to some extent reactive nitrogen species (RNS) retain the role of prima donna in the landscape of reactive species, but the impact of reactive sulfur species (RSS) and their interactions, which altogether gave rise to the concept of reactive species interactome (RSI) (31), has emerged. Similarly, ROS-generating mitochondria have been acknowledged a more central role in the cell response/adaptation to the free radical input.
Thus, the casting and starring roles have evolved and may further change in the future. Moreover, the present scenario appears considerably more complex than the original one, with several main actors playing double, and apparently opposite, roles. New functions have also been attributed beyond this dichotomy, and the impact of at least ROS in regulating gene expression, likely through epigenetic modifications, appears significantly influencing the adaptive response of the cell to changing conditions as well as in response to insult. For instance, oxidative stress changes the promoter occupancy of a multifunctional transcription factor, Cockayne syndrome protein B (CSB) (see next sections), possibly impacting on global gene expression (91).
Oxidative stress also promotes epigenetic changes that derepress the p16INK4A locus, and thereby induces cellular senescence that blocks the regenerative capacity of skeletal muscle stem cells in geriatric mice (49). Senescence is an irreversible cell cycle arrest of metabolically active cells, which has been associated with aging (107). The field of aging has not fully investigated yet these emerging notions. Then indications from progeroid diseases, which are genetic syndromes where aging is greatly accelerated, are providing interesting clues for future studies also on physiological aging.
Aging is a multifactorial process that consists of progressive dysfunction and eventually destruction of every tissue and organ over time (80). Even though multiple molecular hallmarks have been identified (98), such as cellular senescence, mitochondrial dysfunction, and epigenetic alterations, the fundamental mechanisms of physiological aging remain largely unknown. The debate on modern biological theories of human aging is now split between programmed theories that claim aging is regulated by intrinsic mechanisms, and the error theories that emphasize environmentally caused accumulation of damage (73).
For decades, ROS have been closely correlated with this latter category by being considered liable for damage to biomolecules during aging, according to the free radical theory of aging developed by Harman (61). Indeed, overexpression or knockout of key ROS-detoxifying enzymes has been shown to extend or shorten lifespan respectively, in different organisms (164). However, this theory has been challenged by multiple studies reporting that ROS and oxidative stress paradoxically promote longevity (164) and, more recently, by the lack of a beneficial (or even detrimental) effect of antioxidant treatment/supplementation on longevity in epidemiological studies (179). These apparent contradictory observations with the free radical theory of aging have been finally reconciled by the identification of physiological signaling functions of basal levels, or low burst of ROS (66).
In the context of aging, the signaling function of low doses of ROS was identified in inducing an adaptive/preconditioning response that protects from age-related alterations (141), which contrasts with the harmful properties of ROS at high concentrations. Thus, the opposite roles of ROS, which depend on the concentration of these molecules in a process known as “hormesis,” seem to play important roles in aging, and have been extensively reviewed (152). It should be noted that in some studies it has been reported a positive effect of antioxidants on lifespan extension (147). However, it is claimed that these beneficial effects occur in species evolutionarily distant to mammals, whereas positive effects in mammals usually show an increase of mean but not maximal lifespan, thereby suggesting an indirect effect on aging (147).
Based on the mechanistic findings in progeroid Cockayne syndrome (CS), and in the context of the wide changes that the reactive species field is experiencing, we survey here present knowledge on the role of components of the RSI in pathophysiological aging, underscoring missing information that will hopefully be fulfilled in the future.
The Damaging Effect of ROS, RNS, and RSS and Implications in Aging and Age-Related Pathologies
The term “oxidative stress” is commonly associated with ROS/RNS-mediated damage of DNA, proteins, and lipids, while RSS contribution has been overlooked so far. The accumulation of damaged biomolecules was the leading explanation of the aging process at the molecular level (140). The chemical interaction of nitric oxide (NO) with the ROS superoxide (O2 −) leads to the formation of peroxynitrite (ONOO−), a potent damaging RNS (31). Similarly, ONOO− can react with other species to form many additional RNS such as nitrogen dioxide (NO2) or dinitrogen trioxide (N2O3) (31).
The role of RNS in aging has been difficult to assess due to the absence of specific endogenous detoxifying enzymes or scavenger drugs that selectively target RNS, although metalloporphyrins (Fe and Mn) scavenge peroxynitrite, specifically, or as well as ROS (see the specific section for detail). Among additional ROS/RNS-mediated reactions, the formation of RSS is now considered to generate biologically relevant molecules (31). RSS are produced either in oxidative or nitrosative conditions by the reaction of ROS/RNS with thiols, leading to the formation of damaging molecules such as thiyl radicals or disulfides, or in physiological conditions by direct oxidation of the product of cysteine metabolism, hydrogen sulfide (H2S), or of thiols (69).
DNA damage
Accumulation of genetic lesions has long been suggested as one of the major forms of damage responsible for the aging process (98). One of the most common products of oxidative damage on DNA is 8-hydroxy-2-deoxyguanosine (8-OHdG), due to the hydroxyl attack of deoxyguanosine residues. If unrepaired, this base modification is highly mutagenic since it can result in GC to TA transversions. Age-related accumulation of 8-OHdG has been observed to a greater extent in mitochondrial DNA (mtDNA) than nuclear DNA (153).
Whether or not mtDNA damage has a causal role in aging, it is considered a biomarker of aging. MtDNA lesions, mainly 8-OHdG, tend to accumulate not only in response to increasing levels of ROS/RNS but also because they cannot be properly repaired when the DNA repair mechanisms are less efficient, as it is the case in aged individuals, or they are defective like in patients affected by repair-deficient progeroid syndromes. Analogously to proteins and lipids (see protein and lipid damage sections), DNA can also be oxidatively damaged by peroxynitrite, which is a primary trigger of multiple forms of DNA damage, leading to acute cellular injury and/or apoptosis (173). H2S has been also reported as a genotoxic agent which increased, genomic DNA damage when DNA repair was inhibited in vitro (5) (Fig. 1).
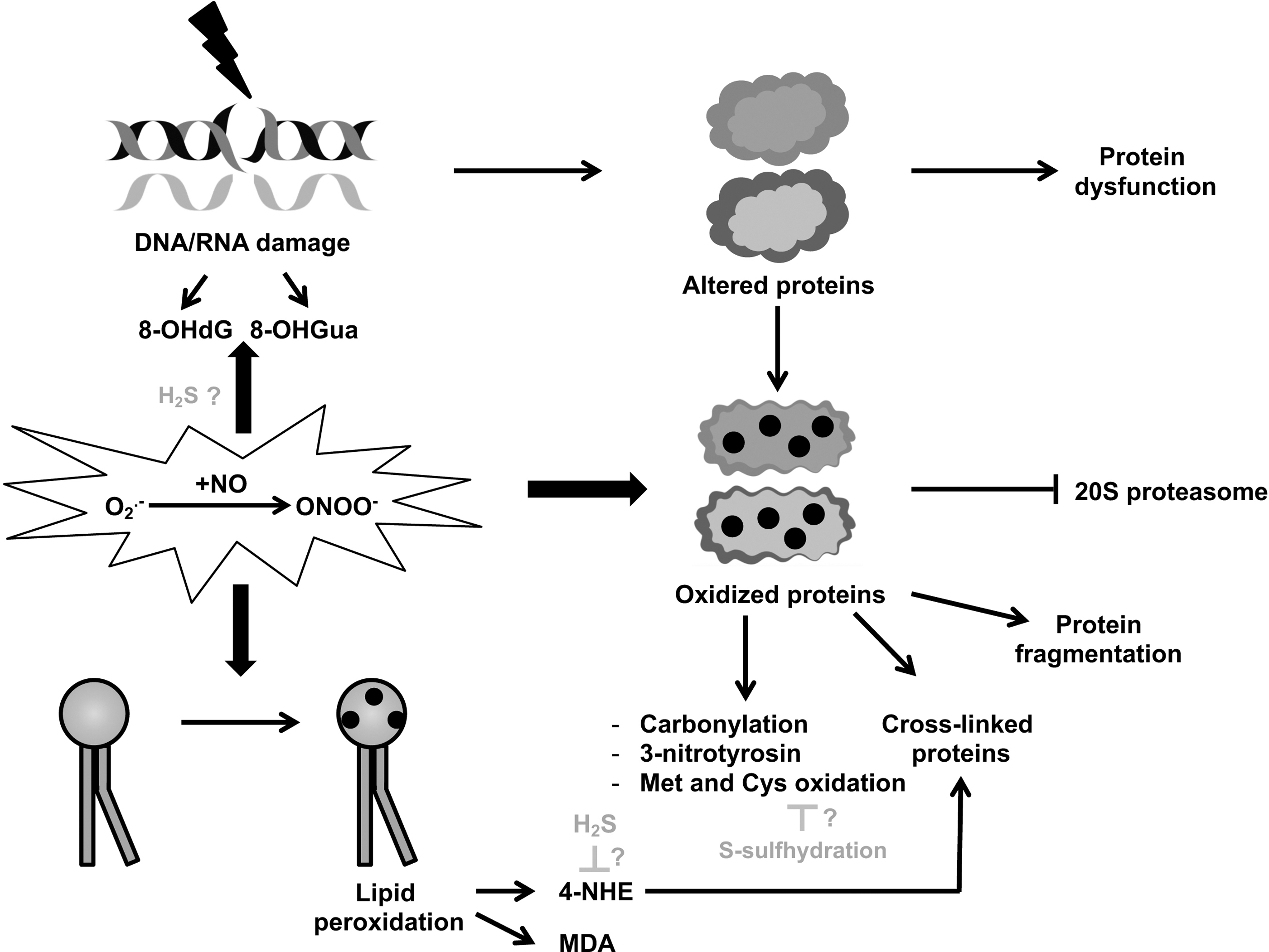
Both DNA and RNA oxidative damages may lead to the production of abnormal proteins, finally resulting in dysfunctional cells that, if not eliminated by apoptosis or put in proliferative arrest by senescence, may compromise tissue homeostasis.
Protein damage
In the literature, a considerable amount of evidence reports oxidative damage to proteins in normal aging and age-related diseases, the basic mechanisms of which have been detailed (168). In contrast to reversible methionine and cysteine oxidation, protein carbonylation (the most frequent type of ROS-induced protein modification) appears irreversible and has therefore been used as a marker for severe chronic oxidative damage (35). Protein oxidation is a downstream effect of ROS that has been reported in progeroid diseases, in particular elevated levels of carbonylated proteins in fibroblasts from the Hutchinson–Gilford progeria syndrome (HGPS) and Werner syndrome (WS) patients (116, 183) as well as CS patients (2), supporting the classic notion of oxidative stress-induced damage in accelerated aging (see next sections).
Due to increased surface hydrophobicity and decreased solubility, oxidized proteins are considered toxic since they can form large aggregates, impairing fundamental cellular processes. For instance, these aggregates can interact with membrane proteins, causing morphological changes and an increase in free Ca2+ that may eventually lead to apoptosis or necrosis (170). Moreover, these aggregates become progressively resistant to proteolytic digestion and irreversibly inhibit the 20S proteasome activity (36) (Fig. 1). Indeed, the intracellular steady-state level of oxidized proteins is determined not only by the rate of ROS/RNS production and antioxidant response but also by the rate of proteolytic degradation, which also depends on age (98).
Regardless of the causing mechanism, it is well established that the level of oxidized proteins increases during aging. On the contrary, protein persulfidation, described as a cytoprotective posttranslational modification capable of preventing cysteine oxidation, has been recently found to decrease with age (200) (see next sections).
NO is ubiquitously and reversibly able to oxidize protein cysteines through an enzymatic process called S-nitrosylation that results in a posttranscriptional regulation of protein function (169). Mice deficient for denitrosylase, the enzyme that removes S-nitrosylation, are considered a model of accelerated aging (143). Similarly, the expression of this enzyme progressively declines in cells undergoing senescence, as well as in aging mice and humans, whereas it is preserved in centenarians (143). Accordingly, aging is associated with an excess of protein S-nitrosylation (78), which becomes itself responsible for cellular damages (143). The biology of NO and the subsequent S-nitrosylation of proteins seem to follow a process of antagonistic pleiotropy, that is, being beneficial early in life and detrimental during aging, displaying temporally opposite processes, in addition to hormetic dose dependence, as it is the case also for ROS.
The most preponderant biological action of RNS, which can be considered their footprint, is protein tyrosine nitration (3-nitrotyrosine) (42). 3-nitrotyrosine increases in the aged brain (68), as well as in different tissues during aging and cellular senescence, as assessed by mass spectrometry (192), suggesting the involvement of RNS-mediated damage in aging. However, it is still unclear whether this mostly irreversible posttranslational modification is just a biomarker or actually participates in mechanisms leading to aging.
To note, ONOO−-mediated protein nitration in most cases results in loss of function (42). These data are in line with the observed loss of proteostasis during aging (98). This phenomenon is particularly noteworthy when mitochondrial respiration (oxidative phosphorylation [OXPHOS]) and antioxidant proteins are targeted, potentially promoting aging by reinforcing the oxidative/nitrosative stress vicious circle (131). Altogether, these results suggest an active role of 3-nitrotyrosine in aging and age-related processes. However, further studies are needed to discriminate whether nitration of specific protein families/signaling pathways drives or protects (see next sections) against the aging process. Finally, little is known about the implication of other RNS-mediated reactions in aging, which will represent a major challenge for future investigations.
Lipid damage
ROS/RNS can additionally inflict damage to polyunsaturated fatty acids in membrane lipid bilayers, determining the formation of aldehydes, which are considered toxic especially for their ability to interact with membrane proteins and thereby interfering with intracellular functions, similarly to oxidized proteins (128). Among aldehydes produced by lipid peroxidation, which is mediated by peroxynitrite (145), malonaldehyde (MDA), and 4-hydroxynonenal (HNE) have been extensively reported (7). MDA is produced at high levels during lipid peroxidation, and is therefore commonly used as a biomarker of oxidative stress (103) (Fig. 1).
4-HNE can affect cell function through its ability to form adducts with proteins involved in signal transduction and gene expression (39) as well as crosslink with oxidized proteins, thereby interfering with their degradation (46). Accumulation of 4-HNE has been previously linked to senescence (45), age-related diseases (52), as well as the progeroid diseases CS (63) and Down syndrome (DS) (8).
Given the age-associated increase in the steady-state concentration of this aldehyde, lipid peroxidation may play an important role in initiating and/or mediating specific aspects of the aging process. Some lipid peroxidation products are believed to contribute to the production of lipofuscin, an intralysosomal yellow-brown pigment that accumulates with age, which is widely considered a hallmark of aging (175). Moreover, previous studies suggested that an increased peroxidation of mitochondrial membrane lipids is responsible for reduced membrane fluidity observed with age (59). Reactive aldehydes are also able to interact with DNA bases, exhibiting genotoxic effects.
Interestingly, 4-HNE-DNA adducts are recognized by nucleotide excision repair (NER) proteins, including CSB that is defective in progeroid CS (100) (see specific section below). Preliminary in vitro studies led the authors to speculate that when these lesions are not repaired due to CSB gene mutation, they may contribute to the accelerated aging observed in CS patients. Given the multiple deleterious effects of 4-HNE accumulation, detoxification strategies by 4-HNE scavenger compounds (including H2S, see next section) have gained interest in the recent years.
In summary, the ROS/RNS ability to interact with, and consequently damage, redox-sensitive biomolecules has been widely used to investigate oxidative stress, whereas future studies should reasonably also include RSS. Given their short half-life, direct quantification of ROS/RNS has been associated with or replaced by the detection of oxidative damage biomarkers. These include oxidized cysteine that generates highly reactive thiyl radicals (RS*) that can in turn catalyze the secondary oxidation of other aminoacids, leading to protein damage (159).
Consequently, a large amount of evidence shows an age-dependent accumulation of oxidatively damaged biomolecules, such as lipids, proteins, DNA, and RNA, in various organs and tissues, rather than the reactive molecules themselves. Detection of RSS per se is particularly challenging because of the apparent overlap of RSS with ROS with standard laboratory techniques (37). Moreover, hydropersulfides are rather considered an antioxidant with cytoprotective effect.
Tissues and Organs Affected by ROS, RNS, and RSS During Aging and Age-Associated Diseases
Damages induced by altered regulation and altered levels of ROS, RNS and RSS are implicated in the aging process per se (see specific section), and also in age-associated diseases. These diseases result from degeneration of tissues and organs with age, where the cardiovascular and nervous systems are particularly affected. Indeed, the aging heart is characterized by structural remodeling associated with ventricular hypertrophy and fibrosis, ultimately leading to functional consequences on filling and ejection (71). These dysfunctions make aging possibly the major risk factor to develop cardiovascular pathologies (144).
Similarly, the age-associated endothelial dysfunction, which is mainly driven by nitrosative stress, has a significant impact on the incidence of cardiovascular diseases (14). A substantial number of chronic heart diseases, including heart failure, rhythm disorders, hypertension, and atherosclerosis, have been associated with oxidative stress (71).
Mechanistically, reactive species have been found to participate in the pathophysiology of arteriosclerosis. Indeed, oxidative modifications of low-density lipoprotein lead to the release of bioactive lipids triggering activation of endothelial cells and expression of adhesion molecules (e.g., VCAM-1) (60). ROS can also, through oxidation of cysteines, directly activate G proteins. In the hypertrophic heart, these proteins mediate cardiomyocyte transduction of factors such as angiotensin II or alpha-adrenergic agonists, to finally participate in ventricular remodeling-dependent heart failure. Similarly, heart failure depends on heart fibrosis and matrix remodeling that can be induced by the ROS-dependent posttranslational activation of metalloproteinases classically secreted in inactive forms (167).
Finally, plasma levels of 8-OHdG correlate with the incidence of cardiac events and are now considered a biomarker of heart failure (162). In cardiomyocytes, the oxidation of thiol components of the ryanodine receptors favors the release of Ca2+ from the sarcoplasmic reticulum, a mechanism that participates in the pathogenesis of heart rhythm disorders (i.e., atrial fibrillation) (188). The reaction of O2 − with the vasodilator NO leads to decreased bioavailability of the latter, vasoconstriction, and eventually chronic hypertension (15). Most of these examples involve damage related to ROS and RNS, however, deregulation of the RSS metabolism with aging could also impact on cardiovascular diseases, as suggested and recently reviewed (84).
During aging, degeneration of the brain is characterized by a decline in cognitive, sensory, and coordination capacities (1). The brain is sensitive to oxidative damage, especially because of its high lipid content, and lipid peroxidation as well as protein oxidation and oxidative damage to DNA have been involved in cognitive decline in aging (18). However, differently from the cardiovascular system, the mechanism(s) by which reactive species affect cerebral aging is/are less clear. Evidences suggest a decrease in the ROS/RNS-dependent signaling of nuclear factor erythroid-derived 2-like 2 (NRF2) that elicits a robust antioxidant response (149). Conversely, ROS can play a role in brain aging by activating signaling pathways related to inflammation or cell death (18), or affecting neurotransmitter receptors, and thereby synaptic communication (18).
Another aspect of cerebral aging is dementia and the increased risk to develop neurodegenerative diseases (67). Neurodegenerative diseases are characterized by neurotoxic aggregations, several of which have been linked to oxidative damage (18). For instance, in Alzheimer's disease (AD), lipid peroxidation reduces membrane permeability and consequently neurotransmission (99). In addition, 4-HNE adducts on the major neuronal receptors, LRP1, inhibit the clearance of beta-amyloid peptides, and therefore trigger their accumulation (119). These adducts also promote the accumulation of the Tau protein, a major alteration in AD (22).
In Huntington's disease, an inherited condition resulting in progressive degeneration of nerve cells, free radicals are directly implicated in protein misfolding and accumulation of the mutant huntingtin (127). To notice, the cytoprotective role of RSS in the nervous system has been documented (199) (see also next section), but further studies are needed to identify whether alterations of RSS-related mechanisms and pathways are implicated in neurodegenerative diseases. Oxidative damage and the related alteration of numerous pathways have been implicated in the development of other age-related pathologies, such as chronic kidney and lung diseases, as well as diabetes and cancer (95), which is not further developed here.
The Signaling Role of ROS, RNS, and RSS with Insight on Age-Related Conditions
The signaling function of reactive species that control cellular processes as diverse as growth, survival, proliferation, and metabolism (157) is largely initiated by ROS. Indeed, the production of O2 − is regulated by various physiological extracellular (endogenous: e.g., cytokine, growth factors; exogenous: e.g., nutrition, exercise) or intracellular cues (e.g., mitochondrial metabolism) via the stimulation of ROS-producing enzymes such as mainly NADPH oxidase and the mitochondrial respiratory complexes (122). Although O2 − may have a signaling function per se, the underlying physiological signaling of reactive species is rather attributed to the more stable hydrogen peroxide (H2O2), a product of the rapid conversion of the O2 − by cytosolic and mitochondrial superoxide dismutase (SOD) (157, 184).
H2O2 is now considered at the basis of redox signaling, suggesting that H2O2 levels must be tightly regulated to sustain this physiological role. Indeed, catalase, glutathione peroxidases (GPX), and peroxiredoxins (PRDX) take care of restoring basal levels of H2O2 to avoid the instauration of deleterious high levels of oxidative stress (166) (Fig. 2).
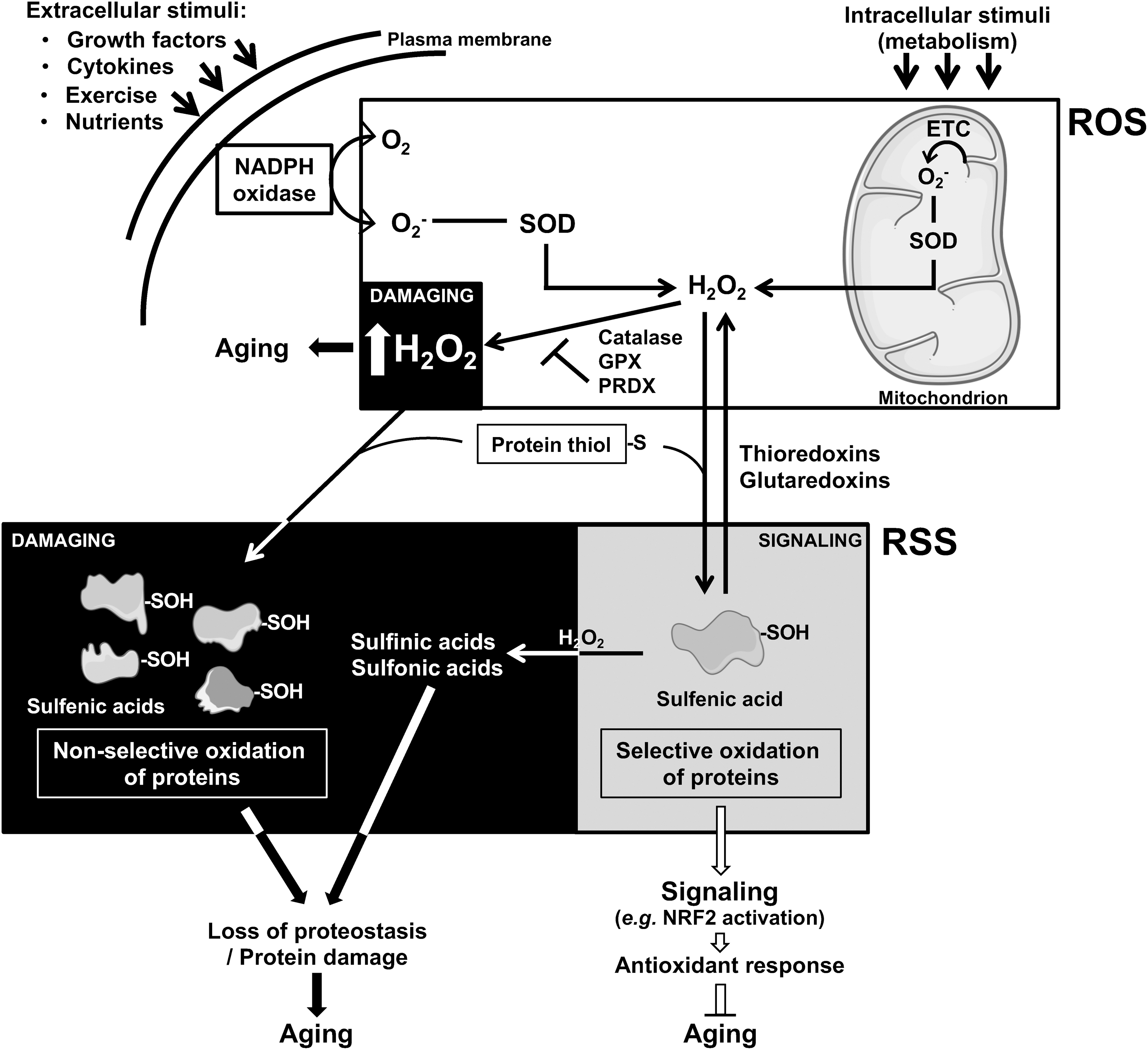
In addition, the physiological modulation of H2O2 levels propagates its signaling by reacting with thiol groups of proteins (cysteines), thereby affecting the allosteric conformation of regulatory proteins (e.g., transcription factors, membrane receptors, enzymes), and potentially impacting on their downstream functions or targets (166). Interestingly, this reversible oxidation of protein thiols induces the formation of sulfenic acid (21), which is an RSS (53). Thus, the signaling function of reactive species starts with ROS and is then mediated by RSS, showing an interesting example of the cooperation between different reactive species, and underscoring the concept of RSI (31) (Fig. 2). Some authors demonstrated that peroxynitrite reacts faster than H2O2 with certain types of protein thiols, and suggest a specific role of peroxynitrite in physiological signaling (134), not further detailed here.
The levels of this specific posttranslational protein modification (oxidation of protein thiols) are intracellularly controlled by the action of thioredoxins and glutaredoxins (65), as it was the case for the abovementioned enzymes that control H2O2 levels. Importantly, further uncontrolled and generally irreversible oxidation of sulfenic acids can generate other RSS, which no longer act as signaling molecules, and rather generate damaged proteins (157). Notably, H2O2-dependent oxidation of cysteines seems to affect some proteins more than others (e.g., proteins carrying more accessible cysteines) (55), suggesting a selectivity in the propagation of the signaling.
As a consequence, deregulation of the H2O2-dependent signaling can potentially lead to (i) the formation of additional damage generating reactive species, and (ii) nonselective oxidation of global proteins, which are both characteristics of age-related biological alterations. Indeed, most components of reactive species signaling pathways have been associated with aging. For instance, the epidermal growth factor (EGF), known to physiologically regulate ROS production, as well as its receptor (EGFR), is downregulated in multiple aging contexts (51). Similarly, an increase of the ROS-producing enzyme NOX2 was reported during aging (148).
More specifically, H2O2 seems to accumulate in various tissues with age, thereby altering the downstream signaling (165). Finally, a recent quantitative proteomic analysis revealed a shift in the cysteine oxidation landscape of old versus young rats, suggesting an alteration of the selectivity of this process with aging (187).
Other reactive species-specific signalings have been also described. Indeed, H2S, which is itself described as a RSS, has long been considered the main mediator of the RSS biological effects (75). H2S was considered a “gasotransmitter” that regulates neuronal and vascular functions, modulates inflammation/immunity, and protects from oxidative stress (cytoprotective effect) (82). However, the lack of a clear mechanism of action and the low concentrations of H2S measured in biological samples did not allow to verify and sustain its role as a signaling molecule (163). In fact, the redox active form of sulfur-containing molecules coexists in the cell in different forms that are composed of free H2S and a pool of reactive sulfur atoms always bonded to another sulfur atom (named the sulfane sulfur pool) (84).
This sulfane sulfur pool is mainly composed of hydropersulfides that are formed either through the oxidation of H2S by oxidized thiols or direct enzymatic processes (117). Mounting evidences suggest that the sulfane sulfur pool rather than H2S, and especially hydropersulfides, mediates the signaling function of RSS (176). The enzymatic production of low-molecular and nucleophilic hydropersulfides such as cysteine persulfides and related glutathione persulfides, which have been demonstrated to be more reductant than their thiol analogues, constitute powerful antioxidants that ensure the vital cytoprotective role of RSS (69).
This cytoprotective antioxidant activity is also ensured by the chemical properties of related anions and radicals derived from hydropersulfides such as persulfide anions and perthiyl radicals (48). For instance, experiments with H2O2 demonstrated that ROS are more efficiently scavenged by glutathione persulfide than by GSH (70). On the contrary, the reaction of H2S with oxidized protein thiol leads to a specific type of posttranslational modification of proteins, called S-sulfhydration, that is the primary mechanism mediating the biological functions of H2S (196). H2S has been also proposed as a scavenger of deleterious 4-HNE adducts, extending its potential cytoprotective properties (90). Conversely, the physiological signaling of RNS remains largely unknown.
The downstream proteins and cellular pathways regulated by reactive species signaling are numerous and include stress-related transcription factors (forkhead box O [FOXO], p53 pathways) as well as transcription factors of the NF-κB, NRF2, and hypoxia-inducible factor 1 (HIF1) pathways [see review Sies and Jones (166) for details]. They also include important cellular regulators of metabolism (GAPDH), energy and nutrient response (adenosine monophosphate-activated protein kinase [AMPK], and mammalian target of rapamycin [mTOR]), and mitochondrial function (uncoupling proteins) (166). More generally, reactive species-dependent signaling targets kinases and phosphatases, as well as ion channels and signal receptors (166).
Reactive species signaling in aging
Importantly, most of these pathways are dysregulated during aging (104, 106, 150, 158) and in progeroid syndromes (via transcription factors) (47, 87, 118). An illustrating example is the NRF2 pathway. The transcription factor NRF2 is negatively regulated by the Kelch-like ECH-associated protein 1 (KEAP1) inhibitor that allows ubiquitinylation and proteasomal degradation of NRF2. The reactive species signaling induces oxidation of highly reactive cysteines of KEAP1, resulting in a conformational change and finally disruption of the KEAP1-dependent ubiquitinylation of NRF2 (83). The subsequent nuclear stabilization permits NRF2 transcriptional regulation and downstream expression of antioxidant target genes (166), through a system that is under the control of thioredoxins (19).
During aging, increased expression of KEAP1 makes the reactive species-dependent activation of NRF2 ineffective, and cells become more susceptible to oxidative stress, as well as to the other hallmarks of aging, such as genomic instability or epigenetic alterations (158). However, further studies are needed to determine whether alteration of the reactive species-dependent physiological signaling is a passenger or a driver of the aging process.
It is thus evident that damage to biomolecules caused by supraphysiological levels of ROS, RNS, and RSS is not the only mechanism by which reactive species impact on aging. First, an increase in the levels of reactive species is not necessarily deleterious. As an example, a mild increase in reactive oxidative species prolongs the lifespan through an adaptive and beneficial signaling response (hormesis) (141).
The primary mechanism by which this reactive species-dependent signaling impacts on aging relies on direct alteration of target cellular pathways. In support of this notion, beneficial oxidant-induced hormetic responses have been reported to decline with age (130). Then, it is now well accepted that reactive species participate in homeostatic regulation as signaling molecules (31), and the alteration of these processes may affect aging. The role of ROS in aging has been extensively reviewed (141, 152, 164) (see also Fig. 3), and we focus here rather on RNS and RSS.
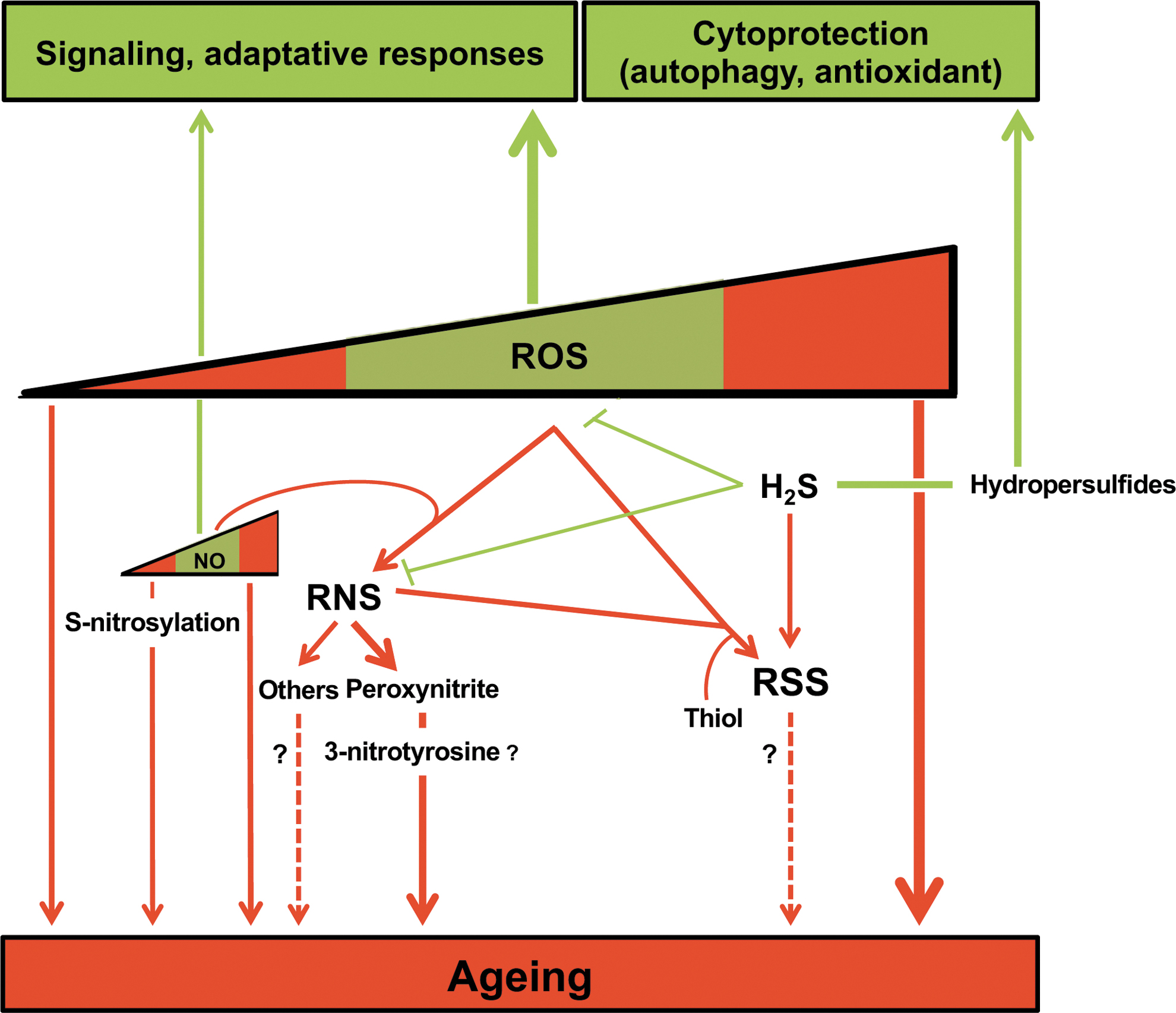
The RNS precursor NO is known to act as a signaling molecule in the cardiovascular and neuronal systems, by contributing to vasorelaxation and long-term potentiation of synapses, respectively (174). During aging, upregulation of neuronal nitric oxide synthase (NOS), an NO-producing enzyme, and the subsequent high levels of NO are associated with impaired proteostasis due to S-nitrosylation, and thereby protein damage as well as age-associated pathology in the brain (174) (see sections above). In the cardiovascular system, the age-dependent increased expression of endothelial NOS is controversial, but alteration of NOS function, known as uncoupling, which results in the decrease of NO bioavailability, could play a role in progressive age-dependent cardiovascular dysfunction (191).
In a few cases, RNS-induced protein nitration improves function, for instance, increased enzymatic activity of antioxidant proteins (e.g., glutathione S-transferase 1 and peroxiredoxin 2) (42), representing a potential protecting effect against aging. Protein nitration has also been demonstrated to participate in phosphorylation signaling cascades by either preventing or maintaining persistent protein phosphorylation after targeting kinases and phosphatases (42) (Fig. 3).
Although RSS as a whole have been shown to influence or regulate several biological processes, the specific function of individual molecules remains elusive, notably during aging. In recent years, efforts have been made to understand the role of hydropersulfides in senescence and longevity (Fig. 3). Interestingly, the levels of H2S and H2S-producing enzymes decline with age (133). The first evidence of lifespan extension by RSS was observed in Caenorhabditis elegans upon GYY4137 (an H2S donor) treatment that promotes the expression of oxidative stress-related genes (132). This effect of RSS seems to contribute to the beneficial antiaging effect of dietary restriction, and is evolutionarily conserved across species (yeast, worm, fruit fly, and rodents) (64).
Finally, a recent study revealed that dietary restriction is associated with increased levels of S-sulfhydration, suggesting a mechanistic role of this protein modification in RSS (hydropersulfides)-mediated lifespan extension that could represent an interesting target for antiaging interventions (200). In the same line, a decrease in the levels of S-sulfhydrated proteins was observed in a longitudinal study of fibroblasts derived from an individual at different times of his or her lifetime, and in old versus young rats (200). Moreover, a recent report demonstrated a decline of the total sulfane sulfur pool during aging of numerous species (136).
The cytoprotective properties of RSS may also reduce oxidative and nitrosative stress, providing a beneficial effect on aging. Indeed, H2S was demonstrated to activate the NRF2 transcription factor (that induces an antioxidant response) (17), and also inhibit the damage of ONOO− on biomolecules in vitro (186). On the contrary, and as mentioned above, RSS produced under oxidative/nitrosative conditions are detrimental during the aging process (54).
In a recent study, treatments with different H2S donors were able to reduce by 50% the number of senescent endothelial cells, without, however, restoring their proliferation capacity (92). This study underscored the interest of studying RSS in aging and age-related processes, not only for therapeutic interventions but also as a tool to identify new regulatory factors/mechanisms. Indeed, the authors unveiled that the reversion of the features of senescence was mediated by two H2S-sensitive splicing factors (SRSF2 and HNRNPD).
ROS and RNS in the Progeroid and Neurodegenerative Disease CS
CS is a devastating progeroid and neurodegenerative disease normally attributed to DNA repair defects since the mutated proteins, either Cockayne syndrome protein A (CSA) or CSB, are involved in the repair of UV-induced DNA damage through the transcription-coupled nucleotide excision repair (TC-NER) pathway (93, 154). However, these proteins and in particular CSB, which is associated with the most severe form of the disease (CS type-II), are also transcription factors, chromatin remodelers, and are present in mitochondria where CSB participates in the repair of oxidized bases in the mitochondrial genome (76, 91). Thus, the impairment of one or more of these functions could be causative of the severe CS clinical defects that cannot be solely explained by the DNA repair deficiency (20, 25, 181).
The possible causal link of reactive species in CS has been revealed by the discovery that cells from patients with the UV-sensitivity syndrome (UVSS), a related genetic disease also due to mutated CSA or CSB, but which is not characterized by precocious aging and neurodegeneration, are not hypersensitive to oxidative stress, whereas CS patient cells are (111).
In this context, we reported that UVSS primary fibroblasts display lower levels of combined ROS and RNS than cells from CS patients with various levels of severity (20). UVSS cells had nevertheless higher ROS/RNS levels than fibroblasts from healthy individuals. Moreover, CS but not UVSS cells carried high levels of the high temperature requirement factor A3 (HTRA3) protease, which leads to degradation of the mtDNA polymerase POLG1, an essential enzyme for mitochondrial function, in turn resulting in lower production of mitochondrial ATP, mtATP (Fig. 4). All these defects appear therefore specific to CS (precocious aging, neurodegeneration), but this is not the case for UV hypersensitivity that is in common between CS and UVSS patients (20). These alterations also pointed to CS as a mitochondrial disorder, a condition that is supported by the observation of multiple mitochondrial dysfunctions in CS (20, 123, 156).
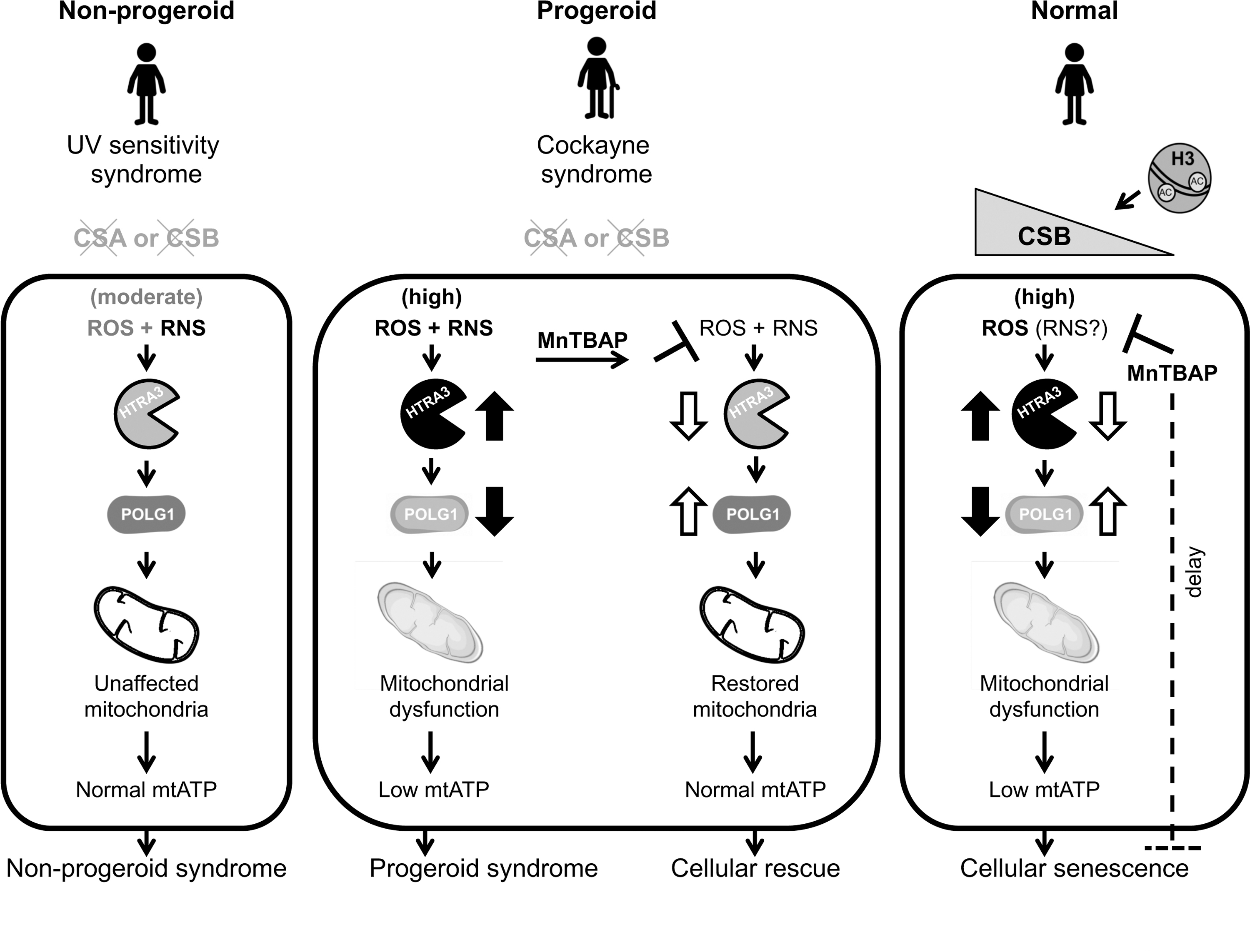
Importantly, reduction of both ROS and RNS levels with the SOD mimetic and RNS scavenger manganese (III) tetrakis [5,10,15,20] 4-benzoic acid porphyrin (MnTBAP) restored HTRA3 and POLG1 levels, as well as mitochondrial ATP production in CS patient cells.
These results indicated that the ROS/RNS imbalance is responsible for alterations specific to CS cells, and provided a possible therapeutic strategy for this disease. Linked to these findings, mitochondrial dysfunction provides a possible explanation for CS pathology, given that dysfunctional mitochondria generate more ROS, which may also increase RNS (in the presence of NO), creating a vicious circle that progressively generates potentially toxic species. This process is possibly responsible for the progressive appearance of defects in CS patients that in most cases appear normal at birth, but progressively develop a variety of clinical symptoms. Progressive deterioration until a pathological state is indeed a classic feature of mitochondrial diseases (56).
Furthermore, diverse levels of ROS/RNS or their downstream effects, and the linked mitochondrial dysfunction could be responsible for the still unexplained large variability of clinical symptoms in CS (93), which is rather unique among progeroid diseases. Indeed, the extent and the progression of mitochondrial dysfunction at any given time and in any given tissue could determine specific pathological states. Finally, high ROS (and perhaps RNS) levels may explain neurodegenerative defects in CS that cannot be solely explained by a DNA repair defect (25) since the brain and the nervous system are normally not exposed to UV light, whereas they are very sensitive to oxidative stress (26).
Several questions remain unanswered. First, how high ROS/RNS levels trigger HTRA3 overexpression and thereby the cascade of events leading to mitochondrial dysfunction in CS? Is this process linked to epigenetic modifications, as it is the case in other contexts (49)? In support of this notion ROS act as players in shaping the epigenetic landscape of the genome by modulating DNA and histone modifications, the expression of noncoding RNAs, and ATP-dependent chromatin remodeling (81). Interestingly, ROS have been pointed out as key epigenetic regulators of senescence-promoting p16INK4A gene in different contexts (49, 146). We hypothesize that this could be also the case for HTRA3, which we have reported to accumulate during senescence of primary human fibroblasts (33). In agreement with this notion, epigenetic modulation of HTRA3 expression by DNA methylation has been shown in some cancers (10).
Second, is HTRA3 a or one of the few major targets of high ROS/RNS levels or are there global gene expression changes in CS? Global transcriptomic changes have been described in CS cells and CS models (115, 185), but to what extent those changes are casually linked to ROS/RNS remains to be elucidated. Moreover, we have evidences that CS fibroblasts undergo extensive DNA methylation changes that affect specific sets of genes and pathways (32), but we do not know yet whether these alterations can be reversed by regulation of ROS/RNS (see also next paragraph).
Third, which levels of ROS/RNS are critical for the abovementioned expression changes, considering that UVSS cells display increased ROS/RNS levels to some extent but no HTRA3 and mitochondrial defects, as well as no precocious aging and neurodegeneration? The hormetic effect of ROS/RNS levels may play a role in the modulation of the epigenetic landscape, but factual assessments are missing to date. This modulation could be also driven by exogenous factors, for instance, several positive effects of physical exercise on the organism are driven by epigenetic alterations, possibly triggered by ROS/RNS (38).
Fourth, what is the specific role of ROS versus RNS, and possibly RSS that have not yet been considered in the equation, in dysfunctional mechanisms in CS? No data are available in this context, which should be investigated with priority in the future.
Fifth, is this condition specific to CS or is this a more global pathogenic model that may also be implicated in regular aging? Growing evidence supports the second scenario, given the role of ROS in epigenetically regulating senescence and other aging-related processes (58).
Oxidative stress is a well-demonstrated feature of CS cells (3, 20, 123, 156). Indeed, some clinical CS symptoms such as neurodegeneration have been attributed to hypersensitivity to oxidative stress (25), and CSA and CSB have been implicated in the control of oxidative stress response (34). Interestingly, the lack of precocious aging and neurodegeneration observed in UVSS patients could be due to the decreased cellular sensitivity to ROS in UVSS compared to CS (111). In support of this notion, a patient with a milder form of CS (CS type-III), which is characterized by late-onset and slow progression, also showed an intermediate sensitivity to oxidative stress, which was larger than healthy controls but smaller than the classical forms of CS (62).
We also reported that the antioxidant response at the transcription level is particularly strong in UVSS compared to CS cells (20). We observed a particularly high expression of peroxiredoxins (PRDX), especially PRDX5, upon roughly comparable levels of oxidases, which may account for a moderate ROS increase in UVSS cells. These data also suggest that ROS/RNS alterations in CS are associated with multiple changes in gene expression, although these data do not indicate causality. Importantly, it has been reported that oxidative stress increases CSB occupancy at promoters, suggesting that CSB regulates to some extent the response to oxidative stress (91).
It remains to be elucidated whether nitrosative stress triggers a response similar to oxidative stress, and whether RSS, mistakenly considered ROS in many the current analyses (37), also play a role in altering gene expression in these conditions. Further studies including cells derived from a large number of patients with CSA or CSB mutations displaying mild phenotypes, and the effect of anti-ROS and/or anti-RNS treatments are necessary to evaluate the single and combined contribution of these species to precocious aging and neurodegeneration in CS.
High levels of reactive species can impair physiological function through damage of DNA, proteins, lipids, and other biomolecules (129). In a recent study, it was demonstrated that cells from CS patients accumulate carbonylated proteins that activate the unfolded protein response (2). Moreover, CS cells display ROS-induced oxidative DNA damage, as a consequence of increased ROS and impaired repair of these lesions (30, 178). Indeed, oxidative stress can generate bulky DNA lesions that are repaired by the NER pathway, and non-bulky DNA lesions that are removed by the base excision repair (BER) pathway (94). Besides their implication in TC-NER, there is evidence that CSA and CSB participate in BER (177).
Neurodegeneration is a major feature in CS and several chronic aging diseases. A recent study revealed an oxidative stress-rich environment and mitochondrial dysregulation in CS patient cerebellum, suggesting a role for ROS and defective mitochondria in neurodegeneration in CS (115). Interestingly, a recent study showed that CSA- and CSB-deficient immortalized fibroblasts display an increase of 8-oxo-purine (8-oxo-Pu) DNA lesions that can be directly generated by peroxynitrite (ONOO−) (85), one of the more toxic RNS. Additional studies are, required to understand the contribution of increased RNS to the damage of cell components in CS.
We demonstrated that CS-specific defects (high HTRA3 levels, POLG1 depletion, lower mitochondrial ATP production) are recapitulated in replicative senescence of normal cells, and are associated with increased cellular and mitochondrial ROS (33). We showed that replicative senescence is triggered by progressive depletion of CSB, the protein mutated in the most severe CS cases (33). We reported that during cell proliferation, CSB inhibits the expression of p21, one of the major effectors of cell cycle arrest (50), by occupying its promoter (p21Waf1 locus). Conversely, upon CSB depletion, the p21Waf1 promoter becomes accessible and p21 expression induces cell cycle arrest and triggers senescence.
It was not clarified whether RNS are implicated in this process, but acute and long-term treatments with the SOD mimetic and RNS scavenger MnTBAP delayed or reduced senescence (33) (Fig. 4). Mismanagement of multiple reactive species, not only ROS, appears therefore as a major driver of the progeroid disease CS, raising the question whether this is also the case for other progeroid diseases, as well as the aging process.
ROS, RNS, RSS in Other Progeroid Syndromes
Other progeroid diseases appear also to display altered generation and response to multiple reactive species. These progeroid diseases are normally attributed to (i) alteration of DNA repair factors (like it was classically the case for CS, see Fig. 5), e.g. the WS where the mutated protein WRN is an helicase involved in the repair of replication-induced DNA damage; [but WRN also acts as a chromatin stabilizer (198)], or (ii) nuclear structure components (i.e., laminin A/progerin [LMNA]) (102), such as in the HGPS, or (iii) unidentified gene-dosage alterations such as in the DS. Indeed, evidence of redox homeostasis dysregulation in HGPS was reported long ago (116), based on accumulation of oxidized proteins in HGPS fibroblasts compared with age-matched controls, and has been confirmed in more recent studies (139, 183).
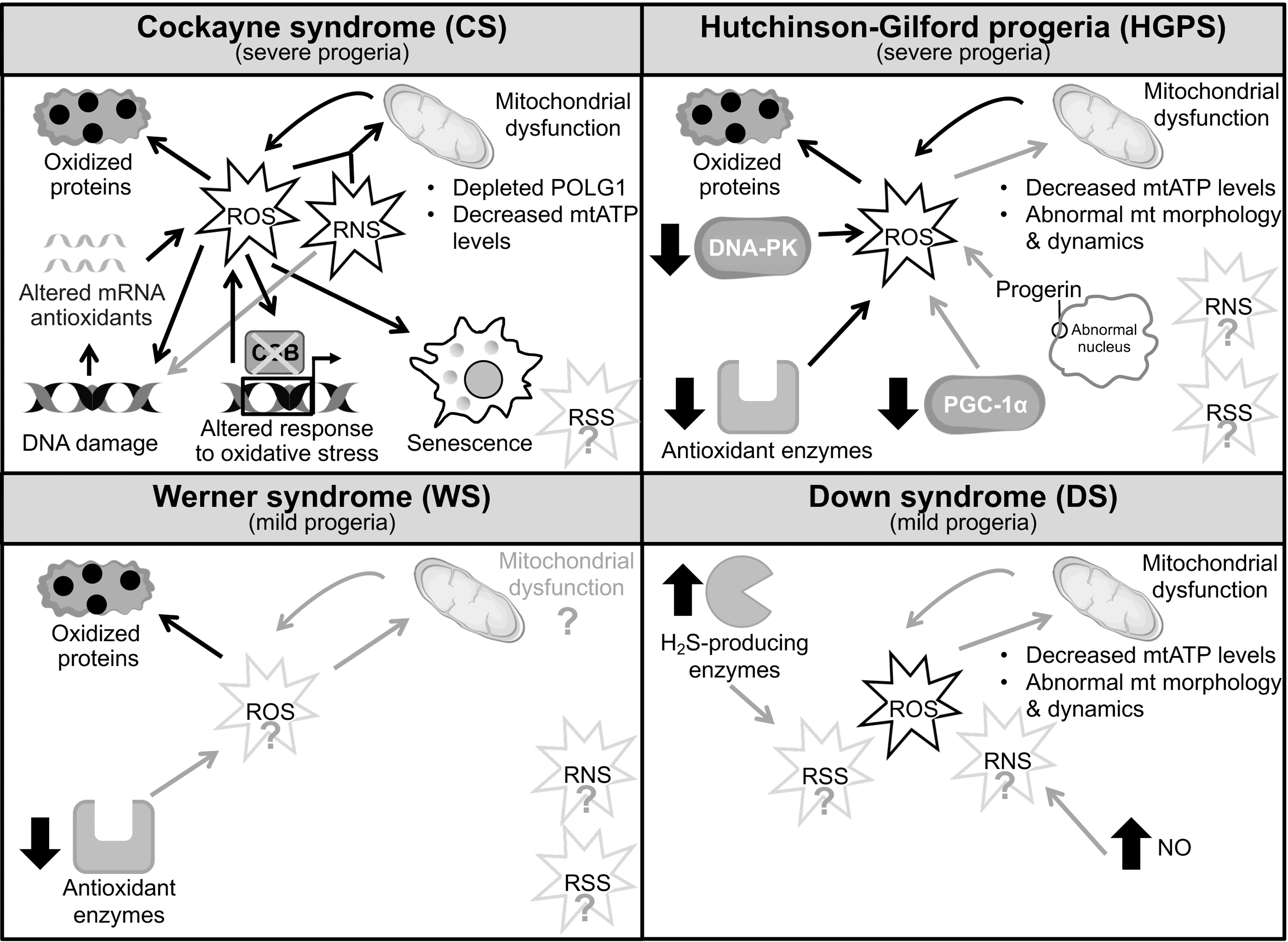
As a major source of ROS, the mitochondrial function has been widely investigated in the context of laminopathy progeroid syndromes such as HGPS. Alteration of mitochondrial function has been increasingly recognized as an important player in progeroid and physiological aging [extensively reviewed elsewhere (72, 86)]. Severe mitochondrial dysfunction has been reported in HGPS fibroblasts (142) as well as in HGPS mouse models (182). Altered mitochondrial function was demonstrated by reduced ATP levels, abnormal mitochondrial morphology and dynamics (189), and a consequent metabolic switch from OXPHOS to glycolysis (110) (Fig. 5). Reduced mitochondrial ATP production and glycolytic shift were also reported in progeroid CS, as we mentioned above (20).
Furthermore, the basal levels of antioxidant enzymes that protect cells against ROS-induced damage were reduced in HGPS fibroblasts (190). Overall, these analyses confirmed the dysregulation of redox homeostasis, pointing to reducing ROS levels in HGPS as a rescue strategy. Rescuing activities (see next section), which were associated with increased proliferation rates and ameliorated cellular progeroid defects, led to the notion that the ROS-generating environment is possibly a more significant factor than nuclear shape abnormalities in the development of premature aging. Again, this notion reminds of CS where oxidative/nitrosative stress appears to be causative of the pathology beyond classic UV-repair defects.
Other experiments suggested that progerin accumulation in HGPS increases oxidative stress through downregulation of the PPARG coactivator 1 alpha (PGC-1α) protein, a central regulator of mitochondrial biogenesis that also regulates the expression of numerous ROS-detoxifying enzymes (6, 189). A reduced expression of PGC-1α and its downstream target genes, including nuclear respiratory factor 1 (NRF1), which also controls the expression of key genes required for mtDNA transcription and replication, was detected in HGPS adipocytes and fibroblasts. However, overexpression of PGC-1α in HGPS cells did not significantly improve the mitochondrial defects or reduce ROS, suggesting that PGC-1α is not a major effector of mitochondrial dysfunction in HGPS (23).
Arteriosclerosis is a major defect in HGPS and it is also classified as a disease of aging. An initial step in atherosclerosis development is dysfunction of endothelial cells. Progerin accumulates in HGPS patients' endothelial cells and also during vascular physiological aging. A recent study reported that endothelial progerin expression increases ROS production as well as the proinflammatory response (11). Progerin expression affects also other factors that may intervene in further altering the redox status or balance. For instance, the double-strand break repair DNA-PK complex, which is composed of the DNA-binding Ku70/80 heterodimer and the catalytic subunit DNA-PKcs, is a downstream target of progerin and is downregulated in HGPS fibroblasts (97). DNA-PKcs plays also a role in the repair of oxidative DNA lesions and participates in oxidative stress signaling events through activation of its kinase activity upon oxidative stress (23).
Altogether, these findings support a link between increased ROS levels, DNA damage, and mitochondrial dysfunction in HGPS, but more in-depth studies should be performed to elucidate the effective contribution of oxidative stress to the premature aging in HGPS patients.
Potential involvement of oxidative stress in WS pathogenesis was suggested by the report of in vivo and in vitro redox abnormalities in WS patients and cells (113, 116, 120) (Fig. 5). A reduced antioxidant defence was also more recently described in WS fibroblasts (161). Interestingly, there is a lack of comprehensive evaluation of the mitochondrial phenotype in WS, since mitochondrial impairment was solely described in WRN-depleted fibroblasts, WS patient-derived cells (41), and hepatocytes from homozygous WRN helicase transgenic mice (28). Conversely, Wrn-null mice did not exhibit a defective mitochondrial phenotype (41, 105), suggesting that the nature of mitochondrial dysfunction (and its links with ROS) in WS pathogenesis should be further examined.
After the first evidences of ROS imbalance in DS (79), an increasing number of studies suggest that oxidative stress plays a central role in the pathogenesis of this mild progeroid disease (Fig. 5). According to the gene-dosage hypothesis, overexpression of some genes encoded by chromosome 21 such as the amyloid precursor protein (APP), copper–zinc superoxide dismutase (SOD1), and beta-secretase (BACE2) can be directly or indirectly associated with oxidative stress observed in DS (125). Indeed, ROS overproduction was detected in DS patient-derived skin fibroblasts (180), primary DS cortical neurons (16), and neurons from DS mouse models (160), and proposed as a major factor in DS pathogenesis (125).
Increased levels of oxidative stress biomarkers measured in DS individuals (74), and mitochondrial dysfunction widely described in DS cells (195), further support the assumption of altered ROS-dependent processes in DS that should be nevertheless addressed in future studies.
These previous studies essentially focused on ROS-dependent redox imbalance, but recent studies highlighted the importance of other free radical species such as RNS and RSS (155). To date, the levels of RNS and RSS have not been investigated in the abovementioned progeroid syndromes. However, higher levels of NO compared with healthy controls have been reported in the amniotic fluid of DS fetuses (29), and in specific brain regions of DS mouse models (57). NO overproduction was found enhanced during the development of clinical dementia in DS individuals (29), suggesting that the RNS levels should be considered in this pathology.
Moreover, the upregulation of H2S-producing enzymes, such as cystathionine-β-synthase (CBS) and 3-mercaptopyruvate sulfurtransferase (3-MST), was recently documented in DS fibroblasts (121) in support of the “Kamoun hypothesis” according to which a toxic overproduction of H2S occurs in DS (77). These preliminary findings provide an incentive for future studies on the detection of RNS and RSS levels in DS individuals and/or in in vitro and in vivo DS models.
In conclusion, oxidative stress is an important phenotypic characteristic associated with progeroid syndromes. However, a direct cause-and-effect relationship between dysregulation of the redox homeostasis and accelerated aging processes has not been strongly established, perhaps with the exception of CS that also implicates RNS.
Pharmacological Modulation of Reactive Species in the Context of Aging and Progeroid Diseases
A proper reduction of ROS and RNS levels, by either scavenging free radicals or enhancing antioxidant defenses, may provide new insights into therapeutic strategies for oxidative stress-related diseases. The beneficial effect of the pharmacological modulation of reactive species in progeroid syndromes has been widely demonstrated in vitro and in vivo. ROS and oxidative stress were efficiently lowered by N-acetylcysteine (NAC) treatment as well as reactivation of NRF2, whose transcriptional activity is impaired in HGPS cells. This treatment resulted in reduced DNA damage and improvement of cellular HGPS defects (87).
Other antioxidant compounds such as a rho-associated protein kinase (ROCK) inhibitor (70) and methylene blue (189) improved the mitochondrial function, whereas RAD001, currently used in clinical trials for HGPS patients (NCT02579044), upregulated catalase activity (101), rescuing the premature aging phenotypes in HGPS fibroblasts. Also sirtuin-activating compounds such as resveratrol (12), which display antioxidant properties as well, and have been extensively characterized as antiaging candidates, have been proposed as an alternative treatment for progeria (96). Moreover, resveratrol reversed some of the clinically relevant phenotypes in a mouse model for the progeroid WS, although it did not extend the lifespan of these mice (88).
Recently, an in vivo study showed that reprogramming HGPS cells to pluripotency, a process that also alters oxidative stress, ameliorates cellular and physiological hallmarks of aging and prolongs the lifespan in an HGPS mouse model (114). This study highlighted a novel but yet poorly understood mechanism of oxidative stress involvement in epigenetic dysregulation in aging.
Progeroid syndromes may provide important clues to understanding the molecular mechanisms involved in normal human aging. Thus, the antioxidant strategies described for the treatment of HGPS, WS, and CS that delivered promising results in both mouse and cellular models have been investigated as antiaging approaches as well as possible treatments for pathologies associated with physiological aging.
Several antioxidants have thus been investigated for their therapeutic potential in clinical trials so far. These include enzymatic antioxidants such as SOD (193), natural nonenzymatic antioxidants, such as vitamins (ex. α-tocopherol), carotenoids, polyphenols, flavonoids (44), and other molecules naturally produced within the human body that chelate redox metals to protect the cells against oxidative stress. Antioxidant treatment with vitamin C restored healthy phenotypes in WS mouse models (105), while decreasing ROS with NAC reduced senescence features (e.g., expression of p16, another senescence trigger) in CSA-deficient primary keratinocytes (30).
One of the early discovered natural ROS and RNS scavenger was melatonin (138) that has attracted a great interest due to its capability to extend lifespan (4) and enhance certain antioxidant enzymes (137). However, there is still no clinical evidence to conclude that melatonin has a role in extending normal longevity. In fact, a positive association between supplementation with pharmacological or natural antioxidant compounds and health beneficial effects has not been demonstrated. Most evidences supporting the beneficial effects of antioxidant supplementation derive from animal studies, which are not necessarily translated to humans.
The results of clinical trials with antioxidants showed that these compounds fail to prevent the progression of ROS-associated diseases, provide a few benefits (27), and exert harmful effects at high doses (13). The reasons for this therapeutic inefficacy in vivo may derive from high renal clearance, low bioavailability, or physiological mechanisms that prevent high tissue concentrations (44). Moreover, the difficulty of measuring antioxidant efficacy in vivo also makes difficult the interpretation of results from clinical trials.
The global inefficiency of antioxidant activity during aging can be attributed to the inhibition of ROS signaling. However, this setback could be also due to the high selectivity of antioxidants for ROS, leaving essentially unchanged the levels of other reactive species (31). The evaluation of beneficial versus detrimental doses of reactive species (ROS alone and in combination with RNS and RSS) is also generally missing in the tested cells and organisms.
Several low-molecular-mass SOD mimetics have been designed to overcome some of the limitations (e.g., large size, antigenicity, and limited cell permeability) associated with the ineffective use of the native SOD enzymes (151). Metalloporphyrins, manganese salen derivatives, Mn cyclic polyamines, porphyrin-related compounds, and other synthetic molecules, as well as natural products, have been discussed in detail (9). Among these, Mn(III) porphyrins and Mn(III) salen derivatives exhibit an SOD-like activity accompanied with peroxynitrite reducing ability (Fig. 6). These compounds resulted protective in a variety of in vitro and in vivo oxidative stress models, involving the generation of superoxide and/or peroxynitrite (124).
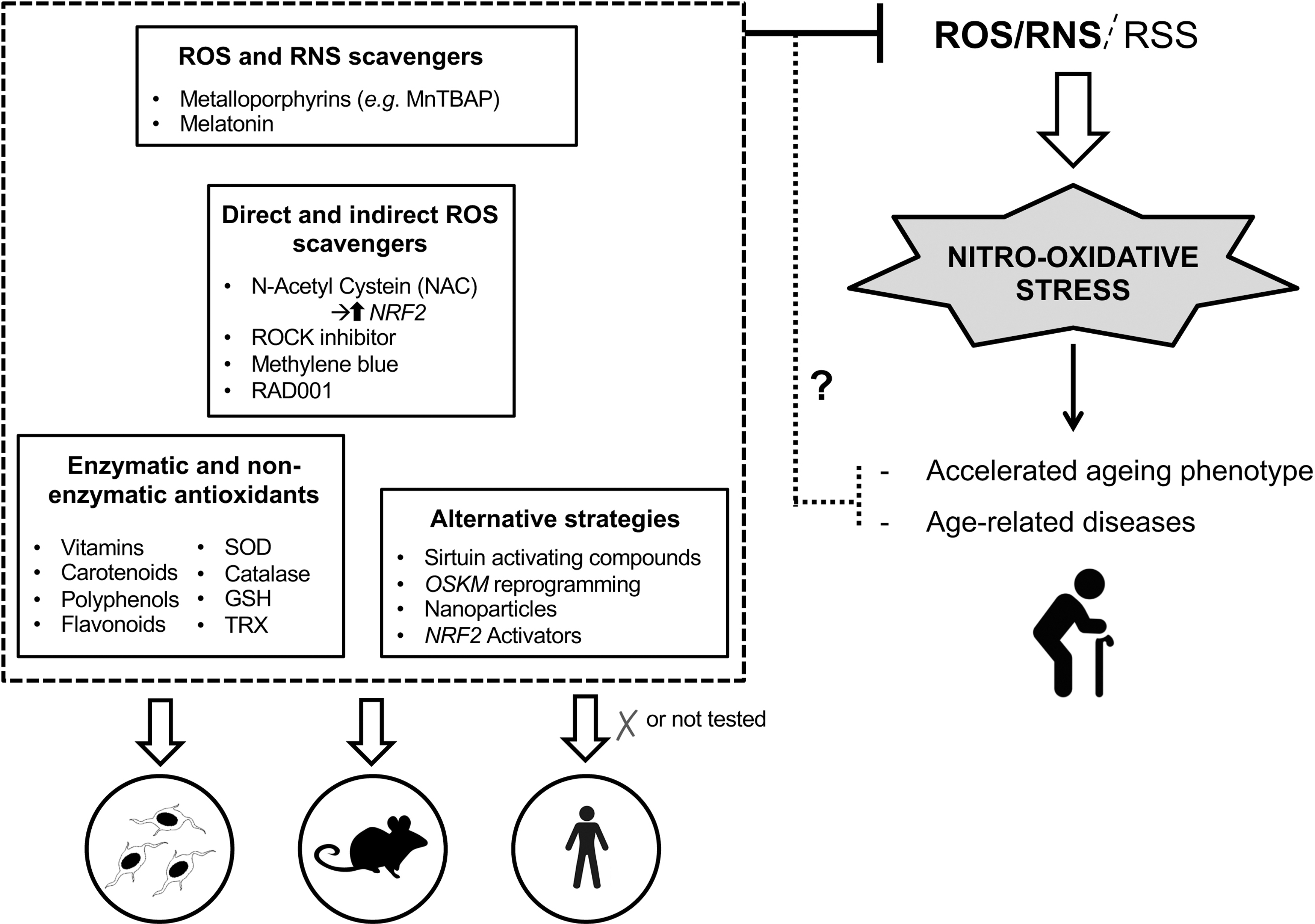
Among these compounds, in the last few years, the effect of MnTBAP has been investigated in age-related processes (33, 89) and progeroid diseases such as CS (20) where, in a context of CSB deficiency it restores HTRA3 and POLG1 levels as well as mitochondrial ATP production in CS patient-derived cells. This compound has been reported as ROS/RNS scavenger in numerous studies (43, 172) and it possesses both SOD and catalase activities. MnTBAP has been shown to be effective in ameliorating in vivo oxidative stress-induced injuries, and extending the lifespan of Sod2 (a ROS-detoxifying enzyme) null mice (109). However, MnTBAP treatment was not able to rescue the neurological phenotype, presumably due to the inability of MnTBAP to cross the blood–brain barrier.
In support of this hypothesis, three salen manganese complexes (EUK-8, EUK-134, and EUK-189) that cross the blood–brain barrier significantly extended the lifespan of nullizygous mice beyond that of untreated or MnTBAP-treated animals, preventing also the emergence of a spongiform encephalopathy (108). The remarkable potential of MnTBAP as a therapeutic has been widely described in mice with obesity (126), amyotrophic lateral sclerosis (40), acute liver failure (43), carrageenan-induced pleurisy (9), and renal fibrosis (194).
The reported prolongation of the lifespan of superoxide dismutase Sod2 deficient mice (see previous paragraph) suggests an active detrimental role of RNS in the aging process (109), and a close interplay between oxidative and nitrosative stress (31). Similarly, the implication of nitrosative stress in the pathogenesis of aging-associated cardiovascular dysfunction was demonstrated after peroxynitrite decomposition with a porphyrin derivative (135). More recently, the scavenging of peroxynitrite was shown to ameliorate signs of aging in different organs (kidney and vasculature) via the modulation of age-associated processes such as cellular senescence and autophagy (171, 197).
The possible role of combined ROS and RNS in age-related processes was confirmed by our recent identification of a causal link between the progressive loss of the progeroid factor CSB and replicative senescence in normal cells, which was delayed or reduced by acute or long-term MnTBAP treatment (33). Additional proofs of the impact of anti-RNS strategies in preventing senescence have been previously identified (24, 89). Indeed, peroxynitrite scavengers were able to prevent senescence induction in endothelial cells after irradiation (MnTBAP) (89) or culture on glycated collagen, a trigger of premature senescence (Ebselen and MnTBAP) (23).
An innovative strategy for scavenging ROS and RNS in vivo is based on the use of nanoparticles (NPs) that may overcome some limitations associated with classical antioxidant compounds. Different types of NPs can directly react with ROS and RNS, mimicking the natural scavenging capability of cellular enzymes (112).
In conclusion, the antiaging therapeutical use of known antioxidant compounds turned out to be paradoxically ineffective, despite many compelling results obtained in vitro as well as in vivo. Significant progress has been achieved in the development of novel antioxidants, given the requirement for compounds that are chemically more effective, exhibit better bioavailability than classical antioxidants, and which can be selectively targeted to particular tissues and intracellular locations. Further investigations are required to verify the beneficial effects of these novel compounds in clinical trials.
Conclusions
Physiological and pathological aging is characterized by dysfunctions that have multifactorial causes. Reactive species generate, and are subject to, a very complex but not unintelligible network of reactions that significantly affect biological processes. Pathophysiological aging and reactive species are related, and despite a solid body of existing evidences, we are aware that we only start to discover and disentangle the cause/effect relationships. This knowledge is of relevance for understanding physiological aging as well as for developing ameliorating or rescue strategies for age-related genetic and non-genetic pathological conditions.
Footnotes
Authors' Contributions
Cl.C., Ch.C., C.F.M., and M.R. wrote the article with support and input from M.R., M.R. supervised the project. All authors discussed the Forum Review Article and contributed to the final article.
Authorship Confirmation Statement
All authors confirm that this article is original, has not been published before, and is not currently being considered for publication elsewhere. All authors confirm that the article has been read and approved by all the authors, and that there are no other persons who satisfied the criteria for authorship but are not listed. All authors confirm that the order of authors listed in the article has been approved by all authors. All authors understand that the corresponding author is the sole contact for the editorial process regarding communication, progress, submission(s) of revision(s), and final approval of proofs.
Author Disclosure Statement
M.R. is the inventor of the following international/PCT patent application that includes the use of MnTBAP as a possible therapeutic strategy for Cockayne syndrome: WO 2015/121459 entitled “Methods for in vitro investigating mitochondrial replication dysfunction in a biological sample, kits and usethereof, therapeutic methods against progeroid-like syndromes or symptoms and screening method for identifying particular protease inhibitor(s) and/or nitroso-redox stress scavenger compound(s).” Except the abovementioned disclosure, M.R. declares no other competing interests. The other authors declare no competing financial interests.
Funding Information
This Forum Review Article was supported by the Agence Nationale de la Recherche (ANR) (project CS_AGE) and the Direction of Applications, Research, and Industrial Relations of Institut Pasteur (DARRI) (IARP-2019).