
Abstract
Aims:
Acute myocardial infarction (MI), caused by acute coronary artery obstruction, is a common cardiovascular event leading to mortality. Nuclear dot protein 52 (NDP52) is an essential selective autophagy adaptor, although its function in MI is still obscure. This study was designed to examine the function of NDP52 in MI and the associated mechanisms.
Results:
Our results revealed that MI challenge overtly impaired myocardial geometry and systolic function, along with cardiomyocyte apoptosis, myocardial interstitial fibrosis, and mitochondrial damage, and NDP52 nullified such devastating responses. Further studies showed that the blockade of mitochondrial clearance is related to MI-induced buildup of damaged mitochondria. Mechanistic approaches depicted that 7-day MI induced abnormal mitophagy flux, resulting in poor lysosomal clearance of injured mitochondria. NDP52 promoted mitophagy flux through the recruitment of Ras-associated protein RAB7 (RAB7) and TANK-binding kinase 1 (TBK1). On protein co-localization, TBK1 phosphorylated RAB7, in line with the finding that chloroquine or a TBK1 inhibitor reversed NDP52-dependent beneficial responses.
Innovation:
This study denoted a novel mechanism that NDP52 promotes cardioprotection against ischemic heart diseases through interaction with TBK1 and RAB7, leading to RAB7 phosphorylation, induction of mitophagy to clear ischemia-induced impaired mitochondria, thus preventing cardiomyocyte apoptosis in MI.
Conclusion:
Our results indicate that NDP52 promotes autophagic flux and clears damaged mitochondria to diminish reactive oxygen species and cell death in a TBK1/RAB7-dependent manner and thus limits MI-induced injury. Antioxid. Redox Signal. 36, 1119–1135.
(Color images are available online).

Introduction
Acute myocardial infarction (MI) remains a major public health threat and culprit for the ever-rising cardiovascular morbidity and mortality. Occlusion of coronary perfusion imposes a devastating consequence, including ischemic insult, necrosis, arrhythmia, myocardial fibrosis, and, ultimately, cardiovascular mortality (23, 39, 44). Although timely coronary arterial reperfusion is considered a practical treatment to reduce infarct size and increase survival, little effective target remedy is readily accessible to block the progress of unfavorable cardiac remodeling and cardiomyocytes death (44, 46). In this context, developing a new therapeutic option is pertinent to improve prognosis suffering from acute MI attack.
Innovation
The main work of this study explores the mechanism of impaired mitochondria in MI-induced heart failure. Our findings suggest that NDP52 prevents cardiomyocyte apoptosis in MI by promoting the interaction between TBK1 and RAB7 and increasing phosphorylation of RAB7. We propose that the activated RAB7 by NDP52 enhances mitophagy in cardiomyocytes and clear ischemia-induced impaired mitochondria, which may provide a new target in the clinical treatment of acute MI.
Fission, fusion, mitophagy, and biogenesis are critical for quality control of mitochondria (2, 7, 45). Mitophagy (selective autophagy of mitochondria) functions as the primary safeguard for mitochondrial homeostasis and integrity (2, 48). The mitophagy process is a highly conserved quality control process to remove damaged mitochondria and recycle long-lived proteins (48). Mitophagy is considered a protective process in various pathological myocardial settings, including cardiac hypertrophy and heart failure (28, 50), although its role in post-infarct hearts is still dubious (16, 17, 22). In particular, the precise regulatory mechanism(s) is lacking regarding the fusion process between mitophagosomes and lysosomes (a.k.a., mitophagy flux) in acute MI.
Clearance of damaged mitochondria is regulated by a cascade of signaling molecules involving PINK1 and parkin (8, 31, 34). Parkin assembled ubiquitin chains on the mitochondrial outer membrane, where autophagy cargo receptors, including nuclear dot protein 52 (NDP52), were recruited in conjunction with TANK-binding kinase 1 (TBK1) (11, 12, 42). NDP52 was initially identified as a xenophagy-specific receptor that is necessary for intracellular bacterial degradation (37, 46).
In this process, NDP52 targeted bacteria to autophagosomes and promoted pathogen degradation by regulating pathogen-containing autophagosome maturation (38, 40). However, the function of NDP52 in MI remains mysterious. Thus, the current research was designed to evaluate the effect of NDP52 on heart function and integrity of mitochondria with acute MI and the potential molecular mechanism(s) involved.
Results
NDP52 overexpression ameliorates myocardial function and improves cardiac morphology
To estimate the possible role of NDP52 on heart function after acute MI, echocardiography was applied to assess myocardial geometry and function, including left ventricular end diastolic diameter (LVEDD), left ventricular end systolic diameter (LVESD), ejection fraction (EF), and fractional shortening (FS). Echocardiography data indicated that MI significantly diminished EF and FS, the effect of which was partially diminished by NDP52 overexpression. Along the same line, LVEDD and LVESD were markedly augmented after MI, and overexpression of NDP52 reversed such an effect. NDP52 alone exerted little effect on LVEDD, LVESD, EF, and FS (Fig. 1B–H). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) and Masson trichrome staining were applied to evaluate cardiac injury. Our finding revealed that interstitial fibrosis and cardiomyocyte apoptosis were significantly elevated after the 7-day MI, and NDP52 overexpression relieved this effect (Fig. 2A–E). Neither MI nor NDP52 overexpression exhibited any effect on resting cardiomyocyte cell length. However, acute MI challenges prolonged time-to-90% relengthening and suppressed maximal velocity of shortening/relengthening (±dL/dt), as well as peak shortening without changing time-to-peak shortening. Though NDP52 only treatment did not exert any significant effect on cardiac function, overexpression of NDP52 overtly attenuated MI-induced cardiomyocyte contractile anomalies (Fig. 2F–K). These findings indicated that cardiac dysfunction induced by MI was partially preserved by NDP52 overexpression. Remarkably, chloroquine (CQ) (a lysosomal inhibitor) mitigated NDP52-offered cardioprotection against acute MI.

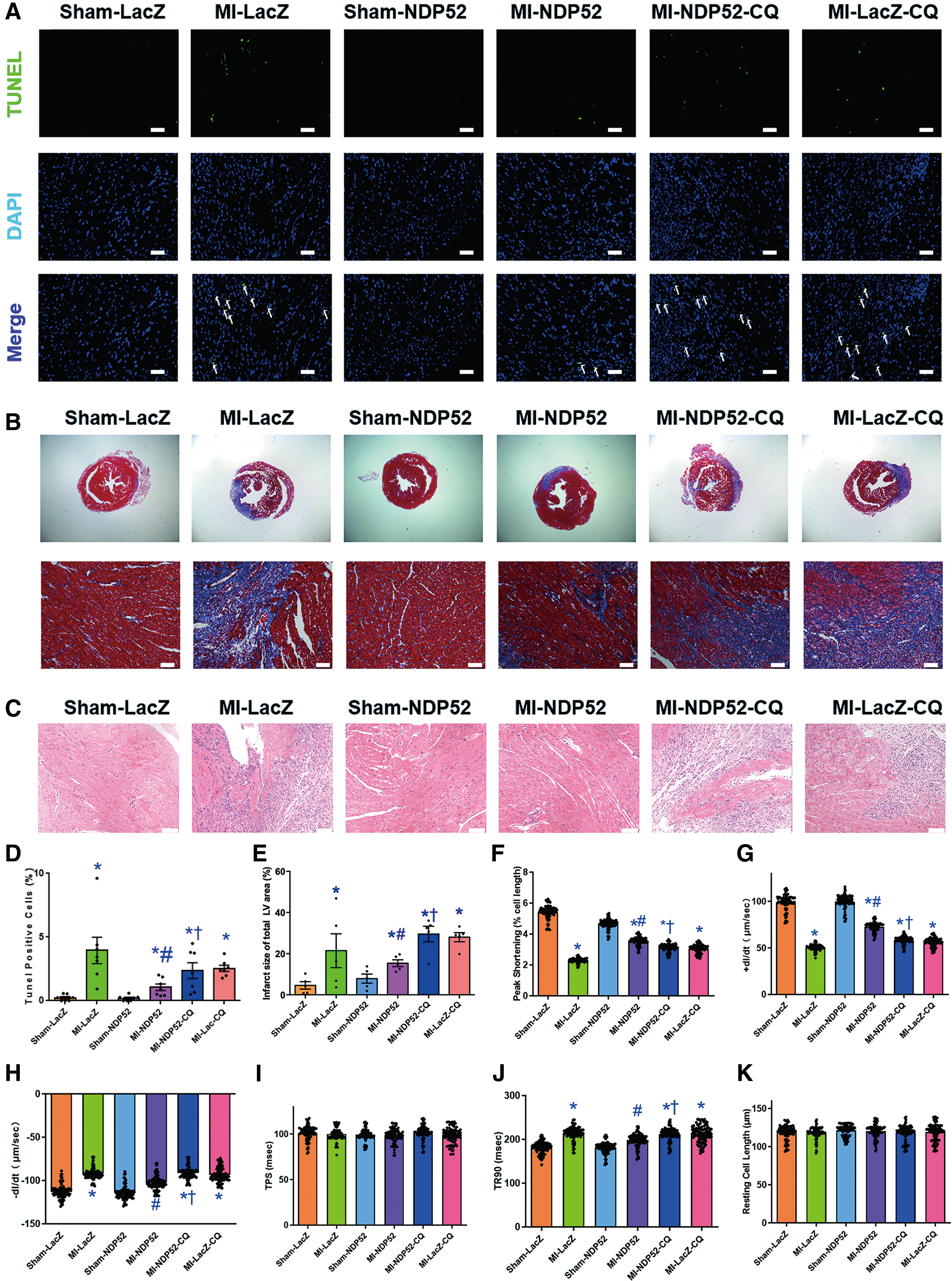
NDP52 overexpression attenuates mitochondrial dysfunction
Considering the crucial function of mitochondria in regulating cardiac function (1), transmission electron microscopy (TEM), tetramethylrhodamine methyl ester (TMRM) staining, and reactive oxygen species (ROS) staining were employed to determine NDP52-offered responses in mitochondrial function. After the 7-day MI, mitochondria presented discernable TEM ultrastructural abnormalities, including deformation, swelling, rupture, cristae fracture in cardiomyocytes, and overexpression of NDP52 attenuated these anomalies of mitochondria (Fig. 3A, E). Mitochondrial membrane potential (MMP) evaluated using TMRM displayed reduced MMP subject to a 9-h anoxia challenge simulating acute MI in vivo. Intriguingly, NDP52 overexpression reversed anoxia-induced collapse in MMP (Fig. 3B, F). Further, 9-h anoxia increased TUNEL-positive cells, and NDP52 overexpression decreased the percentage of TUNEL-positive cells evoked by anoxia (Fig. 3C, G). Besides, ROS staining data showed that NDP52 overexpression mitigated ROS production in neonatal mouse cardiomyocytes (NMCMs) after a 9-h anoxia insult (Fig. 3D, H). Of note, NDP52 alone did not exert notable responses in the ultrastructure of mitochondria, ROS production, or MMP. Notably, NDP52 knocked down by shRNA did not affect the level of MMP, apoptosis, or ROS in NMCMs. These findings suggested that the protective effects of NDP52 on mitochondria after acute MI in vivo or anoxia in vitro were dependent on mitophagy flux.

NDP52 overexpression improves mitophagy flux
The ROS production and cell death induced by damaged mitochondria are closely related to mitochondrial autophagy clearance (3, 27). It is, thus, plausible to speculate that NDP52-evoked improvement in mitochondrial function is due to improved mitochondrial autophagy clearance. The mitophagy activity was examined in cardiomyocytes after anoxia by transfecting NMCMs with Mito-Keima adenovirus. Keima is a dual-excitation radiometric fluorescent protein, and it is PH-sensitive. Mito-Keima indicated the location of mitochondria. Mt-Keima switches to longer-wavelength excitation and more abundant red Keima dots could be observed when mitochondria moved to acidic lysosomes (pH 4.5) after the mitophagy process. Mito-Keima data showed that more mitochondria moved to lysosome after anoxia. NDP52 overexpression promoted mitochondria targeting acidic lysosomes (Fig. 4D, F). Western blot analysis indicated that both MI and anoxia decreased the expression of NDP52. LC3II/LC3I and P62 were significantly augmented after MI or anoxia, and such an effect was abolished with NDP52 overexpression (Fig. 4A, G). These suggested that NDP52 overexpression rescued against acute MI-compromised mitophagy flux.
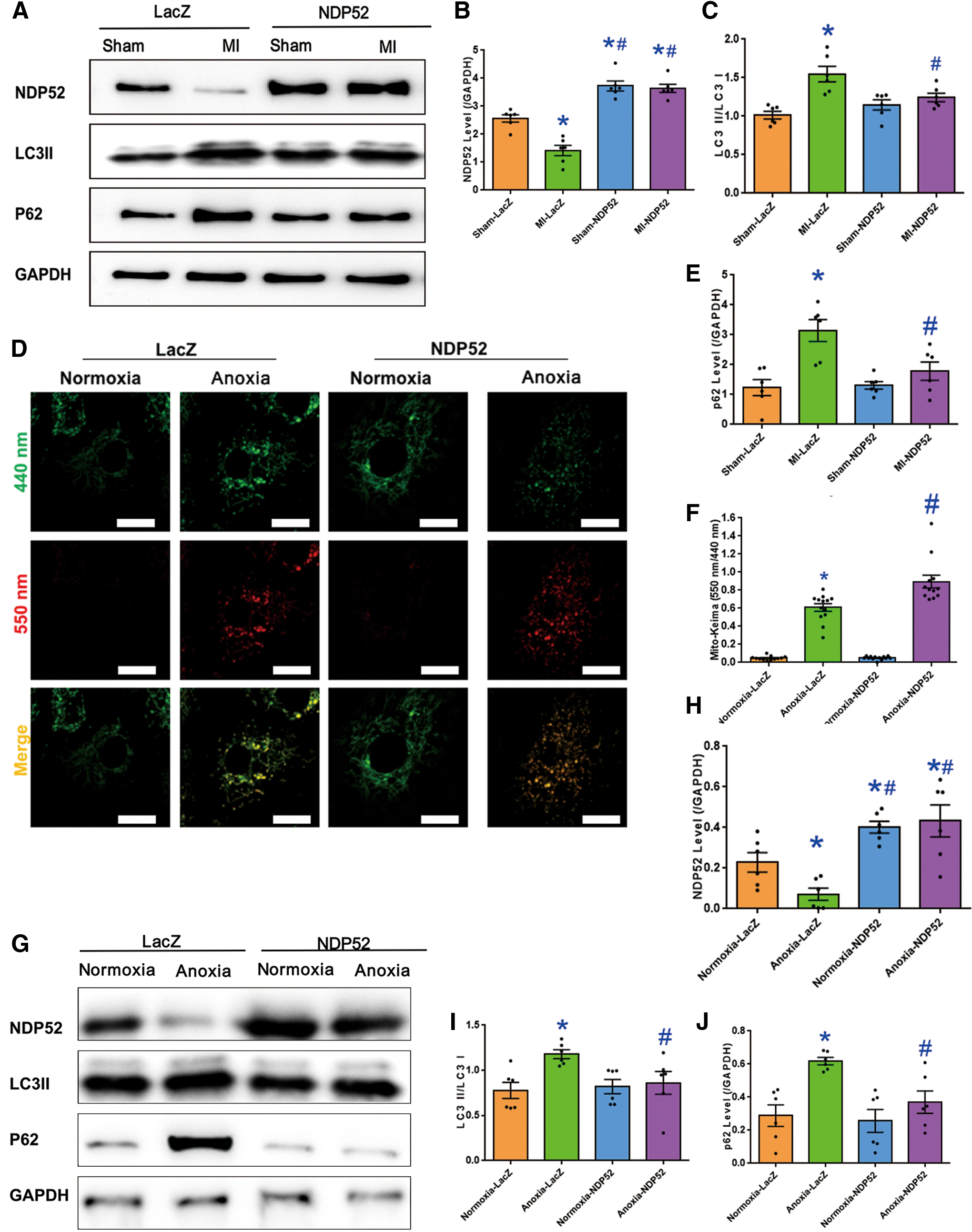
Inhibition of mitophagy flux counteracts NDP52-dependent cardioprotective property
CQ, an autophagy flux inhibitor, was used to test whether the protective effect of NDP52 on long-term hypoxic cardiomyocytes was dependent on mitophagy flux. ROS, TUNEL staining, and TMRM were determined. Our data showed that MMP was decreased by 9-h anoxia, and NDP52 overexpression annulled this damage (Fig. 5A, D). Meanwhile, 9-h anoxia overtly increased ROS production, and NDP52 overexpression attenuated the effect (Fig. 5B, E). Further, 9-h anoxia increased TUNEL-positive cells, and NDP52 overexpression attenuated this effect (Fig. 5C, F). Inhibition of autophagy flux by treatment with CQ mitigated NDP52-dependent cardioprotection without any undesired effect on anoxia cardiomyocytes. These findings suggested that NDP52-dependent cardioprotective effect is likely related to regulation of mitophagy flux.

NDP52 promotes autophagosome–lysosome fusion through recruiting Ras-associated protein RAB7 and TBK1
Immunoprecipitation was employed to examine the possible interacting partners of NDP52 in conjunction with mass spectrometry (IP-MS) (Supplementary Table S1). In H9C2 cells, flag-tagged NDP52 (NDP52-Flag) was overexpressed and was lysed for co-immunoprecipitation (co-IP) with an anti-flag antibody. Liquid chromatography-mass spectrometry (LC-MS) was then employed to determine proteins recovered in the immunoprecipitation. Ras-associated protein RAB7 (RAB7) and several proteins in the autophagy pathway were identified to co-IP with NDP52 specifically (Fig. 6A). To predict the possible interaction between NDP52 and RAB7, the protein interactions by structural matching (PRISM) tool was applied (

TBK1 is essential for NDP52-dependent protection effects on cardiomyocytes
To further discern the role of TBK1 in NDP52-regulated cardioprotective effects against anoxia, TUNEL staining, ROS, and TMRM were applied. The results showed that NDP52 exhibited a protective effect on 9-h anoxia-induced collapse in MMP and a rise in ROS production. The protection of NDP52 on MMP and ROS was nullified by GSK8612 (Fig. 7A, B). Further, the inhibition of TBK1 mitigated NDP52-dependent cardioprotection without any detrimental effects on 9-h anoxia-induced cell apoptosis using TUNEL staining (Fig. 7C). These data implied an essential role of TBK1 in NDP52-dependent cardioprotection.

TBK1 recruited by NDP52 promotes the phosphorylation of RAB7
To further inspect the function of TBK1 and RAB7 in NDP52-dependent protection effect on cardiomyocytes against ischemia, immunostaining was performed to analyze their expression. Interestingly, the expression of neither TBK1 nor RAB7 was overtly altered by MI or NDP52 overexpression (Fig. 8A–D). Western blot results also indicated that MI did not significantly alter the expression of TBK1 and RAB7 in vivo or ischemia (or anoxia) in vitro. However, phosphorylation of RAB7 was decreased in anoxia treatment in vitro and rescued by NDP52 overexpression (Fig. 8E–H). Interestingly, MI reduced the expression of PINK1 and PARKIN, but NDP52 overexpression under MI did not rescue their expression, which implied that the effect of NDP52 on MI was not dependent on the PINK1/PARKIN signaling cascade (Fig. 8E, I and K). In vitro experiments using NMCMs revealed that the inhibitor of TBK1 abolished NDP52-offered benefit on RAB7 phosphorylation. This finding favored the notion that NDP52 protects against acute MI that is likely dependent on the recruitment of TBK1 and RAB7 (Fig. 8J, M). Once recruited, TBK1 phosphorylated RAB7, which was supported by the finding that the increased RAB7 phosphorylation by NDP52 overexpression was reversed by TBK1 inhibitor.

Discussion
Our data revealed that NDP52 was dramatically downregulated in association with abnormal mitophagy flux (poor lysosomal clearance for damaged mitochondria) in mouse hearts after acute MI challenge. This is in line with the earlier report for the presence of deranged mitochondrial and cardiac function on acute MI challenge (19). Ample evidence has suggested a role for mitophagy after acute MI insult, whereas the accumulation of damaged mitochondria with ROS ultimately leads to cardiac injury. NDP52, one of the well-perceived mitophagy adaptors, has been linked to pathogenesis of Alzheimer's disease (13, 18, 21). NDP52 interacts with cargoes and LC3 to direct mitophagy cargoes toward autophagosomes preceding any interaction with mitochondrial RNA poly(A) polymerase to promote mitophagy (4). Besides, by linking autophagosomes to myosin VI, NDP52 promotes autophagosomes fusion with endolysosomes (2). However, the mechanism of NDP52 in the maintenance of cardiac homeostasis is still ambiguous. In our hands, NDP52 overexpression protected cardiac contractile function and recovered myocardial morphology in acute MI.
Data from our study denoted overt cardiac contractile dysfunction after acute MI challenge, in line with earlier findings (2, 41). In our hands, acute MI promoted overt apoptosis, production of ROS, and damage of mitochondria (TEM ultrastructure and MMP) in myocardium, which should underscore acute MI-associated myocardial dysfunction. More importantly, our findings denoted overt changes in mitophagy in acute MI-induced cardiac injury. The highly conserved lysosome-dependent process, mitophagy, facilitated the degradation and recycling of the damaged mitochondria for quality control. Mitophagy dysregulation is related with the whole process of cardiac diseases (5). Mitophagy activity has been reported to vary during the process of MI-induced myocardial abnormalities. In different phases of MI, mitophagy could be either upregulated or downregulated (25). Our data indicated that after the 7-day coronary artery ligation, mitophagy flux was interrupted. This is coherent with previous reports where autophagosomes clearance was impaired in injured cardiomyocytes (24). In our hands, overexpression of NDP52 effectively reduced the number of damaged mitochondria and promoted mitophagy flux, coinciding with the earlier finding of NDP52-regulated mitophagy (15, 20). Findings from our lab and others have shown the necessity of alleviating damaged mitochondria to confer the protection of cardiac function against MI or I/R injury (25, 26, 46). Mitochondria that were too large or small can be harmful to cell homeostasis. Although our study provides some hints toward the role of NDP52 in mitophagy governance, further investigation is necessary to reveal the precise function of NDP52 in the modulation of mitochondria.
This study proposes that NDP52 functions as the bridging molecule for TBK1-induced phosphorylation of RAB7, which promotes mitophagy flux. Multiple studies have consolidated the role of RAB7 in membrane fusion, including formation of autophagolysosomes (or mitophagolysosomes for mitophagy) (9). In MI-induced cardiac diseases, RAB7 was reported to regulate contacts of mitochondria-lysosome and subsequently lysosome-dependent mitophagy flux (9, 12). Loss of RAB7 impaired fusion of autophagosomes and lysosomes, resulting in autophagosome accumulation in the heart and cardiac defect under hypoxia/reoxygenation conditions (40). With mitochondrial damage such as depolarization, both TBK1 and RAB7 are recruited to damaged mitochondria. RAB7 and cargo receptors were perceived as the dynamic physiological targets for TBK1 (12, 30). Our data displayed little difference in the level of TBK1 or RAB7 in response to NDP52 overexpression. However, phosphorylation of RAB7 was clearly evoked by NDP52, which was dependent on TBK1 in hypoxic cardiomyocytes. These results supported a beneficial function of NDP52 in mitochondria through mitochondrial quality control machinery. Bioinformatic analysis, co-IP MS, co-IP, and immunostaining findings further supported the notion that acute MI dampened co-localization of NDP52 with RAB7 and TBK1, resulting in decreased phosphorylation of RAB7. NDP52 overexpression rescued both co-localization of RAB7/TBK1 and RAB7 phosphorylation. TBK1 is capable of phosphorylating RAB7 to promote the downstream fusion process to capture damaged mitochondria for completion of mitophagy (12). Consequently, NDP52 promotes the damaged mitochondria clearance via elevating the activity of RAB7 through TBK1, giving rise to the protection effect of cardiac mitochondria, and heart function in acute MI challenge.
Experimental limitations
First, further studies are needed to decipher the function of NDP52 on mitophagy or mitochondrial function, although we had information on mito-keima, and TMRM staining to evaluate mitophagy and mitochondrial function, respectively. Second, the cellular process related to MI is complicated and can be more than what we have observed. Our study disclosed the possible modulation of NDP52 in mitophagy after MI. Nonetheless, further investigation is warranted for other potential roles of NDP52 in MI, including necrosis, apoptosis, and inflammation. Third, it could be an ideal way to deliver NDP52 after MI attack and evaluate the therapeutic effect in this model. However, the onset of protein expression with adenovirus delivery requires a minimal duration of 16–24 h. It takes around 3 days to reach a peak expression level for the protein of interest (10). To this end, adenovirus with NDP52 was given 3 days before MI. This could be a limitation of the use of adenovirus, and more evidence from pharmacological activation of NDP52 after the MI procedure is needed to elucidate the role of NDP52 in post-infarction treatment.
Our study suggested that NDP52 promotes phosphorylation of RAB7 through recruiting TBK1 to the NDP52-RAB7 complex. This research should provide new perspectives on the function of NDP52, and NDP52 is a potential therapeutic target in the treatment of MI. More research is warranted to fully uncover the function of NDP52 in the pathologic process of ischemic heart diseases and the related molecular pathogenesis. A complete understanding of the physiological role of NDP52 could be beneficial to disclose the novel therapeutic targets in MI.
Materials and Methods
Animals and experimental protocols
All animal procedures were approved by the Institutional Animal Care and Use Committee of Zhongshan Hospital Fudan University (Shanghai, China). Overall, 1.0%–1.5% isoflurane gas was used to anesthetize C57BL/6 mice (8–10 weeks old), and a mechanical animal respirator was applied. Left anterior descending (LAD) coronary artery was visualized by thoracotomy and was ligated with 6–0 silk thread for 7 consecutive days (33). NDP52-Flag adenovirus was purchased from the Obio Technology Corporation (Shanghai, China). For obtaining NDP52 overexpression in cardiac tissue, 3 days before LAD ligation, adenovirus was injected into the intra-myocardium at three different sites of the target ischemic zone by using 29-G needles. Adenovirus carrying LacZ gene (Obio Technology Corporation) was employed as a control (14, 46). In a cohort of NDP52 transfected mice, the lysosomal inhibitor CQ (20 mg/kg/day, i.p.) was injected intraperitoneally for 7 consecutive days immediately after LAD surgery until experimentation (50). A Vevo 2100 system (Visual Sonics, Toronto, Canada) was employed to perform transthoracic echocardiography.
Histological examination and Masson trichrome staining
One hundred milligram per kilogram sodium pentobarbital was used to anesthetize mice, and complete anesthesia was determined by using toe reflex before sacrifice. Hearts were fixed and embedded in paraffin. Masson trichrome staining and hematoxylin and eosin (H&E) were applied to five-micrometer-thick sections (43). Light microscope (Leica, Germany) was used to obtain a single section for further analysis. Image J software was applied to measure left ventricle and collagen-positive areas. The entire left ventricular area was used to normalize the total collagen-positive area for interstitial fibrosis calculation (41).
TUNEL staining
A TUNEL staining kit (KeyGEN Biotechnology CO., Ltd., Nanjing, China) was applied to stain heart slices. Morphology of cell apoptosis was imaged with a fluorescence microscope (Nikon, Tokyo, Japan) in a blind fashion in random fields (6).
Electron microscopy
A buffer (pH 7.4) containing 1% glutaraldehyde, 4% paraformaldehyde, and 0.1 M Na cacodylate was applied in retrogradely perfused hearts. Overall, 2.5% glutaraldehyde and 0.1 M Na cacodylate were used to fix tissues from midsections of the left ventricular wall. A Leica Ultramicrotome was used to cut sections (75–80 nm) before electron microscopic microscopy (22).
Anoxia protocol of NMCMs
The NMCMs were isolated according to the previous protocol (14, 35). The process of MI was simulated by anoxia. Glucose-free Dulbecco's modified Eagle's medium (DMEM) was used to replace NMCMs culture medium, and cells were cultured for 9 h in an incubator (Columbus Instruments) containing 5% CO2 and 95% N2 gas mixture before further experiments. With ad-NDP52 or ad-LAC, NMCMs were treated with GSK8612 (0.5 μM, S8872; Selleck Chemicals, Houston, TX) for 1 h to inhibit TBK1 (36) or CQ (50 μM C6628; Sigma-Aldrich, St. Louis, MO) for 12 h to inhibit lysosomal enzymes (33).
Adenovirus, shNDP52, and transfection
The NMCMs were plated in a 60-mm dish or a 6-well plate 24 h before transfection. The NMCMs were transfected with adenovirus for NDP52 overexpression or lentivirus carrying shNDP52 for NDP52 knockdown with 100 multiplicity of infection (MOI) for 48 h. For observing mitophagy flux, adenoviruses encoding mito-Keima were transfected into NMCMs with 50 MOI. All viruses were purchased from Hanbio Technology Corporation (Shanghai, China). Confocal sections were collected under uniform settings with a Nikon A1 laser scanning confocal microscope.
H9C2 cell culture and LC-MS analysis
H9C2 cells were purchased from the Shanghai Academy of Science (Shanghai, China). Cells were cultured and transfected with NDP52-Flag for 48 h. Cells were lysed under conditions to optimally yield soluble tagged protein immunoprecipitated by using an anti-flag antibody. Samples were then subjected to LC-MS analysis at the Shanghai Biotechnology Corporation (Shanghai, China).
Mitochondrial inner membrane potential (ΔΨ) detection
Image-iT TMRM reagent (1:1000; Thermo Fisher) was adopted to measure the membrane potential, ΔΨ. After being washed three times, the cells were imaged in live cell imaging solution at 37℃. Then, the membrane potential of the cells was observed by confocal microscopy with absorption and emission at 543 and 560–660 nm (32).
ROS detection
2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) fluorescent probe was used to detect ROS. Phosphate-buffered saline prewarmed at 37℃ was used to rinse cells, and 10 μM DCFH-DA were incubated with NMCMs (1:1000, no serum) for 20 min. Serum-free DMEM was used to wash cells three times, and a ProLong Live antifade reagent was added afterward. Pictures were obtained with Leica confocal microscope at 63 × magnification. Image J software was used to measure the DCFH-DA fluorescence (47).
Co-immunoprecipitation and Western blot
Pierce®Co-IP kit (Pierce, IL) was used for co-IP assay following the manufacturer's instructions. Briefly, 100 μg purified anti-NDP52 antibody was coupled with resin and exposed to 1 mg protein samples for 2 h. After elution, the eluted protein samples were detected with Western blot. The use of antibodies was as follows: Rab7 (1:1000; Abcam, no. ab137029), p62 (1:2000; CST, no. 23214), TBK1 (1:1000; CST, no. 38066), NDP52 (1:1000; thermo, PA5-30367), GAPDH (1:5000; CST, no. 5174), pRAB7 (1:2000; Abmart, no. TA3777), LC3B (1:1000; CST, no. 3868), PINK1 (1:1000; CST, no. 6946), and PARKIN (1:2000; CST, no. 4211).
Immunofluorescence
The NMCMs with different treatments were fixed and washed; cells were incubated with goat serum for 1 h, and then they were incubated with anti-LAMP1 (1:200; Abcam, no. ab25630), anti-Rab7 (1:200; Abcam, no. ab137029), and anti-TBK1 (1:200; Cell Signaling Technology, no. 8066) antibodies overnight. Fluorescence-conjugated secondary antibody was then incubated with cells for 1 h. The Leica confocal microscope was used to obtain micrographs.
Adult mouse cardiomyocyte shortening/relengthening
Hearts of adult mice were isolated with the adult mouse cardiomyocyte isolation and shortening/releasing system after anesthesia. After perfusion with collagenase I, adult mouse cardiomyocytes were plated with laminin (5 μg/mL; Sigma Aldrich). A Softedge MyoCam system (IonOptix Corporation, Milton, MA) equipped with an inverted IX70 Olympus microscope was employed to monitor the mechanical properties of cardiomyocytes (29, 49).
Structure-based protein interaction interface analysis
The homo-dimer structure of the coiled–coil domain of NDP52 was predicted by SWISS-MODEL (
Statistical analysis
Data were shown as the mean ± standard error of at least three independent experiments and were analyzed by using one-way analysis of variance (ANOVA), and Tukey's test was used for post-test analysis. A p value <0.05 was considered as being statistically significant.
Electronic laboratory notebook was not used.
Footnotes
Authors' Contributions
M.S., W.Z., Y.B., X.H., and M.A. performed the data collection. H.P., J.R., and Y.Z. conceived the study, as well as drafted and proofed the article. All authors approved the final submission.
Data Availability
The datasets used and/or analyzed supporting the findings of this study are available in this article or the Supplementary Information. Any raw data that support the results of this study are available from the corresponding author on reasonable request.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Natural Science Foundation of China 2017YFA0506001, 82000274, 81900233, 81770261, 81521001, and 91749128 and the Postdoctoral Science Foundation of China 2020M681183.
Supplementary Material
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.