
Abstract
Significance:
Sepsis is a critical clinical syndrome with life-threatening organ dysfunction induced by a dysregulated host response to infection. Despite decades of intensive research, sepsis remains a leading cause of in-hospital mortality with few specific treatments.
Recent Advances:
Toll-like receptors (TLRs) are a part of the innate immune system and play an important role in host defense against invading pathogens such as bacteria, virus, and fungi. Using a combination of genetically modified animal models and pharmacological agents, numerous preclinical studies during the past two decades have demonstrated that dysregulated TLR signaling may contribute to sepsis pathogenesis. However, many clinical trials targeting inflammation and innate immunity such as TLR4 have yielded mixed results.
Critical Issues:
Here we review various TLRs and the specific molecules these TLRs sense—both the pathogen-associated and host-derived stress molecules, and their converging signaling pathways. We critically analyze preclinical investigations into the role of TLRs in animal sepsis, the complexity of targeting TLRs for sepsis intervention, and the disappointing clinical trials of the TLR4 antagonist eritoran.
Future Directions:
Future sepsis treatments will depend on better understanding the complex biological mechanisms of sepsis pathogenesis, the high heterogeneity of septic humans as defined by clinical presentations and unique immunological biomarkers, and improved stratifications for targeted interventions.
Introduction
Toll-
Sepsis is a clinical syndrome with life-threatening organ dysfunction caused by a dysregulated host response to infection (115). In the United States, sepsis develops in more than 750,000 people annually and 210,000 of them die (9, 80). Despite the recent progress in sepsis management, such as early antibiotic coverage, aggressive fluid resuscitation, and vasopressors to maintain hemodynamics, sepsis remains the #1 cause of mortality in hospitals (32, 102, 110). There have been numerous phase 2 and 3 sepsis clinical trials of various therapies for sepsis, including anti-inflammation, anti-cytokine, and immune and coagulation modulations, most of which have failed (37, 77).
Among many possible causes, incomplete understanding of the biological mechanisms of sepsis pathogenesis may have contributed to the failed clinical trials (51, 77, 123). This article critically reviews the animal investigations on the role of TLR signaling in sepsis, the clinical trials testing the therapeutic efficacy of blocking TLR4, and the lessons we learn from these studies.
TLRs: Pattern Recognition and Signaling Pathways
Following their invasion, microbes release various pathogen-associated molecular patterns (PAMPs), such as endotoxin, lipopeptide, and nucleic acid. PAMPs are sensed by the pattern recognition receptors such as TLRs in the host cells and activate the host proinflammatory responses such as leukocyte activation, complement activation, and coagulation activation (21, 51). These host responses are extremely important to contain and eliminate microbe dissemination. However, if the responses are dysregulated and excessive, it can cause collateral damage to the host. For example, too much cytokine production (cytokine storm) can cause septic shock, coagulation activation, and subsequent consumptive coagulopathy such as disseminated intravascular coagulation.
Dead cells release damage-associated molecular pattern (DAMP) that can act on TLRs and perpetuate proinflammatory responses, causing excessive inflammation and tissue damage. Almost at the same time, the anti-inflammatory responses are initiated. These include the neuroendocrine axis of parasympathetic outflow and adrenal glands, immune cell dysfunction or death, and anti-inflammatory cytokine production. The anti-inflammatory pathways are implicated in the enhanced susceptibility to secondary infections during the later stage of sepsis.
Discovery of toll and TLR
In 1985, Nusslein-Volhard identified the Toll gene critical for the embryonic development of the fruit flies Drosophila (7, 8). A decade later, Hoffmann and colleagues found that Toll was essential to innate immunity against pathogen (69). The following year, Medzhitov and Janeway at Yale discovered that a human Toll protein was a transmembrane protein with an extracellular domain consisting of a leucine-rich repeat and cytoplasmic domain, which they coined “Toll-like receptor” (79). Subsequently, several groups discovered that mice with naturally mutated Tlr4 gene (97, 98), either a missense point mutation (Pro→His, C3H/HeJ strain) or null mutation (C57 BL/10 ScCr strain), or with specific Tlr4 gene deletion (TLR4−/−) (50), exhibited insensitivity to endotoxin, demonstrating TLR4 as the sensor for bacterial endotoxin.
TLR signaling pathways
At least 11 human and 13 mouse TLRs have been cloned and they are expressed on various types of immune and nonimmune cells, such as macrophages, monocytes, dendritic cells (DCs), lymphocytes, endothelial and epithelial cells, and cardiomyocytes. TLRs are single-spanning membrane glycoproteins with an intracellular Toll/interleukin-1 receptor (TIR) domain (58).
Based on their locations in the cell, TLRs are categorized into two groups: (i) those anchored on the plasma membranes including TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, which mainly sense lipopeptides, peptidoglycan, lipopolysaccharide (LPS), or zymosan of bacterial and fungi origins, and (ii) those located inside the cell on the endosome membranes such as TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13, which are mainly associated with nucleic acid sensing (103), such as double-stranded (ds) RNA (TLR3) (4), single-stranded RNA (TLR7/8) (45, 73, 131), and DNA (TLR9) (49). Other known nucleic acid sensors are located in the cytoplasm and include the RNA sensors—retinoic-acid-inducible gene 1 (RIG-I) and melanoma-differentiation-associated gene 5 (MDA5), and the DNA sensors—cyclic GMP-AMP synthase and absent in melanoma 2 in the cytoplasm (103).
Following ligand binding, TLRs form dimers and the resulting TIR-TIR complexes trigger the downstream signaling (Fig. 1) through the specific adaptors (84), that is, MyD88 (myeloid differentiation factor 88), TIRAP (TIR domain-containing adaptor protein), Trif (TIR domain-containing adaptor inducing IFN-β–mediated transcription factor), SARM (sterile α- and heat-armadillo-motif-containing protein), and TRAM (Trif-related adaptor molecule).

TLR signaling can be further divided into two distinct but convergent pathways: MyD88-dependent and Trif-dependent pathways. MyD88-dependent pathway is activated by all TLRs with the exception of TLR3. MyD88 pathway leads to activation of the transcription factor nuclear factor kappa B (NF-κB) and mitogen-activated protein kinases. MyD88 recruits IL-1 receptor-associated kinase (IRAK). The IRAK1-TNF receptor-associated factor 6 (TRAF6) complex then activates transforming growth factor-α activated kinase 1 (TAK1). Activated TAK1 then phosphorylates I-κB kinase beta (IKKβ), leading to phosphorylation and degradation of I-κB, which releases the NF-κB p50/p65 subunits and results in the nuclear translocation and DNA binding of NF-κB. Trif-dependent pathway is utilized by TLR3 and TLR4. It induces type I interferon (IFN) and inflammatory cytokines through the transcription factor interferon regulatory factor 3 (IRF3).
PAMPs and DAMPs
TLRs specifically bind to a wide range of pathogens such as bacteria, fungi, and viruses through “PAMPs” recognition (Table 1) (2, 58). TLRs can also act as a stress sensor in response to noninfectious tissue injury and recognize a variety of endogenous danger molecules through “DAMPs” recognition (94).
Toll-Like Receptors and Ligands
CpG, cytidine-phosphate-guanosine; DAMP, damage-associated molecular pattern; ds, double-stranded; HMGB1, high-mobility group box 1; Hsp, heat-shock protein; IFN, interferon; LDL, low-density lipoprotein; LPS, lipopolysaccharide; miRNA, microRNA; mtDNA, mitochondrial DNA; MyD88, myeloid differentiation factor 88; PAMP, pathogen-associated molecular pattern; TIRAP, TIR domain-containing adaptor protein; TLR, toll-like receptor; TRAM, Trif-related adaptor molecule; TRIF, TIR domain-containing adaptor inducing IFN-β-mediated transcription factor.
LPS, one of the best-characterized bacterial ligand, is a wall component of gram-negative bacteria. The extracellular domain of TLR4 forms a complex to act as the LPS-binding site of TLR4 (88, 89). TLR2 senses a wide range of PAMPs—including lipopeptides, peptidoglycan, and lipoteichoic acid from gram-positive bacteria. TLR2 usually forms heterodimers with TLR1 or TLR6. TLR2/6 heterodimer senses diacylated lipopeptides (125), whereas TLR1/2 distinguishes triacylated lipopeptides (127). TLR5 responds to bacterial flagella through flagellin (43). TLR11 responds to the protozoan parasite and uropathogenic bacteria through a profilin-like molecule (137, 138). TLR3 recognizes double-strand RNA, which can activate immune responses to express IFN and cytokines to exhibit antiviral and antibacterial effects. TLR7 and TLR8 recognize single-strand RNAs, as well as imidazoquinoline compounds such as guanine analogues and imiquimod (142, 143). At last, TLR9 recognizes cytidine-phosphate-guanosine (CpG) DNA motifs with unmethylated dinucleotides from bacteria and viruses (11, 96) and mitochondrial DNA (36, 76, 141).
DAMPs are produced by injured cells under both infectious (e.g., sepsis) and noninfectious (e.g., trauma, ischemic injury, and autoimmune disease) conditions (18, 29, 39, 64, 65, 71, 118, 149, 151). Some of the reported examples include heat-shock proteins, hyaluronic acid, glycoprotein 96 (Gp96), heparan sulfate, fibrinogen, HMGB1, RNA, DNA, amyloid β, and oxidized low-density lipoprotein (Table 1). These endogenous pattern molecules, once released into the extracellular space, are sensed by various TLRs and elicit the host innate immune responses.
Severe acute respiratory syndrome coronavirus 2 and innate immune receptor recognition
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a highly transmissible virus and has caused the current global pandemic of acute respiratory disease, named “coronavirus disease 2019” (COVID-19), that threatens the global health and safety (52). SARS-CoV-2, like other coronaviruses, contains the dsRNA that is produced during viral genome replication and transcription (60, 146) and can be recognized by several nucleic acid sensors, including RIG-I and MDA5, in the cytoplasma (70, 109) and/or by TLR3 in the endosome (78, 124). Activation of these innate immune receptors leads to the antiviral type-I IFN signaling (68).
Role of TLRs in Sepsis
Extensive preclinical work has documented the possible role of TLRs in the sepsis pathogenesis. Many of these studies have taken loss-of-function approaches, either in genetically modified mouse models or pharmacological blocking, to manipulate TLR signaling. Others have used TLR ligands to activate TLR signaling. Table 2 summarizes some of these findings.
Role of Toll-Like Receptors in Endotoxin Shock and Bacterial Sepsis
CLP, cecum ligation and puncture; NF-κB, nuclear factor kappa B; WT, wild type.
TLR2
Both animal and human studies support the role of TLR2 in sepsis-induced immune and multiple organ injuries, such as cardiac, endothelial, and neutrophil dysfunction, and coagulopathy (34, 47, 93, 103, 105, 114, 135, 136, 139, 145, 147, 150). For example, peptidoglycan-associated lipoprotein, a natural TLR2 ligand and a ubiquitous gram-negative bacterial membrane protein (72), inhibits cardiomyocyte function (145) and activates endothelial function and coagulation pathways (114). In mouse models of polymicrobial sepsis, TLR2−/− mice have better survival, improved cardiac function, attenuated blood and myocardial cytokine production (147), improved clotting function (135), less immune cell depletion (82, 93), reduced mitochondrial reactive oxygen species production, and improved mitochondrial function (40) when compared with wild-type (WT) mice.
TLR3
TLR3 senses dsRNA and also endogenous RNA released from necrotic tissues and mediates acute inflammation (16). In a bacterial sepsis model, however, knockout of TLR3 confers a survival benefit and improved organ function (28, 35). Both bacterial and host RNAs are present in the circulation of septic mice and could potentially function as the agonists of TLR3. Interestingly, in a double-hit model, treatment with the TLR3 ligand poly(I:C) of animals before cecum ligation and puncture (CLP) enhances host immunity and improves the survival (24).
TLR4
Extensive reports have demonstrated the role of TLR4 in mediating cytokine storm, immune cell impairment, organ injury, and mortality in endotoxin shock or in polymicrobial sepsis (6). Animal endotoxemia induces NF-κB activation (61) that leads to robust myocardial cytokine response and myocardial dysfunction (12). This process involves signaling via TLR4, CD14, IRAK1, MyD88, and Trif. The endotoxin-mediated cardiac dysfunction may be an indirect effect secondary to immune cell activation rather than a direct effect on cardiomyocytes as in vitro treatment with LPS fails to inhibit cardiomyocyte function (120).
Chimeric models suggest that TLR4 in hematopoietic cells is responsible for cardiac dysfunction during endotoxic shock (13, 119, 120). Both MyD88 and Trif play an equally important role in endotoxin shock (31). Studies using tissue-specific MyD88 knockout models demonstrate that both cardiomyocyte- and myeloid-MyD88 play a role in mediating cardiac dysfunction and mortality during endotoxin shock (31). In animal models of bacterial sepsis, the role of TLR4 is complex and may depend on the type and severity of bacterial infection. For example, while TLR4 deletion clearly confers a survival benefit in endotoxin shock (97) or lethal gram-negative bacterial sepsis (105), it offers no survival benefit (26) or even deleterious effect in mild gram-negative or polymicrobial sepsis (105, 140).
These data seem to suggest that host may mobilize different innate immune mechanisms in endotoxemia and bacterial sepsis. Moreover, studies suggest that signaling via MyD88, but not Trif, plays a predominant role in mediating cardiac dysfunction, marked systemic inflammation, and mortality in a lethal model of bacterial sepsis, whereas MyD88 and Trif are equally important in systemic inflammation, organ dysfunction, and death during endotoxin shock (31).
Several possible mechanisms may explain the deleterious effect of TLR4 deficiency in low-grade bacterial sepsis. First, TLR4 is a part of the host immune defense against bacterial invasion. WT mice exhibit more robust neutrophil migratory and phagocytic functions compared with TLR4−/− mice. As a result of bacterial dissemination in the absence of TLR4, TLR4−/− mice have more cytokine production, bacterial load, and higher mortality. Second, TLR4 signaling may play a “preconditioning-like” role during low-grade bacterial infection. LPS pretreatment confers a cardioprotective effect against hypoxic injury (14, 17). Studies have shown that administration of low-dose endotoxin offers protection against both subsequent endotoxin challenges and polymicrobial infection (134). Finally, it should be pointed out that there is substantial difference in endotoxin sensitivity among different species. Controversies exist over whether or how well endotoxin-challenged mice mimic endotoxemia and acute inflammation in humans (22, 113, 116, 133).
TLR5
Bacterial flagellin, a TLR5 ligand, activates NF-κB-mediated inflammatory response and induces myocardial dysfunction (107, 108). In vivo, flagellin administration leads to cytokine storm, increased myocardial neutrophil infiltration, and reversible cardiac dysfunction.
TLR7/TLR8
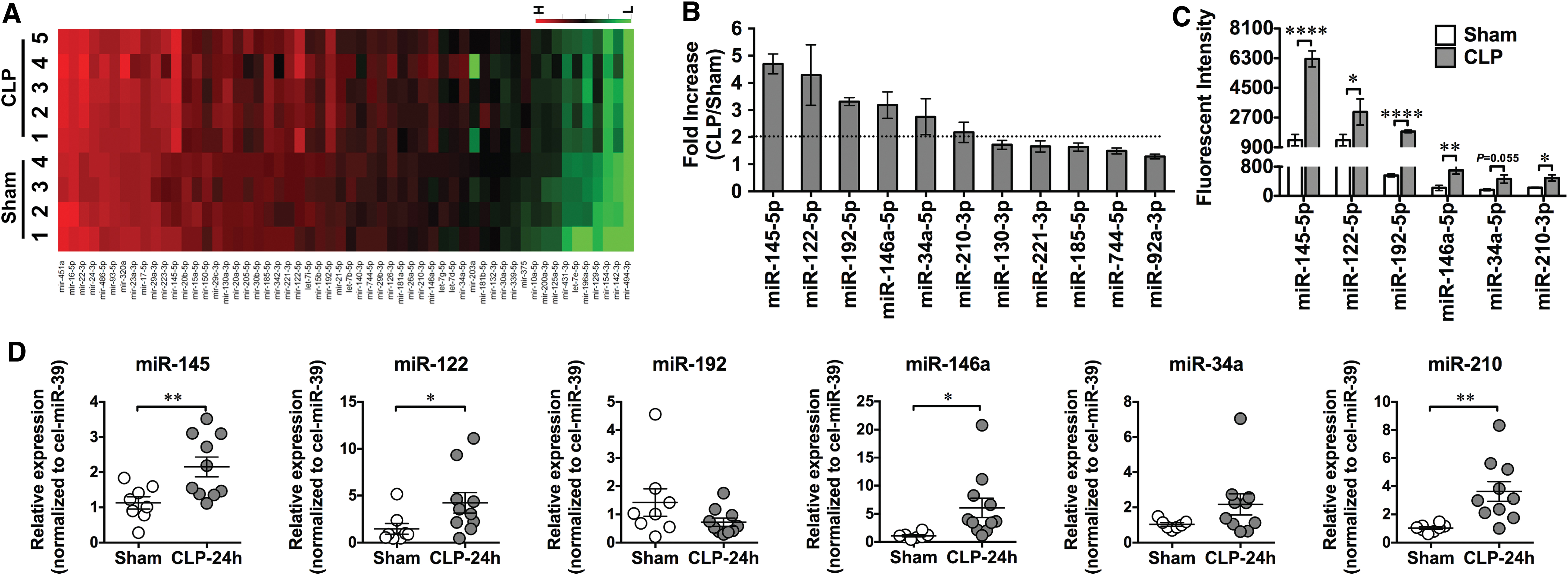
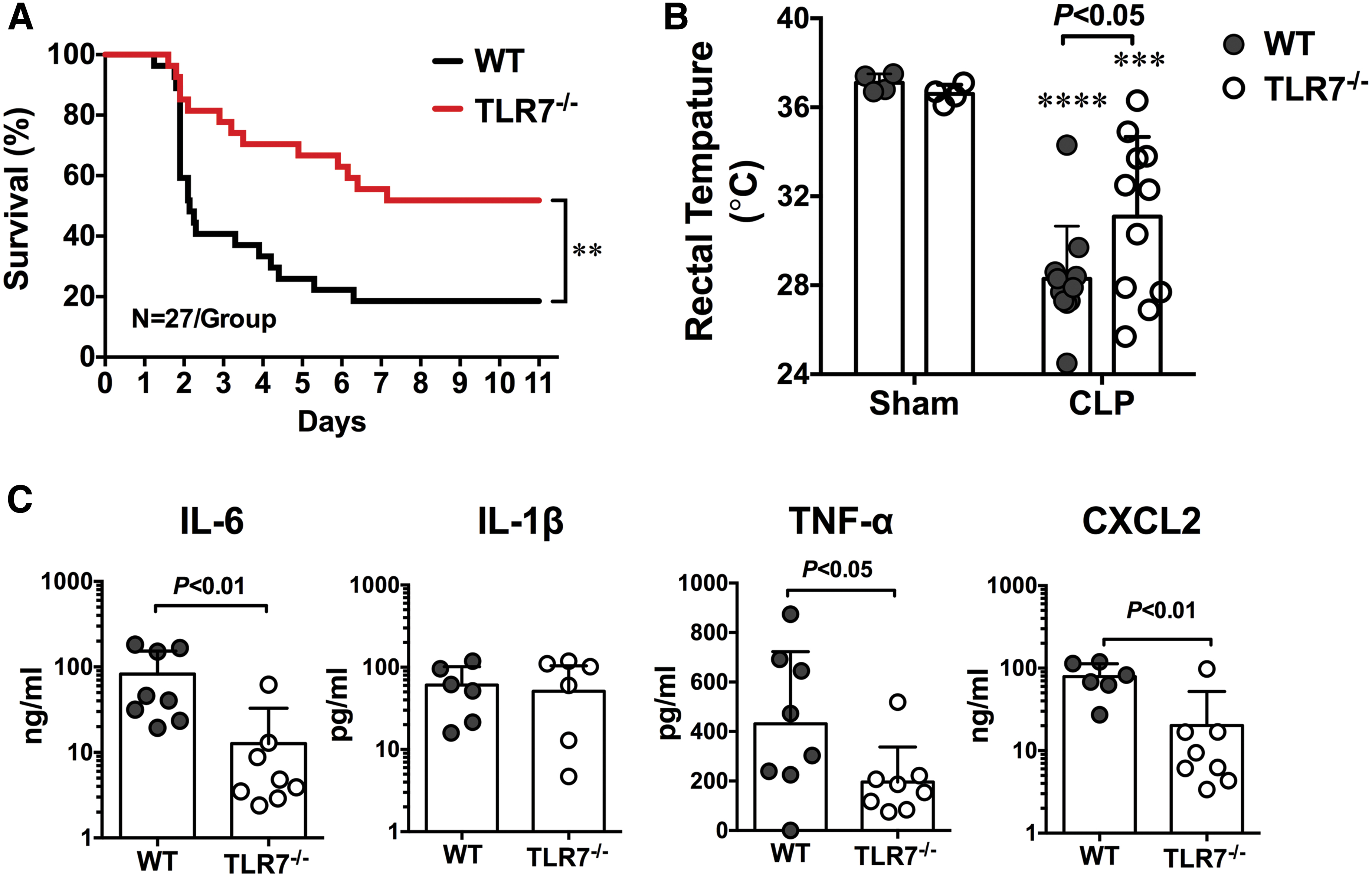
In addition to sensing viral single-stranded RNA, TLR7 may also sense endogenous extracellular RNAs released from injured cells and plays a pivotal role in murine sepsis. In a CLP model of polymicrobial sepsis, studies have found an increased plasma RNA that is closely correlated with sepsis severity (149). Plasma microRNA (miRNA) array revealed upregulation of multiple miRNAs including miR-34, miR-122, miR-145, miR-146a, miR-210 (Fig. 2) (149). Tissue RNA extracts or RNA released from injured cells or miRNA mimics induces proinflammatory cytokine production and complement activation via a TLR7-dependent mechanism (29, 30, 149). Moreover, compared with WT mice, mice deficient of TLR7 had lower plasma cytokines, reduced circulatory shock (lower core temperature), and significantly improved survival (Fig. 3) (54). Finally, similar to humans, septic mice develop sepsis-induced coagulopathy characterized by global clotting dysfunction, severe thrombocytopenia, decreased fibrinogen, and increased plasma tissue factor (TF) and D-dimers (Fig. 4) (135). TLR7−/− septic mice exhibited preserved global clotting function, platelet counts, and near-normal plasma TF concentration (135).

TLR9
DNA and RNA isolated from Staphylococcus aureus and Escherichia coli induce rat cardiomyocyte dysfunction (87). Similarly, CpG-ODN, a TLR9 agonist, inhibits sarcomere shortening of isolated mouse cardiomyocytes. In vivo, CpG-ODN causes myocardial NF-κB activation and cytokine production. Both effects are abolished in TLR9-deficient mice (62). Compared with WT mice, TLR9−/− mice exhibited lower serum inflammatory cytokine levels, higher bacterial clearance, and greater survival after experimental peritonitis induced by CLP. Protection of TLR9−/− mice after CLP was associated with a greater number of peritoneal DCs and granulocytes than in WT controls. Adoptive transfer of TLR9−/− DCs was sufficient to protect WT mice from CLP and increased the influx of peritoneal granulocytes (95). Further studies indicate that host mitochondria-derived DNA may be responsible for TLR9 activation and contributes to sepsis-induced acute kidney injury (128).
Targeting TLRs in Sepsis: A Double-Edged Sword
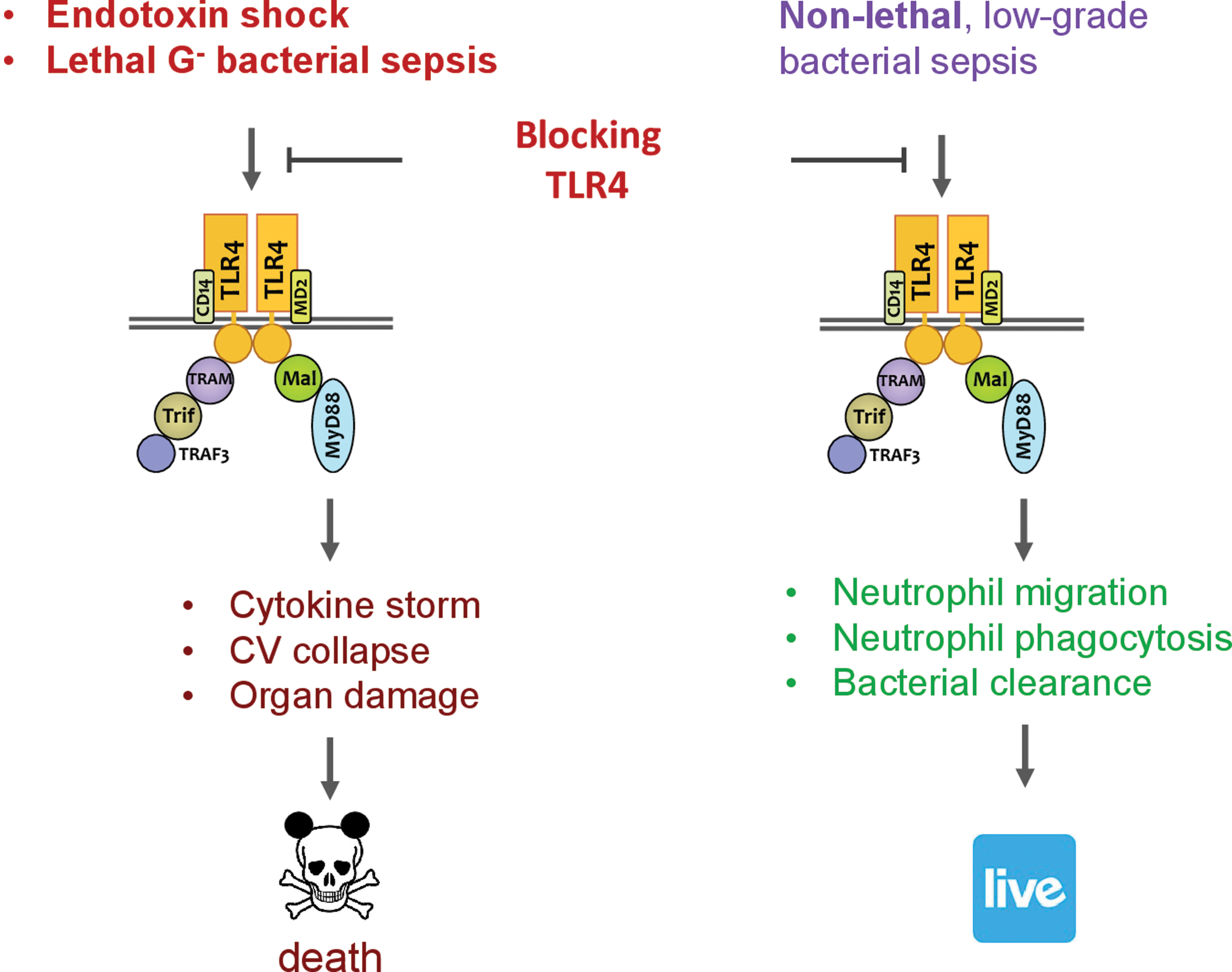
It has long been recognized that systemic inflammatory response is a major contributor to sepsis pathogenesis leading to cardiovascular collapse, multiorgan injury, and mortality. Such systemic response is highly complex and caused by both infectious and noninfectious mediators. Manipulating this process as a therapeutic strategy, while seeming attractive and logical, has proven to be a double-edge sword. One such example is targeting TLR4. Blocking or genetically deleting TLR4 effectively protects animals from endotoxin-induced circulatory shock, cardiac depression, and high mortality (27, 31, 97, 98). Moreover, in a mouse model of lethal gram-negative bacterial sepsis with more than 80% mortality, TLR4 deletion or antibody blocking proves to be beneficial—damping systemic cytokine storm (TNFα and IL-6) and markedly improving the survival of septic animals (Fig. 5) (105). However, as an essential part of innate immunity, TLR4 also plays a critical role in host defense against bacterial invasion. In nonlethal gram-negative bacterial infection or mild-form of polymicrobial sepsis, TLR4 deletion or antibody blocking proves to be deleterious and leads to increased blood bacterial loading, worse cardiac dysfunction, and higher mortality (Fig. 5) (105, 140). Thus, it is evident that sepsis severity is an important factor in determining the outcome of TLR4-targeted sepsis intervention (Fig. 6).

Targeting TLR4: The ACCESS Trials
There have been more than 100 phase 2 and phase 3 sepsis clinical trials (77). The strategies have been to manipulate the systemic inflammatory response by targeting the PAMP or endogenous inflammatory mediators such as TNFα, IL-1, eicosanoids, or platelet-activating factor, or by suppressing immune response or coagulating cascade. One of the most recent clinical trials for sepsis is eritoran. Eritoran is a synthetic lipodisaccharide, with a structure similar to LPS, that binds to MD2-TLR4 and competitively blocks LPS to TLR4 (89).
In a phase 1 trial (75), healthy volunteers were given eritoran before LPS. LPS, at a small dose of 3 ng/kg, induces very robust production of C-reactive protein, TNFα, and IL-6. Similar to animal studies, TLR4 blocking by eritoran in these healthy humans completely eliminated all clinical signs and cytokine production induced by LPS. In a subsequent phase 2 trial (122), a prospective, randomized, double-blinded, placebo-controlled multicenter study, two doses of eritoran were tested: 45 mg versus 105 mg over the course of 6 days, q12 hours, and given within 12 h of sepsis diagnosis.
The study patients had predicted risk of mortality between 20% and 80%. Among a total of 300 septic patients—100 in placebo and 200 in eritoran groups, 28-day all-cause mortality in the placebo group was 33.3%, low-dose group 32%, and high-dose group 26.6%, a substantial (20%) but not statistically significant reduction. Poststratification analysis of APACHE II score on 28-day all-cause mortality revealed that patients with less severe sepsis (score 21) probably did worse with high-dose eritoran compared with placebo. In contrast, patients with high APACHE score 4—most severe sepsis—might have been benefited from eritoran, a finding very similar to what had been seen in animal studies noted above.
It was concluded that the trend toward a lower mortality rate in patients with severe sepsis and high predicted risk of mortality should be further investigated in the phase 3 trial (ACCESS trial, NCT00334828) that involved 1961 septic patients from 197 intensive care units (ICUs) worldwide (86). Like phase 2, it was a randomized, double-blinded, placebo-controlled multicenter study. Based on the phase 2 data, the study only tests high dose at 105 mg total and given every 12 h. The study patients were highly heterogenous in terms of (i) type of infection: G−, G+, mixed, fungi, and other unknown, and (ii) infection sites: mostly in the lungs, abdomen, and genitourinary track. At the end of the 5-year trial, there was no difference in the 28-day all-cause mortality, which was about 27%–28%. The 12-month mortality was also identical between the eritoran and control groups at 40%.
Sepsis Clinical Trials: Lessons Learned
The cause of the failed ACCESS trial might be multifactorial (86): (i) patient heterogeneity: the septic patients enrolled had various severity scores and comorbidities, which could have impacted how patients responded to the treatment; (ii) only a fraction (40.7%) of the septic patients had elevated plasma endotoxin levels. Considering TLR4 as the target, this might explain why some patients failed to respond to eritoran, although a post hoc analysis did not find survival benefit in endotoxin-positive patients either; (iii) lower than anticipated mortality rate in the placebo group. In the course of the trials, the sepsis mortality rate had been gradually decreased from 40% to ∼27%, probably due to the Surviving Sepsis Campaign, which, along with other interventions, might alter the responsiveness to eritoran; (iv) too late for intervention. The time for intervention was within 12 h after initial sepsis diagnosis, which might be too late for intervention as “genie was out of bottle”; and (v) different pathogens. The study patients were infected with gram-positive, gram-negative, fungi, and mixed ones. Each of these pathogens is sensed via different TLRs and thus understandably, blocking TLR4 alone might be less efficacious.
Moving forward, the main challenges facing sepsis research are multiple. The rodent models commonly used in sepsis research are quite different from the septic patients we see in hospital. Unlike young and healthy rodents used in most laboratory research, septic patients are often old with multiple comorbidities, such as diabetes, hypercholesterolemia, hypertension, or other systemic and metabolic diseases, and are often aggressively treated with multiple medications in ICU. These underlying conditions could profoundly impact how the body responds to infection as well as to treatment. Therefore, establishing animal models that closely simulate septic patients is of paramount importance. Septic humans are highly heterogeneous in their clinical presentations of infection (sites, pathogens, severity) and responses to treatments, the underlying comorbidities, the demographics, their genetic makeup and risk factor, and their immune response to pathogen infection. Delineating these complex biological, genetic, immunological, and clinical factors in human sepsis is essential for future sepsis intervention and trial design.
Summary
Sepsis is a deadly clinical syndrome induced by a host's dysregulated immune responses to infection. Acting via pattern-recognition and converging signaling pathways with a set of adaptor molecules, kinases, and transcriptional factors, TLRs play a pivotal role in host defense against microbe pathogens by launching a proinflammatory immune response. Preclinical rodent studies have established the mechanistic role of TLR signaling in sepsis pathogenesis. Targeting these innate immune receptors and manipulating host inflammatory responses in sepsis, while attractive and logical, may yield opposite results depending on the severity of sepsis at the time of intervention. Numerous clinical trials targeting innate immunity, inflammation, and coagulation, including the eritoran ACCESS trial, have failed to demonstrate therapeutic efficacy.
Future work will be needed to better understand the complex biological mechanisms of sepsis pathogenesis, establish animal sepsis models more closely related to human conditions, identify molecular basis—biochemical and immunological risk factors and biomarkers—for clinical heterogeneity of septic patients, and carefully design human trials with clear clinical and immune stratifications and various levels of clinical outcomes.
Footnotes
Authors' Contributions
F.C. drafted the article, L.Z. and B.W. critically edited the article, and W.C. instructed the review contents and finalized the article.
Acknowledgments
We thank all the current and former trainees in our laboratories at the Massachusetts General Hospital/Harvard Medical School and the University of Maryland School of Medicine for their dedication and contributions.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported, in part, by the NIH grants—R01NS110567, R01GM117233, R01GM122908, R35GM124775, and K08HL153784, by the Frontiers in Anesthesia Research Award from the International Anesthesia Research Society, and by the Faculty Research Award from the Shock Society.