
Abstract
Significance:
Hydrogen sulfide (H2S), the third member of the gasotransmitter family, has a broad spectrum of biological activities, including antioxidant and cytoprotective actions, as well as vasodilatory, anti-inflammatory and antifibrotic effects. New, significant aspects of H2S biology in the kidney continue to emerge, underscoring the importance of this signaling molecule in kidney homeostasis, function, and disease.
Recent Advances:
H2S signals via three main mechanisms, by maintaining redox balance through its antioxidant actions, by post-translational modifications of cellular proteins (S-sulfhydration), and by binding to protein metal centers. Important renal functions such as glomerular filtration, renin release, or sodium reabsorption have been shown to be regulated by H2S, using either exogenous donors or by the endogenous-producing systems.
Critical Issues:
Lower H2S levels are observed in many renal pathologies, including renal ischemia–reperfusion injury and obstructive, diabetic, or hypertensive nephropathy. Unraveling the molecular targets through which H2S exerts its beneficial effects would be of great importance not only for understanding basic renal physiology, but also for identifying new pharmacological interventions for renal disease.
Future Directions:
Additional studies are needed to better understand the role of H2S in the kidney. Mapping the expression pattern of H2S-producing and -degrading enzymes in renal cells and generation of cell-specific knockout mice based on this information will be invaluable in the effort to unravel additional roles for H2S in kidney (patho)physiology. With this knowledge, novel targeted more effective therapeutic strategies for renal disease can be designed. Antioxid. Redox Signal. 36, 220–243.
Introduction
Hydrogen sulfide (H2
H2S-Producing Systems in the Kidney
There are three main enzymes responsible for the production of H2S in the kidney, namely (i) cystathionine-gamma-lyase (CSE), (ii) cystathionine-beta-synthase (CBS), and (iii) 3-mercaptopyruvate sulfurtransferase (3-MST) (85). CSE and CBS are expressed in the cytosol. CBS is the first enzyme in the transsulfuration pathway, catalyzing the conversion of serine and homocysteine to cystathionine and water. Cystathionine is then converted to
3-MST is present in both the cytosol and the mitochondria, where it converts 3-mercaptopyruvate (3-MP) to pyruvate while simultaneously yielding additional products, such as cysteine and glutathione (GSH) persulfides, hydropersulfides and polysulfides, and also H2S itself.
A feature of 3-MST that makes it unique at least in the kidney is that there are two alternative sources of 3-MP. One pathway uses
Rather recent publications indicate that both CSE and CBS are expressed in the mouse renal cortex and in particular in the proximal tubules (103, 201). Bos et al. (30) reported that CSE was present in glomeruli, proximal and distal tubular epithelium, and peritubular capillaries in the human kidney. 3-MST is strongly expressed in the mouse proximal tubules and to a lesser extent on the collecting ducts (182) whereas in rat renal tissues strong expression of 3-MST was found in the proximal tubules (136). Interestingly, immunohistochemical data on Human Protein Atlas indicate that CSE is moderately expressed in the proximal and distal tubules whereas 3-MST is weakly expressed in the glomeruli and moderately to strongly expressed in the proximal tubules (Fig. 1). When it comes to CBS, there is controversy about its expression in the human kidney. For example, the Human Protein Atlas (183) reports no expression whereas a recent publication indicated that CBS is strongly expressed in the human proximal tubules (206). Overall, the current published data suggest a strong expression of the three enzymes responsible for H2S biosynthesis in the rodent and human proximal tubules, possibly indicating that H2S plays an important role in water and solute reabsorption of this renal segment.

H2S Degradation Systems
H2S can be degraded or eliminated from the body by a series of enzymatic reactions. Exogenously administered sulfide (S2−) is rapidly oxidized and excreted primarily as thiosulfate (S2O2 3−) and sulfate (SO4 2−) (47, 85). Tissue differences have been noted, with liver converting sulfide mainly to sulfate, whereas the kidney converts it to thiosulfate and sulfate (17). Sulfide is oxidized to sulfite in a two-step reaction in the mitochondria (57, 85). First, sulfide is oxidized by sulfide quinone oxidoreductase (SQR) to yield a persulfide (134). The persulfide is then oxidized by persulfide dioxygenase (ETHE1), to generate sulfite (SO3 2). Sulfite is, in turn, converted to sulfate or thiosulfate by sulfite oxidase (SUOX) and rhodanese (also called thiosulfate sulfurtransferase, TST), respectively (85).
Electrons released in the SQR reaction are captured by ubiquinone and transferred to the electron transport chain, making H2S the first inorganic substrate for the respiratory chain (131). Methylation is a less important mechanism of metabolism and clearance of H2S, which unlike oxidation occurs mainly in the cytoplasm (94, 134). An alternative pathway for sulfide oxidation involves ferric heme–dependent conversion of H2S to a mixture of thiosulfate and polysulfides (130). Finally, H2S can also be metabolized into thiocyanate by a non-reversible reaction catalyzed by rhodanese (139).
Information about the enzymes involved in the mitochondrial oxidation of H2S in the kidney is very limited. SQR has been detected in the kidney (101, 214), mostly in glomerular podocytes and in tubular cells of the medulla (2). ETHE1 and SUOX have also been reported to be present in the kidney (73, 183). Rhodanese activity has been demonstrated to exist in the kidney in many species (9, 80). Its expression in bovine tissue sections was highest in the proximal tubule cells (173), whereas in rats strong expression was documented in tubular cells; however, the glomeruli are moderately stained (180). Impaired rhodanese expression in monocytes was associated with increased global levels of reactive oxygen species (ROS) in the cells, as well as higher mitochondrial superoxide production and predicted mortality in patients with hemodialysis (97). In addition, chronic elevation in serum sulfite levels was proposed to contribute to tissue or organ dysfunction in patients with chronic renal failure (86). A feedback loop between H2S-producing and H2S-degrading enzymes has been reported and it is tissue specific. The 3-MST knockout (KO) mice have significantly higher levels of liver TST, whereas the opposite happens in their heart (137, 148). How the activity and expression of one H2S-producing enzyme affects the levels of H2S-degrading enzymes in renal tissue is unknown. It has been shown in neural tissues that the accumulation of sulfite (a neurotoxic agent leading to encephalopathies) could be due to insufficiency of H2S catabolic pathways such as those described earlier (92). Interestingly, sulfite has been shown to induce ROS accumulation and toxicity in renal cells derived from various species in vitro and patients on dialysis have significantly elevated levels of plasma sulfite (186). Whether this is linked to an impaired ability for H2S catabolism remains to be elucidated. Given the paucity of data for the biological roles of H2S-degrading enzymes in the kidney, the field would benefit from studies aiming at elucidating how SQR, ETHE1, SUOX, and TST affect renal (patho)physiology.
Mechanisms of H2S Signaling
At the molecular level, H2S and other more oxidized H2S-derived species called polysulfides (H2Sn) are able to modify the protein structure and function through S-sulfhydration (also termed persulfidation). S-sulfhydration involves the conversion of a cysteine thiol group (-SH) to a persulfide (-SSH) group. This post-translational modification alters the activity of the modified protein (20). It should be noted that H2S cannot directly interact with cysteine thiols to yield protein persulfides, whereas polysulfides are capable of this reaction. H2S acts on an oxidized thiol of a cysteine residue, for example S-sulfenylated or S-nitrosylated residues to yield persulfides (57). Both stimulatory and inhibitory effects of H2S-mediated persulfidation on protein activity have been described. For example, persulfidation has an inhibitory effect on Keap-1, which allows the release, nuclear translocation of the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf-2), and a higher messenger RNA (mRNA) expression of antioxidant enzymes in mouse embryonic fibroblasts (203). On the contrary, H2S is able to persulfidate and activate MEK1 on cysteine 341, which, in turn, leads to attenuated DNA damage in human endothelial cells (210).
A second mechanism by which H2S signals is the reaction/quenching of reactive oxygen and nitrogen species to form other signaling molecules (20). This mechanism is discussed in more detail later.
Finally, the third mechanism by which H2S signals is via binding to metal protein centers and altering protein or enzyme activity. In particular, H2S binds and inhibits hemeproteins such as the cytochrome c oxidase or the human hemoglobin and myoglobin (151). Nothing is known about this mode of signaling in the kidney warranting further investigation. The main mechanisms via H2S signals at the molecular levels are depicted in Figure 2. Later, we discuss in more in detail what is known regarding these different mechanisms of H2S signaling in the kidney with special emphasis on: (i) the protein S-sulfhydration pattern in the kidney based on additional bioinformatic analysis that we performed on data published earlier this year by Bithi et al. (27); (ii) the interaction between H2S and the ROS and reactive nitrogen species (RNS) producing systems in the kidney.
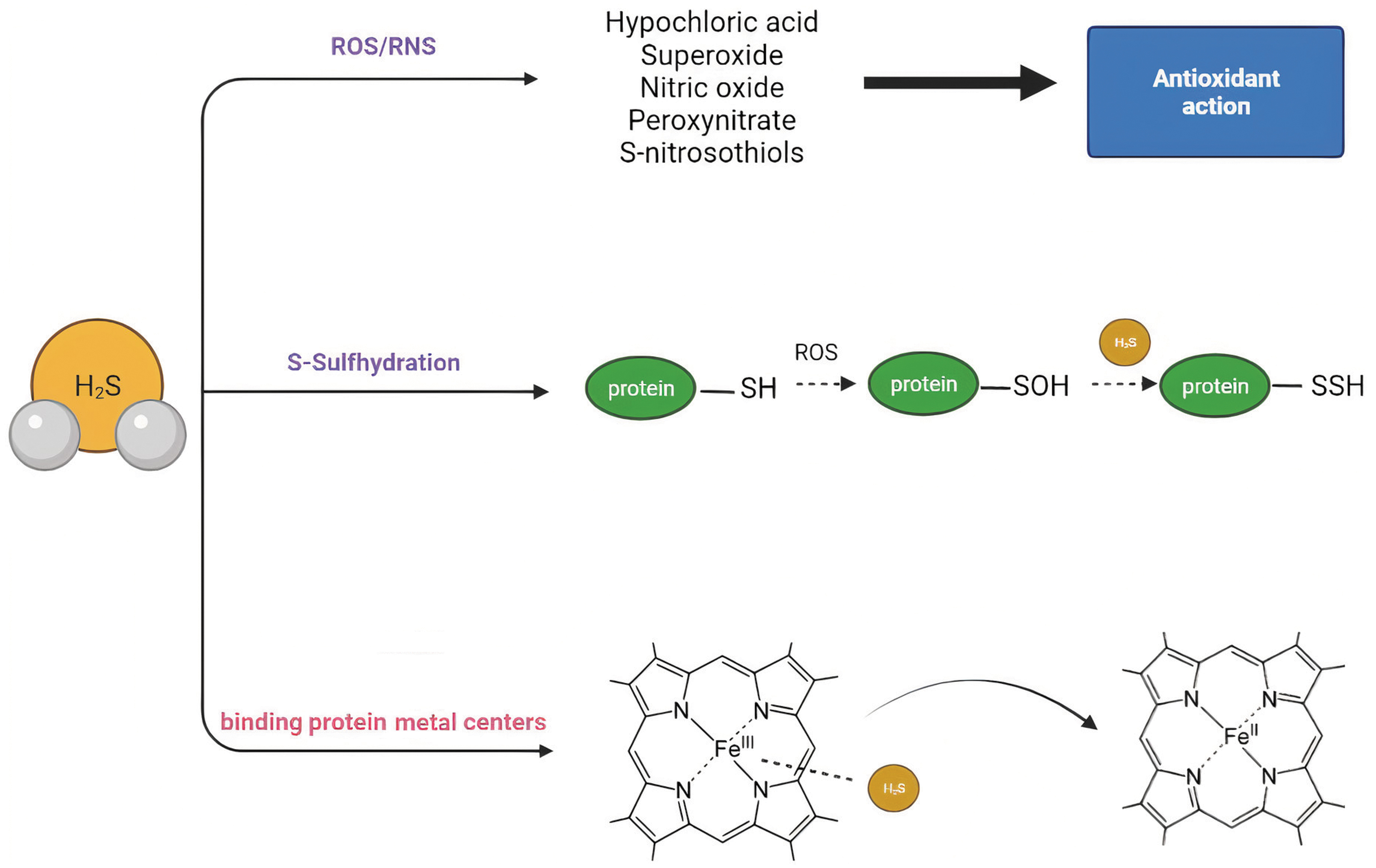
Persulfides, Polysulfides and Interacting Systems
H2S metabolism leads to the formation of oxidized sulfide species that belong to the broader family of what is termed as “bound sulfur” consisting mainly of polysulfides [R-(S*)n-S-R] and persulfides (R-S-S*n −) where R could be any inorganic or organic molecule or even a whole protein (63). The term “bound sulfur” refers to sulfur atoms covalently bound with other sulfur atoms that can easily release back H2S in a reduction reaction in the presence of a thiol-reducing agent; polysulfides refer to the inner [-(S*)n-S] part of bound sulfur, and persulfides refer to the outer (-S-S*n −) part (93).
For many years, it was believed that “bound sulfur” were inactive forms of sulfur; however, growing evidence indicates that it is rather the opposite with many of the biological effects of H2S to be actually due to persulfide or polysulfide activated signaling pathways and not H2S per se (205). Moreover, the formation of persulfides on cysteine residues in proteins has been shown to protect cysteine from irreversible oxidative loss of function by the formation of Cys derivatives that can subsequently be reduced back to native thiols (52).
Therefore, studying the mechanisms leading to persulfide and polysulfide formation is of great importance for understanding better physiological and pathological phenomena in biological systems. The biological role of persulfides and polysulfides in the kidney is largely unexplored, except for a recent study by Bithi et al. (27), which is presented later in more detail. We find it also useful to explain some of the main known mechanisms controlling the production of persulfides and polysulfides in other biological systems with the hope that in the near future the existence and importance of these mechanisms will be also investigated in the kidney. Some of these mechanisms involve:
The enzyme 3-MST and reducing agents (thioredoxin-1, GSH, Cys)
The enzyme 3-MST has a much more complex biochemistry than the mere production of H2S and it has been actually shown to be able to produce Cys-SSH and GSSH in the presence of reducing agents such as of cysteine and GSH, which can play an important role as cellular antioxidants (91). Moreover, 3-MST releases H2S and polysulfides by using 3-MP and reducing agents such as the antioxidant protein thioredoxin-1 (Trx-1) and the dihydrolipoic acid (DHLA) (128).
The enzyme cysteinyl-tRNA-synthetase 2
The persulfide Cys-SSH has been shown to play a crucial role in maintaining mitochondrial biogenesis and cellular bioenergetics and its production heavily relies on the mitochondrial enzyme Cysteinyl-tRNA-synthetase 2 (CARS-2) (7). CysSSH is reductively metabolized to CysSH and HS−. A recent line of research supports the fascinating notion that CARS-2 is an important persulfide synthase that is functionally coupled to protein translation (since it is a transfer RNA [tRNA]-synthetase) and at the same time regulates cellular bioenergetics in mammalian cells (60). Another exciting aspect indicating a coupling between polysulfide production, protein translation, and metabolism is the fact that 3-MST, which is another important poly- and persulfide generator, has also tRNA thiolase activity (58). The role of tRNA thiolation is not fully understood but Damon et al. suggested that tRNA thiolation links translation to stress responses in Saccharomyces cerevisiae (48). Whether a similar mechanism exists in mammals and the functional significance that this might have in the kidney remains to be investigated.
Direct interaction with NO species
There is a significant degree of a direct cross-talk between NO and H2S since according to Cortese-Krott et al. there is a network of cascading chemical reactions that form simultaneously and not independently: nitropersulfide, polysulfides, and dinitrosulfide. These molecules are then able to scavenge, release, or transport NO/nitroxyl (HNO) or sulfide and sulfane sulfur and possibly exert significant biological effects (45).
Overall, not only H2S but also its sulfane sulfur derivative molecules (persulfides and polysulfides) control fundamental biochemical processes, cellular redox homeostasis, metabolism, and mitochondrial function. Therefore, one should consider not only ROS and RNS as important signaling mediators but also the reactive sulfur species, all of which form an interdependent network with multiple levels of interactions (44).
S-Sulfhydration of Proteins in the Kidney and Potential Biological Significance
Only a limited number of studies have determined the identity of S-sulfhydrated proteins in intact organs. Recently, Bithi et al. (27) compared protein S-sulfhydration in the mouse liver, kidney, heart, muscle, brain, and plasma in young and aged mice under control conditions and after dietary restriction. Based on the published data by Bithi et al. we performed a separate analysis of the sulfhydration pattern in the kidney comparing young vs aged mice (summarized in Figures 3 –5). Overall, Bithi et al. found that dietary restriction triggered an upregulation on protein S-sulfhydrated in the kidney that was CSE-dependent.



In young mice (6 weeks old), 1086 S-sulfhydrated proteins were identified in whole kidney extracts. Many of the S-sulfhydrated proteins determined are involved in metabolic pathways, including amino acid biosynthesis and degradation, pyruvate metabolism, carbon metabolism, fatty acids oxidation, tricarboxylic acid cycle, glycolysis/gluconeogenesis, sulfur metabolism, and GSH metabolism. In addition, proteins involved in peroxisome structure, proteasome, and PPAR signaling were found to be S-sulfhydrated. Interestingly, many of the S-sulfhydrated proteins were compartmentalized in the mitochondrial matrix and the lumen of organelles. Moreover, the main enzymes that are responsible for H2S production (CBS, CSE, CAT, 3-MST) and H2S catabolism (SQR, ETHE1, TST) were also S-sulfhydrated.
In old mice (20 weeks old), 1211 S-sulfhydrated proteins were identified in the kidney. Similarly, most of the S-sulfhydrated proteins identified are involved in metabolic pathways.
In an initial attempt to document age-related differences in the kidney sulfhydrome under physiological conditions, we compared the S-sulfhydrated proteins in young and old mice. Data mining of the supplementary material of the published article revealed 199 proteins that were S-sulfhydrated only in the kidney of young mice and 324 proteins that were S-sulfhydrated only in the kidney of old mice, whereas a total of 887 proteins were present in both age groups. These observations suggest that there is a change in the kidney sulfhydrome that occurs with aging. The 199 unique S-sulfhydrated proteins in the young mouse kidney were associated with metabolism, biosynthesis of cofactors, and oxidative phosphorylation.
Interestingly, some of individual proteins are NAD dehydrogenases, enzymes with biological roles associated with the respiratory electron transport chain, and thermogenesis. Analysis of 324 unique proteins in aged mouse kidneys showed significant correlation with 2 pathways, the splicosome and endocytosis pathways. Thirteen proteins are associated with splicosome function and they are involved in RNA polymerase function, mRNA splicing, post-transcriptional modifications, and prevention of exon skipping; 14 proteins are associated with endocytosis, cellular membrane trafficking pathways, and transportation through biological membranes.
Also, proteins associated with kidney fibrosis (transforming growth factor beta 1 [TGF-β1], THBS1) were S-sulfhydrated only in aged mice. In contrast, 3-MST was the only enzyme from H2S-producing enzymes that was not persulfidated in the aged mice.
Overall, examination of the total number of S-sulfhydrated proteins in the kidney indicates that H2S signaling plays an important role in renal physiology via multiple pathways, many of which are related to metabolic processes. This is in agreement with observations that H2S signaling plays a key role in cellular bioenergetics in other tissues and organs (1, 15, 131, 132). Additional experiments are needed to prove the functional significance of S-sulfhydration of individual cysteine residues in the proteins of interest. Later, we discuss the protective role of H2S in pathophysiological conditions through S-sulfhydration of specific molecular targets.
Expression Pattern and Signaling of ROS-Producing Systems in the Kidney
The ROS and RNS are reactive molecules that contain oxygen and/or nitrogen, with a widespread production in living organisms. They are being continuously produced in vivo both enzymatically and non-enzymatically and many of them participate in biochemical reactions and contribute to biological responses (54). However, when overproduced, cellular damage can occur (59). In the kidney, ROS/RNS are important signaling molecules and their levels are tightly regulated; excessive production of ROS/RNS has been linked to many renal pathologies such as chronic kidney disease (CKD), acute kidney injury (AKI), ischemia–reperfusion injury (IRI), diabetic and obstructive nephropathy, and many others (155).
The main type of ROS or other oxidant molecules that play a detrimental role in the kidney are the free radical superoxide and the oxidant hydrogen peroxide. When it comes to RNS, these molecules are derived from the free radical NO and include species such as the nitrogen dioxide [
The ROS/RNS are generated during physiological and pathophysiological conditions and their production can be enhanced by exogenous stimuli, such as smoking, alcohol, certain drugs, air pollution, ionizing radiation, and hyperbaric oxygen poisoning. The endogenous sources consist mainly of complex enzymatic systems and overall they play a more important role in the formation of ROS/RNS.
Some important endogenous sources of ROS in the kidney are the following: (i) nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), (ii) mitochondria, and (iii) xanthine oxidoreductase (XOR).
NOX-derived ROS in the kidney
The NOXs, along with the mitochondria, are the major producers of superoxide and are the principal sources of abnormal cellular signaling in the kidney (141, 161). The NOXs transfer electrons from NADPH, and these electrons react with oxygen to form the free radical superoxide, which is usually rapidly converted to hydrogen peroxide. In the kidney, there are four different isoforms of NOXs, called NOX1, NOX2, NOX4, and NOX5 (13). The expression of these different NOX isoforms has been observed throughout the cortex and medulla of the mammalian kidney, and in particular areas such as in the mesangium, the proximal convoluted tubules, the distal convoluted tubules, the collecting duct, the macula densa cells, and, finally, the renal blood vessels (endothelium and vascular smooth muscle cells) (13). NOX4 has the highest expression in the kidney (62), producing large amounts of hydrogen peroxide constitutively (62).
It has been suggested that the NOX4 isoform is also localized in the mitochondria and not only in the cytosol of the rat kidney cortex (28). Moreover, it has been shown that NOX activity and especially that of the isoform NOX2 can be stimulated by angiotensin II (Ang II) (72) and proinflammatory cytokines such as interferon gamma (IFNγ) (140). Finally, recent reports indicate that the NOX5 isoform may also influence tubular physiology and contribute to glomerulopathies (74).
Mitochondrial ROS
Under normal physiological conditions, the renal mitochondria produce low levels of ROS that participate in signaling processes. In contrast, under pathological conditions, signaling molecules such as Ang II, tumor necrosis factor alpha (TNF-α), high glucose, or ROS from other sources (i.e., NOX) create a positive feedback mechanism that further reinforces the production of ROS from the mitochondria (181). Most of the superoxide anions produced in the renal mitochondria are produced by complex I and complex III of the electron transport chain (155). The levels of mitochondrial-derived superoxide are usually kept low by converting superoxide to hydrogen peroxide via manganese-dependent superoxide dismutase (Mn-SOD; SOD2) in the mitochondrial matrix. This enzyme is crucial for the maintenance of mitochondrial ROS balance and when downregulated, oxidative stress is manifested.
Ablation of superoxide dismutase (SOD) 2 in aged mice leads to accelerated renal cellular senescence and enhanced tubular damage, glomerular sclerosis, and renal interstitial inflammation (157). When ROS generation in the mitochondria continues, uncoupling protein-2 (UCP2) accumulates, which, in turn, triggers an inward proton leak and lower adenosine triphosphate (ATP) production. The impaired ATP production then leads to impaired ATP-dependent reabsorption in renal tubular epithelial cells. When these phenomena are severe enough, the renal tubular epithelial cells undergo autophagy, apoptosis, or necrosis (155).
XOR in the kidney
XOR is generally known as the final enzyme involved in purine metabolism and as a source of ROS in the oxidase form, whereas in the reductase form it can also produce NO via reduction of inorganic nitrate and nitrite (150). We and others have shown that XOR is ubiquitously and very highly expressed in the kidney, implying the very important role that this enzyme plays in renal physiology (96, 143, 149).
In a hallmark study, Ohtsubo et al. showed that XOR is essential for kidney development via a mechanism that involves the enzyme cyclooxygenase-2/COX-2 (143). In particular, XOR KO mice have severe renal dysplasia and die at a very young age. Later studies from the same group revealed that XOR depletion induces renal interstitial fibrosis through aberrant lipid and purine accumulation in the renal tubules (142).
When it comes to renal disorders, excessive elevation of XOR-derived ROS can also be problematic. Early studies have suggested that XOR produces superoxide in post-hypoxic injury of renal epithelial cells or that XOR-derived ROS contribute to renal ischemic injury (67, 147). More recently, we have also shown that there is an elevation of renal XOR-derived ROS in a rat model of renal hypertension and that this elevation could possibly contribute to the development of the hypertensive phenotype (149).
Interplay Between H2S and ROS, RNS
H2S has been shown to play a major role in regulating the levels of ROS and RNS and there is often a significant degree of cross-talk between the systems producing ROS/RNS and H2S in the body (57, 198). For example, H2S can activate the regulator of redox homeostasis Nrf-2 (122). H2S sulfhydrates the Nrf-2 inhibitor Kelch-like ECH-associated protein 1, allowing the translocation of Nrf-2 to the nucleus where it promotes the transcription of antioxidant enzymes (33, 87, 203). Moreover, H2S can exert direct antioxidant properties since it can readily react with the superoxide anion at rates higher than classic antioxidants, such as GSH and cysteine (194). It can react with peroxynitrite, hydroxyl radicals, and hydrogen peroxide (37, 46, 129); however, the physiological significance of these reactions is questionable, since H2S is present in much lower concentrations compared with other cellular antioxidant systems (23). In addition, H2S-producing enzymes have been shown to participate in redox cycling and to contribute to the detoxification from cellular oxidants (135). CSE and CBS are members of the transsulfuration pathway that converts methionine to cysteine, which is required for GSH production, and H2S itself can increase GSH synthesis (85, 90, 174, 191).
The 3-MST can also produce persulfidated species, which have known redox actions (91). Moreover, the H2S-producing enzymes can change the sulfhydration status and the activity of key proteins involved in redox balance, for instance, the Mn-SOD (215); exogenously applied H2S was shown to reduce NOX4 expression (66).
An interplay between XOR and H2S has been recently reported. In a study published last year, H2S and polysulfide donors were able to activate XOR and increase the bioconversion of inorganic nitrite to NO. This H2S→XOR→NO axis seemed to also trigger an increase in vasodilation and in blood flow (146). Considering the fact that renal XOR and bioconversion of nitrite to NO plays a key role in alleviating hypertension in rodent models (61), one could hypothesize that such a positive feedback loop between H2S and XOR also exists in the kidney.
The free radical NO is very selective in its reactive properties and it can go on to form other RNS, including peroxynitrite, which is generated by a reaction of NO with superoxide. The RNS are often observed to interact with cellular components, including thiol groups and unsaturated fatty acids, to promote regulation through processes such as thiol (RSH) or lipid oxidation or other modifications, including nitrosation (RSNO) or nitration (RSNO2) (50). Although some of these interactions are a part of signaling, as RNS levels increase, they actively contribute to pathological processes in the cardiovascular and renal systems.
There are multiple levels of interaction between the gasotransmitters NO and H2S (138). Although, it is well established that NO is required for the angiogenic, vasodilatory and cardioprotective actions of H2S (24, 26, 43, 95), the NO-H2S cross-talk in the kidney has been investigated in only a few studies. Exogenous H2S has been shown to increase endothelial nitric oxide synthase (eNOS) expression and downregulate caveolin-1 levels, facilitating NO production (152). Shirazi et al. demonstrated that the H2S donor sodium hydrosulfide (NaHS) upregulated eNOS expression, whereas it reduced inducible nitric oxide synthase (iNOS) levels in CKD (166); in this model, the renoprotective effects of NaHS were blocked by
Similar results demonstrating eNOS upregulation and iNOS downregulation in response to exogenously administered H2S were obtained in a renal ischemia–reperfusion injury model (79). These findings indicate the existence of a positive interplay between H2S and the constitutive NO synthases, which produce moderate amounts of NO that are required to maintain renal physiological function, whereas H2S interferes which the excessive amounts of NO that trigger nitrosative stress and inflammation in the kidney (65). Apart from the indirect interactions of H2S with the NO-producing enzymatic systems described earlier, there is also a direct interaction. Filipovic et al. demonstrated that the clinically used NO donor sodium nitroprusside (SNP) exerts its beneficial effects by reacting with H2S to generate HNO, a molecule with multiple cardiovascular actions (56).
H2S Donors and Biosynthesis Inhibitors
The increased interest for the biological roles of H2S along with ample evidence for the involvement of H2S in many pathological conditions has led to the development of pharmacological agents that modulate its endogenous levels. There are two main pharmacological approaches, which are based on either (i) increasing H2S levels in biological systems through molecules that are capable of releasing H2S and designated as H2S donors or (ii) inhibiting endogenous H2S production usually through inhibition of one or more of the three H2S-producing enzymes (145).
H2S donors constitute a diverse pharmacological category. There are differences in structure, release rate, mechanism of H2S release, and tissue/subcellular distribution. According to release rate, H2S donors are traditionally classified into fast- and slow-releasing donors. The fast-releasing donors include inorganic salts of H2S, such as NaHS and Na2S, that rapidly dissociate to yield a burst of H2S; however, the slow-releasing donors, with (p-methoxyphenyl)morpholino-phosphinodithioic acid (GYY-4137) being the first-in-class such donor, deliver smaller H2S concentrations over a prolonged period of time. The advantage of the latter category is that these agents yield H2S in a manner that better reflects endogenous production of H2S and protects against possible exposure to toxic concentrations of H2S (177).
Further, various donors with regulated H2S release profiles have been identified, including redox-activated H2S donors, pH-controlled donors, and thiol- and esterase-activated donors (177). These donors, however, have seldom been applied in vivo, and to the best of our knowledge there are no kidney-related studies using these agents. An approach that has attracted significant interest is the synthesis of organelle-targeting compounds, exemplified by donors that preferentially distribute into the mitochondria. [10-Oxo-10-[4-(3-thioxo-3H-1,2-dithiol-5yl)phenoxy]decyl]triphenyl-phosphonium (AP39) is the prototype agent of this class (178).
Another agent worth mentioning is SG1002, an orally bioavailable drug that was tested for safety and tolerability in Phase I clinical trials (64). An interesting approach that has been used by several investigators is coupling of a pharmacophore moiety found in clinically approved drugs to an H2S-donating group to improve efficacy or to reduce the side effects of the original compound (177, 211). The most advanced drug in this class is otenaproxesul (ATB-346), a naproxen-thiobenzamide conjugate. Otenaproxesul has completed Phase 2 clinical testing, and it has shown efficacy along with a marked reduction of GI toxicity (188, 189). Otenaproxesul is scheduled to begin a Phase III trial this year.
Several compounds are available to inhibit endogenous H2S synthesis; however, the majority lack potency and/or selectivity (177, 195). By far, the most frequently used agents are aminooxyacetic acid (AOAA) and propargylglycine (PAG), which were initially described as selective CBS and CSE inhibitors (14, 145). However, AOAA can inhibit direct or indirect H2S production from all three enzymes. AOAA has the ability to inhibit many pyridoxal 5′-phosphate (PLP)-dependent enzymes through direct binding to PLP and is more potent against CSE than CBS, at least in vitro (14).
AOAA is a non-selective transaminase inhibitor (156), so it might also block CAT, which provides 3-MST with its substrate 3-MP. It has, thus, the potential to serve as an indirect inhibitor of 3-MST. A series of structurally related selective, direct 3-MST inhibitors is available that all bear an aromatic ring carbonyl-S-pyrimidone structure (71). Given the drawbacks of currently available H2S synthesis inhibitors (105), studies aiming at unraveling the role of endogenous H2S requires a combination of approaches in addition to pharmacological inhibition, such as genetically modified animals or knockdown approaches (small interfering RNAs [siRNAs]).
H2S in Renal Physiology
Glomerular filtration, reabsorption, and secretion
Several groups have shown that endogenously produced H2S regulates water and electrolyte balance. Dual inhibition of CSE and CBS by intrarenal infusion of AOAA/PPG decreased glomerular filtration rate (GFR), urinary sodium and potassium excretion (197). The effects of H2S on electrolyte handling include both vascular and tubular effects, with the later resulting from inhibition of the Na+/K+/2Cl− cotransporter and attenuation of the Na+/K+/ATPase activity (197).
Endogenously produced H2S also affects water handling. Inhibition of CBS and CSE was reported to lead to increased urine output and reduced urine osmolality (121). These changes occurred concomitantly with downregulation of aquaporin-2 (AQP-2) expression in the inner medulla. Moreover, although unilateral nephrectomy in mice does not affect GFR, increased GFR and proteinuria after nephrectomy is observed in mice lacking one CBS allele (162, 163). These findings highlight the importance of CSE- and CBS in basic renal functions.
Renal blood flow
Inhibition of CSE and CBS for 4 weeks was shown to reduce cortical, medullary, and total renal blood flow in rats (160), whereas intrarenal arterial infusion of NaHS increased renal blood flow (197). Moreover, prolonged inhibition of CSE by PAG in conscious animals significantly decreased blood flow in the renal artery, further confirming that H2S is an important regulator of vascular resistance in the kidney under physiological conditions (133).
In addition, NaHS causes a characteristic redistribution of blood flow in normal rats, reducing renal blood flow in a dose-dependent fashion; however, opposite results were observed in rats that were subjected to cecal ligation and puncture, suggesting that the effects of H2S in the renal circulation are context-dependent (4). The slow-releasing H2S donor GYY-4137 has been demonstrated to reverse the reduction in renal blood flow caused by Ang II (193).
The renin-angiotensin-aldosterone system
The renin-angiotensin-aldosterone system (RAAS) is a key hormone system regulating blood pressure, electrolyte and fluid balance. H2S is known to affect various components of the RAAS system. H2S inhibits angiotensin-converting enzyme activity (100) and increases the methylation of the AT1b gene, reducing AT1 receptor expression (68). H2S also decreases the synthesis and release of renin (119). In line with these findings, CSE overexpression attenuates isoproterenol-induced renin release (119, 120) and pharmacological inhibition of CSE in renal mesangial cells, results in increased angiotensin 1 receptor and ACE expression (200). Overall, H2S suppresses or counteracts the biological effects of the RAAS.
Erythropoiesis
Hypoxia, the lowering of blood oxygen levels, is known to trigger erythropoietin (EPO) production from the peritubular fibroblasts in the kidney cortex to promote proliferation and differentiation of erythrocytic progenitors (83). Several lines of evidence have implicated endogenously generated H2S in EPO production and erythropoiesis. Inhibition of H2S attenuated the hypoxia-induced increase in EPO expression in vitro through downregulation of hypoxia-inducible factors (106). Similarly, CSE KO mice placed under hypoxia displayed reduced renal EPO levels and hemoglobin production (105). Interestingly, EPO levels were elevated in normoxic conditions in CSE KO mice (105), suggesting that CSE-derived H2S differentially affects EPO production depending on oxygen availability.
H2S in Renal Pathophysiology
The importance of endogenous H2S in kidney physiology explains its crucial role in renal diseases. Deregulation of endogenous H2S production and signaling has been reported in several pathological states related to the kidney. Below, we highlight the involvement of H2S in acute and chronic kidney injury, kidney cancer, and diabetes insipidus (DI). A schematic representation of how H2S endogenous production is altered in renal pathophysiological conditions is given in Figure 6. Moreover, a schematic representation of the molecular mechanisms mediating the beneficial effects of H2S donors in kidney injury and renal fibrosis is given in Figure 7.


Acute kidney injury
AKI or acute renal failure is characterized by rapid loss of the kidney's excretory function and is typically diagnosed by the accumulation of end products of nitrogen metabolism, such as urea and creatinine and/or decreased urine output. It is the clinical manifestation of several disorders that affect the kidney acutely (18). Below we discuss the role of H2S in pathological conditions that lead to renal failure, specifically kidney ischemia–reperfusion injury, obstructive nephropathy, pharmacological nephrotoxicity, and sepsis.
Ischemia–reperfusion injury
The role of endogenous H2S production and exogenous H2S supplementation in ischemia–reperfusion injury has been extensively studied in the heart (53, 109). H2S has been shown by us and others to be cardioprotective; the beneficial actions of H2S are mediated by attenuating ROS levels, reducing apoptotic and inflammatory responses, and inhibiting calcium overload, following both NO-dependent and NO-independent pathways (11, 26, 38, 148).
In the kidney, mRNA levels and protein expression of CBS and CSE, as well as endogenous levels of H2S have been shown to be decreased in ischemia–reperfusion injury (69, 199). It has been proposed that the reduction of kidney CBS during ischemia–reperfusion injury is mediated by a decrease in transcription factor Sp1 (Sp1) transcriptional activity (196). In addition, CSE KO mice are characterized by exacerbated renal damage and mortality after renal IRI, which could be due to enhanced production of ROS. Similarly, inhibition of CSE by PAG leads to increased lethality after IRI in mice (30). Treatment with NaHS rescued CSE KO or PAG-treated mice from the injury associated with renal ischemia (30, 69).
In contrast, Marko et al. reported that global CSE deficiency does not deteriorate renal injury, but rather ameliorates the injury, probably due to a reduced pro-inflammatory response (125). It should be noted that in this latter study, an independently generated CSE KO line was used (179), which unlike the most commonly used CSE KO created by the Snyder group (202) is not hypertensive. A recent in vitro study using renal proximal tubular epithelial cells demonstrated that CBS, CSE, and 3-MST expression was increased during prolonged anoxia, but it returned to baseline after reoxygenation. Inhibition of H2S production by AOAA during anoxia increased the apoptotic markers cleaved: caspase-3, tumor protein P53 (p53), and bcl-2 (B-cell lymphoma 2)-like protein (Bax) (55).
The administration of H2S donors before or after ischemia acts beneficially in renal IRI. The majority of these AKI studies have been performed by using inorganic fast-releasing donors. NaHS administration accelerated the recovery of renal function and tubular morphology (69). NaHS also improved kidney function after IRI (4, 112, 168). Several mechanisms have been proposed to mediate the pharmacological effects of exogenous H2S in kidney IRI, with antioxidant pathways being the most prominent. Treatment with NaHS reduced renal malondialdehyde (MDA) concentration, lipid peroxidation, and NOX4 expression (16, 69); increased renal antioxidant enzymes, such as SOD and catalase (16); and increased the levels of reduced GSH (69).
Similarly, the administration of AP39, a mitochondrial-targeted H2S donor, exerted dose-dependent protective effects during renal IRI and inhibited MDA levels (4). Additional protective responses triggered by NaHS involve anti-inflammatory and antiapoptotic pathways (168). NaHS was shown to reduce neutrophil infiltration and cytokine production. NaHS can also upregulate autophagy induced by endoplasmic reticulum stress, via the scavenger receptor A to protect against IRI (112).
IRI is clinically important in the context of renal transplantation, as it can significantly impact graft function and survival and is unavoidable in the peri-transplant period. NaHS drastically increased the survival of renal transplant recipient rats; it also improved syngeneic graft function and decreased graft injury. These effects were associated with reduced expression of apoptotic and inflammatory markers (118). However, most human transplants are allogeneic and thus complicated by the innate and adaptive recipient immune responses, resulting in greater graft injury compared with syngeneic transplantation.
A follow-up study by the same group investigated the role of an H2S donor in mitigating renal IRI in a murine model of allogeneic transplantation after cold organ storage (117). Similar to the syngeneic transplants, treatment with NaHS improved early allograft survival and function, decreased renal injury, and resulted in early levels of necrosis and apoptosis compared with the University of Wisconsin preservation solution alone; however, it did not affect allograft rejection. The effects of the H2S donor coincided with altered gene expression related to renal injury, coagulation, cell stress response, and cellular proliferation. Using microarray analysis, Vuillefroy de Silly et al. demonstrated that CSE and several members of the transsulfuration pathway were downregulated in rat kidney allografts 4 months after transplantation in recipients that received a tolerizing regimen (187).
CSE downregulation occurred early and lasted for at least 3 months compared with syngeneic grafted organs. Although the effect of CSE on kidney graft rejection was not tested, treatment with the CSE inhibitor PAG prevented cardiac graft rejection. Inhibition of CSE presumably participates in the maintenance of the tolerant state by inhibiting interleukin (IL)-12 production. The earlier mentioned results highlight how both H2S donation and H2S synthesis inhibition could be beneficial in different settings. H2S donors used ex vivo on the donor organ can improve allograft function, whereas the inhibition of CSE modulates the immune response to prevent graft rejection.
Nephrotoxicity mediated by pharmacological agents
Many of the cancer chemotherapeutic agents are nephrotoxic. One of the best studied nephrotoxic chemotherapeutic drugs is cisplatin, which causes AKI in 20%–30% of cases (22, 123, 124). It is well known that cisplatin-induced nephrotoxicity involves increased ROS production, inflammation, and renal tubular cell death (12, 124, 144). Given that H2S can ameliorate oxidative stress and inflammation, several groups have investigated the therapeutic potential and the underlying mechanisms of H2S supplementation in chemotherapy-induced nephrotoxicity.
An early study reported that cisplatin administration upregulates the CSE expression in a rat model. When PAG was co-administered with cisplatin, upregulation of CSE was abolished and renal damage was reduced by suppression of inflammation and apoptosis (49). In contrast, subsequent research suggested that CSE (35) or both CSE and CBS levels (113) are decreased by cisplatin treatment. The administration of NaHS reduced oxidative stress, improved kidney function, and ameliorated structural damage caused by cisplatin (3).
Mechanistically, NaHS was shown to activate sirtuin (SIRT) 3 via S-sulfhydration, which leads to improved mitochondrial function and decreased oxidative injury (207). In contrast, results with the slow-releasing donor GYY-4137 have yielded contradictory results. Liu et al. showed that GYY-4137 aggravates cisplatin-induced renal damage by increasing inflammatory response (114). However, a more recent publication demonstrated that the H2S donors NaHS and GYY-4137 mitigated cisplatin-induced nephrotoxicity. The mechanisms underlying these protective effects were related to oxidative stress, as H2S donation suppressed the intracellular ROS by inhibiting NOX activity, which might be related to neutrophil cytosolic factor 1 (p47phox) persulfidation (36).
Polysulfides were also shown to limit cisplatin-induced toxicity. Treatment with polysulfide donor Na2S4 exerted its protective effects by inhibiting ROS/mitogen-activated protein kinase (MAPK) signaling, promoting Nrf-2 translocation into the nucleus, and reducing NOX activation (35). In a follow-up study by the same group, Na2S4, NaHS, and GYY-4137 decreased the production of inflammatory cytokines and ameliorated renal inflammation caused by cisplatin. Mechanistically this effect was linked to S-sulfhydration of signal transducer and activator of transcription (STAT) 3 and the nuclear factor kappa-B (NF-κB) inhibitor (IKK-β), which reduced their phosphorylation, lowering pro-inflammatory gene expression (172).
Obstructive nephropathy
Obstructive nephropathy is a type of renal injury caused by obstruction of the urinary tract. Ureteral obstruction results in renal fibrosis and this, in turn, leads to chronic nephropathy (29).
In a rat model of unilateral ureteral obstruction (UUO), endogenous H2S production was reduced after the ablation of CBS expression (169). Another study in mice demonstrated that kidneys from wild-type mice have reduced expression of CBS, as well as CSE and 3-MST (70). Moreover, CSE gene deletion exacerbates UUO-induced fibrosis and oxidative stress in the kidney and triggers mitochondrial damage and tubular cell apoptosis (70).
Conversely, H2S therapy attenuates renal fibrosis and improves renal morphology and function after UUO (111, 169, 213). The administration of NaHS reduced the renal injury and inflammation caused by ureteral obstruction and restored redox homeostasis (84). Τhe protective action of NaHS was shown to result from the ability of NaHS to evoke a reduction of excessive autophagy by inhibiting the ROS-adenosine monophosphate-activated protein kinase (AMPK) pathway after UUO, whereas NaHS itself could also enhance the endogenous H2S by CSE and CBS (39). The anti-inflammatory effects of NaHS were attributed to the suppression of the NLR family pyrin domain containing 3 (NLRP3) inflammasome activation and to the reduced activation of NF-κB and IL-4/STAT6 signaling (213).
Slow-releasing H2S donors have also been tested in UUO. Treatment with GYY-4137 restricted cortical loss, inflammatory damage, and tubulointerstitial fibrosis in a rat model of obstructive nephropathy (111). It was proposed that the GYY-4137 accelerated recovery of renal function and the mitigation of fibrosis were mediated by interfering with the TGF-β1 pro-fibrotic signaling pathway, as GYY-4137 increased Smad7 expression in the kidney, which, in turn, led to decreased expression of TGF-β1 receptor II (110).
Sepsis-associated kidney injury
Sepsis is the most common cause of AKI, with a frequency of 40%–50% among patients with AKI (19). In a rabbit model of urinary-derived sepsis, a single dose of NaHS attenuated kidney injury, as determined by decreased glomerular deformation, tubular epithelial swelling, and tubulointerstitial infiltration of inflammatory cells (40). In addition, plasma creatinine and blood urea levels were decreased, indicating improved kidney function. These pharmacological effects were accompanied by a downregulation of pro-inflammatory markers and upregulation of the anti-inflammatory cytokine IL-10.
Further, in a swine model with septic AKI with underlying coronary artery disease, a marked drop in CSE expression was observed that correlated with decreased glomerular filtration (127). In a study that investigated the role of H2S in kidney injury in a human cohort, free plasma H2S concentration in patients with sepsis-associated acute kidney injury (SA-AKI) was found to be reduced. This reduction was inversely correlated with plasma markers of renal injury (creatinine and urea nitrogen) (41). However, these findings should be interpreted with caution since free sulfide levels are difficult to be determined in biological samples, because of the presence of a large biological pool of sulfide, bound to a variety of biomolecules (51).
Therefore, future studies should attempt to measure as many as possible of the different H2S pools by using a variety of different techniques that will measure not only free H2S but also other pools such as sulfane sulfur and polysulfides in a similar fashion, as recently indicated (148), The same authors showed that 3-MST expression was downregulated in kidney lysates in mice with lipopolysaccharide (LPS)-induced AKI. NaHS administration attenuated LPS-induced inflammation and oxidative stress and inhibited the expression of Toll-like receptor 4 (TLR4), NLRP3, and caspase-1 (41).
Based on these encouraging preliminary results, further preclinical studies using H2S donors to treat SA-AKI are needed to select the most suitable donor compound for this indication and to determine the dose regimen.
Chronic kidney disease
CKD is defined by persistent urinary abnormalities, structural changes, and impaired excretory renal function, which are suggestive of a loss of functional nephrons. This pathological condition is associated with genetic factors and low nephron number at birth or risk factors leading to nephron loss, such as aging, acute or chronic kidney injuries caused by exposure to toxic agents, and diseases, especially obesity, type 2 diabetes mellitus, and hypertension (158).
A widely used, invasive animal model that simulates CKD is nephrectomy of 5/6 of kidney mass. Reduced expression of H2S-producing enzymes, enhanced oxidative stress, and NAPDH oxidase-4 upregulation have been reported in this model (10). NaHS treatment in rats with 5/6 nephrectomy improved redox balance, reduced apoptosis, and attenuated the expression of genes associated with inflammation and tissue remodeling (166). The NOS inhibition reversed the protective effects of H2S. In addition, NaHS administration improved renal function and survival. These results provide the rationale for further testing of H2S supplementation strategies to delay disease progression in CKD.
Diabetic nephropathy
Diabetes is the most common cause for chronic kidney injury and currently the leading cause of end-stage renal disease. Some of the pathologic findings associated with the diabetic kidney are mesanglial cell expansion, glomerulosclerosis, tubulointerstitial fibrosis, and inflammation. These changes lead to hypertension, proteinuria, progressive reduction in GFR, and end-stage renal disease (8).
Observations in animal models, as well as in human patients indicate that endogenous production of Η2S is reduced diabetes (31, 82, 175). In human patients, Η2S plasma levels were found to be significantly lower in chronic hemodialysis patients with diabetic nephropathy compared with chronic hemodialysis patients without diabetic nephropathy (107). Moreover, a post hoc analysis demonstrated that higher urinary sulfate concentration (a metabolic end-product of H2S) is associated with a more beneficial profile of renal risk markers in patients with diabetic nephropathy (185).
Changes in the expression of H2S-producing enzymes and H2S levels in the kidney have been noted in several animal models of diabetes. In a transgenic model of autoimmune type 1 early Class I diabetic nephropathy, CSE was upregulated whereas 3-MST and CBS were downregulated (184). Renal CBS levels were attenuated in hyperglycemic mice fed a high fat diet (89, 113), whereas the expression of both CBS and CSE was reduced in the kidney cortex of OVE26 mice (type 1 diabetes model) and in db/db mice (103).
In the presence of high glucose levels, H2S serves as an inhibitor of protein synthesis. Reduced H2S levels allow increased protein synthesis through de-repression of an AMPK/mammalian target of rapamycin (mTOR) 1 pathway, leading to renal matrix protein accumulation and renal hypertrophy (103). A study published this year suggested that high fat diet activated insulin receptor in proximal tubular epithelial cells, which, in turn, attenuated endogenous H2S production that led to renal injury (104).
The CBS and CSE levels were also reduced in Akita diabetic mice (99). Using a combination of in vitro and in vivo approaches, Kundu et al. demonstrated that ROS activated matrix metalloprotease-9, which, in turn, leads to downregulation of CBS and CSE expression in the diabetic kidney (99). In studies performed by several independent research teams, renal CSE expression was reduced in streptozotocin (STZ)-induced diabetic nephropathy (153, 204, 212).
Overall, diminished levels of CSE, CBS, and/or 3-MST have been consistently observed in diabetic nephropathy; the enzymes affected depend on the model and the species used. Interestingly, pharmacological inhibition of CSE simulates high glucose-induced glomerular podocyte injury (116). In line with this finding, CSE upregulation after taurine treatment was shown to suppress calcium overload by inhibiting transient receptor potential cation channel, subfamily C (TRPC6) and prevented podocyte injury (209).
Given the reduced levels of H2S present in the diabetic kidney, H2S supplementation strategies present as a reasonable therapeutic approach. Indeed, H2S donors have been shown to prevent or ameliorate many of the pathophysiological changes associated with diabetic nephropathy (structural changes, fibrosis, creatinine clearance, proteinuria, and enhanced oxidative stress) (6, 98, 108, 153, 204, 212).
One suggested mechanism for the beneficial effects of H2S is the reduction of matrix metalloproteinases (MMPs) expression, especially MMP9, which contributes to renal fibrosis and nephropathy (98). In STZ diabetic rats, the expression of several MMP was decreased after NaHS administration; the effects on MMPs coincided with downregulation of TGF-β1 (108). The ability of NaHS to inhibit TGF-β1 expression and ROS levels was confirmed in two other independent studies (108, 204). Additional pathways associated with the beneficial actions of NaHS in diabetic nephropathy include activation of Nrf-2-triggered antioxidant pathways and anti-inflammatory effects mediated by the inhibition of NF-κB signaling (212).
S-propargyl-cysteine (SPRC), an H2S donor, attenuated inflammation and oxidative stress in diabetic kidneys. The SPRC also reduced TGF-β1 signaling and Smad3 phosphorylation (153). Moreover, NaHS reduced high glucose-induced mesangial cell proliferation by blocking MAPK signaling pathways and inhibited the renin-angiotensin system in the diabetic kidney (204, 212). In glomerular epithelial cells, NaHS treatment inhibited high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase, thus preventing matrix accumulation (103). NaHS also inhibited high glucose-induced NOX4 through AMPK activation (102). The antioxidant effects of NaHS in STZ-induced diabetic nephropathy have also been associated with enhanced SIRT1 activity (6).
In summary, H2S donors alleviate the development of diabetic nephropathy by reducing oxidative stress and inflammation, attenuating mesangial cell proliferation, and inhibiting the renin-angiotensin system.
Kidney function and hypertension
Hypertension is one of the most significant risk factors for chronic nephropathy. Several publications, including studies performed in our laboratory, have demonstrated an important role of H2S in blood pressure regulation (25, 88, 126, 167). It has been shown that CSE-derived H2S is an important regulator of blood pressure and vascular resistance in the renal circulation (133, 202). Moreover, CBS heterozygous mice exhibit homocysteinemia, elevated systolic blood pressure, and renal dysfunction. NaHS supplementation decreased eNOS homocysteinylation, upregulated eNOS expression, and reduced the expression of the eNOS inhibitor caveolin-1, leading to improved renal function (152).
In high-salt-induced hypertension in rats, CBS expression and H2S levels in renal tissue are reduced (77, 78). The reduction in CBS levels was mediated by the inhibition of hypoxia-inducible factor alpha subunits (HIF-1α) and it leads to increased activation of the renin-angiotensin system. In a follow-up study by the same group, NaHS administration counteracted the high-salt diet-induced impairment in renal function and structure, and it inhibited excessive collagen deposition (78).
In 2-kidney 1-clip (2K1C) hypertensive rats, NaHS administration restored H2S levels and reduced systolic blood pressure, whereas it upregulated SOD-1 protein expression in the myocardium (115). NaHS also exerted a protective function in renal arterial endothelium in hypertension in a 2K1C model (177). In a different model where mice were rendered hypertensive by Ang II infusion, GYY-4137 reduced renal vascular resistance and reversed the decreased in renal cortex blood flow triggered by Ang II. In addition, GYY-4137 reduced blood pressure and plasma creatinine levels, increased plasma nitrite/nitrate, and reversed the Ang II-induced reduction in CSE and CBS protein in the kidney (193).
In line with the beneficial effects of H2S supplementation in the Dahl salt-sensitive, 2K1C, and Ang II-induced hypertension models, NaHS ameliorated soluble fms-like tyrosine kinase 1-induced hypertension, proteinuria, and glomerular endotheliosis in rats (75). Moreover, early administration of
Interestingly, H2S has the ability to regulate sodium handling by modifying the opening of the epithelial sodium channel (ENaC). ENaC mediates Na+ absorption across epithelial cells in the collecting duct and its overactivation causes hypertension (192). In vitro studies have shown that NaHS prevented H2O2- and advanced glycation end-product-induced activation of ENaC (190, 208). It should be mentioned that the effects of H2S on ENaC have not been confirmed. Finally, NaHS administration decreased renal gene expression of polycystin-1, a marker of polycystic kidney disease that is upregulated in hypertension and other cardiovascular diseases (5). Based on the earlier observations, it is clear that the impact of H2S in physiological blood pressure regulation, as well as in hypertension, is at least partially due to its renal effects.
Renal cancer
Urinary tumors include prostate, kidney, and urothelial carcinoma and are a common type of malignancy. Several studies have demonstrated that H2S-synthesizing enzymes are upregulated in various cancer types. Depending on the tumor type, different H2S-producing enzymes are overexpressed (176). Published data on the role of H2S in renal cancer have yielded conflicting results. In advanced stages of renal clear cell carcinoma, CBS was found to be upregulated compared with benign kidney samples (164). In a different study that compared malignant tissue with biopsies from kidney sites distal from the tumor of the same patient, all three H2S-generating enzymes (CBS, CSE, and 3-MST) were downregulated (32). In vitro experiments in RCC4 cells demonstrated that CSE and CBS upregulation resulted in increased apoptotic cell death and that pharmacological inhibition or silencing of CSE and CBS protected cells from apoptosis (32). However, dual inhibition of CBS and CSE with the non-selective inhibitor hydroxylamine inhibited the proliferation, the metabolic activity, and the survival of renal cancer cell lines with Von Hippel–Lindau deficiency (170).
Hydroxylamine also inhibited tumor angiogenesis and growth of xenografts placed in the chicken chorioallantoic membrane. More extensive experimentation involving a larger number of human biopsies, detailed cancer staging, as well as more specific tools and orthotopic mammalian models are needed to elucidate the role of H2S in clear cell renal cell cancer and other types of kidney malignancies.
Nephrogenic DI
DI is a rare disease characterized by excretion of large amounts of hypotonic urine. Central DI results from a deficiency of the hormone arginine vasopressin (AVP) in the hypothalamus, whereas nephrogenic DI results from resistance to AVP in the kidneys. Nephrogenic DI is usually the result of either an adverse effect of drugs (such as lithium salts) or a hereditary condition due to mutations in the genes encoding arginine vasopressin receptor 2 (AVPR2) or the water channel AQP-2 (42).
Recently, Luo et al. (121) studied the role of H2S in urine concentration. Their findings indicate that the inhibition of CBS and CSE simultaneously via administration of AOAA and PAG increased urine output, decreased urine osmolality, and upregulated AQP-2 expression in the medulla and cortex. In addition, GYY-4137 administration improved urine concentration and prevented the reduction of AQP-2 protein levels in the inner medulla of kidneys from lithium-induced nephrogenic DI. It was proposed that the H2S ability to regulate AQP-2 protein expression occurs via the activation of the cAMP-protein kinase A (PKA) pathway. H2S has been shown to modulate AQP expression and function in the context of pulmonary (21) and brain edema (34), reinforcing the notion that H2S is important for water transport across the cell membrane.
Conclusions: Future Directions
Similar to other organs, endogenous H2S is reduced in several different renal diseases, in both acute and chronic settings. The drop in H2S levels is linked to oxidative stress, although it is unclear whether increased ROS lead to the depletion of H2S or whether it is the attenuation in endogenous H2S that contributes to the disruption of redox balance. Therefore, future studies using genetic models and pharmacological inhibitors of both ROS and H2S are needed to comprehend the interplay between ROS and H2S in the kidney. The functional importance of endogenous H2S in preserving renal function during injury is highlighted by the observations that genetic or pharmacological inhibition of H2S synthesis exacerbates renal injury in a variety of models.
Another important aspect to consider is that studies performed so far have focused exclusively on the expression of H2S-synthesizing enzymes in the kidney. However, how H2S-degrading pathways are affected by renal dysfunction is currently unknown and is worthy of investigation.
Finally, a crucial aspect of H2S biology that remains unexplored is the identity of the molecular targets of H2S in the kidney. For example, the effect of S-sulfhydration or H2S binding to protein metal centers in individual protein targets remains uncharacterized. As far as S-sulfhydration is concerned, an initial report has provided a list containing a broad spectrum of proteins carrying this post-translational modification in the kidney; many of the proteins that are S-sulfhydrated in the kidney are interestingly linked to metabolism.
Based on the findings that renal H2S concentration is attenuated in pathological conditions, restoration of H2S by administration of H2S donors constitutes a therapeutic opportunity. Indeed, administration of (almost exclusively fast-releasing) H2S donors has yielded promising results in preclinical models. The possible indications for H2S donors include preservation of renal grafts, protection from ischemia–reperfusion injury and nephrotoxic agents, and improvement of renal function in CKD.
It should be kept in mind that due to the pleiotropic effects of H2S in the cardiovascular system the improvement in renal function by H2S supplementation might be either direct or indirect. For example, attenuated kidney damage in hypertension models might be due to both vasodilation and reduction in blood pressure, as well as to local effects of the H2S donor in the kidney. H2S donors with slower, controlled release and better renal tissue-targeting properties are expected to be more effective and useful. Clinical trials should be performed to test whether basic science promise can be translated into human therapies. A summary of the future directions and conclusions in relation to targeting H2S signaling in the kidney is given in Figure 8 next.

Footnotes
Acknowledgment
Figures were created using the Biorender software.
Authors' Contributions
M.P., P.Z., and A.P. wrote the article.
Author Disclosure Statement
A.P. is a member of the Scientific Advisory Board of Sulfagenix, Inc. All other authors have no competing interests to disclose.
Funding Information
A.P. is supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “First Call for H.F.R.I. Research Projects to support Faculty members and Researchers and the procurement of high-cost research equipment grant” (Project No. H.F.R.I.-FM17-886 to A.P.) and by the grant “Upgrading the plant capital (PlantUP)” that is co-funded by the European Regional Development Fund (ERDF) and Greek national funds through the Operational Program “Competitiveness, Entrepreneurship and Innovation,” under the call “STRENGTHENING RESEARCH AND INNOVATION INFRASTRUCTURES” (project code: 5002803). P.Z. is supported by H.F.R.I. under the H.F.R.I. PhD Fellowship grant (Fellowship No. 1087).