
Abstract
Significance:
The vasculature responds to the respiratory needs of tissue by modulating luminal diameter through smooth muscle constriction or relaxation. Coronary perfusion, diastolic function, and coronary flow reserve are drastically reduced with aging. This loss of blood flow contributes to and exacerbates pathological processes such as angina pectoris, atherosclerosis, and coronary artery and microvascular disease.
Recent Advances:
Increased attention has recently been given to defining mechanisms behind aging-mediated loss of vascular function and development of therapeutic strategies to restore youthful vascular responsiveness. The ultimate goal aims at providing new avenues for symptom management, reversal of tissue damage, and preventing or delaying of aging-induced vascular damage and dysfunction in the first place.
Critical Issues:
Our major objective is to describe how aging-associated mitochondrial dysfunction contributes to endothelial and smooth muscle dysfunction via dysregulated reactive oxygen species production, the clinical impact of this phenomenon, and to discuss emerging therapeutic strategies. Pathological changes in regulation of mitochondrial oxidative and nitrosative balance (Section 1) and mitochondrial dynamics of fission/fusion (Section 2) have widespread effects on the mechanisms underlying the ability of the vasculature to relax, leading to hyperconstriction with aging. We will focus on flow-mediated dilation, endothelial hyperpolarizing factors (Sections 3 and 4), and adrenergic receptors (Section 5), as outlined in Figure 1. The clinical implications of these changes on major adverse cardiac events and mortality are described (Section 6).
Future Directions:
We discuss antioxidative therapeutic strategies currently in development to restore mitochondrial redox homeostasis and subsequently vascular function and evaluate their potential clinical impact (Section 7). Antioxid. Redox Signal. 35, 974–1015.
1. Historically Explored Mechanisms of Mitochondrial Oxidative Changes in Aging
1.1. Causes of reactive oxygen species production in aging
Increased oxidative stress coincides and contributes to the aging process. The oxidative stress theory of aging postulates that age-associated functional loss (such as vascular function to maintain adequate perfusion) is the result of oxidative damage of proteins, DNA (nuclear and mitochondrial), and lipids, leading to cellular senescence. Cellular senescence is characterized by the senescence-associated secretory phenotype (SASP), whereby cells exhibit increased secretion of interleukins, chemokines, growth factors, matrix metalloproteinases, insoluble proteins, and extracellular matrix (132). This leads to a proinflammatory state with feedforward mechanisms that further increase harmful reactive oxygen and nitrogen species (ROS and RNS), affecting many pathological processes linked to chronic diseases such as cardiovascular disease, respiratory diseases, chronic kidney disease, cancer, neurodegenerative diseases, and muscular disorders (132). Indeed, chronic inflammation may be induced in the elderly population by genetic inflammasome involvement, leading to increased oxidative stress, hypertension, and arterial stiffness (144). In addition, increased apoptosis has been demonstrated in senescent endothelial cells in humans, and vascular senescence can lead to irreversible pulmonary arterial hypertension in both rats and humans (462). The occurrence of SASP and proinflammatory state is variable between individuals depending on genetic, environmental, and behavioral influences, leading to the Geroscience hypothesis, which proposes that the “rate of aging” can be influenced by modifiable factors (132). Specific to this review, we will focus on the role and mechanisms by which oxidative stress hastens vascular aging, resulting in endothelial dysfunction.
The mitochondria are responsible for not only cellular energy production but also the majority of ROS generation (90, 185). During oxidative phosphorylation, the electron transport chain (ETC) utilizes the transfer of electrons between complexes to establish the proton gradient responsible for powering ATP-synthase generating ATP (438). The electrons traversing the ETC end up at complex IV, where they are ultimately utilized in the production of water from oxygen and hydrogen. One to three percent of these electrons do not make it to this step; instead, they leak to prematurely react with oxygen to form superoxide (O2 •−) (113). In senescent endothelial cells (via passaging), the catalytic function of complex IV is reduced by up to 84%, allowing for exacerbated electron leakage with aging (521). Superoxide generation occurs predominantly at complex I (39, 302) during conditions of high proton motive force, NADH/NAD+ ratio, and CoQH2/CoQ ratio (sensor of respiratory chain efficiency), with the latter two occurring more frequently with aging (165, 309, 326).
A significant amount of superoxide from complex I is generated during reverse electron transport, where electrons are shunted back from CoQH2 back to complex I and reduce NAD+ to NADH. This process is highly sensitive to membrane potential (protonmotive force) changes, with a 10 mV decrease leading to a 70% reduction in superoxide production within the mitochondrial matrix, although not affecting cytosolic production (292). Mitochondrial membrane potential decreases with aging, which would theoretically lead to a decrease in superoxide production in aging. This is due to an inherent increase in leakage of protons, reducing the membrane potential with age, as a probable adaptive mechanism to reduce oxidative stress in aging (320, 388, 426). Despite this adaptation, accumulation of ROS dominates aging-associated mitochondrial alterations.
There is an increase in pro-oxidation processes in the aging vasculature (121). With aging, there is a known increase in circulating angiotensin II (Ang II), angiotensin converting enzyme (ACE), and angiotensin receptor 1 (AT1) expression (206, 231, 482). Ang II induces endothelial dysfunction through activation of inflammatory signaling (e.g., VCAM-1, NFκB), activation of COX2-producing vasoactive prostaglandins when bound to AT1 receptors, and activation of NADPH oxidase (NOX) (79, 94, 111, 206, 277, 448, 449, 451, 490, 494, 504). These transmembrane proteins transfer electrons from NADPH to molecular oxygen, leading to the generation of superoxide and hydrogen peroxide (H2O2) (2, 101, 111, 171, 180, 234, 434). NOX 1 and 2 contribute to superoxide production in endothelial cells and endothelial and smooth muscle cells (SMCs), respectively (101, 180, 210, 450). AT1 and NOX activation also lead to endothelial nitric oxide synthase (eNOS) uncoupling, shifting from nitric oxide (NO) to superoxide production and attenuating endothelium-dependent dilation (EDD) (93, 103, 371, 527). In aging, NOX subunits are overexpressed, including the p67 subunit in mouse aorta (119, 135), p22phox subunit in rat mesenteric arteries (199), and p47phox subunit in aging men (107). Collectively, these signaling cascades result in enhanced ROS generation and endothelial dysfunction, whereas inhibition of NOX improves overall EDD in vessels from aging humans and rats (88, 306, 367, 456).
Of the NOX isoforms, NOX4 is considered one of the primary vascular NOXs responsible for endothelial hydrogen peroxide production. NOX4 is further upregulated by hypertrophic signaling and β-arrestin mediated signaling, and along with the other NOXs are increased with age (2, 3, 41, 163, 284, 334). There is current lack of clarity and consensus on the vascular effects of NOX4, with studies showing both deleterious and protective roles (296). The discrepancy in these studies, likely stemming from differences in knockout/overexpression strategies, showcases the tight redox balance required for homeostatic vascular function. Low chronic hydrogen peroxide accumulation can be vasculoprotective, lowering blood pressure if produced acutely as an endothelium-dependent hyperpolarizing factor in response to shear stress.
In NOX4 knockout mice, aortas develop increased inflammation, increased media hypertrophy, reduced heme oxygenase-1 and nrf-2 gene expression, and endothelial dysfunction, including reduced eNOS function and nitric oxide (391, 392). On the other hand, overabundance of hydrogen peroxide chronically can induce autophagy, apoptosis, and reduced vascular function, and it is classically known as a pathogenic mediator of atherosclerosis. In aging, NOX4 may err on the side of promoting endothelial dysfunction, rather than protection (247, 248). Potential positive effects of NOX4 are likely mitigated due to eNOS uncoupling-mediated superoxide production, whereby any effect of NOX4 to active eNOS through protein kinase B (Akt) signaling is potentially counterproductive. Evidence suggests upregulated NOX4 and subsequent chronic hydrogen peroxide exposure in aging causes eNOS uncoupling, reduced nitric oxide production and acetylcholine (Ach)-mediated vasodilation, endoplasmic reticulum (ER) stress via IRE1α-oxidation, and mitochondrial dysfunction (189, 247, 248, 472). Further details on the vascular protective and deleterious effects of NOX4 are reviewed by Salazar (380).
Xanthine oxidase has increased expression and activity with aging and is associated with oxidative stress in several tissues, including the aorta, coronary, and mesenteric arteries (13, 241, 317, 422, 476, 477, 517), and it contributes to superoxide generation in response to increased pressure (204). Although inhibition of xanthine oxidase reverses endothelial dysfunction in hypoxia, hypercholesterolemia, hyperuricemia, and heart failure patients (54, 92, 105, 110), inhibition seems unable to reverse aging-mediated endothelial dysfunction in humans, at least while evaluating peripheral vasculature (122).
The adaptor protein p66shc has been considered one of the master regulators of superoxide production. Its expression, increased in advancing age, is mediated by Ang II signaling, hypertrophy, and alpha-adrenergic receptor (αADR) agonism (78, 150, 166, 237, 409, 438). In addition, p66shc is imported into the intermembrane space of the mitochondria, where it steals an electron from cytochrome c, facilitating its transfer to molecular oxygen to produce superoxide (454), while also inhibiting the transcription of manganese superoxide dismutase (SOD2) through α1ADR/FOXO3A-mediated signaling (166). Genetic deletion of p66shc leads to a 30% increase in lifespan in mice (289). Genetic knockout of p66shc in old murine aortic rings preserves, whereas overexpression reduces EDD to acetylcholine (ACh) and nitric oxide bioavailability (52, 78, 138, 506).
1.2. Reduction of anti-oxidative processes in aging
During youth and into healthy middle age (roughly ages <55), antioxidants quench ROS to maintain redox homeostasis. Superoxide is converted into hydrogen peroxide by SOD2 in the mitochondrial matrix and copper/zinc superoxide dismutase (SOD1) in the intermitochondrial space and cytoplasm. Hydrogen peroxide can further react via the Fenton reaction to form the hydroxyl ion. This is prevented by the conversion of hydrogen peroxide to water and molecular oxygen by catalase and glutathione peroxidase (predominant). Glutathione peroxidase oxidizes glutathione in concert with glutathione reductase to reduce GSH, occurring in a cyclic manner (303, 438). Superoxide can also react with NO to form peroxynitrite (ONOO−), a potent oxidizing and nitrosylating agent.
Vascular aging is associated with a decrease in density and function of SOD1/2 (64, 100, 291, 491, 526), glutathione, glutathione reductase, and glutathione peroxidase (32, 58, 95, 136, 177, 349, 352, 368, 497), which are associated with reduced EDD and aortic stiffening in mice and rats. Positive correlations of aortic catalase concentration and activity with aging have been observed as a potential compensatory mechanism (although insufficient) for increased hydrogen peroxide concentration and vascular sensitivity to hydrogen peroxide (200, 439). In contrast, in aged pulmonary arteries and patients with untreated essential hypertension catalase concentration is decreased (356, 379). Overexpression of catalase, glutathione peroxidase, or SOD in animal models of aging is demonstrated to restore EDD and protect against vascular pathologies such as aneurisms, atherosclerosis, with the added benefit of increased lifespan (42, 82, 84, 127, 276, 343, 489).
Additional antioxidative contributors to maintaining vascular redox homeostasis are sirtuin deacetylases (Sirt), which have been demonstrated to be decreased in aging mice and humans (109, 225, 400). Sustaining eNOS deacetylation status preserves its ability to produce nitric oxide, and shifting to acetylated status with aging-mediated loss of Sirt1 contributes to loss of nitric oxide bioavailability and increased superoxide (255, 281, 329, 366). This loss of nitric oxide is associated with diminished capacity for endothelium- or shear stress-induced vasodilation in aorta, femoral, and middle cerebral arteries that are subject to Sirt1 inhibition (109, 281, 433). Conversely, overexpression of endothelial Sirt1 mediates vasoprotection in aging through nitric oxide-independent effects via enhancing soluble guanylyl cyclase in smooth muscle and reducing AT1 receptor expression (14, 168, 294, 425).
Sirt1 and mitochondrial Sirt3 regulate transcription factors for antioxidant proteins, including FOXO, the transcription factor for SOD2, to suppress the effects of p66shc and reduce ROS (43, 153, 359, 526). Childhood Sirt1 expression is a predictor of adulthood microvascular function, as reduced Sirt1 in young participants in a longitudinal cohort was associated with premature cutaneous microvascular dysfunction in adulthood. This was assessed via maximal response to local thermal hyperemia, post-occlusive reactive hyperemia, and iontophoresis with ACh (369). Many of the beneficial effects of caloric restriction on vascular function are induced via sirtuins as they suppress senescence by delaying telomere attrition, instigating DNA repair and genomic stability, and preserving proteostasis by regulating heat shock proteins (233, 249, 359, 360, 522).
1.3. Effect of estrogen loss on oxidative stress during aging
Aging-associated changes in hormones contribute to increased oxidative stress. Loss of estrogen during the menopausal transition to postmenopause is associated with elevated risk for cardiovascular disease. Postmenopausal females may exhibit coronary microvascular dysfunction, defined by endothelial dysfunction, pathologic smooth muscle tone, and increased oxidative stress (9). The loss of estrogen is one mediator of this disease due to the loss of its antioxidative properties. Estrogen modulates NOX to reduce superoxide production in rat in vivo mesenteric microvessels (91, 505).
Loss of estrogen diminishes nitric oxide-mediated flow-mediated dilation (FMD) in young ovariectomized rats while exhibiting elevated nitrotyrosine (oxidative stress marker) and reduced SOD protein (213). Ovariectomized rats given hormone replacement therapy (HRT) show restored SOD and nitric oxide-mediated FMD. Estrogen also activates telomerase, limiting mitochondrial ROS (mtROS) production via the subunit telomerase reverse transcriptase (TERT) (239). The activation of mitochondrial estrogen receptor-β leads to increased S-nitrosylation (SNO) of proteins, such as proteins that have a role in the homeostatic balance of β-adrenergic function.
The mitochondrial prooxidant effects on EDD in aging promote pathologic cardiovascular remodeling and hypertension, whereas reduced perfusion and chronic ischemia have an impact on morbidity and mortality. Therefore, understanding these intricate interactions is vital for understanding proper therapeutic management and development. How these aging-dependent vascular changes impact mechanisms for vasorelaxation, including flow-mediated, potassium channel-mediated, and βADR-mediated relaxation as well as parallel processes such as mitochondrial fission and fusion, are the focus of the remainder of this review.
2. Vascular Mitochondrial Fission/Fusion in Relation to Mitochondrial Redox Homeostasis with Aging
2.1. Mitochondrial dynamics and dysfunction with aging-induced oxidative stress and hyperglycemia
Mitochondria are dynamic organelles, changing shape and function as a direct result of the surrounding molecular environment. Within other tissues and organs, the mitochondria are traditionally known as energy hubs, producing ATP through oxidative phosphorylation. However, within the vasculature, mitochondria act as a signaling hub rather than for energy as endothelial cells are highly glycolytic (229, 438). The mitochondria are integral in maintaining a balance between production of ROS and signaling actions of nitric oxide, which can affect vasodilative balance as alluded to in later sections. In this capacity, the mitochondria act as a signaling hub, with structure and function playing an important role in vascular health and disease (438).
Mitochondrial dynamics are delineated by biogenesis and mitophagy. On the one hand, biogenesis results in production of new mitochondria characterized by a highly filamentous and networked structure that is viewed as “fused.” Conversely, damaged mitochondria tend to exhibit a more diffuse, punctuated structure, caused by a process termed fission, which is mediated by dynamin-related protein-1 (DRP-1) and mitochondrial fission protein-1 (Fis-1). Damaged mitochondria either fuse with healthy mitochondria—mediated by mitofusin 1 or 2 (Mfn-1 or 2) or optic atrophy 1 (Opa-1)—or are encapsulated and degraded via mitophagy. Oxidative stress alters cultured endothelial mitochondrial structure, transitioning from a more structured, tubular, networked state (e.g., fusion), to a more fragmented, disrupted, or punctate state (e.g., fission). Aging-related changes to antioxidant versus prooxidant expression and function and their relation to mitochondrial dynamics, mitophagy, FMD, and adrenergic-mediated dilation are illustrated in Figure 1.
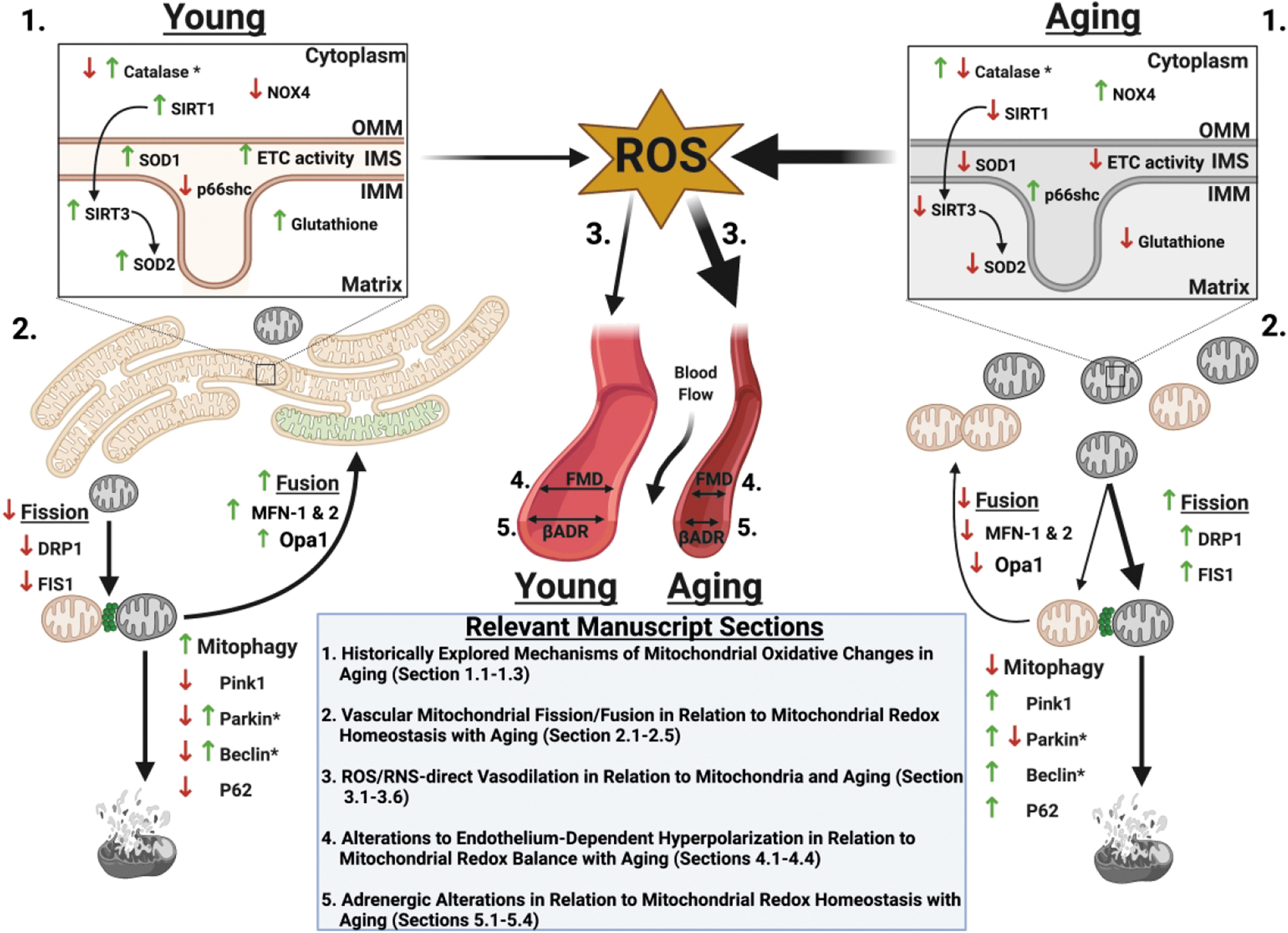
Mitochondrial dynamics during health exist in a homeostatic balance between fission and fusion, the disruption of which is implicated in cell senescence, quiescence, and aging processes (423). Indeed, Jendrach et al. demonstrated in human vascular endothelial cells serially cultured to induce senescence, thereby mimicking aging, that mitochondrial structure is altered in aging via reduced fusion and fission events; aging was associated with mitochondrial DNA (mtDNA) damage and loss of mitochondrial membrane potential opening of the mitochondrial permeability transition pore (mPTP), resulting in the release of cytochrome C and enhancing further oxidative stress (205).
Hyperfusion is a protective response to boost mitochondrial respiration and counteract cell stress. A more connected (fused) network of mitochondria allows for a more closely linked network of mitochondrial respiratory complexes, maximizing energy production and signaling (351). However, excessive hyperfusion via Mfn-1 overactivation may be associated with induction of senescence, and DRP-1 repression may induce ATR-mediated DNA damage response.
Some level of fission appears necessary, as senescent endothelial progenitor cells exhibit reduced Fis-1 with restoration of expression, thereby reducing ROS and restoring youthful morphology, bioenergetics, and angiogenic potential (480). Overly reduced DRP-1 function, while inducing hyperfusion, also reduces Mfn-1 and 2 and Opa-1 expression and processing (295). On the other hand, disruption of this interconnected mitochondrial network with hyperfission is linked to aberrant respiratory complexes and also excessive ROS (197, 419).
In endothelial cell senescence, hyperfission dominates. In senescent rat aortic endothelial cells (induced by Ang II), mitochondrial fission is upregulated alongside the pro-inflammatory phenotype (293). One mechanism for increased senescent endothelial fission is enhanced activity of DRP-1 due to loss of protein disulfide isomerase A1 (PDIA1). PDIA1 acts as a thiol reductase for DRP-1, without which DRP-1 becomes sulfenylated at cysteine 644, leading to activation with increased mitochondrial fragmentation and ROS production, further induction of senescence with reduced angiogenesis, and reduced endothelium-dependent vasodilation to ACh (223). DRP-1 may also be redox regulated by SNO-mediated activation, although this notion is controversial (34, 71). DRP-1 is negatively whereas Opa-1 is positively regulated by Sirt 3 deacetylation, which is downregulated in aging (300, 382).
Reductions in rat aortic NO bioavailability (which occurs in aging) also reduce the amount of fused mitochondria, while increasing mitochondrial fission (290). These aging-induced balance shifts toward hyperfission have been observed in our lab, with RNA sequencing showing reduced Mfn-1 and enhanced DRP-1 in aging rat coronary microvessels (unpublished observations).
Changes in mitochondrial structure and function are impacted by their surrounding environment. Exposure to ROS imparts damage to mtDNA, with excessive oxidative stress impairing oxidative phosphorylation and subsequently increasing the amount of ROS produced in a feedforward manner (16). As it relates to fission/fusion balance, these alterations have consequences for dilative ability of the vasculature to maintain proper patency for oxygen and metabolic and demand and for processes of aging-associated cardiovascular pathologies.
2.2. What are the functional ramifications of age-associated changes in mitochondrial dynamics?
Reductions in vasodilation in aging have been attributed to excessive mtROS production as a result of dysfunctional mitochondria, including dynamics of fission/fusion balance. As described in subsequent major sections of this review, aging is associated with skewed balance of ROS and vasoprotective nitric oxide bioavailability (108, 460, 461), in part due to altered mitochondrial dynamics, with reductions in vasodilation to shear stress (e.g., exercise or FMD) and adrenergic agonism in older adults (298, 303, 314, 347, 455, 460). For now, we describe associations of altered fission/fusion balance and their functional consequences on vasodilative capacity and classic pathologies.
Evidence from animal models indicate that aging is associated with a general decrease in mitochondria content within endothelial cells, coinciding with decreased mitochondrial respiration and increased mitochondria superoxide generation and hydrogen peroxide (438). These maladaptive alterations with aging are associated with decreased messenger RNA (mRNA) expression of mitochondrial biogenesis, and reductions in Mfn-1 and 2 (401, 459, 460). Functionally, aging-induced increased DRP-1-induced mitochondrial fragmentation increases mtROS generation and reduces EDD to ACh (223, 458).
Exposure to high glucose, mimicking insulin resistance with aging (independent of adiposity) (120, 378), is associated with increased ROS, thus associating exposure of ROS to changes in mitochondria shape. Using a photo-activatable mitochondrial reporter within the endothelium in mice both in vivo and ex vivo, Durand et al. demonstrated that acute exposure to high glucose (via tail vein injection or cultured primary cells) resulted in fragmented, disrupted mitochondrial networks relative to normal glucose conditions (117). Type 2 diabetes mellitus (T2DM) and manipulation of glucose concentrations (mimicking aging-increased insulin resistance) induces mitochondrial fission and reduces microvascular endothelial function (226, 408, 440). Within the brachial artery and in resistance arterioles, the presence of T2DM is associated with increased ROS, increased mitochondrial membrane potential, and increased protein markers of mitochondrial fission. These hyperfission changes in mitochondrial dynamics are associated with reduced vasodilation to ACh within resistance arterioles (223). Interestingly, administration of mitochondrial-targeted antioxidants improved FMD and ACh-induced vasodilation and decreased ROS (152, 375). Although T2DM and exposure to high glucose induced mitochondrial fission, Tanner et al. (440) demonstrated that exposure to low glucose also is associated with increased mitochondrial fission in cultured endothelial cells, whereas both pharmacological and genetic disruption of mitochondrial fission in low-glucose exposed resistance arterioles improved microvascular endothelial function (ACh-induced vasodilation). Although the specific role for aging in-it-of-itself (vs. aging-associated hyperglycemic stress) within this vasodilative context has yet to be fully elucidated, particularly within the microvasculature, it does provide evidence of mitochondrial dynamics driving both conduit artery and microvascular endothelial function. Therefore, further studies to directly link aging-mediated mitochondrial dynamic dysfunction to vasodilative function are highly warranted.
Similar to endothelial cells, mitochondrial dynamics drastically impact vascular smooth muscle (VSM) function, which is reviewed elsewhere (70, 283). Liu et al. demonstrated in rat mesenteric arteries and thoracic aorta that administration of phenylephrine (α-adrenergic agonist) and potassium resulted in mitochondrial fission within VSM cells, whereas inhibition of mitochondrial fission reduced vasoconstrictor responses to phenylephrine and potassium (263). Interestingly, aging favors α-adrenergic constriction in coronary vessels as described in Section 5 (19). In a similar light, Chen et al. (62) demonstrated that inhibition of mitochondrial fission reduced the vasoconstrictor response of mesenteric arteries and thoracic aorta in rats to endothelin-1, which is also increased in aging (155, 424).
There are implications for a role for mitochondrial hyperfission in aging-associated cardiovascular pathologies. Wang et al. (481) demonstrated that reduction of mitochondrial fission protein DRP-1 decreased VSM migration and reduced ROS, implicating a role for mitochondrial hyperfission in atherosclerosis. Interestingly, Mfn-2 has been demonstrated to inhibit VSM proliferation. Mfn-2 mediates apoptosis independent of its role within mitochondrial dynamics, and it is critical for the apoptotic response to hydrogen peroxide in VSM cells (hydrogen peroxide being an important pathologic mediator of atherosclerosis) (167, 401). However, Mfn-2 expression has been shown to be reduced in mouse and rat atherosclerosis and hypertension (66). Cardiac ischemia–reperfusion injury is associated with downregulation of Opa-1 in a DRP-1 hyperfission-dependent manner (67, 328), and Opa-1 and Mfn-1 and 2 have been shown to be protective against the development of heart failure from pressure overload (280, 355).
2.3. Vascular mitochondrial mitophagy
Over the past decade, the role of mitophagy in vascular structure and function in health and disease has become a popular area of research. The most well-characterized mitophagy signaling cascade is the PTEN-induced kinase 1 (Pink1)-Parkin-mediated pathway. Emerging evidence has demonstrated that mitophagy may be activated independent of Parkin-mediated mitophagy. More recent work has shown that these distinct signaling pathways are unique to the environmental stressors that mitochondria are exposed to, as well as distinct to the tissue studied (285).
2.4. Receptor-mediated mitophagy
The outer mitochondrial membrane expresses receptors that are specific for mitophagy, described as containing LC3-interacting regions, which function to dock with LC3 for canonical autophagy degradation. Currently, three receptors have been described as such: NIX, BNIP3 (BCL2/adenovirus e1B 19 kDa interacting protein 3), and FUNDC1 (FUN14 domain-containing protein 1). De-phosphorylation of threonine and serine residues allows for FUNDC1 to link with LC3B and undergo canonical autophagy (65, 262). Hypoxia appears to be the primary driving factor that activates FUNDC1-mediated mitophagy, and this mitophagy pathway may be particularly important within the vascular responses to hypoxia (e.g., ischemia–reperfusion). Zhang et al. demonstrated that hypoxia elicits FUNDC1-mediated mitophagy, evidenced by elevated mitochondrial proteins and the reciprocal marker of autophagy, p62, in FUNDC1 knockout mice, and no change in LC3-I expression (524). Further, platelets (which are exposed to varying levels of oxygen throughout the vasculature) from FUNDC1 knockout (KO) mice do not demonstrate formation of autophagosomes containing mitochondria. These responses are autophagy-dependent, as deletion of Atg5 resulted in no mitophagy activation in response to hypoxia in platelets.
Functionally, exposure to coronary ischemia–reperfusion increased mitophagy in wild-type (Cre negative) but not FUNDC1 platelet-specific KO (524). Although the specific role of FUNDC1-mediated mitophagy has been described in other organs and tissues, the specific role within the vasculature and whether this is altered with aging is unclear, representing a ripe area for future investigation. Fundamental questions regarding the role of FUNDC1 need to be addressed. Specifically, is FUNDC1-mediated mitophagy altered with disease or aging? In vascular-specific diseases such as peripheral arterial disease (PAD), the primary culprit of symptoms is a mismatching of oxygen delivery to oxygen demand, rendering tissues hypoxic. Do aberrant deviations in FUNDC1 and mitochondrial function play a role in the development and progression of PAD?
The aforementioned mitophagy-related receptors BNIP3 and NIX are also oxygen sensitive. Both BNIP3 and NIX express LC3 interacting region and interact with LC3 via phosphorylation of various serine sites to degrade mitochondria via canonical autophagy. In old mouse aortas (aged 27–28 months), no BNIP3 protein expression differences were found relative to young mice (242); however, protein expression of NIX was increased in older mice (18–19 months) basally and in response to serum starvation relative to young mice (457).
Interestingly, administration of trehalose (2% in drinking water) increased protein expression of BNIP3 (242). Within these studies, mitochondrial morphology of the older mice aortas was not examined. From a cell perspective, BNIP3-mediated mitophagy is important for maintenance of homeostasis in response to experimental hyperglycemia in the cerebral vasculature (207). Together, these findings indicate that receptor-mediated-mitophagy within the vasculature may be impaired with advancing age and in the presence of cardiovascular risk factors. Future investigations into the specific roles of receptor-mediated mitophagy are warranted.
2.5. Non-receptor mediated mitophagy
The most widely described mitophagy pathway is Pink1-Parkin mitophagy. Broadly, under non-stressed conditions, Pink1 is shuttled into the mitochondria, where it is subsequently degraded. Pink1 and Parkin act in concert with mitochondrial fission to encapsulate and degrade damaged mitochondria. On mitochondrial outer membrane depolarization, Pink1 accumulates on the outer membrane and recruits the E3 ubiquitin ligase Parkin. Parkin ubiquinates the damaged, depolarized mitochondria, which are then encapsulated by an autophagosome that fuses with an autolysosome containing acidic hydrolases, thereby degrading the mitochondria. Loss of Parkin function or aberrant actions of regulated (S-nitrosylated) Parkin results in the inadequate clearance of damaged mitochondria and may trigger further mitochondrial fission or fragmentation (74, 269, 479). Parkin plays a crucial role in mitochondrial dynamics. The role of mitophagy within premature hypertension and vascular aging has recently been succinctly reviewed elsewhere (390).
In relation to vascular aging, LaRocca et al. (242) demonstrated that within the aorta of old mice, protein expression of Parkin is reduced relative to young mice. The reduction in Parkin was paralleled with an increase in superoxide production and an increase in aortic stiffness (aortic pulse-wave velocity). Interestingly, Tyrrell et al. (457) demonstrated that aged mice demonstrated increased levels of Parkin within the aorta, coinciding with reduced mitochondrial function and increased mitophagy.
The differences in Parkin expression in these two studies may be due to the selected age range, as LaRocca et al. (242) studied mice that can be considered very old (27–28 months) whereas Tyrrell et al. (457) were interested in mice transitioning from middle age to old (18–19 months). Activation of autophagy with trehalose (2% drinking water) normalized Parkin expression, whereas spermidine did not influence levels of Pink-1 or Parkin in older mice fed a low-fat diet. Interestingly however, when mice were administered a western style diet (high fat, high sugar), spermidine offset the hypercholesteremic-induced increases in Parkin, and it preserved mitochondrial respiration within the aorta (457). Future investigations should examine the association between changes in mitochondrial proteins such as Pink1, Parkin, BNIP3, FUNDC1, and NIX and alterations in mitochondrial structure as they relate to functional outcomes.
3. ROS/RNS-Direct Vasodilation in Relation to Mitochondria and Aging
3.1. Age-related decline in nitric oxide with increased mitochondrial hydrogen peroxide signaling
In healthy adult individuals, nitric oxide plays a pivotal role in endothelium-dependent regulation of coronary vasodilation and blood flow (213). In the aging population, the endothelium has diminished control over vascular tone (496). This loss of endothelial function is induced by a reduction in nitric oxide availability via diminished flow-induced endothelial nitric oxide production (99, 213, 299) and/or amplified nitric oxide scavenging due to oxidative stress (45, 267, 299). In addition, nitric oxide generated in response to local hypoxia from red blood cell nitric oxide synthase (RBC-NOS) and RBC ATP-purinergic receptor agonist signaling-induced nitric oxide are reduced with aging due to reduced red blood cell deformability and oxidative stress (236, 335, 364, 365).
Indeed, oxidative stress is a primary player in declining endothelial function in aging; excessive production of superoxide in the aged vasculature leads to an increase in nitric oxide scavenging as superoxide and nitric oxide combine to produce the cytotoxic free radical peroxynitrite in mice (31), rats (130, 172, 463), and humans (367). Combination of superoxide and nitric oxide to produce peroxynitrite involves a diffusion-limited reaction that occurs approximately three times faster than the dismutation of superoxide to hydrogen peroxide by SOD (178, 308).
Flow-induced hydrogen peroxide generation is not diminished in advanced age (213), but rather is excessively enhanced in an aging rat model (322). Beyer et al. utilized a series of inhibitors/scavengers of vasodilative precursors (indomethacin for COX mediated prostacyclin synthesis, L-NAME for eNOS, and PEG-Catalase for hydrogen peroxide) and evaluated vasodilative function in human coronary and adipose microvessels across the human lifespan (30). The mechanism of FMD evolves throughout life. In children (0–18 years old) and young adults (19–55 years old), prostacyclin PGI2 and nitric oxide are the main vasodilative mediators, respectively. With onset of coronary artery disease (CAD) in older adults (>55 years old), the FMD mediator switches to hydrogen peroxide independent of age, and total vasodilative effect is diminished irrespective of aging, effects also seen in rodents (213). Supplementation of exogenous low-dose DETANONOate (nitric oxide donor, mimicking youth) in CAD vessels reduces the mtROS and hydrogen peroxide-FMD response, whereas eNOS blockade in young vessels (mimicking age/CAD) induces compensatory hydrogen peroxide-FMD (30). Collectively, this evidence demonstrates a plasticity in vasodilator mechanisms that changes throughout the human lifespan.
Hydrogen peroxide (as well as superoxide and peroxynitrite) can serve as an endothelium-derived hyperpolarizing factor that acts through calcium-activated potassium channels to elicit FMD (256, 267, 303, 324, 487). It is thought that eNOS-generated superoxide may be a primary contributor to hydrogen peroxide generated for FMD (414). Against the dogma that ROS are intrinsically harmful, it appears that they function as a compensatory mechanism in aging (and CAD), at least initially, leading to reduced nitric oxide bioavailability although with less efficacious vasodilation (303). Hydrogen peroxide may be the least damaging of the ROS, as it cannot quench nitric oxide and may confer some benefit. In some cases, hydrogen peroxide has been shown to improve nitric oxide production, upregulate eNOS expression and function, and enhance nitric oxide-FMD by stabilizing sGCβ1 mRNA to increase cGMF production (50, 51, 114, 279, 446).
Peroxynitrite may have a beneficial function, as it is a nitric oxide donor, acting as a reserve pool for nitric oxide (303). However, these potential beneficial effects are limited or reversed when concentrations exceed buffering capacity due to antioxidant protein dysfunction and pro-oxidant signaling, as discussed in the first section, as well as by enhanced toxic hydroxyl radical formation. In some cases, hydrogen peroxide can exhibit vasoconstriction (303). Overall, these lessons in the complexity of redox balance for vasodilative function and perfusion capacity teach us that the aim for future therapeutics should be to restore youthful nitric oxide-mediated FMD as opposed to merely inhibiting ROS, as ROS serve as a beneficial compensatory mechanism (although insufficient).
3.2. ACh and sodium nitroprusside-mediated vasodilation reduced in age
Impairment of EDD in aging is further evidenced by reduced relaxation to the eNOS agonist ACh in mice (97, 157, 252, 511), rats (72, 299, 463, 496), pigs (353), and humans (318, 431, 432). Proper dose-dependent dilation to ACh indicates an intact, healthy endothelium, whereas vasoconstriction to ACh may point to endothelial dysfunction (447). In contrast, endothelium-independent relaxation to sodium nitroprusside (SNP) is not commonly compromised (244, 431, 432, 463), which suggests that SMC-mediated dilation to nitric oxide is preserved in advancing age. Oxidative stress appears to trigger this age-related loss in endothelial function as well, as the simultaneous decreasing nitric oxide and increasing superoxide levels result in deteriorating nitric oxide-mediated signaling. Scavenging of superoxide via SOD transiently reestablished normal endothelial function in a transgenic mouse model of aging (97).
3.3. mtROS uncouple eNOS in age, shift to superoxide instead of nitric oxide production
The mitochondria play an incredibly important role in vascular stasis, regulating ROS levels via a complex interplay of enzymes as well as non-enzymatic antioxidants (12). When this mitochondrial regulatory network is imbalanced, vascular deficits follow. For example, the uncoupling of eNOS by mtROS plays a pivotal role in the shift from nitric oxide to superoxide in advancing age (307). In addition to direct mtROS release and NOX, uncoupled eNOS is one of the primary sources of endothelium-derived superoxide (99, 172). Tetrahydrobiopterin (or BH4), an essential cofactor in the catalysis of nitric oxide from
These are example of ROS-induced ROS release (RIRR), which can also trigger dysfunctional mitochondria and/or NOX to produce additional superoxide (85, 87, 531). In RIRR events, a slow accumulation of ROS serves as the trigger for the ensuing burst of ROS released on depolarization of the mitochondrial membrane potential (530, 531) and opening of mPTP (86). This phenomenon can be adaptive, as in instances of culling injurious organelles or cells, or maladaptive, leading to the death of otherwise healthy mitochondria and cells (531). The opening of mPTP can also directly stimulate the release of superoxide from the mitochondrial matrix; this efflux of superoxide from a subset of dysfunctional and/or activated mitochondria can prompt nearby mitochondria to do the same, leading to even higher levels of circulating ROS (85).
Uncoupled eNOS can also lead to endothelial dysfunction in both coronary and peripheral vessels (87). SOD2 plays a vital role in the dismutation of superoxide to hydrogen peroxide and the regulation of cellular redox homeostasis (387). In a transgenic mouse model of aging involving SOD2, amplified levels of mtROS and mtDNA damage were found in SOD2+/− mice compared with wild-type littermates; the resulting eNOS uncoupling in these mice was evidenced by severely impaired vasorelaxation (491). Transgenic SOD2−/− mice only survive roughly 10 days to 3 weeks due to a number of physiological ailments, including extensive mitochondrial injury within cardiomyocytes (245, 257). Similar studies have shown that endothelial dysfunction associated with eNOS uncoupling by mtROS can be prevented by the mitochondrial-targeted antioxidant mitoTEMPO (102). In addition, SOD2-overexpressing mice were moderately spared from eNOS uncoupling-induced vascular dysfunction (394).
3.4. FMD by nitric oxide or hydrogen peroxide influenced by mitochondria signaling (ROS)-dependent exocytosis of endothelium-derived extracellular vesicles
Mitochondrial generation of both superoxide (267) and hydrogen peroxide (48) directly contribute to flow-induced vasodilation and are upregulated in response to shear stress. In human coronary and other resistance arterioles, mitochondrial-derived superoxide and hydrogen peroxide are required for the formation of endothelium-derived extracellular vesicles (eEV), which bind with the cell membrane and release their contents into the circulation (140). These eEV can be beneficial, assisting with protein, lipid, and microRNA transport. However, the presence of high levels of eEV in blood serum has been linked to endothelial dysfunction (36), likely through the contents contained within the eEV, including ceramide and plasminogen activator inhibitor 1, which can cause arterioles to shift from nitric oxide to hydrogen peroxide production, inducing further endothelial dysfunction (140). Indeed, exogenous introduction of ceramide to healthy human arterioles induces mtROS production, leading to a diseased phenotype (139).
3.5. Mitochondrial respiratory dysfunction correlates to endothelium-dependent (ACh) but not independent (SNP) vasodilation in aging, likely through free radical-linked mechanism
Mitochondrial respiration also plays a role in vasodilatory responses. In experiments involving human skeletal muscle feed arteries, vasodilation via endothelium-dependent agonist ACh is drastically reduced in vessels from older patients and directly correlated via linear regression to reduced mitochondrial respiratory function within the vessels in an age-dependent fashion (346). Vascular mitochondrial oxidative respiratory capacity, as measured by state 3 mitochondrial respiratory complex I as well as complex I + II, is significantly higher in vessels from young when compared with elderly subjects (345, 346). Similar to the EDD deficits, this mitochondrial respiratory dysfunction in older patients is significantly correlated with endothelium-dependent (ACh) but not endothelium-independent (SNP) vasodilation, again suggesting normal smooth muscle control of dilation to nitric oxide even in the elderly population. Concomitantly, mitochondrial levels of superoxide are highest in the older patients.
3.6. FMD attenuation in aging correlates with increased S-nitrotyrosine
The attenuation of FMD observed in advanced aging is correlated with increased nitrotyrosine, an oxidatively modified amino acid and marker of oxidative stress usually linked to peroxynitrite. In humans, excessive levels of nitrotyrosine were found in brachial artery endothelial cells of older men; deficits in flow-mediated vasodilation were strongly correlated with increased levels of nitrotyrosine (107). Similar age-linked increases in nitrotyrosine have been described in rat arteries (81, 427, 463). In aortas isolated from aged rats, decreased nitric oxide, increased eNOS activity, superoxide overproduction, and subsequent peroxynitrite formation all combined to increase nitrosylation of tyrosine within mitochondrial SOD2 in a dynamic example of the complex interplay of ROS within the aging vasculature (463). In addition, overexpression of nitrotyrosine and flow-induced deficits have also been linked to estrogen levels, as ovariectomized rats show similar levels of peroxynitrite-induced nitrosylation when compared with aged rats (213). Scavenging of peroxynitrite via FeTMPyP, a synthetic porphyrin complexed with iron, significantly improved endothelium-dependent relaxation in aging rats (427). Antibiotic treatment has also shown beneficial antioxidant effects in aortic oxidative stress of old mice, as shown by decreased pro-inflammatory cytokines, decreased nitrotyrosine, and lower levels of superoxide (46).
4. Alterations to Endothelium-Dependent Hyperpolarization in Relation to Mitochondrial Redox Balance with Aging
4.1. Description of normal vasodilative endothelium-dependent hyperpolarization
Various factors mediate endothelium-dependent hyperpolarization (EDH), starting in the endothelial membrane and conducting through to the VSM cells to facilitate vasodilation. When endothelial intracellular [Ca2+] is increased due to shear stress activation of mechanosensitive transient receptor potential vanilloid type 4 channel (TRPV4), calcium-dependent potassium channels (SKCa, calcium-sensitive small conductance potassium channels [SKCa] and calcium-sensitive intermediate conductance potassium channels [IKCa]) cause hyperpolarization and K+ efflux, thereby stimulating VSM Na+, K+-ATPase and inward rectifying potassium channels (KIR) (147). The calcium increase to initiate this pathway can also activate eNOS to produce nitric oxide; however, this is diminished with aging and instead is instigated by ROS such as hydrogen peroxide to activate the endothelial KCa channels (361).
Hyperpolarization and calcium signaling are propagated through connexin gap junctions to the VSM, which together with hyperpolarization from other VSM potassium channels (voltage-gated potassium channels [Kv], ATP-sensitive potassium channels [KATP], and calcium-dependent large conductance potassium channels [BKCa]) lead to closing of voltage-gated calcium channels (VGCCs), reducing intracellular calcium and leading to vasodilation (147 –149, 203). ACh- and FMD-mediated vasodilation can be blocked in part by inhibiting endothelial potassium channels with apamin (SKCa) and charybdotoxin (IKCa and BKCa), highlighting the critical role of potassium channels in the signaling of vasodilation. These pathways are of unique importance, as they allow axial conductive signaling for coordinated modulation of tone and blood flow of a large area of vascular network. Therefore, alterations due to pathology may be especially consequential. More details on EDH mechanisms can be read in the comprehensive reviews by Feletou and Garland and Dora and depicted along with influences of aging and oxidative stress in Figures 2 and 3 (126, 147).

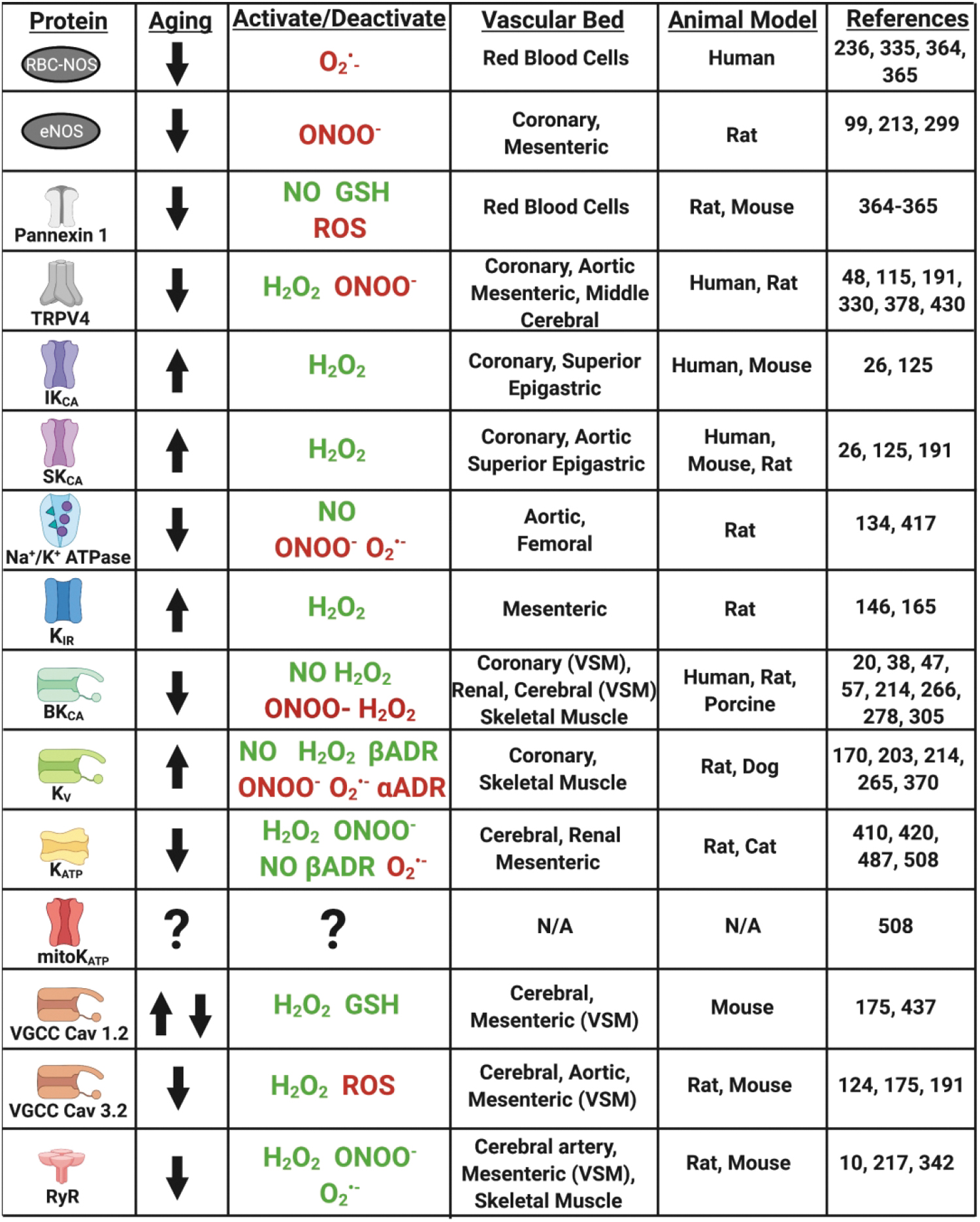
4.2. Effect of ROS and RNS on EDH
Mitochondrial superoxide, hydrogen peroxide, and peroxynitrite demonstrate vasodilator actions through activation of hyperpolarization pathways, and therefore there is a reliance on this in aging. This effect is partly mediated by TRPV4, which can be activated by hydrogen peroxide but is inhibited by peroxynitrite (48, 330, 428, 430).
Superoxide generated through xanthine oxidase and peroxynitrite has been shown to inhibit Kv channel activity, which can be restored by SOD whereas hydrogen peroxide is able to activate Kv channels (170, 265, 370). In rat cerebral arterioles, peroxynitrite inhibits BKCa channels (47, 266). In porcine coronary arteries and rat renal microvessels, nitric oxide and hydrogen peroxide were shown to activate BKCa (20, 305, 306). On the contrary, hydrogen peroxide has also been shown to close BKCa channels in porcine renal artery patch clamp experiments (38). This discrepancy likely indicates a direct inactivating and indirect activating effect of hydrogen peroxide on BKCa channels.
Superoxide decreases whereas hydrogen peroxide, peroxynitrite, and nitric oxide enhance KATP function in renal, cerebral, mesenteric, and coronary vasculature (420, 487). The KIR channel is activated by superoxide in rat mesenteric artery (146). These complex effects are likely due to thiol modification of key cysteines, including glutathionylation (510). Further details on how oxidative stress affects potassium channel function are available in various reviews (59, 69, 170, 179).
4.3. Mitochondrial depolarization-mediated vasodilation
Mitochondrial depolarization induced by ROS generates calcium sparks that have vascular effects (501). In a study by Katakam et al., ROS-dependent (diazoxide) and -independent (BMS-191095) activators of mitochondrial depolarization were utilized in wild-type, lean, and insulin resistant obese rats to elucidate a pathway for mitochondrial depolarization-mediated vasodilation in cerebral artery VSM (217). In the VSM, diazoxide and BMS-191095 lead to activation of mitochondrial KATP channel activation and depolarization. In mitochondrial microdomains adjacent to the sarcoplasmic reticulum, ryanodine-sensitive calcium channels become activated, releasing calcium sparks that activate plasma membrane BKCa channels, leading to K+ efflux and hyperpolarization to close VGCCs to decrease overall intracellular [Ca2+] and facilitate vasodilation (217). Activation of the ryanodine receptor occurs due to ROS-mediated post-translational modification (hydrogen peroxide-, superoxide-, or peroxynitrite-mediated) (10, 217, 342). However, since BMS-191905 also activated this pathway, the authors conclude that there is a secondary ROS-independent mechanism for mitochondrial depolarization-mediated vasodilation.
In obese insulin resistant rats, there is impairment of calcium spark release due to ER stress, impaired activation of BKCa, and plasma membrane ATP-sensitive K+ channels. In aging, the functional release of calcium sparks from ryanodine receptor activation with caffeine is reduced, owing to reduced coupling of the ryanodine receptor with the T-type calcium channel CaV3.2 in mouse mesenteric arteries (124). It is unknown how aging affects mitochondrial KATP function, although there are indications of dysfunction since the potassium cycle is impaired with decreased intramitochondrial potassium in aging rats (508).
4.4. Effect of aging on EDH
During certain phases of pathology or aging with active lifestyle (i.e., “healthy aging”), hyperpolarization-mediated vasodilative mechanisms can increase in function to partially compensate for lack of nitric oxide-mediated signaling (25, 313, 403, 404). However, in diabetes and studies of sedentary aging, it appears that elevated oxidative stress including increased superoxide, hydrogen peroxide, and peroxynitrite overall culminates in reduced EDH function (125, 126, 142, 265, 525). For instance, in aged rats, EDH-mediated relaxation from ACh or TRPV4 activator GSK1016790A was reduced compared with young rats; the response in young rats is reduced to old levels by inhibiting TRPV4 and SKCa (191). In old age, Feher et al. found that although local responses to bradykinin remain intact with age, the conductive axial response (spread hyperpolarization and dilation at distant sites) was diminished in coronary arteries of humans, and local conductive responses were due to enhanced SKCa and IKCa activity, with similar findings observed in mouse models (26, 125). Similarly, KIR function is increased with aging, with no change in KIR mRNA (176).
On the other hand, endothelial TRPV4 expression and calcium signaling, an initiator of the hyperpolarization vasodilation response, are reduced in aging but can be restored by lentiviral induced overexpression in aging rat mesenteric arteries (115). Expression and function of the BKCa channel is diminished in aging mesenteric, skeletal muscle, and coronary (but not cerebral) vasculature, leading to inhibited vasorelaxation (57, 214, 278). However, KV1.5 channel protein expression does not change with age, although inhibition leads to increased myogenic tone in aging versus youth, indicating increased KV1.5 function with aging in skeletal muscle arterioles (214). Direct inhibition of VSM cell Na+/K+ ATPase leads to constriction in young, but not old rats, indicating potentially reduced contribution for the Na/K ATPase in EDH-mediated vasodilation in aging (417). It is known that ROS-mediated glutathionylation of the β1 subunit inhibits Na+/K+ ATPase function and is reversible by nitric oxide (134).
In addition, KATP channels are dysfunctional during aging, as the necessary subunits Kir6.1 and SUR2B are downregulated and protein kinase A (PKA)-mediated activation is attenuated (although activation with nicorandil and direct PKA activation leads to no age-related differences) (508). The Cav3.2 T-type VGCC, involved in the ryanodine receptor/BKCa-mediated hyperpolarization and dilation response, is also downregulated in aging, including in coronary arteries, and can be inhibited by hydrogen peroxide and ROS (175, 190). Alternatively, the L-Type Cav1.2 channel expression and function in aging varies greatly from study to study, being either over- or under-expressed depending on the arterial bed being studied. In coronary artery VSM cells, Cav1.2 expression and current is not affected by aging in rats but is decreased in posterior and middle cerebral arteries and increased in mesenteric arteries in mice (175). The Cav1.2 channel can be glutathionylated and activated by hydrogen peroxide and oxidized glutathione (437).
Considering ROS are important factors in EDH, yet excessive oxidative stress tends to have inhibitory effects on EDH, this implies that a fine homeostatic balance is required for ideal vasodilative performance. The EDH is also a minor contributor to relaxation during β-adrenergic-mediated vasodilation through simultaneous activation of Kv and KATP channels (170, 203, 410), and α-adrenergic agonism can close Kv channels (203). Further, βADR activation leads to a transient increase of ROS, which can activate L-type calcium channels and induce alterations in calcium signaling (11, 37). Therefore, EDH dysfunction in aging may also contribute to βADR dysfunction. In addition, the fine redox balance necessary for functional EDH extends to intrinsic βADR function. In all, mitochondrial dysfunction plays a critical role in the signaling of vasodilation, and alterations of mitochondrial function with aging have significant implications for vasodilative function.
5. Adrenergic Alterations in Relation to Mitochondrial Redox Homeostasis with Aging
5.1. Description of adrenergic receptor homeostatic shift with aging
The expression and function of adrenergic receptors varies with tissue location and branching order, and they are influenced by redox status of endothelial cells causing adrenergic dysfunction in aging. The major role of these receptors on vascular tissue, specifically, is to mediate vasorelaxation or vasoconstriction via agonism typically in response to circulating catecholamines. The β-adrenergic receptors β1, β2, and β3 activate G-stimulatory (Gs) protein to activate adenylate cyclase, which, in turn, converts ATP into cyclic AMP (cAMP), leading to activation of PKA to phosphorylate the myosin light chain kinase, resulting in inactivation of the myosin light chain, and finally inducing vasodilation (96, 466).
On the other hand, α1ADR activates Gq alpha subunit and phospholipase C to hydrolyze phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol triphosphate (IP3), which, in turn, activates its receptor in the ER to release calcium and induce vasoconstriction. The α2 receptors induce vasoconstriction via activation of G inhibitory (Gi) protein, thereby inhibiting adenylate cyclase.
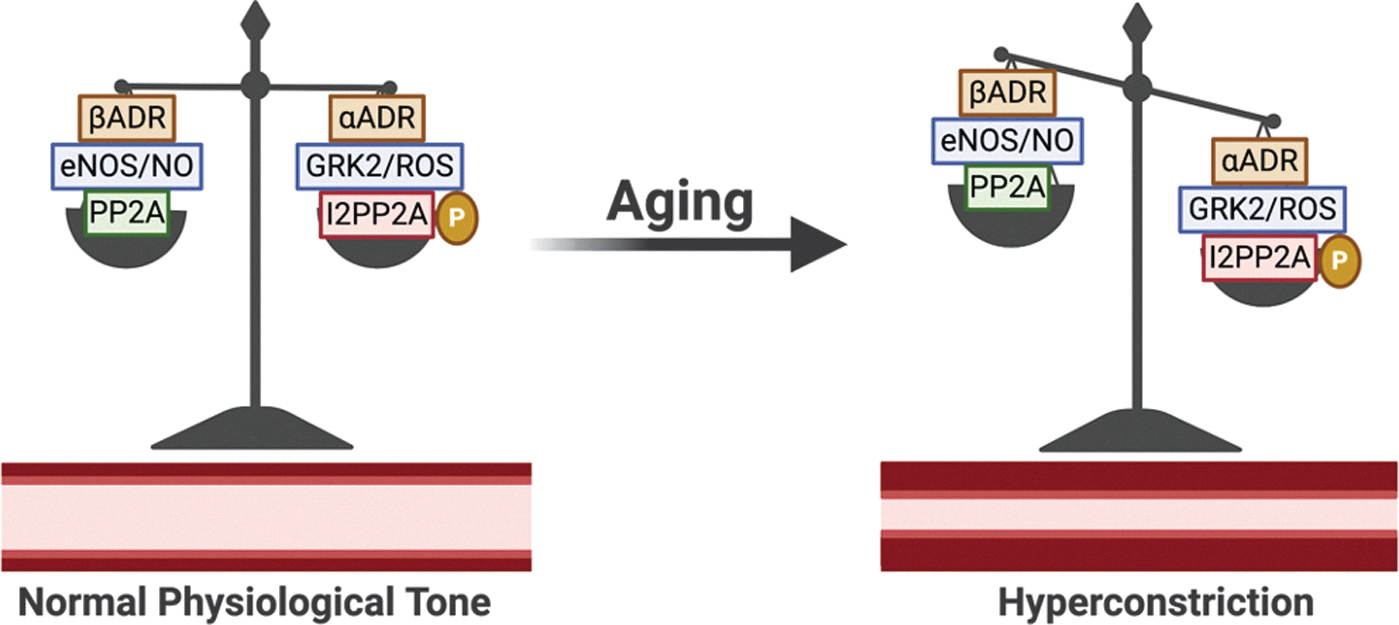
In the coronary vasculature, endothelial cells express predominantly β1 whereas the SMCs express predominantly β1 and α1 receptors (19). As branching order increases, relative expression of β1ADR and α1ADR decreases whereas β2ADR, β3ADR, and α2ADR increase. In healthy vessels, the balance favors β vasodilation to adrenergic agonism; however, with aging or disease, this balance shifts to favor αADR-mediated vasoconstriction (19, 376). This is, in part, due to the decline in function and expression of βADRs with age in many tissues, including the aortic (270), mesenteric (142, 143), and coronary (376) vasculature, but increased in aging cerebral vasculature (211, 389, 395).
Coronary and aortic α1ADR, on the other hand, do not display changes in expression with age, promoting a propensity for hyperconstriction (19, 104, 169), whereas α1ADR expression or function with aging decreases in skeletal muscle and renal artery, although skeletal muscle is paradoxically hyperconstricted with aging (258, 348, 377, 500). The changes in vascular adrenergic function with age are irrespective of alterations of downstream signaling. βADR dysfunction with age leads to insufficient cAMP synthesis, whereas exogenous stimulation of elements downstream from the receptor stimulates normal cAMP production (209, 218, 271, 272, 398). Age-related βADR function is not due to G-protein switching from stimulatory to inhibitory (218, 272, 397), suggesting dysfunction of the βADR itself or with coupling, rather than downstream signaling messengers.
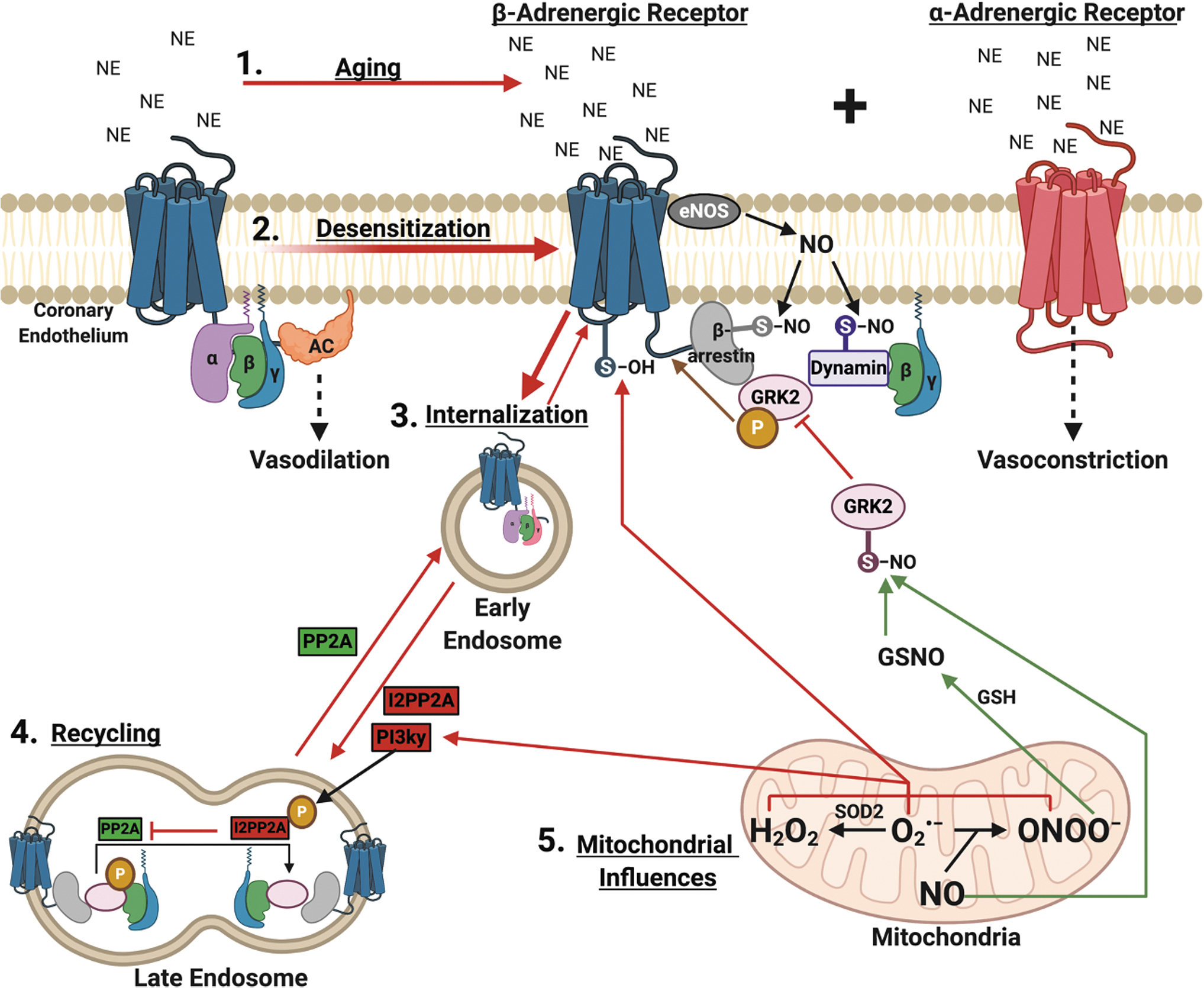
5.2. Effect of aging on adrenergic receptor regulation through desensitization, internalization, and recycling
Current consensus is that functional adrenergic changes in aging are mediated by regulatory proteins and post-translational modifications at the level of the βADR, which can be influenced by aging-induced redox changes. On agonism in young vessels, a protein complex including either G-protein receptor kinase (GRK) or β-Adrenergic Receptor Kinase, and β-arrestin associates with the βADR to cause desensitization, blunting the ability of agonists to increase cAMP. This regulatory system is upregulated in aging, alongside increased circulating catecholamines (61, 341). Interestingly, GRK2 (the most commonly implicated GRK) increases 3.6- and 1.5-fold in the cytosolic and membrane fraction, respectively, with β-arrestin increasing 1.6-fold in aged (24 months old) Fischer 344 rat aortas (145, 396).
On receptor-agonist binding, GRKs associate with the β receptor, leading to receptor phosphorylation, thereby sterically hindering the G protein coupling and causing G stimulatory protein dissociation (28). The β receptor can also be phosphorylated independent of agonist (or at very low agonist concentration) binding by PKA and PKC. Both of these mechanisms are negative feedback regulators since their activation is cAMP-dependent (251). In addition, GRK-mediated phosphorylation recruits β-arrestin to the receptor-GRK complex, further causing steric hindrance and desensitization (251). β-arrestin primes the β-receptor for dynamin-mediated internalization into clathrin-coated endosomes for storage, trafficking to lysosomes for degradation, or eventual recycling to the plasma membrane (112, 128, 250, 273).
These pro-desensitization and internalization processes exist in a homeostatic balance with the process of resensitization, where βADR receptors are dephosphorylated at the plasma membrane or predominantly at the endosome, then trafficked back to the plasma membrane ready to receive agonistic signals and induce vasodilation (411, 466, 516, 519, 520). Resensitization is mediated by phosphatases, namely protein phosphatase 2A (PP2A) (235). This homeostatic balance between desensitization/internalization and resensitization favors the former in aging, not only because of increased GRK2 and β-arrestin expression, but also due to inhibition of resensitization. The dephosphorylation action by PP2A is inhibited by the endogenous inhibitor of PP2A, I2PP2A. I2PP2A is activated by PI3Kγ-mediated phosphorylation (357, 466). The Ang II activation of the AT1 receptor activates PI3Kγ-mediated pathways and increases with aging (181, 240, 332, 333).
Oxidative stress has been shown to activate PI3Kγ signaling in the diabetic rat cardiomyocyte model (high glucose co-culture) (418). Together, aging-induced Ang II and ROS signaling could, in part, explain the eschewed balance toward βADR phosphorylation, desensitization, and, ultimately, hyperconstriction with diminished agonist-relaxation. Inhibiting PI3Kγ in peripheral vessels rescues endothelium-independent vasodilation through induced L-type calcium channel dysfunction and also preserves β-adrenergic receptor function in cardiomyocytes from heart failure rats, although the ability to preserve βADR function in the endothelium by PI3Kγ inhibition still needs confirmation (55, 56, 311, 321, 331, 350, 412, 467, 468).
5.3. Effect of redox status on adrenergic receptor regulation
The processes mentioned earlier can be influenced by redox status to affect βADR function and location. The βADR and its regulatory proteins can be oxidized/nitrosylated by ROS/RNS. A major known regulator of desensitization/internalization is SNO post-translational modification (83, 466); SNO of β-arrestin and dynamin favor desensitization/internalization and inhibit αADR mediated vasoconstriction (4, 323, 337, 464, 478). In contrast, SNO of GRK2 prevents desensitization/internalization and is the major post-translational regulator against desensitization/internalization (275, 493). Studies utilizing exogenous SNO agents find βADR localization to the plasma membrane, with decreased β-arrestin localization, and enhanced vasodilation suggesting that SNO of GRK2 supersedes pro-desensitization/internalization SNO processes (493). In addition, PKA is constitutively activated by SNO and facilitates cross-talk by increasing cAMP to induce vasodilation in “β-adrenergic-like” signaling (49).
Of equal importance is the process for denitrosylation of these proteins by nitrosoglutathione reductase. Knockout of nitrosoglutathione reductase reduces vascular tone, whereas inhibition improves β-adrenergic function, suggesting that a fine balance of SNO is necessary for ideal βADR function (27, 68, 129). These proteins are nitrosylated, in part, via nitric oxide produced by eNOS, which is dysfunctional in aging (511). Therefore, it is possible that aging-mediated reduction of eNOS function, increased superoxide (which siphons nitric oxide to form peroxynitrite), and reduced nitric oxide bioavailability reduce the ability to maintain the nitrosylated status of these proteins, causing a shift favoring adrenergic receptor desensitization/internalization.
Although it has been known that oxidative stress negatively impacts functional vasodilation, its role in βADR physiology has only recently being investigated. The β1–3ADRs contain cysteine residues that are susceptible to oxidation, whereas only β1–2ADRs have extracellular tyrosine residues susceptible to nitration (254). Unpublished data from our lab in female Fischer-344 rats suggest that percent vasorelaxation to β1 agonist norepinephrine directly and positively correlates with nitric oxide concentration and inversely correlates with superoxide and hydrogen peroxide, but not peroxynitrite concentration, in an aging-dependent manner (unpublished observations). Exogenous superoxide/hydrogen peroxide incubation in young rat vessels completely abrogates β1ADR receptor agonist (dobutamine and norepinephrine) mediated vasodilation (116).
Exogenous peroxynitrite also blunts vasodilation to S-nitrosocysteine (nitrosylating agent) by oxidizing cysteine or nitrosylating tyrosine recognition sites (253). Peroxynitrite forms S-nitrosoglutathione with glutathione presence, providing a potential pathway for SNO of GRK2 and inhibition of desensitization/internalization (or dynamin/β-arrestin promoting desensitization/internalization). This pathway is likely reduced with aging as is GSH (379). Of interest, S-nitrosoglutathione also inhibits NOX and could, therefore, also reduce oxidative stress (379). Phosphorylated STAT3 is a positive transcriptional regulator of cardiomyocyte β1ADR that also mediates transcription of antioxidant (SOD2), proangiogenic (VEGF), and antiapoptotic proteins, and it exerts noncanonical actions to reduce ROS production, regulate mitochondrial complex 1 function and mPTP opening (523, 532). In addition, STAT3 can become activated on agonism of the β1ADR (523). This provides another regulatory link between β1ADR and ROS, and of interest STAT3 is known to be reduced with aging and therefore contributing to endothelial dysfunction, although many of these listed actions of STAT3 need to be confirmed in the vasculature (532).
Although oxidation tends to attenuate βADR vasodilatory function, nitrosylation has a protective effect. Exogenous S-nitrosoglutathione (a nitrosylating agent) is able to mediate nitrosylation of GRK2, inhibiting phosphorylation and subsequent desensitization and internalization of the β2ADR even during isoproterenol agonism, with subsequent cAMP production (275). Frame et al. showed that pre-incubation with SNP (a nitrosylating agent and nitric oxide donor) improves β2ADR function to vasodilate in vivo arcade and terminal arteries of the hamster cheek pouch tissue (137). Specifically, SNP pre-incubation “uncovers” distinct pools of βADR receptors to allow for increased potency at the picomolar range (although with reduced efficacy at the micromolar range).
In addition, in the study by Frame et al., a culture system and fluorescence imaging (fluorescence resonance energy transfer analysis) were used to show the influence of nitrosative and oxidative stress on internalization of the β2-adrenergic receptor. Exogenous superoxide production increased internalization of the β2ADR. Desensitization/internalization was blocked with SNP and dynasore (dynamin inhibitor of endocytosis) incubation; GRK2 was seemingly nitrosylated by nitric oxide, whereas dynamin-mediated endosome formation was blocked by dynasore. The concentration response and fluorescence data suggest that with this treatment, some of the β2ADR were protected from internalization (i.e., not internalized) and remain functional at the plasma membrane. It was hypothesized that preventing desensitization and endosome formation would improve βADR function in the classic dose ranges (10−9–10−4 M). Instead, SNP/dynasore pretreatment altered the pharmacodynamics by improving isoproterenol potency but reducing efficacy. The explanation is that increased cellular RNS state also facilitates dynamin and β-arrestin nitrosylation, which are pro-internalization processes. The balance between GRK2 nitrosylation/dynamin inhibition and dynamin/β-arrestin nitrosative activation leads to the functional changes seen. It is speculated that SNP shortens the time for β2ADR dephosphorylation with increased cycling of phosphorylation state, uncovering dilation potential at lower doses (10−14–10−11 M).
To summarize, βADR density and function are reduced in aging vasculature, leading to diminished vasodilatory capacity and hyperconstricted tone. Adrenergic regulatory proteins are subject to regulation by ROS/RNS. Therefore, aging-mediated alterations of redox balance may influence the homeostatic balance of desensitization/internalization and resensitization. This is supported since changes in ROS/RNS with age correlate with β1ADR function and the results of Frame et al. show that exogenously supplying ROS (mimicking aging) or RNS (mimicking youth) influences desensitization/internalization of the β2-adrenergic receptors (although in HeLa cells, not endothelial cells) (137). These complex interactions are illustrated in Figure 4, including pathways for how ROS/RNS may influence βADR function in what we name for the first time the ROS/RNS-βADR Desensitization and Internalization Axis. Further experimentation in young versus aged endothelium, mimicking studies such as by Frame et al., are needed to establish the validity of this axis, which could represent several novel opportunities for therapeutic targeting for diseases with adrenergic pathologies (137).
5.4. Clinical consequences of aging-induced adrenergic alterations
These alterations in adrenergic homeostasis with aging that favor αADR hyperconstriction manifest in clinical pathologies such as CAD and coronary microvascular disease (CMD), also known as cardiac syndrome X (19, 376). CMD presents in a majority of aging postmenopausal women with chronic angina due to microvascular hyperconstriction, as opposed to atherosclerotic blockage seen typically in men. Diagnostically, CMD is suspected when a female patient has a chief complaint of chest pain but there is no obvious obstruction on a coronary angiogram. Clinically, CMD is defined as having coronary flow reserve (CFR) ≤2.5, endothelial dysfunction with constriction to ACh, or <20% coronary dilation to nitroglycerin. The CFR may be calculated via dobutamine (β1ADR agonist) or adenosine-induced stress echocardiography, highlighting the adrenergic ties in CMD (348), although other imaging modalities (positron emission tomography, computerized tomography) are more often employed clinically with various merits and cons to each (348).
The CMD has limited guidelines and treatment effectiveness. The European Society of Cardiology recommends symptomatic relief with nitrate, β-blocker, or calcium channel blocker (443). However, such a strategy does not address the root cause of the pathology, that is, hyperconstriction; rather, it focuses on anti-ischemic protection via reduced inotropism and oxygen conservation. Further, β-blockers may actually potentiate dysfunction by blocking βADR-mediated dilation. The information in this section as well as the proposed ROS/RNS-βADR Desensitization and Internalization Axis (Figs. 4 and 5) provides numerous potential targets to restore functional βADR such as attenuating aging-induced mitochondrial-derived oxidative stress, inhibiting proteins involved in the βADR desensitization and internalization process, or activation of βADR recycling.
In CMD, such a strategy is of significant clinical interest considering that up to 75% of women and 15% of men with angina have no obstructive coronary pathology, representing 90,000 cases per year with a greater chance of major adverse cardiac events (MACEs) than asymptomatic women (3.0%–8.2% 5-year mortality for normal to nonobstructive disease) (75). The CMD represents significant societal economic cost burden, similar to that of obstructive disease (∼$750,000 vs. ∼$1,000,000 lifetime cost burden), driven by repeat angiography, increased anti-ischemic therapy costs, and increased hospitalization, thus showcasing the significance of this pathology and the need for comprehensive management strategies (407). More details on CMD can be found in the Women's Ischemia Syndrome Evaluation study (9).
6. Aging-Mediated Mitochondrial/Endothelial Dysfunction and Current Therapies: Effect on Major Adverse Cardiac Events
mtDNA4977 deletion, also referred to as the “common deletion,” is responsible for key subunits of mitochondria respiratory complexes that, if damaged, result in impaired respiration and production of ROS (488). mtDNA damage is associated with the presence and progression of atherosclerosis, resulting in impaired mitochondrial structure and function to ultimately produce excess ROS, which, if left unchecked, further produces ROS (RIRR) (514, 515). The relation of mtDNA damage and atherosclerosis has been recently reviewed elsewhere (469, 513), and higher mtDNA damage is associated with impaired vascular function in peripheral blood mononuclear cells (133).
In relation to aging, accumulation of mtDNA4977 deletions within cardiac tissue (e.g., left ventricle) over time can result in pathological cardiovascular consequences (6, 77, 133, 286). In patients with CAD, increased levels of mtDNA4977 deletion in peripheral blood are associated with MACEs and all-cause mortality, suggesting that accumulation rather than the presence of the common deletion alone is a crucial factor in predicting adverse outcomes (35, 470, 471). Further, mtDNA copy number may also decrease with pathologies. Indeed, for every one standard deviation reduction in mtDNA copy number, Wang et al. (484) demonstrated that the risk of CAD increases 1.14-fold in peripheral blood leukocytes. Further, Koller et al. demonstrated that lower mtDNA copy number in peripheral blood leukocytes was associated with an almost twofold increase in risk for peripheral artery disease and all-cause mortality (230).
This evidence suggests that mtDNA copy number is an independent risk factor for future CAD (484), vascular dysfunction, and all-cause mortality. On the contrary, Vecoli et al. (470) demonstrated that changes in mtDNA copy number in peripheral blood alone did not predict MACE or all-cause mortality; rather, changes in mtDNA copy number coupled with increased mtDNA4977 deletion increase the risk for MACE. In further support of this notion, data from the VA Normative Aging Study demonstrated that peripheral blood mtDNA copy number is associated with increased mtDNA damage, independent of chronological age (106). The link between mitochondrial dysfunction and MACE is further propagated by the finding that in patients with mitochondrial diseases, there is a 2.4-fold greater risk for MACE, and 14-fold increased risk of all-cause mortality (319). Taken together, mtDNA damage accumulation and decreased mtDNA copy number significantly increase the risk for future MACEs.
Current pharmacologic strategies to alleviate symptoms of angina include the use of nitrates, a strategy that has been employed since 1876 when nitroglycerine was first used to treat stable angina (441). Nitrates work by activating NO-cGMP signaling pathways. Rapid relief of acute angina symptoms is accomplished by peripheral venous dilation-mediated reduction of preload and left ventricular wall stress, lowering afterload and systemic blood pressure, reduced vasospasm, and epicardial coronary artery dilation. Overall, this leads to decreased myocardial oxygen demand and improved myocardial flow. Long-acting nitrates that may be taken for angina prophylaxis are second-line therapy to β blockers and calcium channel antagonists.
The major limitation to nitrate therapy is tolerance with rebound angina, necessitating carefully planned dosing regimens (441, 442). Tolerance occurs within 12–24 h of use; therefore, nitrates are currently indicated for symptomatic relief only. The causes of tolerance are due to exhaustion of cofactors, downregulation and inactivation of enzymes, and, most importantly, paradoxical induction of oxidative stress (161, 441). Superoxide formation in response to chronic nitrate therapy uncouples eNOS, decreasing NO availability and eventually causing further endothelial dysfunction. Another pharmacologic, nicorandil, is without the problem of tolerance and has the additional benefit of activating KATP channel-mediated dilation (441). Nicorandil has been shown in the Japanese Coronary Artery Disease (JCAD) Study to significantly reduce deaths from all causes, cardiac deaths, and fatal (but not nonfatal) myocardial infarction events (186). However, there is no conclusive evidence for improved clinical outcomes over currently indicated therapies utilizing either nitrates or nicorandil alone or in combination, and combination therapy may actually increase MACE (232, 435, 441, 442). Therefore, although these therapies are useful for providing symptomatic relief, it is incumbent to develop alternative strategies without these limitations that are superior to current standards and to encourage healthy preventative evidence-based habits.
7. Emerging Therapeutic Strategies to Improve Relaxation of Aging Vessels
Therapeutic targeting to dissuade desensitization/internalization and/or bolster resensitization of the β-adrenergic receptor would have impactful implications on the treatment of adrenergic-related pathologies, such as asthma, chronic obstructive pulmonary disease, muscle wasting, heart failure, and endothelial dysfunction seen in CMD. Such a strategy may have a benefit over conventional treatments, such as direct agonism (salbutamol effects on β2ADR for asthma, isoproterenol effects on β1 and β2ADRs for CMD, etc.), as these treatments can further potentiate desensitization/internalization by facilitating recruitment of GRK2, β-arrestin, and PI3Kγ to the receptor. Further, therapeutic approaches to encourage boosting nitric oxide availability and signaling, such as for FMD, is of benefit while considering the cardio and vasculo-protective effects of nitric oxide and that hydrogen peroxide signaling coincides with atherosclerotic development and aging pathogenesis.
Several approaches to address mitochondrial dysfunction-mediated oxidative stress and vascular pathology are currently being investigated with the philosophy that since arterial endothelial dysfunction is a precursor to cardiovascular disease, early therapeutic intervention may stave off premature aging-induced cardiovascular disease. These interventions are presented in Table 1 as a theoretical construct for which current therapeutic investigation and development in the field have been recently focused. Table 1 does not necessarily indicate current medically indicated treatments. The most effective method to ameliorate aging-associated vascular pathology is to focus on preventative measures, of which simple exercise and dietary considerations (such as the Mediterranean diet) may be most easily achieved and effective (1, 227, 228).
Therapies to Reduce Reactive Oxygen Species and Restore Cardiovascular Function in Aging
ACE, angiotensin converting enzyme; ADR, adrenergic receptor; ADSC, adipose-derived stem cells; AT1, angiotensin receptor 1; BM, bone marrow; DRP-1, dynamin-related protein-1; EDD, endothelium-dependent dilation; eNOS, endothelial nitric oxide synthase; F, female subjects only; HASMC, human aortic smooth muscle cell; HMVEC-L, human lung microvascular endothelial cells; HRCEC, human retinal capillary endothelial cell; HUVEC, human vascular endothelial cell; iPSC, induced pluripotent stem cell; KO, knock out; LDL, low-density lipoprotein; LLC, Lewis lung carcinoma; LV, left ventricular; M, male subjects only; Mfn, mitofusin; mitoQ, mitochondrial-targeted ubiquinone; MSC, mesenchymal stem cell; NO, nitric oxide; NOX, NADPH oxidase; Opa-1, optic atrophy 1; PASMC, human pulmonary artery smooth muscle cell; R, review; RVSMC, rat vascular smooth muscle cell; Sirt1, sirtuin deacetylase 1; SOD, superoxide dismutase; SOD2, manganese superoxide dismutase; SVF, adipose-derived stromal vascular fractions; TERT, telomerase reverse transcriptase; wt, wild type.
7.1. Diet modification to preserve vascular function in aging
Diet supplementation with antioxidant Vitamins (B6, B12, C, D, E) shows promising results in aging rodents to restore EDD, although studies in humans are inconsistent (421). Vitamin E supplementation reduces superoxide in heart failure to restore rat aortic ACh-mediated relaxation; this effect was also seen in postmenopausal women brachial artery FMD (22). Vitamin B12 (folic acid) improved skeletal muscle blood flow in aged patients (372). In contrast, the Women's Angiographic Vitamin and Estrogen trial showed that vitamins (E and C) do not rescue postmenopausal EDD (219). Metanalysis has since found that vitamin supplementation alone cannot reduce MACE (310). Deficiency, on the other hand, is associated with endothelial dysfunction, and inclusion of a “healthy diet” (as opposed to mere vitamin supplementation) including vitamins (fruits, vegetables, nuts) shows favorable outcomes (76, 202, 287, 372, 415).
The most effectively studied “healthy diet” is the Mediterranean diet. In a recent review, Shannon et al. describe the ways in which the Mediterranean diet has been shown to positively impact hallmarks of aging, such as genomic instability, senescence, telomere attrition, and mitochondrial dysfunction (406). Omega 3 polyunsaturated fatty acids (fish oil) has been shown to decrease mitochondrial fission proteins DRP-1 and Fis-1, increase fusion proteins Mfn-2 and Opa-1, promote uncoupling via uncoupling protein 2, and induce mitochondrial tubular morphology, culminating in reduced mtROS production (358). The mitophagy activator trehalose, found in Mediterranean diet foods such as mushrooms, crustaceans, baker's yeast (bread), and seaweed restores nitric oxide bioavailability, reduces oxidative stress and aortic inflammatory markers, and it restores EDD and endothelial-independent dilation in aging mice with similar results in human (216, 243). As mitophagy eliminates damaged organelles as sources of ROS, redox homeostasis can be restored. From a clinical standpoint, large-scale meta-analysis of randomized control and observational trials shows that the Mediterranean diet reduces diastolic and systolic blood pressure, decreases odds of developing hypertension, reduces polypharmacy, and lowers risk of coronary/cerebrovascular events and all-cause mortality, stratified based on the level of adherence and length of the diet (80, 405, 475).
Polyphenols (blueberries, strawberries, red grapes, murtilla berry, blackberries, garlic, tea, etc.), flavonoids (apples, spinach, etc.), theanine and catechins (green and black tea), cyanidine 3-glucoside (cardamom), and selenium (nuts) confer antioxidative properties that protect endothelial function and restore EDD and FMD in aging mice (33, 141, 182, 183, 194, 196, 208, 215, 259, 315, 336, 385, 474, 485, 495, 499, 503). As reviewed by Kicinska and Jarmuszkiewicz, flavonoids are capable of targeting mitochondria to promote fission over fusion, modulate antioxidant enzyme response to ROS (including SOD2, catalase, glutathione peroxidase), and reduce complex 1 and 3 ROS generation, thereby activating mitochondrial biogenesis, skewing mitochondrial dynamics toward fusion, and modulating Pink1/PARKIN-mediated mitophagy (222). In settings of aortic calcification and murine endothelium, the flavonoid quercetin can inhibit DRP-1 activation by blocking phosphorylation, likely by inhibiting PKCδ (63).
Blueberry anthrocyanins increase vasodilator nitric oxide, promote and rescue eNOS and SOD function, protect against high-glucose mediated vascular damage, and decrease pro-oxidant and vasoconstrictor functions of ACE, NOX4, xanthine oxidase-1, and low-density lipoprotein levels (193 –195, 354). Meta-analysis of 39 prospective cohort studies by Micek et al. spanning ∼1.5 million individuals found that a flavonoid-rich diet (anthrocyanins, catechin, flavan-3-ols) significantly lowers the relative risk of cardiovascular disease in a dose-dependent manner (288). Further, high flavonoid intake reduced cardiovascular, cancer-related, and all-cause mortality in a cohort (1063) of older (>75 years) women (201).
Red grape skin and red wine contain the anti-aging polyphenol compound resveratrol, which is hypothetically responsible for the “French Paradox.” French citizens have been found to have a lower risk of cardiovascular disease than Americans, despite having similar smoking tendencies, low exercise, and Western diets high in saturated fats (131). Resveratrol rescues mesenteric endothelial function in diabetes mellitus, obesity, and prevents aging-induced vascular pathology if consumed starting during youth in mice (60). These effects are mediated by increases in Sirt1-dependent on peroxisome proliferator-activated receptor δ (PPARδ). Drinking water supplemented with red wine polyphenols (RWP) given to rats at age 16 weeks improves EDD of mesenteric arteries to ACh at week 40 (88, 154). The RWP treatment also reduced oxidative stress, normalized peroxynitrite and eNOS levels, normalized aging-dependent increases in pro-oxidant NOX subunit p22phox, nox1, arginase 1 (competitive inhibitor of eNOS), and AT1 receptor expression similar to that of young levels.
Resveratrol can itself elicit vasodilation response via nitric oxide signaling from ERK signaling and guanylyl cyclase activation and smooth muscle relaxation from BKCa channel activation in porcine retinal arteries, and it even confers retinal ganglion protection from ocular hypertension (53, 312). The benefits of resveratrol also extend to the renal system; resveratrol dilates renal arteries via increased eNOS and nitric oxide synthesis, improving naturesis and chronically limiting angiotensin pressor effects on the renal artery (158 –160). Resveratrol also has potential implications for sarcopenia, as injection improved muscular microvascular perfusion and blood volume, also through eNOS and nitric oxide (483). These findings in animal models have been shown to translate in humans, including evidence for increased cerebrovascular and cognitive function, improved nitric oxide-mediated FMD in postmenopausal women, and cardioprotection during CAD (17, 164, 221, 274, 338, 363, 416, 445, 498, 529).
Despite these promising results, further epidemiologic scrutiny through meta-analysis concludes that the health-promoting effects of resveratrol have not yet been definitively confirmed (73). Omidian et al. found 16 clinical trials that reported that resveratrol mediated increases in glutathione peroxidase concentration, but it neither increased SOD nor significantly reduced the markers of oxidative stress (327). Although in human studies it was determined that resveratrol improves systolic blood pressure, in a meta-analysis by Weaver et al., resveratrol did not improve diastolic pressure or FMD as seen in animal studies, and improvement is greater in pure resveratrol compound studies versus wine drinking (486). These conflicting meta-studies suggest that additional large, prospective, randomized, double-blind, and placebo-controlled studies with standardization of protocol are necessary to properly delineate the effect of resveratrol in aging.
An important confounding phenomenon to note is the metabolism of these phenolic compounds. Resveratrol is quickly metabolized and its bioavailability is only <1%. Further, it has been calculated that the red wine necessary to obtain sufficient resveratrol for cardiovascular benefit would be 50 L per day (73). Therefore, it is no surprise that in a prospective cohort study by Semba et al. of older adults (>65 years) there was no predictive association between urine resveratrol concentration and cardiovascular events or all-cause mortality (402). For pure pharmacologic resveratrol compound, the proper dosing regimen is unclear and based on animal-to-human dose conversions (73).
7.2. Exercise to preserve vascular function in aging
Aerobic exercise has been shown to be vasculoprotective in aging. In aged mice, routine wheel running is associated with restored endothelium-dependent and -independent vasodilation in carotid arteries, reduced aortic nitrotyrosine oxidative stress, increased SOD activity, and reduced NOX activity and expression (119). In rat ex vivo arterial tension assays, Huang et al. found that endothelium-derived hyperpolarizing factor-mediated relaxation in aging is restored on regular aerobic exercise (191). Even if initiated late in life, aerobic exercise is able to improve coronary microvascular function, prefusion, and EDD, and it reverses diastolic dysfunction in aged rats; these benefits are translated in humans (44, 187, 304). In humans, consistent aerobic but not resistance exercise is found to enhance EDD and reduce endothelin-1-mediated vasoconstriction in both middle-aged and elderly adults (98, 123, 212, 384, 399, 465). This enhanced vasodilation allows increased skeletal muscle microvascular blood flow in active versus sedentary older adults (198, 386). It is thought that exercise-induced recovery of vascular dilative function extends to cerebral arteries and has positive impacts on reducing cognitive decline during aging (21). Related to mitochondrial function, exercise has been shown to reduce fission via reduced DRP-1 activation and promote fusion via enhanced Mnf-2 and Opa-1 in various tissues and pathological states (436), but these studies have not, to our knowledge, explored the setting of aging vasculature.
The positive effects of exercise on preserving endothelial function seem to provide less benefit to postmenopausal women, possibly due to estrogen deficiency (men with low testosterone also exhibit greater pronounced endothelial dysfunction) (297). With estrogen (and testosterone) deficiencies, oxidative stress increases, whereas BH4, an essential eNOS cofactor, is depleted. Exercise training in postmenopausal women can confer restoration of endothelial function only coinciding with estradiol (but not placebo) therapy, indicating its necessity for exercise-induced vascular adaptation. Therapies to activate estrogen receptors without the risks associated with estradiol, such as resveratrol, have so far come up short in providing similar restoration of function with exercise as seen in men (although resveratrol improves basal FMD) (338). These sex-specific disparities, difficulty in patient adherence regarding advice for diet and exercise, and patient populations unable to exercise, warrant investigation into other restorative strategies.
7.3. Pharmacologic mitochondrial targeted antioxidation
MitoTEMPO and related compound MitoTEMPOL are scavengers of mitochondrial-specific superoxide. They have been shown to reduce endothelial oxidative stress, improve phenylephrine-induced contraction and ACh-induced dilation in rat aortic rings, and can restore mitochondrial fission/fusion balance in mice with endothelial dysfunction and oxidative stress (8, 40, 224, 226, 325, 344). Mitochondrial-targeted ubiquinone (mitoQ) is a compound that includes a conjugation between the antioxidant ubiquinone and the lipophilic cation decyl-triphenylphosphonium and associates with the mitochondrial inner membrane. After serving in its antioxidant capacity, respiratory complex II serves to restore mitoQ's potential for reduction, therefore allowing for cyclic antioxidant activity, a limitation of previous antioxidants studies such as vitamins E and C.
A study of old mice given oral mitoQ by Gioscia-Ryan et al. demonstrated the protective effects of mitoQ on age-associated endothelial dysfunction in mice (152). In this study, mitoQ facilitated a reduction of aortic mitochondrial superoxide production and oxidative stress as evidenced by electron paramagnetic resonance spectroscopy and reduced nitrotyrosine levels, respectively. This was accompanied by a restoration of NO, as treatment with L-NAME (eNOS inhibitor) yielded less reduction in ACh-mediated vasodilation in the old group than the young or old+mitoQ group (although aging-mediated reduction in eNOS activity itself was not restored by mitoQ). The EDD assessed with ACh revealed significantly increased vasorelaxation in the old + mitoQ group to near youthful levels. Exogenously producing acute mitochondrial oxidative stress with rotenone led to ∼25% reduction in EDD in the old but not young or old+mitoQ mouse carotid artery, indicating mitoQ's ability to confer resistance to acute oxidative stress. The protein profile showed an aging-associated increase in p66SHC (prooxidant) with decreased SOD2 (antioxidant), COV-IV (mitochondrial mass marker), and PGC-1α (mitochondrial biogenesis marker), all restored to youthful levels with mitoQ, which indicates that the maintenance of mitochondrial homeostasis is crucial to maintain vasodilative capacity.
Age-associated aortic stiffness with aging, as evaluated with Pulse-Wave Velocity, was attenuated with mitoQ supplementation in association with restored elastin expression (relative to youth) without affecting aging-related increases in collagen or proinflammatory cytokine expression (151). The authors speculate that this reduction in aortic stiffness was mediated by attenuation of ROS-mediated stimulation of αADR hyperconstriction (374). In addition, mitoQ also reverses diastolic dysfunction as assessed by reduced left ventricular relaxation constant Tau, left ventricular end diastolic (LVED) pressure, and LVED-volume relation (473).
Additional studies with mitoQ have been replicated in human skeletal muscle feed arteries, showing similar capacity to restore mitochondrial homeostasis with corresponding improvement in EDD. Unlike the Gioscia-Ryan et al. study (152), mitoQ was able to restore eNOS (increased phosphorylated eNOS to eNOS ratio) in human skeletal arteries with mitoQ-mediated EDD improvement ablated by the eNOS blockade (347). In a randomized triple-blinded clinical trial of 55 participants, mitoQ (20 mg/day for 6 weeks) or placebo was supplemented in patients 60–79 years of age with impaired (brachial artery) endothelial function. At the clinical endpoint, brachial artery FMD was 42% higher in the mitoQ group compared with the placebo with concomitant reduction of aortic stiffness and oxidized low-density lipoprotein (global oxidative stress marker) (375). More details on mitochondrial and vascular dysfunction, mitoQ, future directions, and detailed current gaps in research can be found in the recent review by Rossman et al. (374).
7.4. Modulation of TERT
The importance of telomerase in aging is well known, as its role is to prevent telomere shortening during DNA replication, the function of which is diminished with aging and is a cause of senescence phenotype. TERT, the catalytic subunit of telomerase, limits ROS production in both the cytosol and mitochondria. It is trafficked from the nucleus to the mitochondria during times of peak ROS production by heat shock protein 90 and Akt, whereby it inhibits ROS production through a currently unknown mechanism. The trafficking fate of TERT is dependent on phosphorylation of different amino acid residues; nuclear import via phosphorylation of serine 227 mediated by Akt, nuclear export via tyrosine 707 mediated by Src, and overall activation via serine 823 are also mediated by Akt. With this knowledge, strategies to influence the trafficking of TERT, as well as TERT overexpression and loss of function, have been used experimentally to study its effects on vascular function.
The non-canonical functions of TERT have recently been summarized in a review by Rosen et al., including resistance to apoptosis in somatic cells, mtDNA protection, antioxidation, and improvement of mitochondrial function (373). Although TERT cannot function to preserve mtDNA by preserving telomeres, which mtDNA does not have, reduction of ROS in the mitochondria can reduce the damaging “hits” that the mtDNA takes. Therefore, TERT can indirectly protect mtDNA, which has been confirmed in overexpression models of TERT. Overexpression or targeting the shunting of TERT to the mitochondria also leads to increased glutathione (reduced form), non-oxidized peroxiredoxin, SOD2, and diminished ROS (373). On the other hand, TERT knockout or excluding TERT from the mitochondria leads to increased ROS with increased susceptibility to apoptosis through caspase and Bax signaling, reduced SOD2, and reduced ATP production (373).
The effects of TERT on redox balance have direct implications for vascular function. Inhibition of TERT with BIBR-1532 in vessels from healthy rats leads to a switch in the flow-mediated vasodilator agent nitric oxide to hydrogen peroxide, mimicking the CAD and aging phenotype (29, 30). Interestingly, vascular TERT is downregulated in both CAD and aging. As described earlier, Ang II-mediated AT1 receptor agonism leads to increased ROS and endothelial dysfunction with reduced vasodilator capacity (7). However, mice overexpressing TERT are protected from reductions in vasodilator capacity from Ang II infusions seen in wild-type, and TERT knockout significantly reduces FMD in response to ANG II. In addition, loss of TERT function is associated with reduced peak dilation to flow and ACh in mice, with worsening FMD in subsequent generations.
Exogenous peptides incubated with aging rat coronary microvessels that block nuclear import of TERT (increasing mitochondrial TERT), as well as incubation with TERT activator, AGS 499, improve ACh as well as β1ADR (dobutamine and norepinephrine) concentration responses compared with the scrambled peptide control (453). On the other hand, young rat vessels incubated with trafficking peptides that shunt TERT away from the mitochondria by blocking trafficking to the mitochondria abrogate vasodilation from β1ADR agonists and ACh (453).
The TERT expression and function can be influenced therapeutically. Cycloastragenol (CAG) is a molecule purified from the root of Astragalus membranaceous indigenous to China and used in traditional Chinese medicine for nearly 2000 years. The CAG increases telomerase activity, is an antioxidant, has anti-inflammatory properties (reversing aging-associated immune system changes), and has been used in a variety of other ways: as a diuretic to reduce blood pressure, as an expectorant, to improve bone density, to reverse hyperglycemia, for hepatoprotection, and to reduce hyperlipidemia (173, 174, 264, 381).
The TERT can also be activated in human aortic SMCs and pulmonary microvascular endothelial cells by supplementation with resveratrol found in red wine or red grape skin (192). Interestingly, TERT activation by resveratrol seems to be dependent on an axis, including the activation of nicotinamide phosphoribosyltransferase and Sirt4. Healthy human atrial and adipose arterioles normally vasodilate during incubation with Angiotensin 1–7, but this does not occur in vessels from CAD patients. However, if incubated overnight with angiotensin 1–7, nitric oxide-dependent FMD is restored in CAD-derived vessels through the activation of telomerase (118). In addition, PPAR-γ inhibition with pioglitazone improves telomerase activation by 2 × in mouse aorta and confers protection against hydrogen peroxide-induced oxidative stress and apoptosis (492). Perhaps the most potent known activator of telomerase is estrogen, with loss of telomerase as a potential factor contributing to aging-associated CMD exhibited preferentially in women (9, 23, 24, 162, 239, 383). Although HRT can restore telomerase activity, risks of oncogenesis and stroke are typically thought to be too much of a limitation (246). Although alternative strategies discussed earlier have shown to be safe, since TERT inhibits apoptosis and has a role in tumor development, it is important to weigh the risks and benefits when utilizing these therapeutic options (18).
7.5. Cellular therapy to preserve and recover vascular function in aging
Adipose-derived stromal vascular fraction (SVF) has antioxidant properties that, when injected systemically (via tail vein) in aged female rats, restores β1ADR density and agonist-driven vasodilation (via dobutamine and norepinephrine) in coronary microvasculature (376). Markers of diastolic function (Tau, E/A, E/e′, and Isovolumic Relaxation Time) are restored to youthful levels by SVF alongside an improved CFR, a measure of microvascular function (220, 376). Once cleared of adipocytes, isolated adipose-derived SVF represents a heterogenous population of cells, including microvascular endothelial cells, perivascular cells, fibroblasts, mesenchymal stem cells, regulatory and natural killer T-lymphocytes, B-lymphocytes, dendritic cells, and macrophages. Injected SVF cells are able to incorporate into the vasculature (and myocardium to a minimal extent), where they are thought to exert their effects through paracrine and perhaps some direct cell–cell interactions (282). Although unknown in SVF, antioxidative benefits are conferred through direct mitochondrial transfer to endothelial cells, often mediated by connexin 43 gap junctional connections in mesenchymal cells from other cell sources (188, 261, 340, 413, 512).
The SVF cells in vascular tissue express CD11b; therefore, the macrophage population is postulated to have a role in these observed effects (301). However, injected SVF-CD11b+ cells alone are insufficient to rescue function, indicating that the potential requirement of synchrony from other cells or paracrine mechanisms is required. Exosomes from mesenchymal stem cell sources, including bone marrow, umbilical cord, and adipose SVF-derived stem cells, have antioxidant capabilities, possibly due to the delivery of antioxidant proteins such as glutathione and glutathione peroxidase (15, 89, 184, 260, 268, 444, 502, 507, 509, 518, 528). Therefore, one possible mechanism for SVF-mediated restoration of adrenergic function is that SVF-derived exosomes deliver antioxidant proteins to recipient endothelial cells, whereby the restored youth-like homeostatic redox balance dissuades thiol-oxidation and protective nitrosylation, thus preventing βADR desensitization and internalization as happens with aging (Figs. 4 and 5). Therapy with SVF also alters FMD in aging from hydrogen peroxide-mediated to peroxynitrite-mediated, indicating minimal increase in nitric oxide production with SVF treatment (452). This effect is too minimal to directly influence dilation, but enough to combine with superoxide to form peroxynitrite to then influence FMD.
It is the hope that SVF is an immunologically safe, autologous therapeutic option that can holistically address vascular pathology by targeting oxidative stress, endothelial and adrenergic dysfunction, and pathologic smooth muscle tone and hyperconstriction. Importantly, SVF from old donors seems to be less effective than from young donors at rescuing vascular and cardiac function (5). Since patients with these cardiovascular pathologies are usually aged, this hurdle may need to be overcome using strategies to invigorate the cells before systemic injection. Interestingly, transfection of elderly donor SVF with TERT nucleoside-modified mRNA to increase SVF TERT has the effect to improve SVF secretory function, thereby improving regenerative capacity (316). This finding is typical in the field of stem cell therapeutics, where therapies that are promising in rodent models are not as efficient in clinical translation, leading researchers to investigate the potential of “priming” cell populations before therapeutic delivery.
Despite this hurdle, SVF or SVF-derived subcellular populations or exosomes may still be of advantage over other strategies due to its ability to preferentially target the vasculature. If antioxidation provides a pathway to increase βADR functionality, this is, of course, a benefit in the vasculature to improve cardiac perfusion (which is reduced by ∼43% in aging) to prevent chronic ischemia culminating in the angina of CMD, for example. However, if βADR receptors are similarly increased in the myocardium by therapies that do not preferentially target vasculature, this could be a detriment to patients with heart failure, where a subsequent increase in ionotropic potential could increase oxygen demand outpacing supply, worsening function. This outcome is only a postulation and should be considered as these mitochondrial antioxidative therapeutic strategies are developed.
8. Conclusion
Homeostatic regulation of perfusion is essential to properly match oxygen and nutrient delivery to tissue demand and waste removal needs. Chronic imbalance in the heart vasculature leads to chest pain as transient ischemic angina pectoris, thereby elevating risk for eventual severe acute imbalance and leading to myocardial infarction. The pathological emergence and progression of these imbalances, including coronary artery and microvascular disease, are driven by the aging process. In this review, our major objective was to discuss the pathological alterations of the mitochondria during aging, namely a trend toward fission/fusion imbalance and reduction of mitophagy that is both caused by and contributes to reduction of antioxidative regulation of oxidative stress. These mitochondrial alterations lead to dysfunctional vasodilation and ability to maintain appropriate patency, often leading to hyperconstricted tone. A common theme is the aging-dependent accumulation and reliance of mitochondrial-derived ROS signaling in the place of vasoprotective signaling by nitric oxide.
In Figure 1, we summarize the aging-associated changes to mitochondrial and cellular antioxidant and pro-oxidant enzymes as well as the ETC that lead to (and/or are a consequence of) increasing mtROS. The damage caused by this accumulation leads to mitochondrial dysfunction and a favor toward fission rather than protective fusion or mitophagy processes, all of which is associated with reduced flow and βADR-mediated vasodilative efficacy. In Figures 2 and 3, the signaling cascades initiated by intraluminal flow are shown to alter with aging by favoring mitochondrial-derived hydrogen peroxide as opposed to nitric oxide or prostaglandin-mediated signaling in youth. In addition, receptors and potassium channel expression and function in the endothelial hyperpolarization pathway are altered with aging, due in part to post-translational modifications by mitochondrial-derived ROS, which further affect dilation capacity. In Figures 4 and 5, we illustrate how aging affects the balance between βADR and αADR-mediated dilation and constriction, with emphasis on desensitization, internalization, and recycling with their mitochondrial influences, that is, how mitochondrial-derived ROS or nitric oxide can directly affect receptor trafficking through βADR thiol oxidation, PI3Kγ activation, or GRK2 nitrosylation.
Traditionally, aging has been referred to as an unmodifiable risk factor of chronological aging. However, the Geroscience hypothesis suggests that biological aging and the so called “rate of aging” can vary between individuals owing to genetic, environmental, and pathological influences. The central question is whether this “rate of aging” can be manipulated for therapeutic targeting to protect against aging-mediated pathologies such as atherosclerosis and coronary vascular and microvascular disease. Current therapies such as β blockers may not take into consideration vascular adrenergic trafficking changes that occur in advancing age. Nitrates can offer acute symptom relief but are unable to provide meaningful long-term mortality and MACE benefits due to tolerance and paradoxical induction of oxidative stress and endothelial dysfunction.
Therefore, we discussed emerging therapeutic strategies and targets such as drugs to attenuate mitochondrial dysfunction-induced oxidative stress such as mitoQ and mitoTEMPOL, CAG to increase TERT function, SVF, and recent data explaining the anti-oxidative and vasculoprotective effects of diet and exercise supplementation, all summarized in Table 1. Future directions should continue to elucidate aging-mediated vascular changes to find additional therapeutic targets, including the role and potential manipulation of nitrosative and oxidative modifications. Studies should also provide more extensive and higher-powered clinical trials with standardized protocols for the discussed therapies with emphasis on symptom management, mortality, MACE, and superiority over current clinical standards. When these antioxidative and aging-reversal strategies should begin is a question of importance scientifically and ethically. Should the emphasis be at the start of disease presentation? Beforehand, and if so, when? In all, the current direction of regenerative medicine described is providing exciting avenues to achieve a meaningful benchmark to bedside translational care.
Footnotes
Authors' Contributions
E.P.T., W.H., A.B., and A.J.L. created a conceptual outline; E.P.T., W.H., and J.B. wrote the article; E.P.T. and G.R. made all figures/tables; E.P.T., W.H., J.B., G.R., A.B., and A.J.L. edited and approved the article text, figures, and tables.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors acknowledge funding from National Institutes of Health (AG053585 to A.J.L., T32GM089586 to A.B., R01HL133029 to A.B.), Department of Defense (W81XWH-19-RTRP-IDA and W81XWH-13-2-0057 to A.J.L.), the Gheen's Foundation (A.J.L.), and AHA 20POST35050017 (W.H.).