
Abstract
Significance:
Nonalcoholic fatty liver disease (NAFLD) is a hepatic and systemic disorder with a complex multifactorial pathogenesis. Owing to the rising incidence of obesity and diabetes mellitus, the prevalence of NAFLD and its impact on global health care are expected to increase in the future. Differences in NAFLD exist between males and females, and among females depending on their reproductive status. Clinical and preclinical data show that females in the fertile age are more protected against NAFLD, and studies in postmenopausal women and ovariectomized animal models support a protective role for estrogens.
Recent Advances:
An efficient crosstalk between the liver and adipose tissue is necessary to regulate lipid and glucose metabolism, protecting the liver from steatosis and insulin resistance contributing to NALFD. New advances in the knowledge of sexual dimorphism in liver and adipose tissue are providing interesting clues about the sex differences in NAFLD pathogenesis that could inspire new therapeutic strategies.
Critical Issues:
Sex hormones influence key master regulators of lipid metabolism and oxidative stress in liver and adipose tissue. All these sex-biased metabolic adjustments shape the crosstalk between liver and adipose tissue, contributing to the higher protection of females to NAFLD.
Future Directions:
The development of novel drugs based on the protective action of estrogens, but without its feminizing or undesired side effects, might provide new therapeutic strategies for the management of NAFLD. Antioxid. Redox Signal. 35, 753–774.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a heterogeneous disorder characterized by an excessive fat deposition in the liver (>5% macrovesicular steatosis), which is not caused by an excessive alcohol consumption (36). NAFLD encompasses a wide spectrum of liver disorders ranging from simple steatosis, a benign and reversible accumulation of lipids in hepatocytes, to nonalcoholic steatohepatitis (NASH), a more severe form of the disease (197). Unlike NAFLD, NASH is accompanied by liver inflammation and ballooning, which ultimately results in fibrosis, cirrhosis, or hepatocellular carcinoma (49). Throughout the last decade, NAFLD has become the most common cause of both chronic liver disease and liver-related mortality, affecting nowadays millions of people worldwide (120). The prevalence of the disease shows differences among continents fluctuating between 15% in Africa and >30% in South America and Middle East, with an incidence of 25% in Europe and North America (197). Globally, the prevalence continues to grow, making this disease a major public health problem worldwide (58, 197).
NAFLD is closely associated with abnormalities in glucose and lipid metabolism, and thus highly prevalent in obese and diabetic patients (29, 197). Insulin resistance and body fat distribution, particularly excessive visceral fat, play determinant roles in NAFLD pathogenesis (162). Indeed, NAFLD has been considered the hepatic manifestation of metabolic syndrome (MetS), and has been shown to contribute to the development of atherosclerosis and coronary heart disease (10).
Initially, the “two-hit hypothesis” was the prevailing model for NAFLD. According to this theory, increased hepatic fat accumulation secondary to sedentary lifestyle, high-fat diet (HFD), and insulin resistance act as the “first hit,” sensitizing the liver to further insults acting as “second hit.” The “second hit,” including key factors such as oxidative stress, mitochondrial dysfunction, or proinflammatory cytokines, contributes to inflammation and fibrosis, leading to hepatic injury (36). Although this hypothesis is supported by some animal models of obesity, it became rapidly evident that this view is too simplistic to recapitulate the complexity of the human NAFLD, characterized by multiple parallel factors (oxidative stress, insulin resistance, inflammation, mitochondrial dysfunction), acting synergistically in genetically predisposed subjects to promote NAFLD development and progression, which lead to the currently accepted “multiple-hit hypothesis” (28).
Considering that animals evolved in a nutritionally poor environment, females have been subjected to higher evolutionary pressures to adapt their energy balance to the needs of reproduction and to the environmental conditions (179). In fact, the same hormones involved in female fertility also regulate energy metabolism, ensuring that sex only occurs when reproduction is possible. Estrogens, and particularly, 17β-estradiol (E2), the major female sex hormone, are the greatest exponent of this link between energy metabolism and reproduction (134). E2 effects are mediated by three estrogen receptors (ERs): estrogen receptor α (ERα), estrogen receptor β (ERβ), and the intracellular, transmembrane G protein-coupled estrogen receptor (GPER). The two classical ERs (ERα and ERβ) act as ligand-activated transcription factors that reside in the cytosol and translocate into the nucleus upon ligand binding to regulate gene expression. In addition, estrogens bind to plasma membrane-associated subpopulations of ERα and ERβ, or GPER, thereby activating a variety of rapid intracellular signaling cascades (124).
Research has traditionally considered male and female organisms as equivalent, and most preclinical and clinical studies have been carried out in one sex (mainly males), and the results extrapolated to the other. This historical over-reliance on male physiology has delayed the understanding of key sex influences on health processes and outcomes. Fortunately, the National Institutes of Health (NIH) promoted policies in the last decade to account for sex as a biological variable in their funded studies, including both male and female animals in preclinical studies as well as women in clinical trials. These efforts have resulted in a growing number of publications addressing sex differences across different disciplines and fields (9). However, compared with other metabolic diseases, our understanding of sex differences in NAFLD is still limited.
On this background, our aim is to summarize and critically discuss the current knowledge about the molecular mechanisms by which sex, and particularly, estrogens may influence NAFLD pathogenesis. To this aim, we have focused this review on the current knowledge of sex differences in the crosstalk between liver and adipose tissue, both organs playing a key role in NAFLD pathogenesis and being deeply influenced by sex hormones.
Clinical and Preclinical Evidence of Sexual Dimorphism in NAFLD
The prevalence, incidence, and severity of NAFLD are higher in men than in premenopausal women (14, 99). In general, men are twofold more likely to die from chronic liver disease and cirrhosis than are women. Liver transplant occurs less commonly in women than in men, with variable disease outcomes based on etiology (69). After menopause, the prevalence of NAFLD between men and women becomes comparable or even greater in women with worse outcomes than in men (14, 159). These observations suggest a protective role for ovarian hormones in NAFLD. In this regard, among postmenopausal women with NAFLD, the age of menopause and time from menopause (i.e., a longer duration of estrogen depletion) are associated with increased risk of hepatic fibrosis (103).
This fact has been related with impaired metabolic traits often found in postmenopause, such as dyslipidemia, visceral obesity, and type-2 diabetes mellitus (38). Interestingly, hormone replacement therapy (HRT) has been reported to decrease NAFLD prevalence supporting the hepatoprotective role of E2 (14, 40, 54). However, the use of oral contraceptive pills in menstruating women (119, 148), and gender-affirming hormone therapy in transgender (7, 11, 172), has reported conflicting results regarding E2 therapy and the prevalence of NAFLD and metabolic abnormalities.
More consistently, several studies have observed an association between NAFLD occurrence and low E2 levels. Indeed, pre- and postmenopausal women without NAFLD have higher serum levels of E2 than NAFLD patients (68). In addition, several hypogonadism conditions as well as the use of antiestrogens have been associated with higher NAFLD prevalence. For instance, tamoxifen and aromatase inhibitors, both used in breast cancer treatment to decrease the activity or levels of E2, respectively, have been associated with higher NAFLD (34). Women with Turner's syndrome, a rare congenital X monosomy that leads to underdeveloped ovaries and infertility, show higher NAFLD incidence (106). Nevertheless, it cannot be ruled out that obesity and insulin resistance, which are also commonly present in this syndrome, may account for NAFLD rather than low E2 levels (106).
Polycystic ovary syndrome (PCOS), the most common endocrine disorder in premenopausal women, is also associated with NAFLD (109). Moreover, PCOS women exhibit significant difference in the values for circulating E2 between patients with NAFLD and those without the disease, although higher levels of androgens in PCOS women could also contribute to hepatic steatosis (68). Thus, although clinical studies analyzing several E2-deficient conditions or using different hormonal interventions also provide some evidence for the hepatoprotective role of E2, potential confounders are commonly present, which requires further caution in the interpretation of the data.
Most experimental NAFLD studies using genetic animal models or altered diets find that disease is more severe in males, recapitulating the main feature of clinical NAFLD (44, 75, 174, 183). The animal model of HFD-induced NAFLD is arguably the most resembling to the clinical features of the human disease (174). Genetic (e.g., leptin-receptor deficiency) and other experimental models such as methionine-choline-deficient diet, however, have limited clinical relevance. Despite the particularities and limitations of each model, most studies support the existence of sex differences, with females showing less severity or better metabolic features associated with the disease. The protection of females against the metabolic consequences of hypercaloric diets is lost with ovariectomy and rescued by E2 treatment, revealing the key role of E2 (27, 166). This is also supported by the increased steatosis observed in aromatase knockout (ARKO) mice, which are unable to synthesize E2 from androgen precursors (37, 76).
Several studies using ER knockout mice have addressed the question of which is the main ER driving the protective effects of E2. Most studies support a robust role for the classical ERα (73, 74), whereas studies using ERβ knockout (BERKO) mice or ERβ-selective agonists in ovariectomized or ARKO mice (37, 56, 152) have reported controversial results for ERβ. Interestingly, studies using liver-specific estrogen receptor α knockout mice (LERKO) showed no differences between LERKO females and wild-type controls in hepatic insulin resistance and liver steatosis (73, 130). In contrast, LERKO male mice showed impaired whole and hepatic insulin resistance associated with HFD feeding (67, 200). These findings suggest that in females, the hepatic protection against NAFLD might be driven by ERα in nonhepatic tissues, whereas in males, in which E2 levels are lower, liver ERα retains higher metabolic influence over hepatic insulin signaling. Nevertheless, considering the higher levels of E2 in females, and the fact that liver express all ER subtypes, the question whether other compensatory mechanisms might be masking the relevance of hepatic ERα in LERKO female mice needs to be addressed.
Recently, GPER has also been implicated in the protective effects of E2 against metabolic disorders, but it is not free from conflicting findings either. GPER-deficient female mice fed with a HFD showed lower high-density lipoprotein (HDL)-cholesterol along with increased hepatic steatosis compared with males (137). In addition, Sharma et al. observed impaired metabolic profile in GPER knockout (GPERKO) male mice, with higher visceral fat, impaired insulin sensitivity, and increased proinflammatory cytokine levels (166). Interestingly, a recent study from the same authors showed that G1, a selective GPER agonist, improved metabolic abnormalities in ovariectomized female mice fed a HFD (167).
In contrast to these studies supporting a beneficial metabolic role for GPER against hypercaloric diet, Wang et al. found that GPERKO female mice were protected from HFD-induced obesity and insulin resistance (185), and other authors did not find significant differences in body weight or fat mass in GPERKO mice, even if fed with a HFD (85). The observed disparity in the results from different studies using ER-specific knockout models underscores the limitation of these models to study the complexity of the mechanisms underlying the protective action of E2.
Despite some controversial results in animal models, in general most clinical and preclinical data support a role for E2 in the protection observed in premenopausal women against NAFLD and its comorbidities. In this review, we focus on several master regulators of lipid metabolism and oxidative stress across liver and adipose tissue, for which a sexual dimorphism has been reported, and therefore, contribute to explain the higher resistance of premenopausal women to NAFLD development. As depicted in Figure 1, all these signaling proteins and metabolic traits influenced by sex are interconnected in a scenario that outlines the crosstalk between liver and adipose tissue. Importantly, small biases exerted by sex at these regulatory points might be magnified along this circular interconnection. This perspective, in which sex hormones and particularly estrogens are able to fine-tune metabolism simultaneously acting on multiple targets, highlights the therapeutic power behind estrogenic signaling.
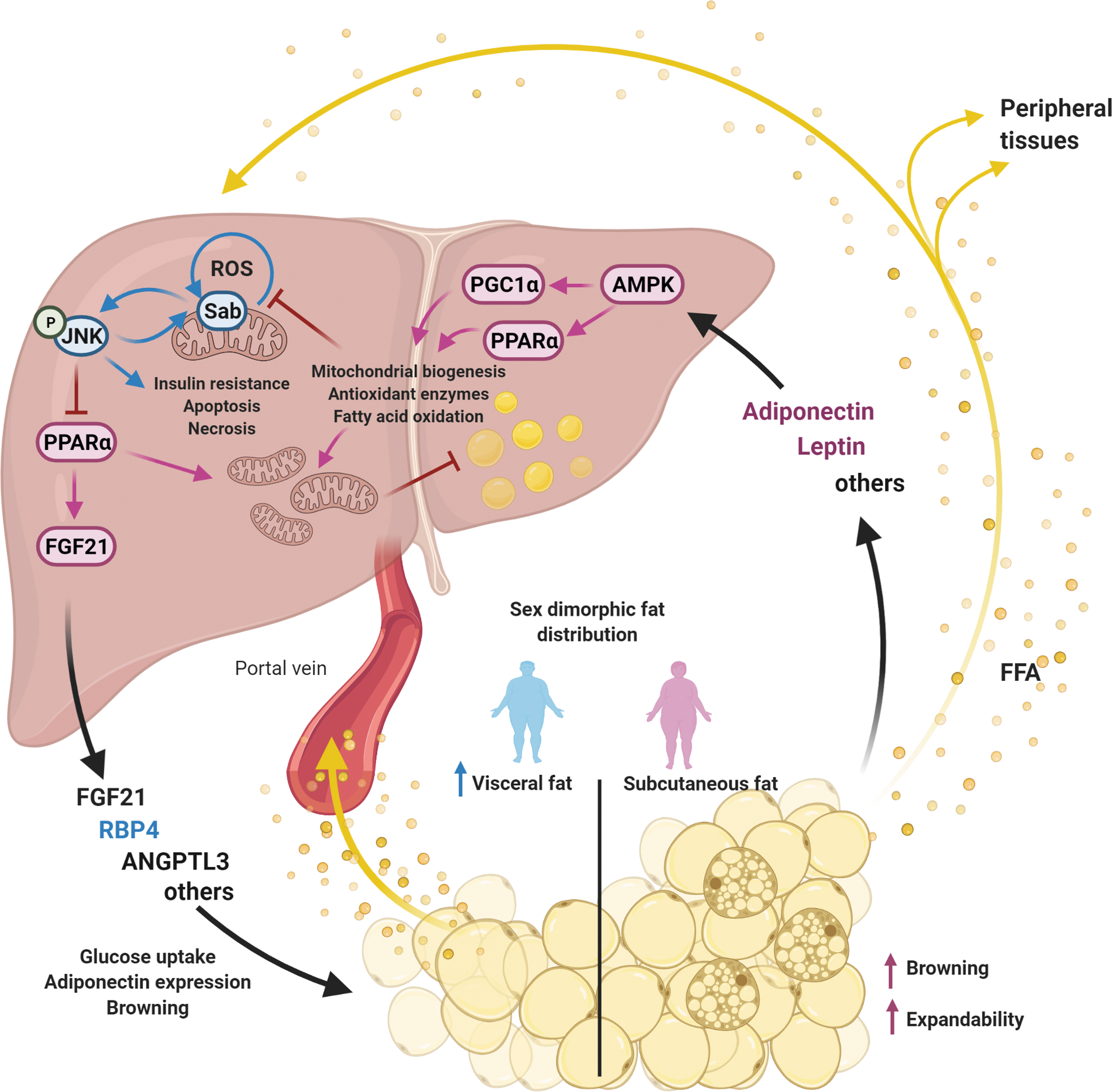
Sexual Dimorphism in Hepatic Adenosine Monophosphate-Activated Protein Kinase Signaling Pathway
Adenosine monophosphate-activated protein kinase (AMPK) has gained much attention for its ability to coordinate multiple metabolic pathways, including hepatic lipid metabolism (31). AMPK is a heterotrimeric kinase, which is activated by low-energy status, and restores energy balance through inhibition of ATP-consuming processes and promotion of ATP-generating processes (63). In liver, its activation results in the inhibition of lipogenesis and the increase in fatty acid oxidation by mechanisms that operate both in the short- and long term (Fig. 2).

AMPK activity is reduced by inflammation, obesity, and diabetes, all of them being considered risk factors for MetS and NAFLD (170). Accordingly, decreased AMPK activity occurs in many genetic rodent models with a MetS phenotype, including ob/ob mice (leptin deficient), fa/fa rats (leptin receptor deficient), and male Zucker Diabetic Fatty (ZDF) rats (leptin receptor deficient with a mutation in the insulin promoter) (156). In all of these rodent models, treatment with the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) improved insulin resistance and glucose homeostasis (70, 151). In addition, mice on NASH-inducing diets exhibited reduced AMPK activity, and studies with liver-specific AMPK knockout (LAKO) mice have demonstrated that loss of AMPK amplifies diet-induced NASH pathology, including increased liver damage, fibrosis, and cell apoptosis (199). Considering all the supportive evidence for the antisteatotic activity of AMPK, it raises the question of whether the protection against NAFLD of females may be mediated by this energy sensor.
Interestingly, female rats supplemented with fructose in drinking water (10% w/v) show higher activation of AMPK compared with males, resulting in decreased expression of Srebf1 (183). The authors of this study found a marked increase of the enzyme fructokinase in females, but not in male rats, resulting in a higher consumption of ATP by this enzyme, which in turn increases adenosine monophosphate (AMP), necessary for AMPK activation (183). Furthermore, evidence for the interaction between estrogen signaling and AMPK pathway in different tissues and cell types has been reported. For instance, E2 has been shown to activate AMPK in muscle (154), endothelial (194), pancreatic (177), cardiac (180), and hepatic cells (145).
With regard to the molecular mechanism linking estrogen signaling and AMPK, both nuclear ER (ERα and ERβ) have been reported to modulate (129, 194), and even directly interact with AMPK (116, 133). Curiously, a study from Pedram et al. showed that the ERα agonist PPT was able to prevent liver steatosis only in wild-type and the membrane-only ER mice (MOER), but not in estrogen receptor α knockout (ERKO) mice, suggesting that the membrane-driven signaling pathway of ERα is necessary for the lipid lowering properties of estrogens (145). These antisteatotic effects of membrane-associated ERα were dependent on AMPK inhibition of sterol regulatory element binding protein-1c (SREBP-1c), downregulating lipogenic genes. Of note, these results suggest that the development of a membrane-specific ERα agonist may have therapeutic potential to prevent liver steatosis without the engagement of proliferative or other side effects of nuclear ERα.
Besides membrane-associated ERα, GPER is bound to plasma and endoplasmic reticulum membrane, from where it mediates the nongenomic effects of estrogens. Interestingly, GPERKO female mice on HFD increase hepatic fat content to a greater extent than do their male counterparts, reinforcing the role for the membrane-driven arm of estrogenic signaling in the control of lipid metabolism (137). Development of new drugs selectively targeting membrane bound ERs would be a valuable tool to evaluate the advantages, in terms of side effects, of activating these receptor pools in mice models in which all their ERs are intact.
Sexual Dimorphism in Mitochondrial Biogenesis and Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1
In the liver, mitochondria play a critical role in carbohydrate and lipid metabolism, being essential for hepatic gluconeogenesis as well as lipogenesis and fatty acid oxidation (150). The peroxisome proliferator-activated receptor gamma coactivator 1 (PGC1) is a master regulator of mitochondrial biogenesis and function. PGC1 family comprises peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α), peroxisome proliferator-activated receptor gamma coactivator 1β (PGC1β; also known as PERC), and PGC-1-related coactivator (PRC; also known as PPRC1) (114). Their versatile actions are achieved by interaction with different transcription factors in a tissue-specific manner.
In the liver, PGC1 levels are increased under the fasted state, when marked metabolic adaptations occur, such as activation of mitochondrial metabolism, gluconeogenesis, fatty acid oxidation, Krebs cycle, ketogenesis, and reactive oxygen species (ROS) detoxification (20, 48, 117). Both PGC1α and PGC1β promote mitochondrial biogenesis mainly through their stimulatory effects on nuclear respiratory factors 1 and 2 (NRF1 and NRF2), estrogen-related receptor-α (ERRα), and transcriptional repression of ying yang 1 (YY1) (21, 32, 33). In turn, NRFs upregulate mitochondrial transcription factor A (TFAM), a nuclear-encoded transcription factor essential for replication and expression of mitochondrial DNA (mtDNA) (34). Thus, PGC1α and PGC1β orchestrate the expression of mitochondrial proteins encoded by mitochondrial and nuclear DNA, increasing the metabolic capacity of Krebs cycle, fatty acid oxidation, and oxidative phosphorylation (OXPHOS) in the hepatocyte (150).
Alterations in mitochondria structure and function have been described in fatty liver from NAFLD patients, with impaired electron transport chain activity and altered mitochondrial metabolism, resulting in increased oxidative stress and inflammation (147, 163). Studies in NAFLD mice models have found a strong association between impaired PGC1α activity and increased susceptibility to fatty liver (48). The deficit in PGC1α decreases coactivation of NRF1- and NRF2-dependent expression of mitochondrial and antioxidant genes, leading to dysfunctional electron transport chain and ROS accumulation (160). In contrast, PGC1α overexpression in rat hepatocytes results in reduced levels of triglyceride accumulation both in vivo and in vitro (132).
Similarly, mice overexpressing PGC1β in liver are protected from NASH development when fed with a HFD (18). In addition to increasing mitochondrial biogenesis and antioxidant defenses, PGC1β increases fatty acid oxidation through the coactivation of peroxisome proliferator-activated receptor α (PPARα) (18) while, paradoxically, it coactivates master regulators of lipogenesis (liver X receptor α [LXRα], SREBP-1c, and carbohydrate-responsive element-binding protein [ChREBP]), which promote the synthesis of unsaturated fatty acids and the export of triglycerides, thus conferring an additional level of protection against NASH (150). In fact, PGC1β-deficient mice on a HFD show hepatic steatosis and dyslipidemia, which cannot be compensated by intact PGC1α (171).
Sexual dimorphism in mitochondrial biogenesis and function has been reported in several human and rodent tissues, with females showing larger mitochondria and higher levels of mtDNA, respiratory chain proteins and mitochondrial enzymes associated with greater oxidative capacities (60, 61, 142). In liver, female rodents have higher electron transport system protein content, elevated respiratory capacity, lower mitophagy, and produce less mitochondrial ROS (164), consistent with increased levels of PGC1α and PGC1β (20, 61). This sexual dimorphism in mitochondrial recruitment, PGC1α/β-driven gene expression, and its relationship to hepatic lipid handling is summarized in Figure 3.

Moreover, ovariectomy has been shown to impair mitochondrial biogenesis and function in liver, effects that were reversed by E2 supplementation (62). A study from Besse-Patin et al. showed that heterozygous liver-specific disruption of Ppargc1α gene caused higher oxidative damage in females when placed on a HFD. Based on in vitro observations in hepatocytes overexpressing PGC1α, the authors suggested that PGC1α coactivation of ERα is required for estrogen-dependent expression of antioxidant genes (20). They also found that compensatory increases in PGC1β could prevent oxidative damage associated with complete loss of PGC1α in female mice. In agreement, previous work in HepG2 cells also showed that E2 effects on mitochondrial biogenesis markers are more sensitive to knockdown of PGC1β than PGC1α (62). Although further research is needed to fully understand the modulatory action of sex hormones in the program of mitochondrial biogenesis, the current knowledge supports a role for PGC1α and PGC1β in the sex differences in the metabolic capacities of mitochondria.
Sexual Dimorphism in the Sustained c-Jun N-Terminal Kinase Activation Loop
Mitogen-activated protein kinases (MAPKs) signaling pathways are activated in response to several physical or chemical stresses such as drugs, toxins, DNA damage, cytokines, or nutrient availability, and lead to adaptive responses by means of changes in cell proliferation, apoptosis, inflammation, cytokine production, and metabolism (98). Among the MAPKs, c-Jun N-terminal kinase (JNK) has been shown to play a significant role in acute and chronic liver injury (25, 165). In fact, JNK deficiency in mice prevents the development of obesity and insulin resistance caused by hyperphagia or the consumption of a HFD (77).
Many important physiological processes are regulated by gene expression through JNK-dependent phosphorylation of AP1 transcription factors. Under physiological conditions, JNK activation is transient since it is attenuated by concomitant activation of nuclear factor-κB (NF-κB) survival genes (191). Contrarily, sustained JNK activation in pathological processes correlates with liver injury and metabolic dysfunction (71).
In recent years, the mitochondrial-dependent mechanism underlying the sustained JNK activation has been deciphered using liver acute injury models (188 –190), and is depicted in Figure 4. In brief, activated JNK (P-JNK) binds to a protein located at the outer mitochondrial membrane, named SH3-domain-binding protein 5 (Sab or Sh3bp5). Binding of P-JNK to Sab initiates an intramitochondrial signaling pathway that leads to inactivation of Src (SRC proto-oncogene, nonreceptor tyrosine kinase), a kinase involved in mitochondrial function regulation. As a result, mitochondrial respiration and electron transport are impaired, leading to mitochondrial ROS release and MAPKKK, mitogen-activated protein kinase kinase kinase (MAP3K) activation.
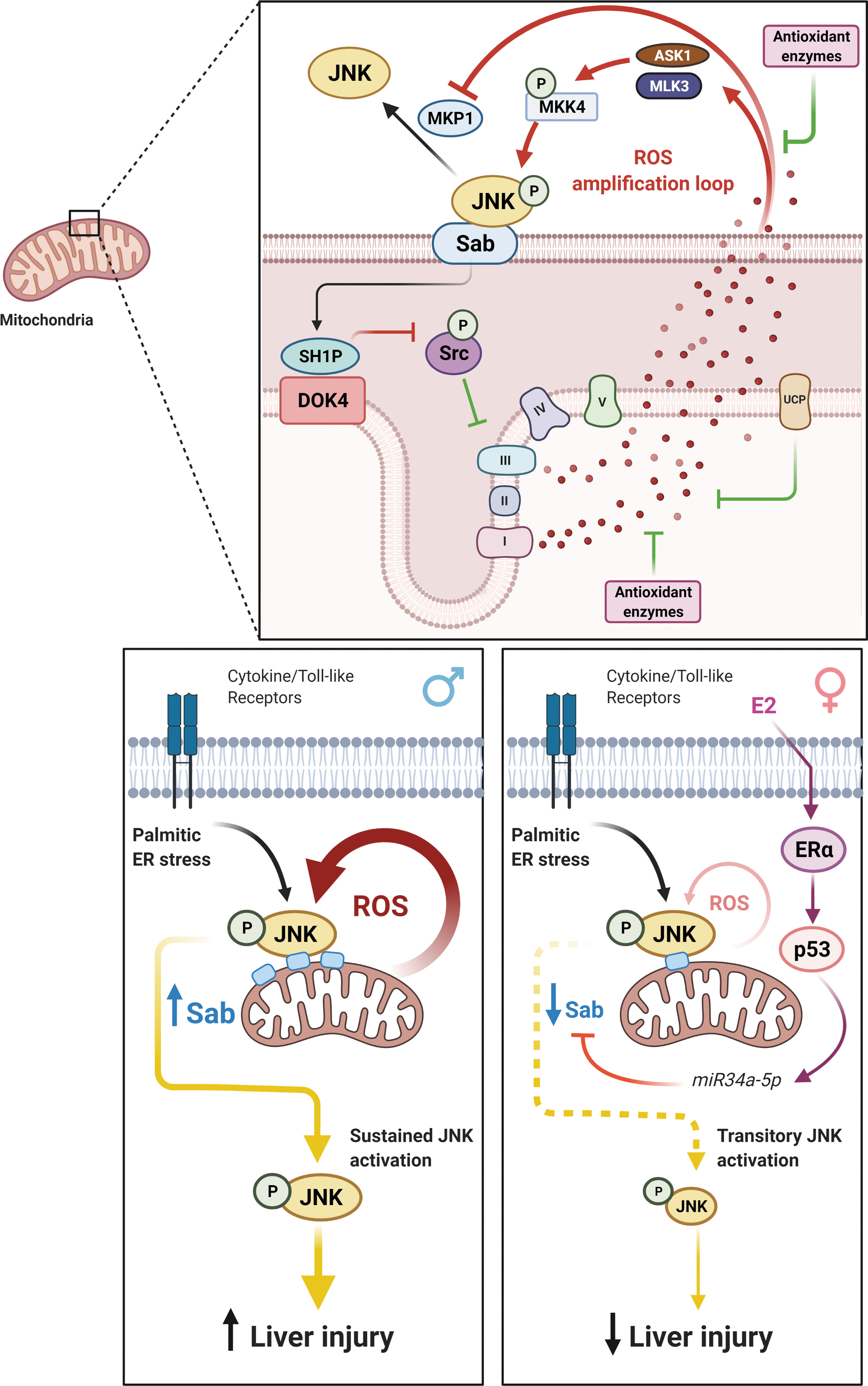
Altogether, MAP3K and increased ROS contribute to the sustained level of JNK activation (191). The prolonged JNK activation can promote apoptosis by inducing BH3 members of B-cell lymphoma 2 (BCL2) family expression, or by direct activation of proapoptotic and/or inhibition of antiapoptotic BCL2 family members (191). In addition, the increased ROS production can overwhelm mitochondria and induce the opening of mitochondrial permeability transition pore (MPT), mediating either apoptotic or necrotic cell death (191).
Recently, the JNK amplification loop has been shown to be influenced by sex. Primary hepatocytes from female mice and woman are more resistant to several JNK-dependent acute liver injury models (acetaminophen, tunicamycin, galactosamine/tumor necrosis factor [GalN/TNF], palmitic acid), and exhibit lower sustained JNK activation and mitochondrial translocation, without differences in total JNK (JNK1 and JNK2) compared with those from males (46, 187, 189). Although earlier work from Du et al. pointed for sex differences in reduced glutathione (GSH) recovery as a potential cause for the lower JNK activation in females, a recent work from Win et al. elegantly demonstrated how estrogenic-dependent repression of Sab protein in females was responsible for their protection against acetaminophen-induced injury (187). In their study, translation of Sab messenger RNA (mRNA) was shown to be inhibited by miR34a-5p, which is in turn induced by the ERα/tumor protein p53 (p53) axis (Fig. 4). In agreement, they also found elevated levels of p53 and low levels of Sab in premenopausal women, the inverse profile in age-matched men, and no differences in Sab expression between men and women at postmenopausal age (187). Although this reciprocal relationship between p53 and Sab expression levels in mouse liver is also present in human, more studies are needed to determine whether this mechanism is relevant to human's sex dimorphism in susceptibility to liver injury.
Sex Influence on PPARα
PPARα is the main PPAR isotype expressed in the liver and plays a major role in energy homeostasis, by regulating fatty acid uptake, fatty acid β-oxidation, ketogenesis, bile acid synthesis, and triglyceride turnover (32). In addition to its role in the regulation of metabolism, PPARα is thought to have anti-inflammatory and antioxidant effects through complex regulation of NF-κB (19). PPARα natural ligands are endogenous lipids such as n-3 polyunsaturated fatty acids (PUFAs) and their derivatives (eicosanoids, oxidized phospholipids), while synthetic ligands include the class of hypolipidemic drugs fibrates, xenobiotic agents, and plasticizers (136). Experimental n-3 PUFA supplementation prevents liver steatosis and inflammation induced by HFD, with underlying mechanisms involving enhanced PPARα signaling and diminished NF-κB activation (182).
Ovariectomy in rats is associated with increased steatosis, which occurs through a decreased expression of Pparα and an increase in the transcriptional activity of genes encoding SREBP-1c, with these effects being prevented by E2 replacement (143). In agreement, several studies have reported sex-dependent interactions on the response to dietary fatty acids in PPARα target genes. For instance, Morise et al. found that the upregulation of PPARα target genes induced by alphalinolenic acid (ALA)-rich diet occurred only in female mice, reinforcing the idea of a crosstalk between E2 and PPARα on lipid regulatory pathways (140). In PPARα-deficient mice, both sexes developed similar low steatosis when fed on an ALA-rich diet (PPARα stimulated). Interestingly, when PPARα-deficient mice were fed on a saturated fatty acid-rich diet (PPAR nonstimulated), the extension of hepatic steatosis and adiposity was lower in females, suggesting the existence of additional protective mechanisms capable to compensate for the PPARα deficiency in females (140).
Although these findings support a partial participation of PPARα in the mechanisms underlying estrogenic protection against hepatic steatosis, the use of PPARα synthetic agonists has reported controversial results regarding the effectiveness and safety in females. Fenofibrate has been used for decades for the control of dyslipidemia and weight loss through PPARα mechanisms involving fat mobilization by increasing fat catabolism in the liver (35, 126). Several studies have reported that female rats are less responsive than males to various effects of fibrates, including increased liver weight, PPARα expression, and peroxisomal oxidation (87). In agreement, Jeong et al. showed that reductions in weight gain and adipose tissue mass by fenofibrate occur in male and ovariectomized female mice, but not in females with active ovaries, suggesting that the action of PPARα on obesity models may be diminished by ovarian factors (88). Results from the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial are also in this line, suggesting lower effectiveness of fenofibrate in women with a trend toward negative effects in this sex (66). However, the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial found similar cardiovascular event reduction in both sexes, even showing a more pronounced improvement of lipoprotein profile in women than in men (42).
Although these trials have focused on lipid profile, insulin resistance, and cardiovascular risk, all factors closely related to MetS and NAFLD, more focused studies on NAFLD are needed to assess the efficacy of fenofibrate to limit steatosis and decipher the interaction between E2 and therapies targeting PPARα (105, 144).
The Hepatokines Fibroblast Growth Factor 21, Retinol-Binding Protein 4, and Angiopoietin-like Protein 3
The liver has been shown to affect the lipids and glucose metabolism by hepatokines released into the blood, and NAFLD seems to be associated with altered hepatokines production (107). Among these hepatokines, fibroblast growth factor 21 (FGF21) deserves special mention, since it has been attributed a role in ketogenesis (12), insulin sensitivity (79, 200), glycemia (1), and obesity (156). Serum FGF21 is secreted mainly by the liver, although expression of this metabolic hormone has also been reported in adipose tissue, central nervous system, and other tissues (176).
FGF21 regulates carbohydrate and fatty acid metabolism, and is induced in liver by fasting and ketogenic diets. One of the main targets of FGF21 is white adipose tissue, in which it induces glucose uptake, browning, and adiponectin secretion, with this tissue mediating many of the systemic effects of FGF21 (23, 79). In this regard, the capacity of FGF21 to ameliorate plasma triglycerides, hepatic steatosis, and liver injuries disappears in adiponectin knockout mice, although the blood glucose reducing effects are preserved (115). Paradoxically, although FGF21 function significantly contributes to lower body weight and enhances insulin sensitivity, circulating FGF21 levels are increased in obesity and NAFLD patients, suggesting the existence of state of “FGF21 resistance” or compensatory mechanisms underlying these pathological conditions (128).
There are few studies supporting a sexual dimorphism in circulating FGF21 levels (97). In children, a sex difference in FGF21 levels has been reported with girls having higher FGF21 levels (22), while no significant differences have been found in many reports between sexes in adults (97). Despite these findings, a sex-specific role for FGF21, however, cannot be excluded. In fact, sex-specific associations of FGF21 levels with femoral intermedia thickness, HDL levels, blood pressure, and brown adipose tissue (BAT) activity have been reported (72).
In mice, Bazhan et al. demonstrated sex differences in the expression of hepatic Fgf21 in response to fasting and refeeding (16). In support of a role for E2 in FGF21 regulation, Hua et al. found that liver Fgf21 expression correlates with endogenous E2 levels during the estrous cycle, but also with exogenous E2 treatment (80). Likewise, Allard et al., using ER- and Fgf21-deficient female mice, reported that treatment with E2 at pharmacological doses increases liver Fgf21 expression and circulating levels, through an ERα-dependent mechanism, with an increase in energy expenditure, at least partially, dependent on FGF21 (3).
Retinol-binding protein 4 (RBP4) is a 21 kDa plasma protein that binds and transports vitamin A (Retinol) in the blood, and is produced by liver and adipose tissue (112). This hepatokine has been suggested to provide the linkage between obesity and insulin resistance. Indeed, plasma RBP4 levels have been demonstrated to be associated with the degree of glucose intolerance (196); the degree of adiposity, especially abdominal obesity (65, 89); and more generally, with components of the MetS (50).
Serum RBP4 concentration varies by sex, generally being lower in females than in males, both among adults and children (13, 196). Several studies hypothesized that sex hormones play a role in the sexual differences observed in RBP4. In this regard, a study conducted in adolescents showed that plasma RBP4 concentrations correlated with obesity and cardiovascular risk factors predominantly in boys, with testosterone as an independent predictor of RBP4 concentrations in this sex (113). In contrast, RBP4 levels are increased after menopause (6) and negatively related to estrogen in obese women (111).
The angiopoietin-like proteins (ANGPTLs) are secreted factors structurally similar to angiopoietins; among them, ANGPTL3, 4, and 8 influence lipid metabolism by binding circulating lipoprotein lipase and antagonizing its activity (8, 43). Within ANGPTLs, ANGPTL3 is exclusively produced by the liver, in particular by the hepatocytes, so it is fully considered as a hepatokine and given its central role in modulating circulating cholesterol and triglycerides levels, is potentially involved in the intrahepatic fat accumulation (43, 96, 139). Higher expression levels of Angptl3 have been observed in male obese spontaneously hypertensive (SHROB) rats (44). However, human studies have not observed significant sex differences in ANGPTL3 levels (153). Interestingly, a recent study showed that ANGPTL3 and -8 were both decreased in females following a high PUFA diet, whereas no response was found in males (94). Therefore, further research is needed to understand how sex and diet interaction influence these hepatokines and NAFLD risk.
Sexual Dimorphism in Adipose Tissue
Adipose tissue cooperates with liver regulating lipid handling and maintaining whole-body energy homeostasis (125). Although the capacity for lipid storage of adipose tissue is considerable, the western pattern of nutrition, characterized by an excess of calories, can surpass the expandability of the tissue, leading to adipose tissue dysfunction, inflammation, and proinflammatory adipokine release that results in increased lipid flux to the liver and other organs, contributing to the development of insulin resistance (51).
Sex hormones have a strong influence on the physiology of adipose tissue and, therefore, shape the interaction between the liver and this tissue, which contributes to explain the sex differences in the development and progression of NAFLD and its comorbidities (Fig. 1). Here, we review the recent literature on two prominent traits of adipose tissue influenced by sex steroids that translate into differences in metabolic pressure exerted from adipose tissue to the liver: fat distribution and adiponectin secretion.
Sexual Dimorphism in Fat Distribution
During the development of obesity, not all fat accumulation is equal. It is well known that abdominal obesity, especially visceral adipose tissue (VAT), plays an important role in the development of metabolic diseases independent of generalized obesity (110). Compared with subcutaneous adipose tissue (SCAT), VAT depots display a greater secretory capacity and a more proinflammatory profile. Furthermore, the release of free fatty acids from VAT occurs directly into the portal blood exposing the liver to higher fatty acid concentrations (78). From a sex perspective, this is of utmost importance, given that men have almost twice as much visceral fat as women for any given fat mass value (164). As represented in Figure 5, women accumulate more fat in the gluteofemoral SCAT depot showing predominantly a pear-shaped fat distribution, whereas men display an apple-shaped fat distribution characterized by an excess of abdominal VAT (83). This sex-dimorphic pattern of fat distribution is dependent on sex steroids, as evidenced by the switch from gluteofemoral to more abdominal fat accumulation during menopause transition (57).
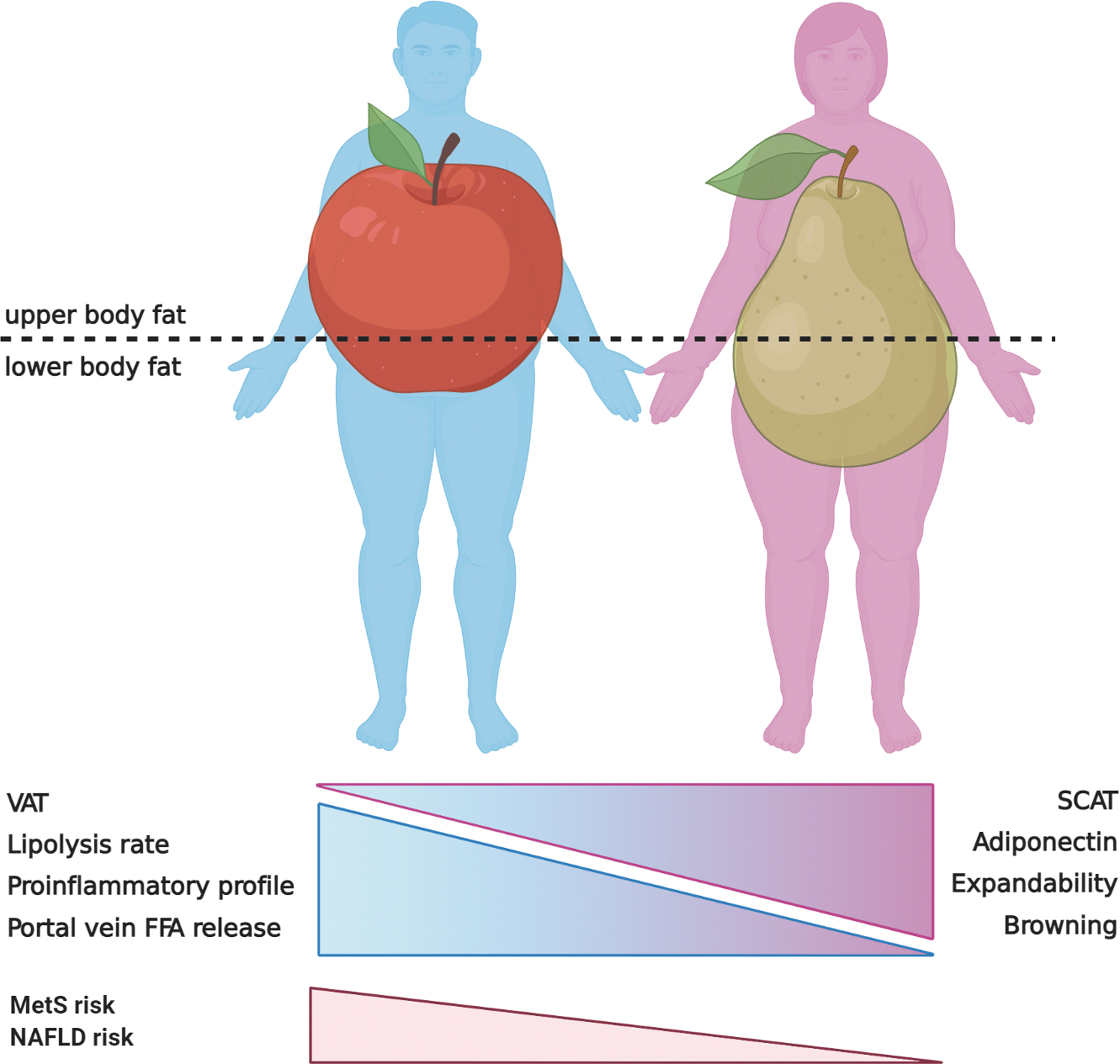
Aside from the anatomical location, VAT and SCAT have important metabolic differences that can impact liver and other organs. VAT is characterized by a predominance of β-adrenergic receptors compared with α2 subtype, which makes it extremely sensitive to catecholamine-induced lipolysis. In contrast, SCAT lipolysis rates are ∼50% lower, have decreased response to catecholamine, lower density of adrenergic receptors, and diminished hormone-sensitive lipase (HSL) expression (127). VAT and SCAT not only differ in lipolysis rate, but also in the production of proinflammatory adipokines, which is also greater in VAT (127). For instance, in obese patients, interleukin 6 (IL-6) concentration is 50% greater in the portal vein than in the radial artery (55).
In addition, the expandability, that is, the capacity to store safely great amounts of fat in the tissue preventing ectopic fat accumulation in other organs, is higher in SCAT compared with VAT (184). Expansion of adipose tissue occurs either by an increase in volume of pre-existing adipocytes (hypertrophy) or by recruitment of new preadipocytes (hyperplasia). Studies in rodents fed on HFD have shown that VAT expands predominantly by adipocyte hypertrophy, while SCAT expands by hyperplasia (186). Although both processes contribute to fat storage during weight gain, adipocyte hyperplasia leads to smaller adipocytes with higher insulin sensitivity, and thus, a healthier way to expand fat, compared with hypertrophy, which results in hypoxia and adipocyte dysfunction (93). Therefore, even considering that VAT accounts for <10% of total body fat, the higher amount of VAT in men compared with women increases the flux of fatty acids and proinflammatory adipokines to the liver, which enhances the risk of NAFLD and liver injury in men, especially in obesity conditions.
In addition to constitutive brown adipocytes in classic BAT depots, brown adipocytes can appear in white adipose tissue in response to cold temperature and β-adrenergic stimulation (157). These cells are different from the classical brown adipocytes, and are known as beige (or BRITE, for brown in white) adipocytes. Brown or beige adipocytes have the ability to oxidize great amounts of metabolic substrates (fats and glucose) to produce heat by a mechanism consisting in uncoupling OXPHOS in mitochondria via uncoupling protein 1 (UCP1) expression. Thus, activation of thermogenesis in brown or beige fat functions as a sink for glucose and fatty acids and therefore, it could have protective effects against nutrient-overload-induced insulin resistance and MetS, harboring an interesting therapeutic potential (157).
Recent studies in mice have shown that E2 can modulate sympathetic innervation in white fat, providing females with a higher capacity to recruit thermogenic brown adipocytes in various white adipose depots (102, 138). Similar findings have been reported in humans by analyzing the expression of UCP1 in adipocyte precursor cells from perirenal adipose tissue (21). In fact, several studies using positron emission tomography have suggested that the amount and activity of brown or beige fat is higher in women than in men (41, 149).
Sexual Dimorphism in Leptin and Adiponectin Secretion
Besides the effect of sex hormones on fat distribution and metabolic traits of the tissue, sex steroids regulate the expression of certain adipokines, extending sexual dimorphism to adipose tissue endocrine function. Sex differences have been reported for many adipokines, including leptin, adiponectin, chemerin, omentin, vaspin, lipocalin-2, glypican-4, and others [reviewed in Valencak et al. (181)]. Among these adipokines, we focus our discussion on adiponectin due to its undeniable key role in the adipose–liver crosstalk.
Adiponectin is the most abundant adipocyte-derived hormone, and is the only one that has a positive role in hepatic and systemic insulin sensitivity, being reported to attenuate liver inflammation and fibrosis (64). Liver expresses both subtypes of adiponectin receptors (AdipoR1 and AdipoR2), and adiponectin stimulation results in the activation of AMPK and increased expression of PPARα target genes. In liver, adiponectin decreases gluconeogenesis and stimulates glycolysis and fatty acid oxidation, which contribute to its antidiabetic and antisteatotic effects (26). Clinical studies have demonstrated that serum adiponectin is lower in NAFLD patients than in healthy controls, and is inversely associated with fat content and insulin resistance in liver (59). Actually, low adiponectin levels generally predict steatosis grade and the severity of NAFLD; however, to what extent this is a direct effect or related to the presence of more severe insulin resistance or obesity remains to be addressed (53).
Women have greater adiponectin levels than men despite having a relatively great percentage body fat (39, 95). Furthermore, sex differences in adiponectinemia persist in men and women pair-matched for age, body mass index, visceral fat, and insulin sensitivity (39). This sexual disparity in adiponectin levels initiates in the puberty, with males showing decrease in adiponectin levels in association with the rise of androgens (24). The role of androgens is further supported by the increase in adiponectin levels in castrated mice and its reversion by testosterone treatment (141). In addition, testosterone was found to decrease adiponectin levels in culture media of 3T3-L1 adipocytes (30, 141). In contrast, in vitro studies analyzing the effects of E2 on adiponectin expression in adipocytes have reported inconsistent results (17, 30, 146). Accordingly, ovariectomy in rodents (5, 141) or menopause in women (91) does not significantly alter adiponectin levels either.
Studies about circulating adiponectin levels as a function of age in premenopausal and postmenopausal women have reported conflicting results (86). Whereas plasma concentration of adiponectin inversely relates to VAT mass in premenopausal women, some studies have reported a weak correlation in peri- and postmenopausal women (104). Circulating adiponectin increases with age, with postmenopausal women having greater levels than premenopausal women (138a). A recent study reported that low serum adiponectin levels are associated with increased risk of developing MetS in both pre- and postmenopausal women, whereas higher levels of the hormone were significantly associated with a lower risk of incidence of MetS in premenopausal, but not in postmenopausal women (2). It is tempting to speculate about the reasons of this apparent discrepancy between the increased adiponectin and the worsened insulin sensitivity observed in postmenopausal women. In the first part of this review, we discussed a series of key factors involved in lipid metabolism and oxidative stress that have shown positive association with estrogens. With the cessation of ovarian function and the subsequent drop in estrogen levels at menopause, the liver may become more refractory to the insulin-sensitizing action of adiponectin, especially considering that many of these factors influenced by sex hormones belong to adiponectin pathway (AMPK, PPARα, PGC1α).
Although the hypothesis of E2 as a sensitizing factor for adiponectin needs further testing, it could explain why HRT has been shown to improve insulin sensitivity in postmenopausal women (122, 158), despite no studies have reported an increase in circulating adiponectin levels (168, 175). In addition, studies in ovariectomized female as well as male mice have reported hepatic insulin-sensitizing effects of E2 treatment (200). Altogether, these data suggest that estrogens potentiate adiponectin sensitivity in liver, contributing to improve the insulin-sensitizing action of adiponectin. However, further studies providing more robust evidence are needed to demonstrate whether these assumptions are real or just speculations.
Therapeutic Perspectives of Sexual Dimorphism in NAFLD
Although great efforts have been dedicated to the understanding of the NAFLD pathogenesis, there are currently no approved medications for the treatment of the disease, with the guidelines recommending lifestyle modifications such as weight loss or drugs targeting the associated comorbidities such as insulin resistance or dyslipidemia (108). Like other MetS-associated diseases, NAFLD has an important sex dimension that needs to be considered in several key aspects, including the development of therapeutic strategies. First, the development of new drugs should take into consideration the sexual disparity in the pathogenesis of NAFLD and NASH to achieve sex-tailored treatments with respect to the dosage and guidelines for administration. Second, a better understanding of the physiopathological peculiarities of NAFLD in premenopausal women, with lower prevalence of the disease than men or postmenopausal women, may contribute to inspire new therapeutic interventions.
Considering the preclinical evidence that suggests E2 exerts protection against liver steatosis and fibrosis, it is tempting to consider HRT as a preventive therapy for NAFLD in postmenopausal women. In fact, clinical trials in postmenopausal women using HRT have shown decreased insulin resistance, a well-known risk factor for NAFLD (54, 155). In addition, other studies in diabetic postmenopausal women receiving HRT have reported decreased levels of serum transaminases, indicative of lower liver injury (135). Although HRT improves several conditions associated with menopause, such as osteoporosis and endothelial function, several side effects such as thrombosis or breast cancer risk have been described in the long-term users (182). Moreover, HRT cannot be implemented in men as a therapy for NAFLD or its comorbidities due to its undesired feminizing effects. Therefore, more selective therapeutic strategies to target the beneficial effects of estrogenic signaling, without the full pleiotropic action of E2, are being intensively studied (Fig. 6).
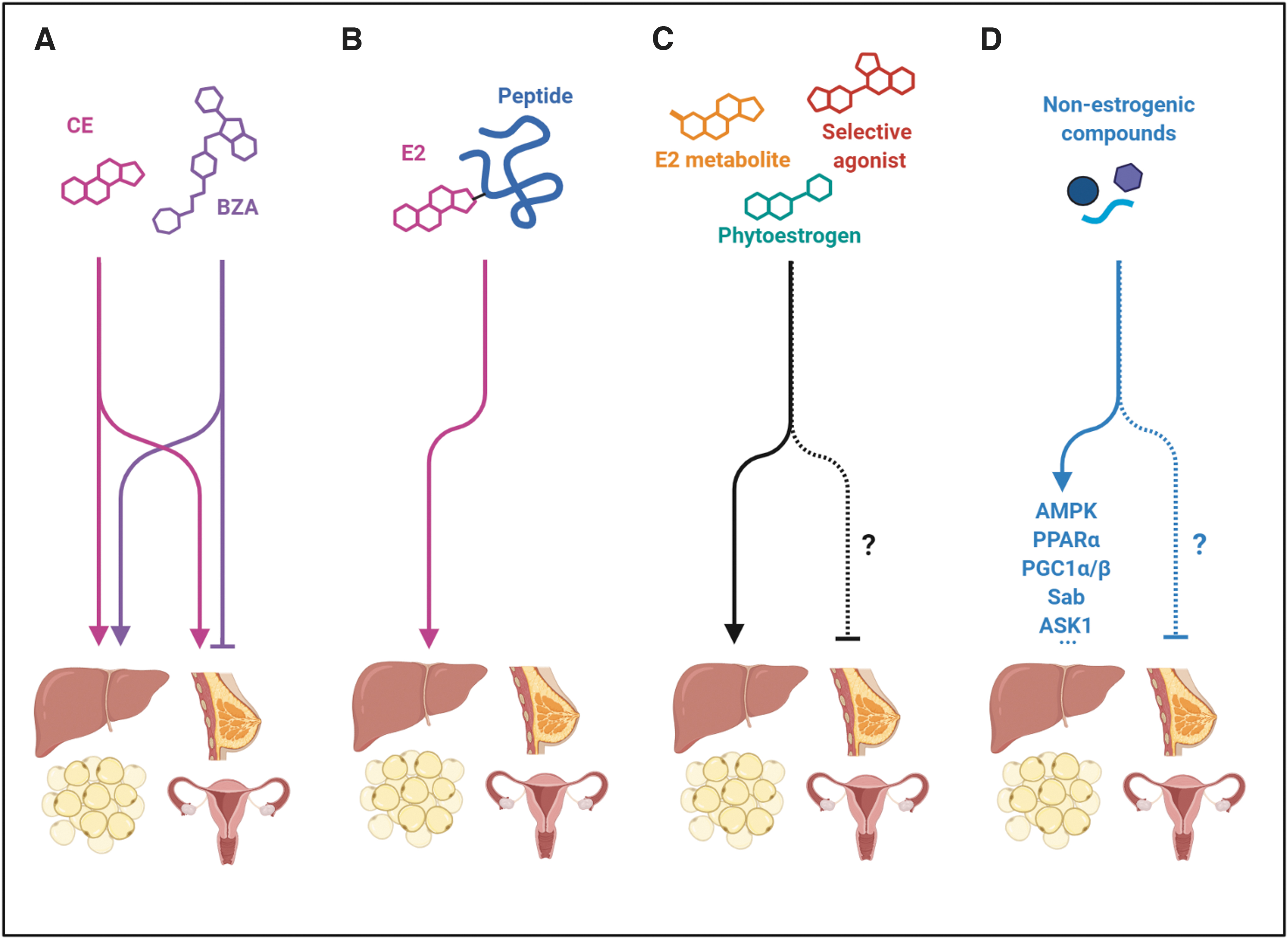
One of these potential strategies could be the use of selective estrogen receptor modulators (SERMs) able to interact with ERs, each with a distinct agonism/antagonism degree at different target tissues. However, the use of tamoxifen and toremifene, SERMs, indicated for the treatment of ER-positive breast cancer is associated with increased prevalence of NAFLD (161, 195). A second-generation SERM, raloxifene, has shown beneficial effects ameliorating the progression of liver fibrosis in a NASH model of ovariectomized mice (123). Nevertheless, although raloxifene is used to treat osteoporosis in postmenopausal women, no studies have reported hepatic metabolic benefits in women, and even a rare case report showed aggravation of steatosis in patients with underlying NAFLD (131).
A third-generation SERM, bazedoxifene (BZA), exhibits estrogen antagonistic activity in breast and uterus, and estrogen agonistic activity in bone, thus preventing osteoporosis while protecting the breast and uterus from estrogenic stimulation (193). A new therapeutic strategy, named tissue-selective estrogen complex (TSEC), consisting in the combination of conjugated estrogens (CEs) with BZA, has reported to provide all the benefits of CE treatment and protection of breast and uterus without using progestin (161). Initial studies have shown that this combined therapy has beneficial effects in lipid profile, reducing low-density lipoprotein-cholesterol and increasing HDL-cholesterol levels (169). In ovariectomized mice, CE/BZA or BZA alone attenuated ovariectomy-induced weight gain, decreasing body and hepatic fat accumulation (100). The reduction in hepatic steatosis after CE/BZA treatment was achieved by stimulation of fatty acid oxidation and repression of lipogenesis (15, 100). There are ongoing clinical trials to address the effects of combined CE/BZA to prevent metabolic disorders in obese postmenopausal women.
Another interesting strategy is targeting estrogens to selective tissues by conjugating the molecule with a peptide. For instance, a glucagon-like peptide 1 (GLP1)–estrogen conjugate was shown to improve MetS abnormalities with higher efficacy than GLP1 alone, and without the hallmark side effects of estrogen in male and female mice (52, 178). This new pharmacological approach has appreciable therapeutic promise considering the multitude of possible combinations of bioactive peptides and small molecules.
Phytoestrogens have also been explored for many years as a safer estrogen supplementation for menopausal women. So far, isoflavones are the most studied phytoestrogens. Black soybean has been shown to have some benefits, including improvement of cholesterol metabolism, insulin resistance, and oxidative stress in mice fed a high-cholesterol/fat diet (90). The most studied isoflavone, genistein, has been shown to improve liver function and attenuate NAFLD in several animal models (192). Beneficial effects of other phytoestrogens, such as daidzein, quercitrin, or oxyresveratrol, have also been reported against liver steatosis and insulin resistance (82, 101).
The reported evidence of the useful effects of phytoestrogens mainly focuses on animal experiments, with fewer studies addressing the effects of these compounds in humans. Based on the beneficial effects of genistein observed in animal models, genistein has been further explored as a promising clinical drug for NAFLD treatment. A randomized controlled trial showed that oral supplementation with 250 mg of genistein for 8 weeks reduces insulin resistance, oxidative stress, and inflammation along with an improvement of fat metabolism in patients with NAFLD (4). Furthermore, two clinical trials have reported beneficial effects of genistein on lipid profile in MetS patients (84, 173). Additional clinical trials exploring the long-term effects and possible side effects of genistein are necessary.
The use of pure agonists, targeting a selective ER subtype, is another interesting strategy to find suitable candidates for NAFLD treatment, which has been extensively studied in animal models. To date, beneficial effects on NAFLD/NASH have been reported for ERα (37), ERβ (198), and GPER (167) agonists, acting through diverse pathways. These results are in agreement with the idea that each ER subtype has, somehow, a contribution to NAFLD, contrasting with ER knockout studies, where not all ERs have shown a clear involvement. As previously mentioned, ER knockout studies must be interpreted with caution in the context of estrogenic signaling since compensatory activation of intact ERs, or even undiscovered compensatory mechanisms, could mask the deficiency of a specific ER subtype (130).
Considering the liver as the main site where E2 is metabolized, another interesting strategy is to investigate the activity of estradiol metabolites in liver function (108). In fact, two biologically active metabolites derived from E2, 2-hydroxyestradiol and 2-methoxyestradiol, have been recognized to have antifibrotic activity in liver, acting at lower concentrations than E2 (118). Given the lower affinity that ERs exhibit for these metabolites and the low effective concentration, these E2 by-products are interesting candidates for the development of NAFLD/NASH therapeutics. However, further studies are needed to understand the mechanisms by which E2 metabolites protect from liver fibrosis.
Another therapeutic strategy based on the sex differences in NAFLD, but without the use of estrogen-like compounds, would be the use of molecules targeting the same effectors of estrogen signaling in metabolic homeostasis; for instance, molecules such as activators of AMPK or PPARα. In fact, many antidiabetic or lipid lowering drugs, such as metformin or fenofibrates, are commonly used for the treatment of comorbidities associated with NAFLD; however, their effectiveness against NAFLD or NASH is limited, underscoring the need to find new therapeutic agents more specific to liver disease (47).
Among these therapeutic targets, the sustained JNK activation loop is worthy of mention (191). While direct inhibition of JNK does not seem a good target as it would interfere with the physiological aspects of JNK signaling, the elements of the sustained JNK activation loop, which mainly regulates metabolism and cell death, are an interesting alternative. Drugs upregulating antioxidant genes or antioxidant supplementation could be a strategy to dampen mitochondrial ROS and the sustained activation of JNK (45). Chemical inhibitors of apoptosis signal-regulating kinase 1 (ASK1), one of the key MAP3K involved in the activation loop, have also reported promising results in preclinical and clinical studies, reversing liver fibrosis in NASH patients (121).
Another approach showing efficacy in decreasing the activation of JNK and the resulting cellular injury involves blocking the interaction between JNK and Sab, which can be achieved by using the chemical compound Antcin H or a peptide blocking the docking site on Sab (33, 81). Finally, it is worth mentioning that other drugs, not based on the sexual dimorphism, are also being tested for the treatment of NAFLD and NASH. Hopefully, in the near future, new drugs or combined therapies with greater efficacy will emerge for the treatment of NAFLD and NASH.
Conclusions
Sex hormones shape the crosstalk between liver and adipose tissue, contributing to explain the lower risk of NAFLD seen in premenopausal women. Sex differences are reported along pivotal master regulators of lipid metabolism and oxidative stress, key determinants in the pathogenesis of NAFLD and its progression to NASH. These sex differences provide novel targets to inspire the development of new therapeutic strategies that may have an impact on the prevention or treatment of NAFLD in postmenopausal women and men. Further research allowing a more in-depth understanding of estrogen signaling is necessary to design new drugs that harbor the protective capacity of estrogens, avoiding their side or feminizing effects.
Footnotes
Authors' Contributions
All the authors drafted the work and revised it.
Author Disclosure Statement
All the authors declare that there are no conflicts to disclose.
Funding Information
This work was supported by FEDER/Ministerio de Ciencia, Innovación y Universidades, Agencia Estatal de Investigación (SAF2016-80384R) of the Spanish Government.