
Abstract
Significance:
Sepsis is a major public health concern, with high mortality and morbidity, especially among patients undergoing trauma. It is characterized by a systemic inflammatory response syndrome (SIRS) occurring in response to infection. Although classically associated with pathogens, many patients with SIRS do not have infection. The variability of the disease course cannot be fully explained by our current understanding of its pathogenesis. Thus, other factors are likely to play key roles in the development and progression of SIRS/sepsis.
Recent Advances:
Circulating levels of damage-associated molecular patterns (DAMPs) seem to correlate with SIRS/sepsis morbidity and mortality. Of the known DAMPs, those of mitochondrial (mt) origin have been of particular interest, since their DNA (mtDNA) and formyl peptides (mtFPs) resemble bacterial DNA and peptides, and hence, when released, may be recognized as “danger signals.”
Critical Issues:
mtDAMPs released after tissue injury trigger immune responses similar to those induced by pathogens. Thus, they can result in systemic inflammation and organ damage, similar to that observed in SIRS/sepsis. We will discuss recent findings on the roles of mtDAMPs, particularly regarding the less recognized mtFPs, in the activation of inflammatory responses and development of SIRS/sepsis.
Future Directions:
There are no established methods to predict the course of SIRS/sepsis, but clinical studies reveal that plasma levels of mtDAMPs may correlate with the outcome of the disease. We propose that non-pathogen-initiated, mtDAMPs-induced SIRS/sepsis events need further studies aimed at early clinical recognition and better treatment of this disease.
Introduction
At least 1.7
The results of the second International Sepsis Definitions Conference defined sepsis as systemic inflammatory response syndrome (SIRS) associated with presumed or confirmed infection (14). “Severe sepsis” indicated additional development of life-threatening organ dysfunction, and “septic shock” implied the additional development of hemodynamic instability. However, the symptoms of sepsis are often non-specific and highly variable, in many cases depending on susceptibility, physiologic reserves, and reaction of the host.
The “classic” signs are tachypnea, temperature >38.3°C (or <36°C), and increased heart rate. Changes in non-specific markers of inflammation, such as white blood cell count >12 × 109/L or leukopenia <4 × 109/L, presence of immature white blood cell forms, or elevated C reactive protein levels, are also common. These changes can then be followed by organ dysfunction and septic shock (71). On the molecular level, there is commonly a so-called “cytokine storm” (30). This is classically described as being initiated by pattern recognition receptors (PRRs) sensing pathogenic patterns such as lipopolysaccharides. However, equally important may be PRR sensing of endogenous molecules such as mitochondrial damage-associated molecular patterns (mtDAMPs), which can turn on the same signal cascades, leading to vast pro-inflammatory cytokine production followed by organ damage, coagulation, and apoptosis, and the vicious circle is completed (15).
In 2016, the summary from the third International Consensus Definitions for Sepsis has proposed a new definition, named Sepsis-3, and described it as “life-threatening organ dysfunction caused by a dysregulated host response to infection” (108, 109, 112). The new definition discarded the use of the term “SIRS” (a diagnostic standard for the past 25 years) in the identification of sepsis and eliminated the term “severe sepsis.” The authors recommended the use of Sequential Organ Failure Assessment (SOFA) scoring to assess the severity of organ dysfunction in a potentially septic patient. The SOFA scoring has often been used in clinical trials. Due to its complexity, however, it is sometimes reduced to the so-called “quick (q)SOFA” score, which includes three clinical parameters: respiratory rate ≥22/min, change in mental status, and a systolic blood pressure ≤100 mmHg.
Nonetheless, these new definitions have limitations, particularly in the early detection of sepsis. Hence, they have encountered criticism (13, 103). It is still not clear whether SIRS criteria or qSOFA score has more prognostic accuracy for in-hospital mortality, thus this needs more research. The Sepsis-3 definition neglects the early (clinical) identification of sepsis before the development of organ failure. Moreover, it relies on descriptions of body responses with no clear biochemical identification of the causes of sepsis. Last, many health care facilities still use older sepsis definitions as drivers of emergency room and intensive care unit protocols.
Damage-Associated Molecular Patterns
Although there has been considerable improvement in the understanding of sepsis pathogenesis in recent years, its classical definition—an exacerbated inflammatory response to pathogens—cannot fully explain some of the abnormal physiological alterations and the unpredictable course of the disease. Moreover, 30% of patients with sepsis-like syndromes are never proven to have infections (48, 94, 130, 131). Therefore, much attention has been turned to other factors that can contribute to the evolution and persistence of the inflammatory state in patients with sepsis.
During infection, the inflammatory response is initiated by recognition of exogenous microbial ligands known as pathogen-associated molecular patterns (PAMPs), by specific PRRs. The PRRs are predominantly expressed on immune cells, but they also exist on resident cells in tissues or even vascular endothelial cells (ECs) (91), and their activation induces the production of pro-inflammatory cytokines, typically beginning with tumor necrosis factor alpha (TNF-α) and interleukin-1 (IL-1) (83). After Matzinger's suggestion that the immune system is also triggered by endogenous “danger” signals released by the body's own injured tissues, many of these endogenous ligands were found to promote immune response (81).
These danger signals, known as DAMPs, are passively released by dying or damaged cells, although some DAMPs can be actively secreted as well. Similar to PAMPs, they can activate the immune system through the activation of classical PRRs. Also, several other non-PRRs recognize DAMPs, including ion channels and G-protein-coupled receptors (89). Activation of both PRRs and non-PRRs by DAMPs promotes sterile inflammation, which is independent of pathogen infection (37).
Numerous endogenous molecules have been identified as DAMPs, and some of them have been shown to have a role in the pathogenesis and prognostic of sepsis. For instance, the first identified DAMP, the high-mobility group box 1 (HMGB1) (133), acts as a late mediator of sepsis (121). High plasma levels of HMGB1 are associated with severity and mortality in sepsis (4, 43). In a murine model of sepsis, targeting HMGB1 increased survival and improved clinical outcomes (118). Heat-shock proteins (HSP), also defined as DAMPs, have a role in the pathophysiology of sepsis as well. Extracellular HSP60 and HSP72 levels are significantly higher in patients with septic shock, and these increased HSP levels have been shown to correlate with worse outcomes (140).
Mitochondrial Damage-Associated Molecular Patterns
Mitochondria are cellular components that produce energy in the form of adenosine triphosphate (ATP). The ATP itself is one of the DAMPs. Injury-induced release of mitochondria and their content increase local ATP levels that can enhance bacterial killing by macrophages in sepsis, acting through P2X7 and P2X4 receptors (18, 19). Mitochondria also produce reactive oxygen species (ROS), which can kill invaders such as bacteria.
Evolutionist Sagan (100a) originally suggested that roughly 2 billion years ago, an anaerobic eukaryotic cell engulfed an aerobic prokaryote, forming an endosymbiotic relationship with the prokaryote, thus creating the organelle that we now recognize as a mitochondrion (26, 100a). Originally, mitochondria contained many molecules common to bacteria simply because they originated from bacteria, but they have adapted to living within cells by exporting most of their genetic material to the nucleus.
Under physiological conditions, mitochondrial components are contained within the cell and although they can be released by cellular injury (62) there was no evidence of active release of mtDAMPs until we recently showed that they can be secreted in extracellular vesicles by monocytes stimulated with lipopolysaccharide (62).
Our group has been interested in DAMPs originating from mitochondria and in their role in injury-related sepsis. We and others have been studying mtDAMPs for more than 10 years and here we overview recent research on mtDAMPs. Though several components of mitochondria may act as DAMPs, including transfactor A, mitochondrial (TFAM), ATP, cardiolipin, cytochrome c, succinate, and mtRNA (72), we will focus on two particular mitochondrial molecules that remain very similar to those of bacteria and thus are of great interest to our group: mitochondrial DNA (mtDNA) and particularly the less recognized mitochondrial N-formyl peptides (mtFPs). We will discuss their ability to induce immune stimulation, immune suppression, and sepsis-like responses. Although mtDAMPs' role in innate immunity is well established (137), their possible function in adaptive immunity has not been well investigated.
mtDNA and Its Sensors: PRRs
Human mtDNA is a circular, double-stranded, highly unmethylated CpG motifs-containing molecule that comprises 37 genes. These genes are encoding 2 ribonucleic acids (RNAs), 22 transfer RNAs, and 13 subunits of the mitochondrial respiratory chain complexes (3). In contrast to nuclear DNA, mtDNA has conserved low-methylated CpG motifs, characteristic of bacterial and viral DNA, and thus can be recognized by classical “foreign” DNA-sensing PRRs such as Toll-like receptor 9 (TLR9), NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome, and stimulatory of interferon genes (STING) (27, 38, 47, 49).
In “resting” cells, TLR9 is located in the endoplasmic reticulum (70) but after activation by CpG DNA it is translocated to the membranes of endosomes and later to lysosomal compartments, where it binds its ligand, CpG DNA, and recruits the intracellular adapter protein myeloid differentiation factor-88 (MyD88). This is followed by activation of nuclear factor-kappa B (NF-κB) and mitogen-activated protein kinases (MAPKs) and induction of inflammatory responses (2, 67). TLR9/MyD88 also activates IL-1 receptor-associated kinase 4 (IRAK4), which activates IRAK1, IRAK2, TNF receptor-associated factor 3, and inhibitory-κB kinase α and finally triggers interferon regulatory factor 7 to induce type I interferon (58, 110).
Activation of NLRP3 inflammasome by mtDNA leads to recruitment and activation of caspase-1, which controls maturation and release of interleukins such as IL-1β and IL-18 whose pro-inflammatory activities regulate host responses to infection and injury (107). And finally, endoplasmic reticulum-resident STING is activated by cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which is generated after interaction between mtDNA and peri-mitochondrial cyclic GMP-AMP synthase (cGAS). The cGAS-cGAMP-STING signaling pathway leads to interferon regulatory factor 3-dependent expression of type I interferon, known to have antiviral activity (74).
mtFPs and Formyl Peptide Receptors
Unlike eukaryotic cells and similar to bacteria, mitochondria require an N-terminal formyl methionine (fMet) residue to initiate protein synthesis. Formylation of the methionyl group is catalyzed by formyl transferases (Fmt) using formyl tetrahydrofolic acid (formyl-THF) as the formyl donor. In eukaryotes, most of the proteins have their formyl group removed after translation by peptide deformylases (36). Nevertheless, fMet is the amino acid coded by the AUG start codon; therefore, nearly all proteins in the prokaryotic domain have fMet as their N-terminal amino acid (75).
In mitochondria, formylated peptides are synthetized by mitoribosomes located in the mitochondrial matrix. There is no leader sequence in the mitochondrial mRNA to guide mitoribosome binding. Instead, the binding of N-terminal fMet to the mRNA start codon is the initiator of translation, justifying the essential role of fMet in mitochondrial peptide synthesis (6). Thus, mtFPs can be potent chemoattractants for immune cells and platelets that express formyl peptide receptors (FPRs) (20, 101, 104), although, as we have shown, only some mtFPs act in this way (55) as discussed later.
The FPRs are a group of well-conserved G-protein-coupled receptors that play an important role in host defense and inflammatory responses. In humans, three FPRs have been identified, FPR1, FPR2/ALX, and FPR3. These receptors are found in a variety of cells, with the highest expression in neutrophils for FPR1 and FPR2, and monocytes/macrophages for FPR3 (143). The FPRs are activated by a variety of structurally and chemically unrelated ligands, and N-formyl-peptides are the only ligand group common to all three human receptors (17, 46, 143).
FPR1 was the first FPR to be cloned and sequenced and it is the most studied receptor among the three of them (22). The primary ligands for FPR1 are bacterial and mitochondrial N-formylated peptides, which are bound with an affinity that is 100-fold higher than the identical non-formylated versions of those peptides. Based on studies with FPR1 agonists, N-formyl-methionyl-leucyl-phenylalanine (fMLF, also called fMLP), a prototypic representative of bacterial chemotactic factor, was identified as the most potent one (111). Gene-knockout (KO) and single nucleotide polymorphism studies revealed that FPR1 can play a dual role in the pathogenesis of various diseases (128). At the cellular level, polymorphonuclear neutrophil (PMN) stimulation via FPR1 induces release of calcium from intracellular stores as well as chemotaxis, degranulation, ROS, and cytokine production (22, 23), all of which will be discussed later.
Compared with FPR1, FPR2/ALX is more promiscuous and binds fMLF with a much lower affinity (45). This suggests that it may become activated at, rather than at a distance from, inflammatory sites. FPR2 is also capable of binding to lipids and lipid-associated proteins, such as lipoxin A4, serum amyloid A, and oxidized low-density lipoprotein (LDL) (46, 69). Depending on the ligand, FPR2 can induce either pro-inflammatory or anti-inflammatory effects. This dual effect may be mostly due to different dimerization states of the receptors after ligand binding (16).
FPR3 is the least studied FPR, and its physiological function remains poorly understood. Contrary to FPR1 and FPR2, FPR3 is not expressed in PMN. FPR3 is also relatively insensitive to formylated peptides and the endogenous 21–amino acid acetylated amino-terminal peptide F2L is its most specific ligand described to date (84, 95).
The FPRs have also been identified in many species, including rodents, rabbits, and horse, with distinct functional responses to formylated peptides observed (143). In mice, their genome encodes at least seven functional genes for FPRs (125). Gene Fpr1 is clearly the murine equivalent of human FPR1, based on homology, cell expression, and physiological function (44). Human FPR2 has two murine equivalents: gene Fpr2 encodes the ALX receptor specific for lipoxin A4, and gene Fpr3 encodes a receptor, which binds formylated peptides, serum amyloid A, and other similar ligands (46, 69).
It is possible that the murine receptor encoded by the gene Fpr2 also mimics the human FPR3, since both bind F2L (35). Despite the high homology between mouse and human FPR1, murine Fpr1 binds fMLF with a 100-fold lower affinity than does the human receptor (44) and the affinity seems to change considerably depending on the mouse strain (unpublished data). Nevertheless, murine Fpr1 is the primary receptor for N-formylated peptides in mice (44).
mtDAMPs in Pathophysiology of SIRS/Sepsis
Similar to bacteria that may reach the circulation, mtDAMPs may be found in blood after organ injury, trauma, or even a major surgery (102, 124, 147). When mtDAMPs are released into circulation, they are recognized by the immune system, driving inflammatory responses (22, 134) and there are increasing numbers of reports indicating that mtDAMPs play an important role in human diseases (7, 39).
We and others have shown that patients undergoing trauma have higher levels of plasma mtDNA compared with healthy subjects (65, 102, 141, 147). Increased plasma mtDNA levels are usually observed within 90 min after a serious injury, such as traffic collisions or falls, and they originate from liver injury, muscle crush injury or bone fracture, and may stay elevated for at least 24 h after trauma (147). The levels of mtDNA in the blood of such patients could be thousands of times higher compared with healthy controls (147). Damage to cells that are rich in mitochondria, such as liver cells, may release more mtDNA into circulation. Plasma mtDNA concentration has been found to be significantly elevated on the first day after trauma and much higher in the non-survivors group (141).
Severe trauma is often followed by uncontrolled inflammation (i.e., SIRS) and end-organ damage, with close associations between mtDNA levels and post-injury complications (50, 65, 113). Plasma levels of circulating mtDNA are also significantly increased in severe sepsis/septic shock patients and are much higher than in patients with postoperative inflammation or after traumatic injury (105, 124, 141). Plasma concentration of mtDNA was proposed to be an independent predictor for post-traumatic SIRS (40).
The concept that mtDNA catalyzes sterile inflammation after trauma currently seems to be well accepted but has not yet led to changes in clinical management. The aims would be to confirm mtDNA as a predictor of severity of SIRS/multiple organ failure to optimize the clinical treatment and possibly find therapeutic agents preventing inflammatory effects of mtDNA. Importantly, mtDNA might be a better biomarker than lactate concentration or even SOFA score for predicting the mortality of patients with sepsis after admission in the emergency room (64), hence further studies are of importance.
Contrary to mtDNA, only a limited number of publications can be found on the role of mtFPs in sepsis. Wenceslau et al. suggested that, by acting through FPRs, mtFPs promote inflammation and vascular dysfunction, contributing to the development of sepsis (135). Using a rat model, they showed that mtFPs causes sepsis-like syndrome and cardiovascular collapse as well as hyperthermia, vascular leakage, blood clotting, and lung injury (136). They also applied mtFPs intratracheally and observed PMN migration to the lungs assisted with tracheal, bronchial, and bronchiolar contraction, which was all blocked by a selective antagonist of FPR2, WRW4 (Trp-Arg-Trp-Trp-Trp-Trp-NH2) (139). They concluded that mtFPs released during bacterial infection and trauma/SIRS may induce FPR activation, followed by sepsis-like symptoms and leading to acute lung injury (ALI).
Dorward et al. clearly demonstrated increased levels of mtFPs in bronchoalveolar lavage fluid (BALF) and serum of patients with acute respiratory distress syndrome (ARDS), and they documented FPR1-dependent PMN activation and chemotaxis, both in vivo and in vitro (23). They evaluated whether mtFPs play a role in the regulation of pulmonary migration by applying formyl-methionyl-isoleucyl-threonine (fMIT) to the lungs of wild type (WT) and Fpr1-KO mice. Compared with untreated WT mice, they showed that WT mice treated with cyclosporin H (CsH; FPR1 antagonist) or Fpr1-KO mice had a decreased inflammatory response and reduced PMN migration toward the lungs and, hence, lesser lung injury.
They further validated these results in a mouse sterile lung injury model, in which they intratracheally administered hydrochloric acid to the lungs of WT and Fpr1-KO mice and found a reduced number of PMN in BALF of Fpr1-KO mice, although the total number of circulating PMN was unchanged (23). The authors concluded that FPR1 plays an important role in PMN migration toward the lungs and thus in lung damage in sterile lung injury and suggested that FPR1 is a potential therapeutic target in sterile ALI.
The mtDAMPs can also induce strong inflammatory responses and sepsis-like symptoms. We have studied their roles and found that many of the signaling pathways that mtDAMPs activate are similar to those activated in SIRS/sepsis. In our systematic approach to recognizing the role and effects of mtDAMPs, we investigated them in both clinical and laboratory settings.
We confirmed the presence of mtDAMPs in patients with trauma, showed exaggerated inflammatory responses triggered by mtDAMPs in animals treated with mtDAMPs, and validated these observations in in vitro experiments. In the latter experiments, we identified the individual roles of mtDNA and mtFPs by blocking their specific receptors or using synthetics mitochondrial f-peptides. Our main findings are outlined in Figure 1 and will be delineated in the next sections. We will show how our studies, though still ongoing, help shed light on the complex pathology of SIRS/sepsis.

mtDAMPs Induce Systemic Inflammation and Organ Damage
In general, mtDAMPs that we applied in our experiments were prepared by using frozen liver samples. First, we prepared mitochondria; then, we sonicated them as described elsewhere (96, 147). For in vivo experiments, mtDAMPs prepared from 10% of a whole liver were administered intraperitoneally unless otherwise noted. Thus, mtDAMPs contained mtFPs, mtDNA, and non-formylated proteins.
Early on, we showed that mtDAMPs applied via the tail vein (i.v.) of rats induced ALI, including oxidative damage as observed by immunohistochemistry (Fig. 2A, B). Other markers of inflammatory responses, that is, increased pulmonary albumin permeability, edema, pulmonary IL-6 accumulation, and PMN infiltration into the airways, were also detected (Fig. 2C–H). Moreover, i.v. administration of mtDAMPs increased levels of the pro-inflammatory cytokines, TNF-α, and IL-6 (Fig. 2I, J) in BALF. Matrix metalloproteinase-8 (MMP-8) levels were increased in rat lungs (Fig. 2K) and liver (Fig. 2L), consistent with increased PMN infiltration into lungs (Fig. 2H) (147). These data confirm the potentially harmful pro-inflammatory effects of mtDAMPs in animal models and parallel clinical observations.

The results from another study suggested that mtDNA might contribute to the initiation of sterile SIRS and lung injury in rats through the activation of the TLR9/NF-κB axis and the induction of pro-inflammatory cytokine expression (145).
mtDAMPs Stimulate PMN
The role of MAPK signaling pathways in the production of inflammatory cytokines is well characterized, and MAPK activities correlate with SIRS/sepsis severity (29, 90, 144). It has also been documented that pro-inflammatory cytokine and MMP levels in BALF and blood are upregulated in sepsis and ALI (33, 116). The MMPs comprise a family of proteases that are widely expressed in developmental, physiological, and pathological processes. They also play broad functions in inflammation and tissue repair (92), often by virtue of their ability to activate other mediators.
Other studies strongly suggest that MMPs are involved in the pathogenesis of sepsis (68, 126, 129). MMP-8 levels in serum are significantly higher in patients with severe sepsis than in healthy controls and higher in non-survivors than in survivors (68). The PMN activation linked to MMP release has also been implicated in the development of sepsis-induced ALI, and MMP inhibition has improved survival in some animal models of sepsis (117). The PMN activation also results in degranulation of MMPs with subsequent extracellular matrix and basement membrane degradation (76). This process has also been implicated in the development of sepsis-induced ALI (117).
Following on from that prior work, we showed that mtDAMPs, indeed, activate p38 and p42/44 (ERK1/2) MAPKs in human PMN (Fig. 3A, B) (147). Moreover, we showed that mtDAMPs induced MMP-8 release from PMN (Fig. 3C, D), which was inhibited by antibodies against FPR1 or by the FPR1 antagonist, CsH. Again, this indicated that mtFPs present in mtDAMPs played a key role in that process. When activated with mtDAMPs, PMN also produce IL-8, a pro-inflammatory chemokine (Fig. 3E, F).
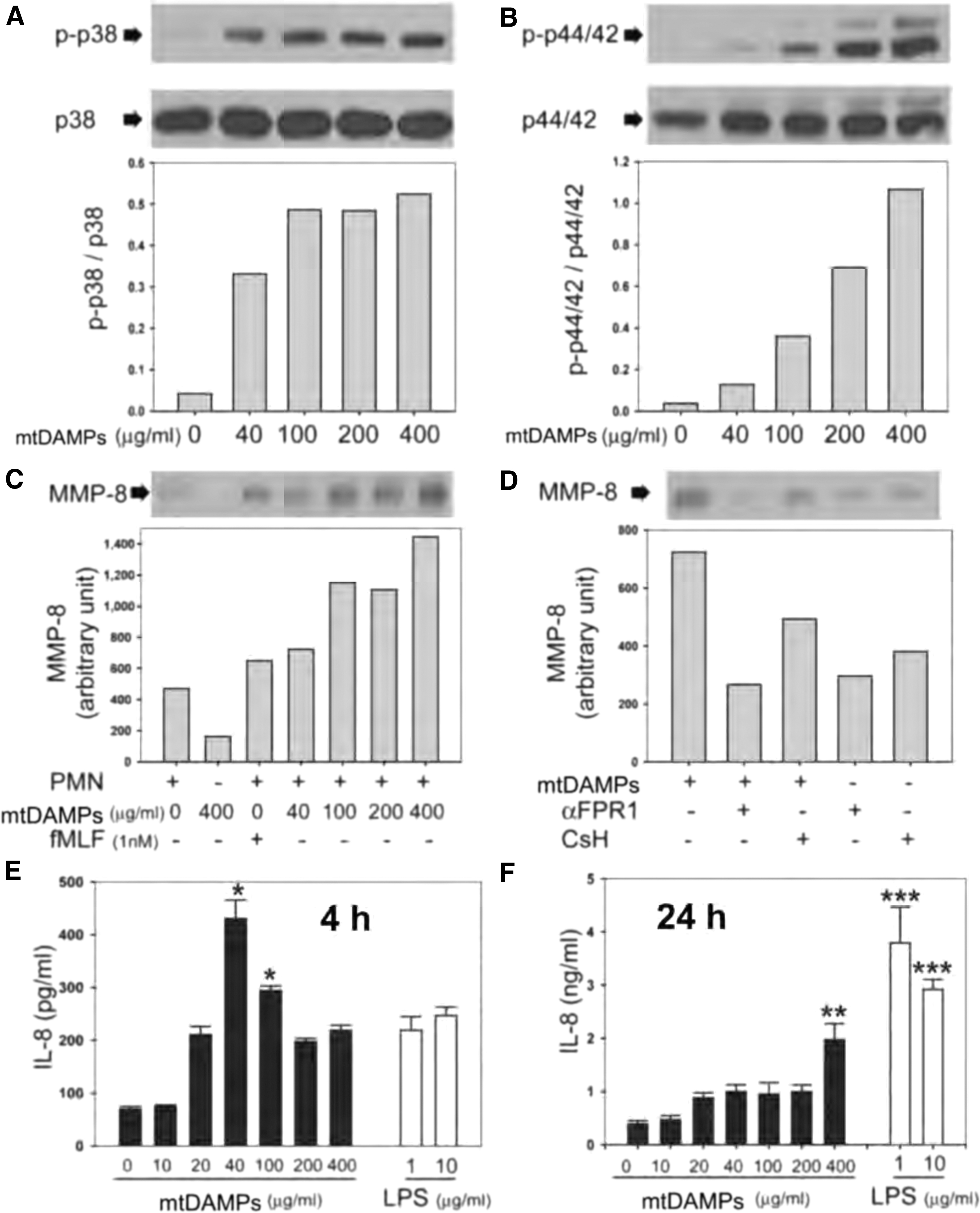
Additional experiments showed that mtDNA or unmethylated CpG oligodeoxynucleotides (ODN; short synthetic DNA fragments that are common in mitochondrial and bacterial DNA, and that can activate TLR9) did not induce the production of IL-8, unless fMLF was applied at the same time. Similarly, mtFPs alone did not elicit IL-8 production, indicating that both mtFPs and mtDNA are required to stimulate production of this important chemokine (147). Phosphorylation and thus activation of p38 and p42/44, which can proceed or follow ROS formation, also triggers neutrophil extracellular trap (NET) formation (41, 59).
mtDNA Induces NETs
The formation of NETs was originally described by Brinkmann in 2004 (10) and has subsequently been investigated by his (8, 9) and other groups (57, 97, 99). Pathogen-activated release of PMN granule proteins such as myeloperoxidase, neutrophil elastase, and cathepsin G occurs along with the release of chromatin. Together, they form extracellular net-like structures that engulf and kill bacteria (10, 127).
Over the past few years NETs have been identified as important participants in the development of sepsis and post-surgical systemic inflammation (12, 21, 106). The NETs can play a dual role in inflammation and other pathophysiological conditions, being either beneficial (i.e., by trapping and eliminating bacteria, fungi, or viruses from the circulation or at infective sites) or detrimental (i.e., by eliciting harmful inflammation and tissue or organ damage in conditions such as sepsis) (88, 100, 123).
Our and another group's data suggest that NETs are formed not only as a defensive response to invading pathogens but also as a response to sterile triggers, such as mtDAMPs (52, 82, 114).
Specifically, we have demonstrated that mtDNA induces human PMN to undergo “NETosis” (Fig. 4A, B) (52). We further showed that this NET formation was TLR9-dependent, as it was completely blocked by the inhibitory ODN TTAGGG that blocks TLR9 signaling (5, 52, 56). It remains to be determined whether mtDNA is the only component of mtDAMPs involved in inducing this effect or whether there are also roles for mtFPs or other mtDAMPs in NETs formation.
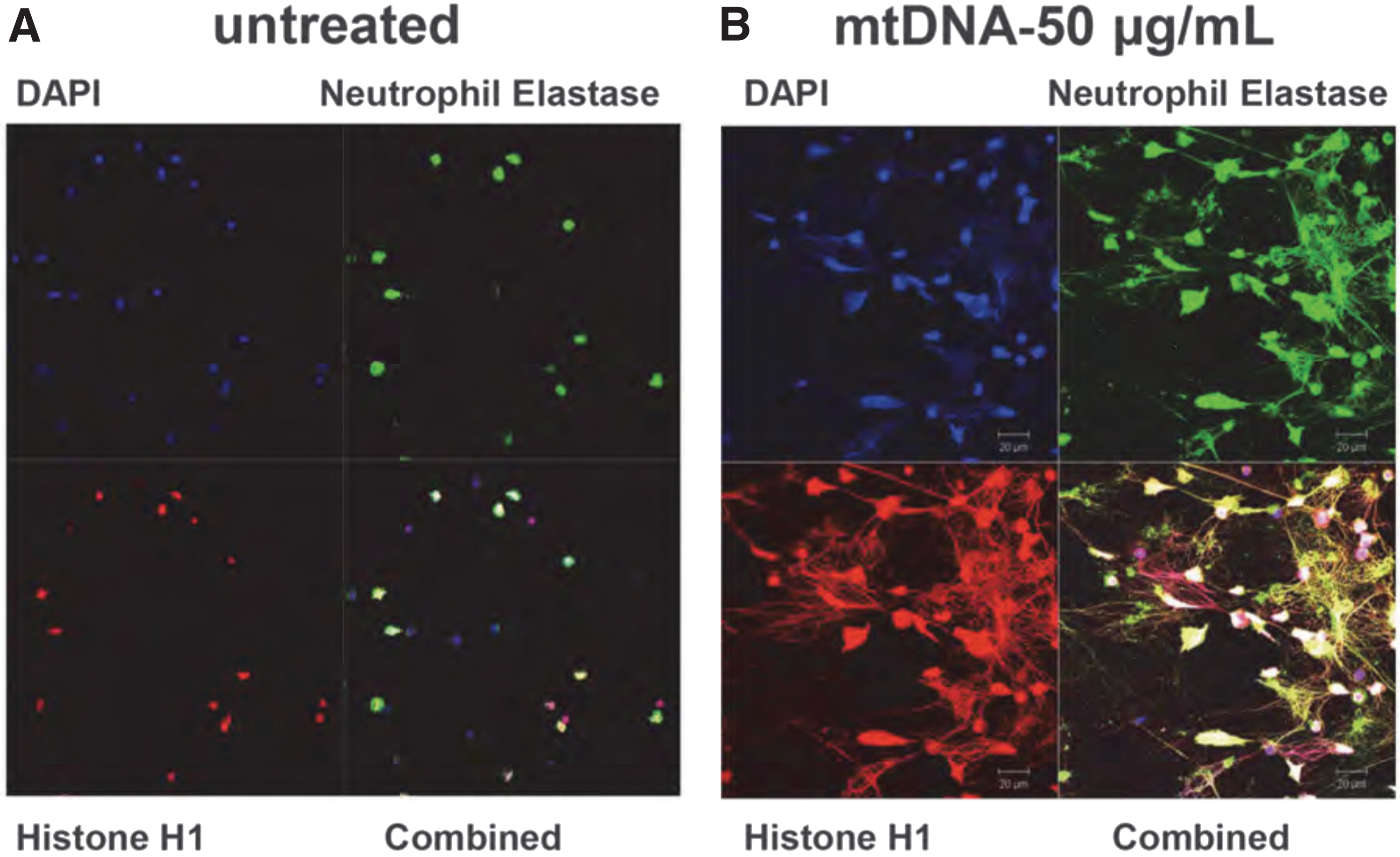
The mtDNA released by tissue/cellular damage after injury or infection (98) has also been reported to induce NETs with a variety of other clinical secondary effects. These include deep vein thrombosis that may contribute to organ injury (34). Moreover, NET formation itself may further increase mtDNA levels. Interestingly, ex vivo generation of NETs has been shown to be downregulated in PMN isolated from patients with sepsis compared with controls (42). It is not clear whether in vivo NETs formation in these patients is also impaired due to sepsis, and whether therefore those PMN kill pathogens less effectively via NETs. Thus clearly, further investigation is necessary in this area.
mtDAMPs Increase EC Permeability
Although sterile SIRS and sepsis may have different etiologies, their pathophysiology overlaps. Thus, each can result in multi-system organ dysfunction and potentially death (28). One of the common characteristics of those conditions is disruption of vascular endothelial barrier function. This can lead to loss of plasma volume, circulatory collapse, and shock or to organ dysfunction due to increased extracellular fluid or tissue edema (135, 136, 138). Thus, vascular leakage may occur early as a cause or later as an effect of such physiologic dysfunction.
Nonetheless, EC dysfunction has commonly been proposed as an early biomarker of sepsis (79, 115). Both bacteria and mtFPs can signal via FPRs expressed on vascular ECs, causing increased EC permeability and thus tissue hypoperfusion (54, 93). However, ECs also express other innate immune receptors, including TLR9, that can be activated by pathogen DNA or mtDNA, inducing inflammatory pathways such as NF-κB and MAPKs. These can modulate coagulation and EC permeability, all of which are involved in the pathology of sepsis (60).
Indeed, we demonstrated that mtDAMPs increased the permeability of EA.hy926 ECs, a cell line derived from the human umbilical cord EC (Fig. 5A) (25, 119). This effect was inhibited by treating mtDAMPs with proteases (Fig. 5B), suggesting that proteins present in the mtDAMPs play an important role in regulating vascular endothelial barrier function. The addition of PMN to this system did not have any effect on mtDAMPs-induced permeability change (Fig. 5C). We did show a role for mtDNA in EC permeability. Chloroquine, which blocks endosomal acidification and thus inhibits TLR9 response to DNA (132, 142), inhibited mtDAMPs-induced EA.hy926 permeability by 50% (Fig. 5D).
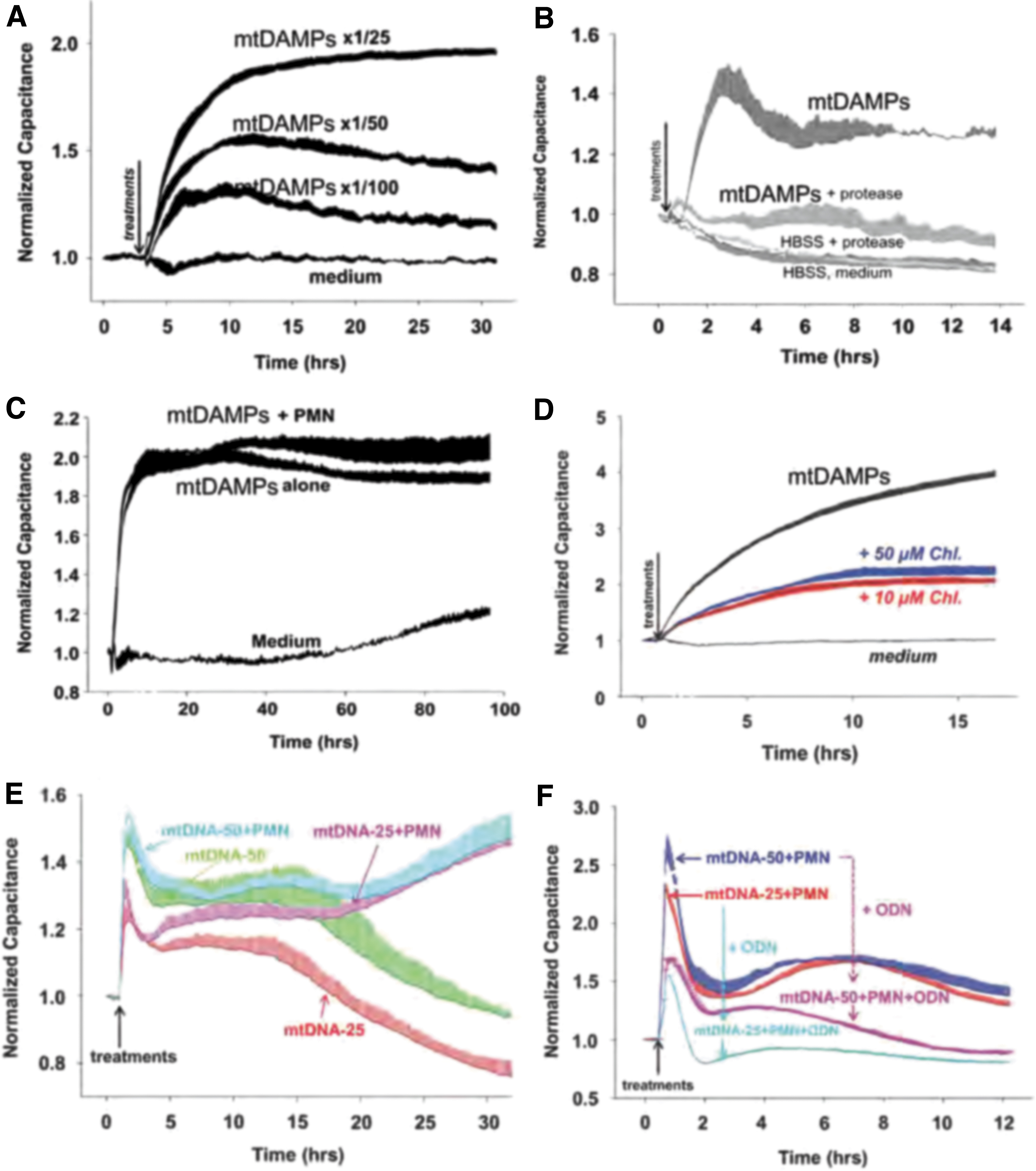
The mtDNA alone also stimulated EC permeability, but contrary to mtDAMPs, co-incubation with PMN here prolonged mtDNA-induced EC permeability for several hours, suggesting a PMN-EC crosstalk (Fig. 5E). The mtDNA-induced EC permeability was blocked by the TLR9-specific inhibitory ODN TTAGGG, again implicating TLR9 in this process (Fig. 5F). Surprisingly, mtFPs had no effect on EA.hy926 EC permeability. This corresponded to a lack of fMLF effects on these cells and indicates an absence of FPRs (119).
We assume that our data, contradictory to those published (136, 138), originate from the phenotype of EA.hy926 cells that we used. This differs from that of primary ECs, which express FPR1 and respond to fMLP and mtFPs by undergoing cytoskeletal changes and increasing their permeability (131). In addition, we observed mtDAMPs-triggered phosphorylation of p38 and p44/42 (ERK1/2) MAPKs and calcium mobilization in EC (119). These pathways can be also involved in stimulating EC permeability. Finally, upregulation of HMGB1 expression, which we showed for the first time in EC exposed to sterile inflammation by mtDAMPs (119), implicates mtDAMPs acting via HMGB1 in an autocrine or paracrine environment that may increase adhesion and endothelial permeability.
mtFPs in Human PMN Activation
To evaluate their activity as agonists acting on human PMN via FPR1, we synthesized f-peptides based on the N-terminal 6–9 amino acids of the 13 human mitochondrial proteins encoded by mtDNA. These short peptide sequences share ∼70% amino acid identity between humans, mice, and pigs (by our unpublished analysis). To study the inflammatory ability of the native proteins, we therefore studied calcium depletion, chemotaxis, and other PMN functions induced by the action of mtFPs on PMN-FPR1 (55).
Interestingly, only 5 out of the 13 human mtFPs induced calcium depletion from PMN endoplasmic reticulum (ND6>ND3>ND4>ND5>COX1) and chemotaxis was induced by the same mtFPs in the same order of potency (Fig. 6A–C). This order strongly correlates with “BLOcks SUbstitution Matrix 62” (BLOSUM62) score, based on similarity to fMLF. BLOSUM62 score estimates sequence divergence of peptides or proteins based on evolutionary conservation (24). Here, analysis of PMN activation by mtFP closely reflected their evolutionary similarity to bacterial f-peptides and confirmed the minor divergence of human mtFPs from fMLF. Considering this low level of divergence, we think that in addition to their role in infection, mtFPs may play important roles in the activation of innate immune responses to injury and initiation of tissue repair.

mtFPs Induce ROS Production
Mitochondria are recognized as a major source of intracellular ROS generation (87), and mitochondria-derived ROS induce different biological responses depending on their levels. Low, moderate, and high concentrations of ROS generate metabolic signaling, inflammasome activation, and apoptosis/autophagy, respectively (31). By stimulation of nucleotide-binding domain leukine-rich repeat (NLR) and NLRP3 inflammasome, ROS induce secretion of pro-inflammatory cytokines such as IL-1β and IL-18 (1, 31, 47, 122).
We have also found that some of the six to seven residues of N-terminal human mtFPs we have synthesized also induce PMN ROS formation (unpublished data). In contrast, mtDNA has no effect on ROS generation (62). Interestingly, some synthetic oligonucleotides containing CpG-DNA that have been reported to activate TLR9 may also cause ROS production (63, 85). Whether ROS generation by other mechanisms induces higher expression of genes related to TLR9 pathways needs to be further investigated. However, it is clear that mtFPs released from the tissues/cells after injury may induce additional ROS production by intracellular mitochondrial machinery.
FPR Desensitization Enhances Susceptibility to Secondary Infection
The role of mtFPs in clinical medicine has yet to be fully elucidated. Recently, however, we have shown that the release of mtDAMPs, especially mtFPs from tissues/cells after injury, can serve as an important PMN stimulus, causing them to migrate toward injury sites. This likely reflects both protection against bacterial invasion and the initiation of clearance of cellular debris required as a first step in healing (51). However, the binding of mtFPs to PMN FPR1 leads to desensitization and internalization of multiple PMN chemokine receptors, thus limiting the number of PMN available to migrate toward secondary infection site (Fig. 7A) (51, 53).
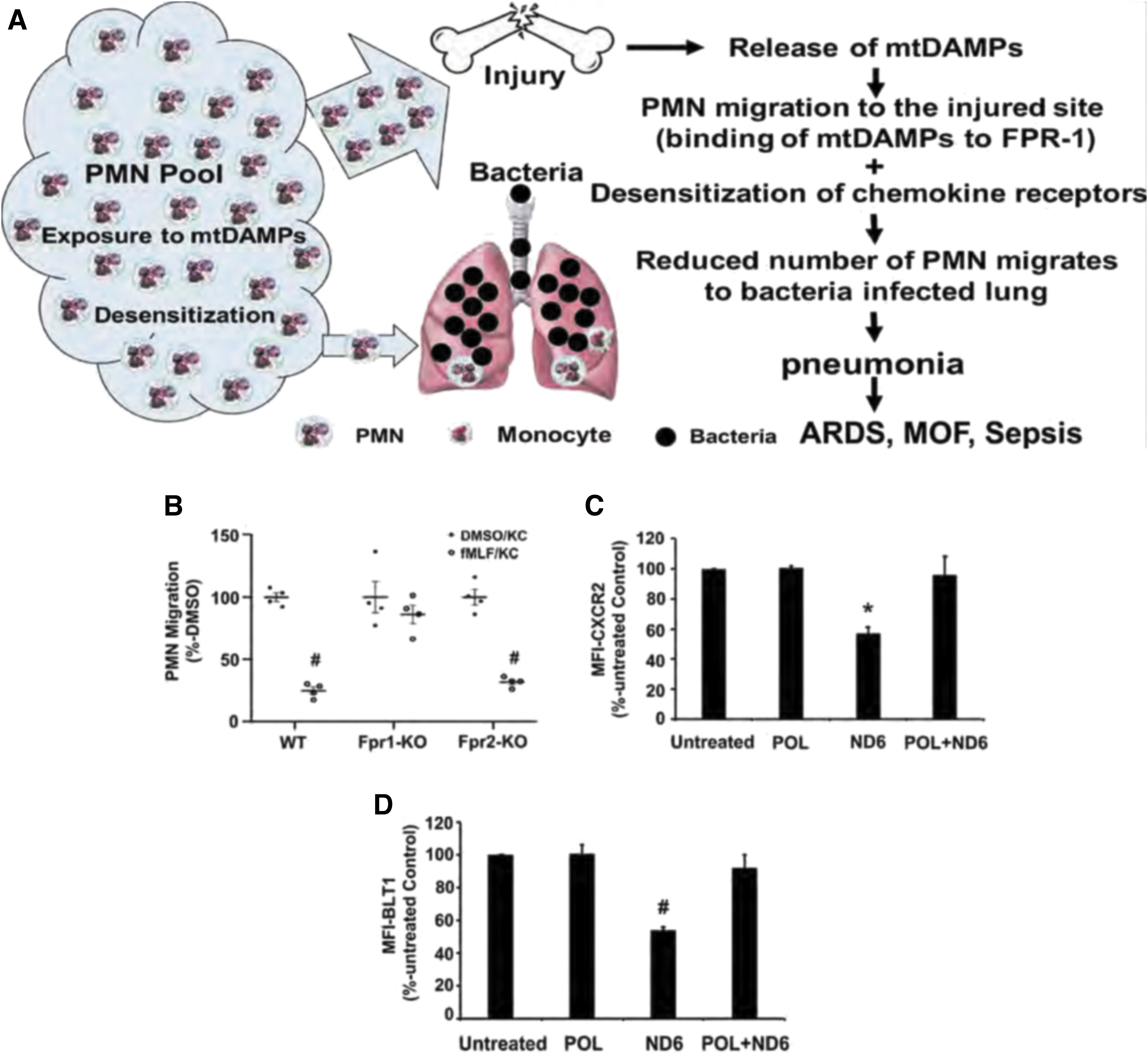
The exposure of mouse PMN to fMLF activates FPR1 and then internalizes FPR1 and other chemokine receptors by homo- and heterologous receptor desensitization/internalization, reducing their availability on the cell membrane (22, 66). Accordingly, we have shown that the treatment of PMN isolated from WT and Fpr2-KO mice, but not from Fpr1-KO mice, with fMLF significantly reduces their migration toward the mouse chemokine KC (keratinocyte cell-derived chemokine, CXCL1) (Fig. 7B) (51).
The PMN from Fpr1-KO mice did not undergo chemokine receptor desensitization after exposure to FPs. Similarly, the exposure of human PMN to ND6, which potently mimics in vivo injury by activating FPR1 (55), reduced cell surface expression of C-X-C motif chemokine receptor 2 (CXCR2) (Fig. 7C) and of leukotriene B4 receptor-1 (BLT1) (Fig. 7D). POL7200, a specific antagonist of FPR1, reverted ND6 effects and abolished ND6-induced receptor desensitization and internalization (51). These results indicate that mtFPs may both activate and desensitize various PMN functions.
To recapitulate, mtFPs are released by injury. Although they attract and activate PMN, they may simultaneously suppress PMN chemotaxis toward infective sites where resident monocytes may have already encountered PAMPs and responded by secreting chemokines. Therefore, although counter-intuitive, blockade of FPR1 may prevent desensitization of other receptors and hence improve overall detection and management of pathogens. This may potentially diminish SIRS and protect the host against secondary infection after either tissue trauma or primary infection.
Both trauma and sepsis increase the risk for secondary infection (61, 86, 120). In search for possible future clinical applications, we have used well-established animal injury/lung infection mouse models (53). Administering mtDAMPs intraperitoneally (11) to emulate a physical injury, we have followed intratracheal administration of bacteria to mimic lung inoculation (53). Assuming that FPR1 plays an important role in the impaired bacterial clearance in the lung we delivered the FPR1-antagonist, CsH, which reduced the number of PMN that migrated toward mtDAMPs (injury sites) and improved bacterial clearance in the lung by preventing chemokine receptor internalization (Fig. 8A) (51). This approach will require careful timing and dosing of FPR1 antagonists to obtain beneficial effects in patients with trauma.
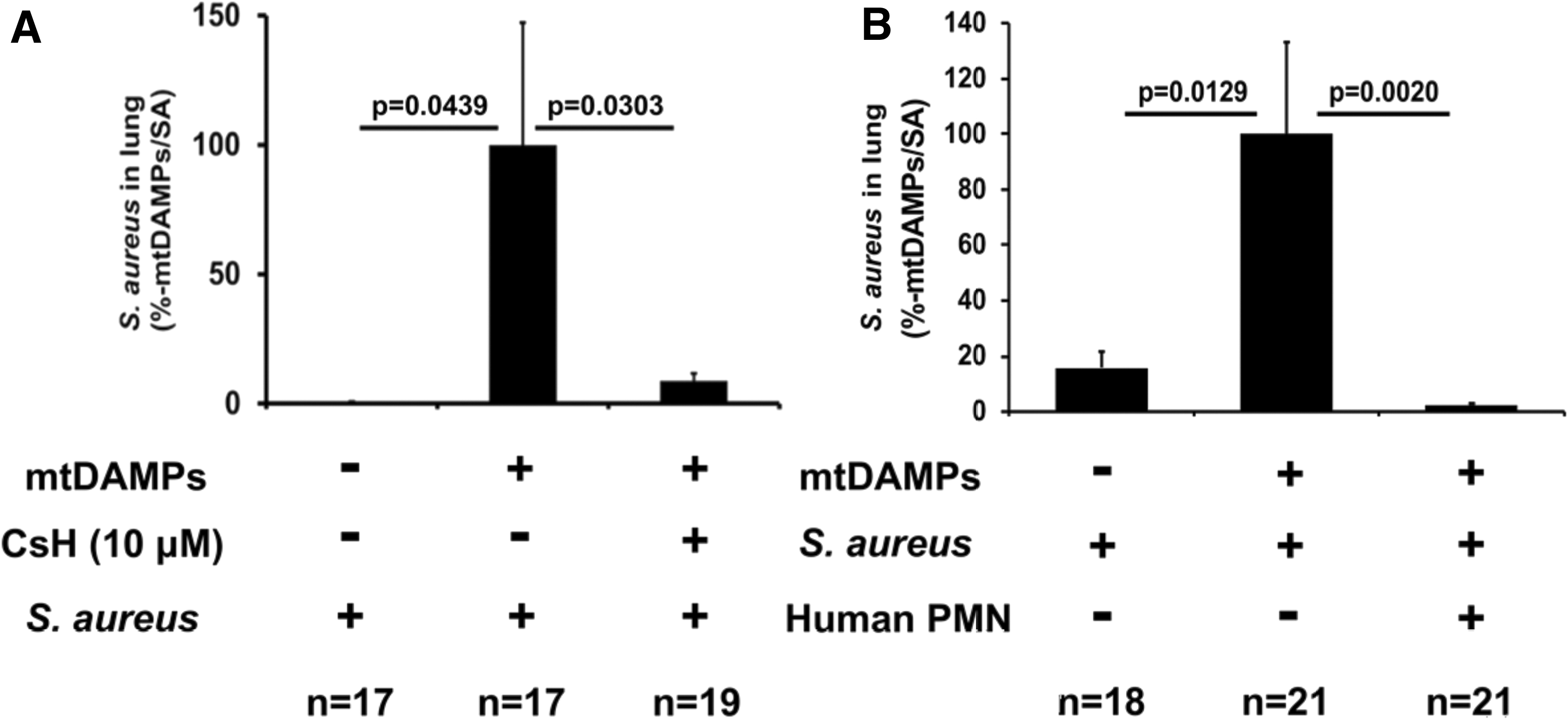
In another approach, to increase the number of PMN in bacteria-infected lungs after injury (hence presumably high levels of circulating mtFPs) we intratracheally administered exogenous human PMN and mitigated pulmonary infection (143). Exogenous PMN did not cause any damage to recipient animals in this study, whereas it dramatically alleviated pulmonary infection (Fig. 8B) (146). Again, this approach would require extensive additional work before attempting application in humans, but we presume that exogenous PMN could be beneficial for immune-compromised hosts (i.e., elderly) even where mtDAMPs do not play a major role as a primary condition.
Recently, we have shown that the binding of mtDNA to PMN TLR9 significantly reduces PMN migration toward chemokines (62). Since injury increases mtDNA in circulation, it will also contribute to susceptibility to sepsis by these means. Thus, both mtDAMPs, mtDNA and mtFPs, might worsen bacterial infection via reducing PMN chemotaxis in cases where tissue damage is induced by injury, chemotherapy (114), primary septic events (62), and after major surgeries (102).
Summary
The presented data clearly indicate that multiple components of mtDAMPs, at the very least mtFPs and mtDNA, are involved in the regulation of innate immunity. They are recognized as “non-self” and “danger signals,” much the same as pathogens such as bacteria and viruses.
After being released due to tissue trauma or primary inflammatory events, including organ damage resulting from SIRS/sepsis, mtDAMPs can trigger various signaling pathways in tissue resident cells and immune cells. Most notable among these are PMN, where mtDAMPs induce chemotaxis, as well as activate ROS generation, pro-inflammatory cytokine production, degranulation, cytoskeletal rearrangements, and NET formation. The exposure of PMN to mtDAMPs also induces desensitization of chemokine receptors. These responses, although depending on the intensity and circumstances, directly impact the occurrence, morbidity, and mortality in SIRS/sepsis.
Future Directions
We agree with other groups who emphasize three essential aspects of the approach to sepsis: early recognition and classification of severity, prevention of and support for organ dysfunction based on an optimal oxygen delivery, and treatment of the cause by control of the infective “source” (13, 103).
We believe that our studies on mtDAMPs both contain important knowledge regarding the fundamental mechanisms of SIRS/sepsis that can help in early disease recognition and also propose specific molecular solutions to aspects of inflammation that may be useful in clinical settings. Due to the similarities of PMN responses to PAMPs and DAMPs, immunological responses to those molecular patterns can be difficult to separate and identify.
Still, investigating new approaches may allow insights into this problem, making for a better clinical understanding of SIRS and sepsis. Both mtDNA and mtFPs can be measured and used as biomarkers, thus helping target patients who may benefit from specific treatment of downstream signaling pathways, such as TLR9, NLPR3, STING, or PRR1.
The reduction of plasma mtDNA and mtFPs by various methods, such as using DNase (to eliminate mtDNA effects) or deformylases (to remove a “formyl tag” from formylated proteins/peptides that makes them recognizable as bacteria-like “non-self”), might be considered as therapies for sepsis-like symptoms (77, 80). Also, specific receptor-blocking antibodies or new and potent FPR1 antagonists that can prevent binding of mtFPs released by injury to FPR1 may increase the number of PMN available to combat secondary infections via other chemokine receptors, making pneumonia, sepsis, and ARDS after trauma potentially preventable. All these approaches will require careful evaluation before they can be used clinically.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The work was funded by NIAID/NIH, grant number 1R03AI135346-01 (Kiyoshi Itagaki) and by the Department of Defense (DoD), grant number W81XWH-16-1-0464 (Carl J. Hauser).