
Abstract
Significance:
Systemic autoimmunity affects 3%–5% of the population worldwide. Systemic lupus erythematosus (SLE) is a prototypical form of such condition, which affects 20–150 of 100,000 people globally. Liver dysfunction, defined by increased immune cell infiltration into the hepatic parenchyma, is an understudied manifestation that affects up to 20% of SLE patients. Autoimmunity in SLE involves proinflammatory lineage specification in the immune system that occurs with oxidative stress and profound changes in cellular metabolism. As the primary metabolic organ of the body, the liver is uniquely capable to encounter oxidative stress through first-pass derivatization and filtering of waste products.
Recent Advances:
The traffic of immune cells from their development through recirculation in the liver is guided by cell adhesion molecules (CAMs) and integrins, cell surface proteins that tightly anchor cells together. The surface expression of CAMs and integrins is regulated via endocytic traffic that is sensitive to oxidative stress. Reactive oxygen species (ROS) that elicit oxidative stress in the liver may originate from the mitochondria, the cytosol, or the cell membrane.
Critical Issues:
While hepatic ROS production is a source of vulnerability, it also modulates the development and function of the immune system. In turn, the liver employs antioxidant defense mechanisms to protect itself from damage that can be harnessed to serve as therapeutic mechanisms against autoimmunity, inflammation, and development of hepatocellular carcinoma.
Future Directions:
This review is aimed at delineating redox control of integrin signaling in the liver and checkpoints of regulatory impact that can be targeted for treatment of inflammation in systemic autoimmunity. Antioxid. Redox Signal. 36, 367–388.
Introduction
The immune system is the body's defense against pathogenic attack, and is governed by a dynamic interplay among cell adhesion molecules (CAMs), integrins, chemokines, reactive oxygen species (ROS), and endosomal trafficking. Dysfunction in these factors can lead to systemic autoimmunity, which is a loss of tolerance for self-antigens (131, 132).
Affecting 3%–5% of the global population, systemic autoimmunity represents diseases in which the immune system attacks its own cells, tissues, and organs (107). Immune cells circulate through the body's organs using cell surface proteins such as integrins and CAMs. These proteins are present both on the immune cells themselves and on the endothelial cells within the organs, and act to localize immune cells into compartments where they can promote immune attack or immunosuppression (111, 115). The integrins and CAMs that govern immune cell infiltration into and out of an organ are regulated by intracellular signaling processes, including ROS.
The integrins and CAMs, which mediate immune cell margination into the liver, are modified through the signaling of ROS, through endosomal trafficking, or both. The molecular “switches” that regulate endosomal trafficking include Ras-like protein from rat brain (Rab) GTPases, some of which contain the redox-sensitive motifs GXXXGK(S/T)
Redox-Sensitive Rab GTPases Control Cell Surface Expression of Receptors That Mediate Signal Transduction and Adhesion
The redox-sensitive Rab GTPases from Figure 2 control the recycling of cell surface proteins and signaling molecules that are implicated in immune functions and hepatic dysfunction.
CD, cluster of differentiation; EMT, epithelial to mesenchymal transition; MHC II, major histocompatibility complex II; Rab, Ras-like protein from rat brain; STAT3, signal transducer and activator of transcription 3; TfR, transferrin receptor; CD71.
The liver is an organ that serves both an immunomodulatory role and an antioxidant role, dampening the immune response from first-pass metabolism and reducing oxidative stress both within the liver and throughout the body. Immune dysfunction and inflammation are associated with states of increased oxidative stress (138) and altered expression of CAMs and integrins (53). Taken together, these represent critical targets of autoimmune regulation and therapeutic interventions. In this review, we explore the mechanisms that govern immune cell infiltration, which promote liver dysfunction in the setting of inflammation and autoimmunity. Driven by pathologic inflammation and oxidative stress, autoimmune-mediated liver disease can be treated either by addressing the underlying inflammatory state driving liver dysfunction or by enhancing our body's own antioxidant systems to reduce oxidative stress.
Integrin- and CAM-Based Communications Between the Liver and the Innate and Adaptive Immune Systems Shape Autoimmunity
Our immune system consists of an innate response and an adaptive response. The Innate Immune System is composed of barrier surfaces, such as the skin, which physically prevent pathogens and foreign substances from entering our bodies. Neutrophils, dendritic cells, and macrophages can phagocytose pathogens resulting in a nonspecific, generalized immunity to pathogens. The Adaptive Immune System picks up where the innate immune response leaves off to mount a specific attack against pathogens that break through the barriers of innate immunity. Immune cells circulate throughout the body, continuously surveilling different organs by using cell surface markers such as integrins, CAMs, and chemokines to enter and exit organs, including the liver (19) (Fig. 1).
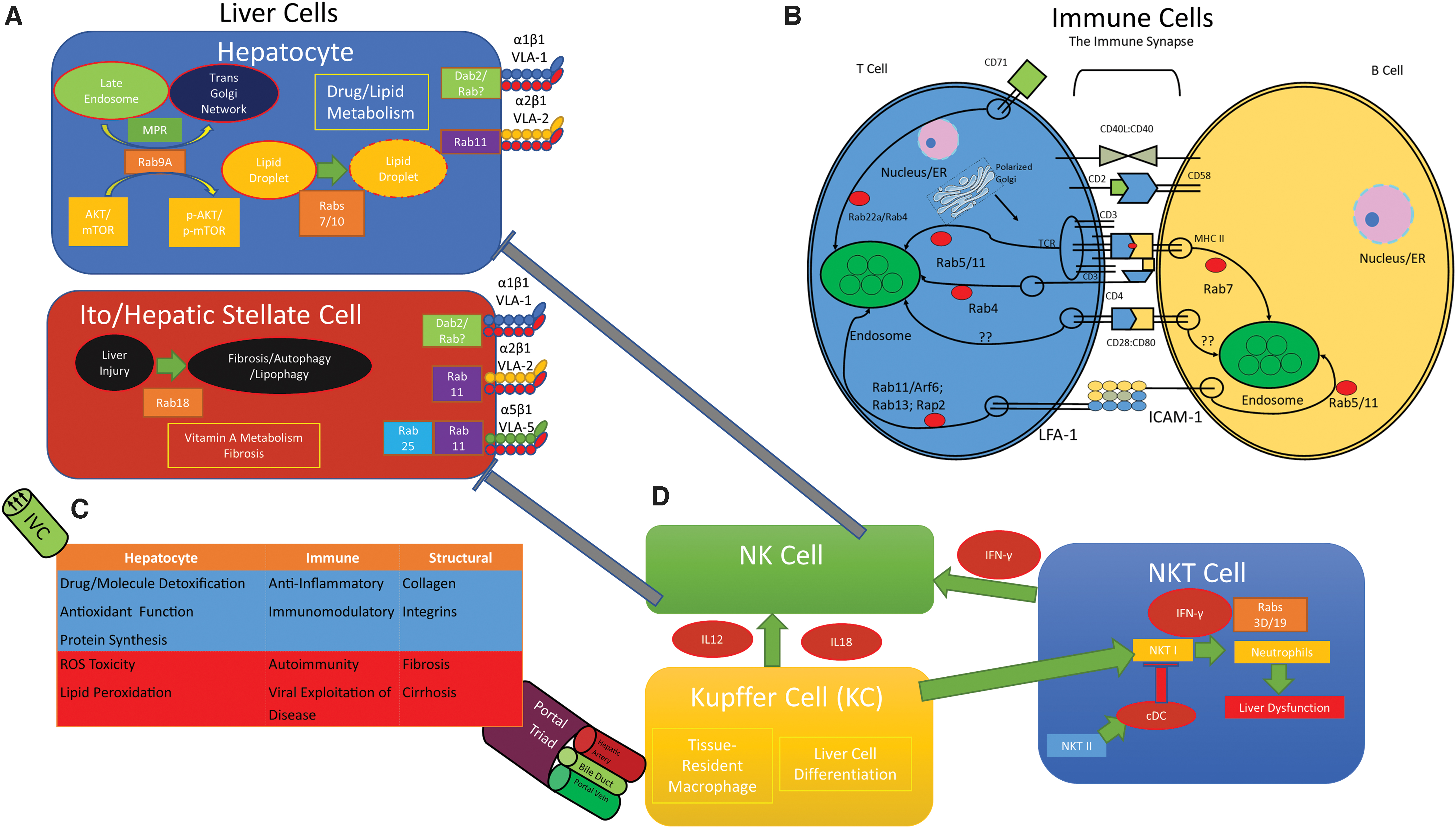
When a pathogen breaches the barriers of the innate immune response, antigen presenting cells (APCs), such as dendritic cells, phagocytose the pathogen and migrate to the nearest lymph node. Cell surface markers, such as integrins, mediate APC migration to the nearest afferent lymphatic channel, while also processing the pathogen onto major histocompatibility complex II (MHC II) for presentation onto its cell surface (173).
The matured APC interacts with integrins located near high endothelial venules (HEVs) at the nearest lymph node to maximize contact with circulating lymphocytes of the adaptive immune response. Specifically, CD11c+ dendritic cells control the surface expression of lymphocyte-binding surface proteins, such as L-Selectin, ICAM-1 (intracellular adhesion molecule-1), and chemokines CCL19 [chemokine (C-C motif) ligand 19], CCL21 [chemokine (C-C motif) ligand 21], CXCL12 [chemokine (C-X-C) ligand 12], and CXCL13 [chemokine (C-X-C) ligand 13] (111). Upregulation of these proteins forces entry of circulating naïve lymphocytes to promote the formation of the immune synapse (IS), which primes the adaptive immune response to facilitate a less generalized and more specific attack against the foreign pathogen (115).
The handoff from innate to adaptive immunity requires the formation of the IS between APCs and T cells. In the IS, integrins act to latch an APC together with a T cell to facilitate a long-term binding between both cells. The engagement of the T cell receptor (TCR) by the MHC II/antigen complex facilitates an interaction between lymphocyte function-associated antigen 1 (LFA-1) and ICAM-1 by promoting the movement of LFA-1 (αLβ2; CD11a/CD18) on T cells and ICAM-1 on APCs toward the TCR/MHC II/antigen complex (39).
As the TCR is crosslinked by MHC I or MHC II, the adhesiveness of the cell–cell anchoring molecule LFA-1, an integrin important in the migration and differentiation of T cells in the immune response, is increased (39, 84). The adhesiveness of an integrin is regulated by both conformation (the affinity of the integrin to bind a ligand) and valency (clustering of integrins on the cell surface) (84).
The central region of the IS, known as the c-SMAC, or central supramolecular activation cluster, is composed of the TCR, CD2 [important as a checkpoint for IS formation and has both adhesion and costimulatory roles (34)], CD4 (on helper T cells) or CD8 (on cytotoxic T cells), and CD28 [a costimulatory protein (1)]. The peripheral region of the IS, called the p-SMAC, or peripheral supramolecular activation cluster, is composed of LFA-1, which binds to ICAM on APCs to strengthen the IS (38).
Central to the cell–cell interactions discussed above are the integrins (like LFA-1), which are transmembrane heterodimeric cell surface proteins, consisting of an α or β chain (32). Integrins act as molecular “hooks and loops,” which mediate intercellular synaptic communication. Extracellular domains of integrins are important in facilitating cell adhesion both to neighboring cells and to the extracellular matrix (ECM) (76). Intracellular domains of integrins are important in converting external stimuli into intracellular signaling cascades (75).
ECM proteins share an integrin-binding motif, Arg-Gly-Asp (RGD), which allows for integrin-expressing cells to bind to the ECM with great strength. Those integrins that mediate cell–cell interactions bind counter-receptors, such as ICAMs (important in cell–cell synapse formation), and vascular cell adhesion molecules (VCAMs; expressed on vascular cells to promote cell adhesion to the vascular endothelium) (75). The tight interaction between extracellular and intracellular domains allows for two types of signaling, referred to by Danen as inside-out and outside-in signaling (32).
Inside-out signaling describes signaling that occurs from intracellular processes, which impact integrin conformation and affinity by altering integrin cytoplasmic tails (32). These changes in integrin strength and specificity are mediated by talins and kindlins, which bind to and separate integrin tails that lead to integrin activation (114).
Outside-in signaling describes the binding of ligand to an integrin and the effects of ligand binding on integrin clustering. Amplification of signal transduction is efficiently mediated by the formation of a growing adhesion plaque (32): Ligand binding to the extracellular domains of integrins recruits adapter proteins to the intracellular domains of the integrins. As a result, intracellular signal transduction is enhanced by the presence of more linkage to the cytoskeleton. These interactions occur as a result of cell–ECM interactions, however, and not necessarily through interactions with soluble signaling molecules (50). These signaling events are the hallmarks of the adaptive immune response.
A combination of inside-out and outside-in signaling takes place within T cell activation. After exposure of an innate immune cell to pathogen, homeostatic chemokines, such as CXCR4 [chemokine (C-X-C motif) receptor type 4], CXCR5 [chemokine (C-X-C motif) receptor type 5], CCR6 [chemokine (C-C motif) receptor type 6], and CCR7 [chemokine (C-C motif) receptor type 7], help in APC and lymphocyte migration into a lymph node (41). The interaction between pathogen and APC mediates the leukocyte adhesion cascade (84), which facilitates movement of primed innate immune cells out of the general circulation and into lymphatics. This process involves selectin-dependent rolling, chemokine-triggered activation, and integrin-dependent adhesion to the endothelial cells of the vasculature.
Inflammatory chemokines mediate the transition of neutrophils and monocytes from rolling to arrest adhesion. Upon binding to chemokine receptors, chemokines increase the adhesiveness of integrins for their ligands on the endothelium. Chemokines generated by the endothelial cells induce arrest of rolling leukocytes through the binding of leukocyte integrins to immunoglobulin superfamily of proteins, such as ICAM-1 and VCAM-1 (84). These chemokines are not soluble—they are immobilized onto the endothelium (156).
Study of the liver integrin and CAM landscape challenged previously hypothesized theories about hepatic immunity. As the liver has a dual blood supply (portal vein and hepatic artery), the blood converges into sinusoids located within the hepatic architecture (10). Sinusoids lack tight junctions and do not express “typical” endothelial markers such as PECAM-1 (platelet endothelial cell adhesion molecule-1), CD34, VE-cadherin, P-selectin, or E-selectin (92, 155, 165). Based on the lack of expression of these markers, it was hypothesized that invading leukocytes were physically trapped in the hepatic circulation.
Human liver sinusoids upregulate vascular adhesion protein-1 (VAP-1) during an immune response (10). VAP-1 is typically found in the cytoplasmic vesicles of endothelial cells, and is a cell surface-mounted CAM with enzymatic/catalytic activity. It catalyzes the conversion of soluble primary amines into their aldehyde counterparts (148). VAP-1 is found in hepatic sinusoidal endothelial cells, and it acts to mediate T helper type 2 (TH2) adhesion (while α4β1 [very late antigen-4; VLA-4] mediates T helper type 1 [TH1] adhesion), suggesting a role in liver inflammation (91) (Fig. 3).
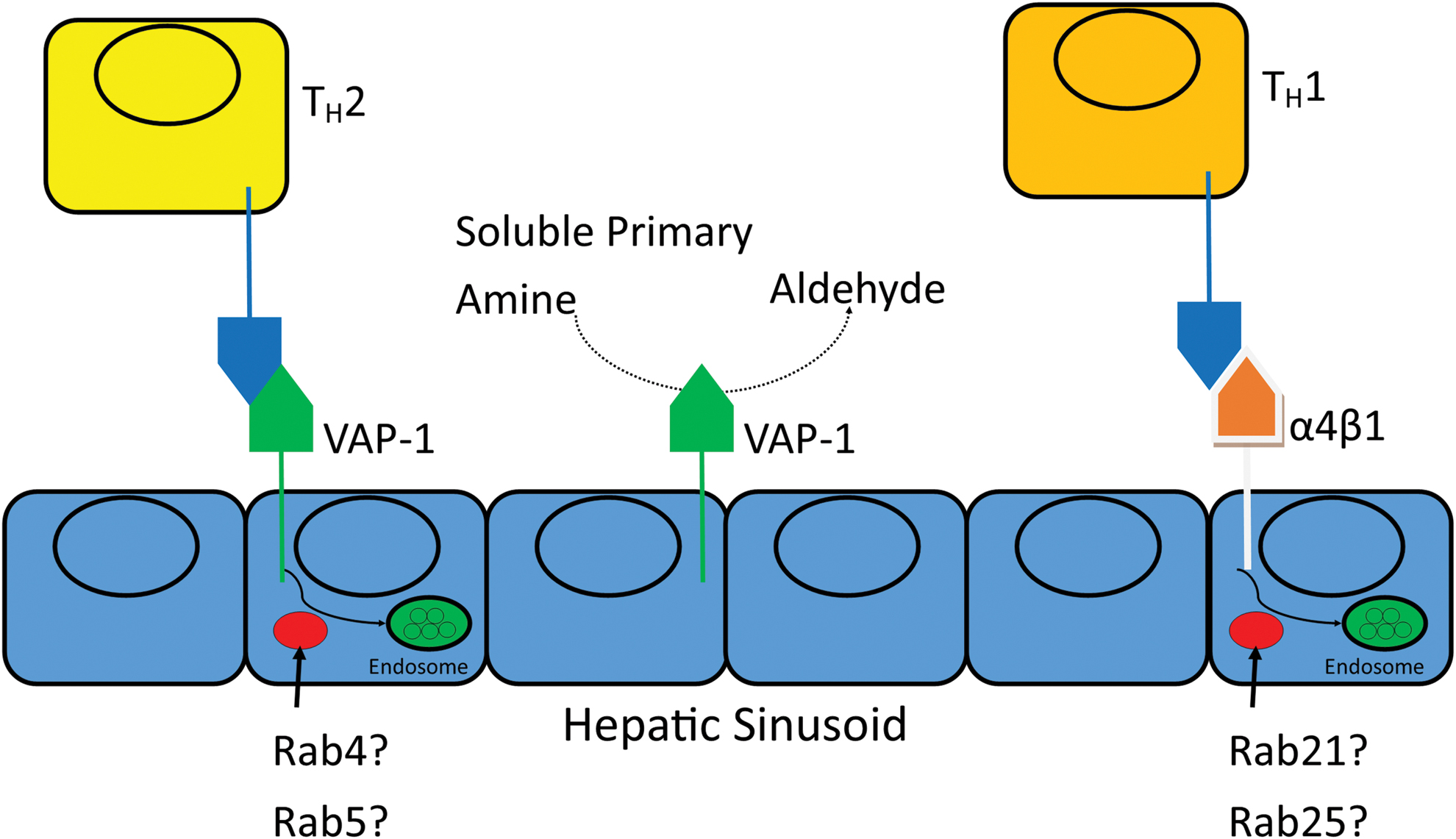
Patients with liver disease have increased levels of VAP-1 (89), and increases in soluble VAP-1 correlate with Non Alcoholic Fatty Liver Disease severity in humans (184). VAP-1 mediates T cell recruitment, and acts as an amine oxidase to produce oxidants and as an adhesion molecule for circulating lymphocytes in the liver (10). Our understanding of liver integrin expression is limited, but the CAM VAP-1 has been theorized as a potential therapeutic target (147). Treatment with semicarbazide-sensitive amine oxidase (SSAO) inhibitors, which interfere with the catalytic activity of VAP-1, results in an antitumor effect on liver cancer through the downregulation of CD11b+ Gr-1+ myeloid-derived suppressor cells (94).
In addition, glucocorticoids have been used to manage liver inflammation for decades and are classically used for treatment of autoimmune hepatitis (AIH) (195). Whether steroids may be involved in regulating VAP-1 expression is unknown. The classic model of glucocorticoid-mediated anti-inflammation involves the suppression of the proinflammatory transcription factor (nuclear factor kappa B) NF-κB (97). Glucocorticoids preferentially decrease the expression of ICAM-1, the cell adhesion ligand for the LFA-1 integrin found on lymphocytes. The treatment of patients with glucocorticoids causes a transient leukocytosis, as the inhibition of integrins causes a demargination of leukocytes that are bound to vascular endothelium (29). Since integrin expression on cell surface can also be regulated by endosomal trafficking enzymes, this remains an open area of research interest.
Redox Mechanisms Control the Expression of CAMs and Integrins via Endosomal Traffic
The integrins, CAMs, etc. which facilitate the immune response are all regulated by endosomal trafficking. The endosome is a complex structure responsible for the targeting of endocytosed materials to a variety of different areas within the cell. The endosome and its associated recycling pathways act to return many of the proteins, lipids, and membranes that were endocytosed back to the surface of the plasma membrane (54).
Endocytosis can occur via clathrin-dependent or clathrin-independent pathways. Clathrin-dependent endocytosis involves adapter protein recognition of the cytoplasmic domains of the plasma membrane proteins of interest. These adapter proteins package the internalized proteins into clathrin-coated vesicles (27). Clathrin-independent endocytosis is not as well studied and is an open area of investigation (151).
After either clathrin-dependent or clathrin-independent endocytosis, the internalized material finds its way to the early endosome, which is where sorting takes place. Sorting determines the fate of these endocytosed materials. The efficient regulation of this sorting and recycling represents targets for physiological activity and for diseases, which may have dysfunctional endosomal sorting pathologies.
Rab GTPases are critical to the endocytic recycling necessary to maintaining cellular homeostasis. In brief, these small guanosine triphosphate (GTP)-binding proteins function as molecular switches to regulate endocytic recycling. They are important in vesicle carrier formation, intracellular vesicle movement, and fusion of vesicles with target membranes. When bound to GTP, they are in an “on” state, where they carry out their biochemical functions. This is carried out by guanine nucleotide exchange factors, which replace guanosine diphosphate (GDP) with GTP. The autohydrolysis of GTP to GDP leads to switching “off” of enzyme function (54).
The cell surface expression of the transferrin receptor (TfR; CD71), important in iron homeostasis, is regulated by endosomal trafficking through the dynamic interplay of a variety of Rab GTPases. After binding to its ligand transferrin, CD71 is rapidly internalized to early endosomes (59). Studies of CD71 in a Chinese hamster ovary cell line found that CD71 is trapped in Rab22a-containing vesicles, and overexpression of Rab22a prevented its recycling (105).
HTLV-I-related endogenous retroviral sequence (HRES-1)/Rab4, a GTPase homologous to the Rab4 family of GTPases, regulates the expression of CD71, in addition to CD4. Curiously, the overexpression of Rab4 increases CD71 expression on the surface of cells, an observation that is opposite to the activity of Rab22a (120). HRES-1/Rab4 is also involved in the maintenance of mitochondrial homeostasis involving the mitophagy regulating protein Drp1, and it is responsible for reducing the expression of the zeta chain of CD3 by promoting endocytic recycling for degradation in the lysosome (45, 120), making this an enzyme of unique and varying function (16). HRES-1/Rab4 is also overexpressed in human lupus T cells, and its activity in lupus pathogenesis is an open area of investigation (45).
Trafficking of integrins is mediated by the small GTPases Rab and ADP ribosylation factor (Arf) (33). Rab GTPases have endosomal compartmentalization, which suggests unique functions for each GTPase (162). Rab5 and Rab21 are known to endocytose integrins from the plasma membrane into early endosomes (130). Arfs, on the contrary, are important in integrin recycling back to the plasma membrane and focal adhesion dynamics (137).
T cell activation causes endosomal recycling of the TCR to increase or decrease TCR expression on the T cell (140). Once the TCR binds to antigen and MHC, the centrosome and Golgi apparatus reorganize to shift protein production and microtubule assembly toward the intracellular side of the newly formed IS (150). While the endocytosis and recycling occur through both clathrin-dependent and clathrin-independent mechanisms, which response predominates is unknown.
A variety of proteins are known to recycle the TCR, such as flotillins, which are not necessary for TCR internalization, but are required for recycling (26). T cell activation causes the internalization of the zeta subunit of the TCR complex in a clathrin-independent manner. The vesicles containing TCRζ are positive for flotillin-2, a protein that is necessary to target certain membrane proteins to their final locations and in regulating Rab11a-dependent recycling of integrins such as α5- and β1-integrins (73). Deficiency of flotillin-2 is associated with decreased lung metastases in mouse breast cancer animal models (7). The sorting of the endocytosed TCRs from early endosomes back to the cell surface is regulated by GTPases, such as Rab4 and Rab5 (199).
MHC II expression in B cells is regulated by Rab7, a GTPase involved in early to late endosomal fusion (8). While the binding of CD28 on T cells to CD80 on APCs is critical to immune activation, precise mechanisms involving their recycling and cellular trafficking are unclear. Mechanisms governing CD2 recycling, along with its ligand, CD58, are also unclear.
In the p-SMAC, LFA-1 and ICAM-1 are also recycled by GTPases. LFA-1 is recycled (129) through Rab11/Arf6 (42), Rab13 (123), and Rap2 (164) endosomal trafficking. ICAM-1 recycling to the early endosome is regulated by Rab5 in circulating T cells and both Rab5/Rab11 in neutrophils (42, 169).
Unlike cell surface proteins, cytokines are synthesized, loaded into vesicles/endosomes, and exocytosed into the extracellular space. Cytokine release via recycling mechanisms is not well understood. The Golgi apparatus in T cells is polarized toward the APC, helping to secrete both cell surface proteins and secreted cytokines out of the cell and directly into the synapse (150). Two pathways exist for cytokine secretion: synapse-directed and nonspecific multidirectional export. The Rab GTPases Rab3d and Rab19 are expressed along with the secretory vesicles containing interleukin 2 (IL-2) and interferon gamma (IFN-γ). Some compartments containing interleukin (IL-4) also localize with Rab37 (74).
Endosomal aberrations and deficiencies in intracellular trafficking can promote the pathogenesis of systemic lupus erythematosus (SLE), a systemic autoimmune disease. In SLE patients, plasmacytoid dendritic cells (pDCs) have increased IFNα production in response to toll-like receptor 7 (TLR7) activation but decreased IFNα production in response to toll-like receptor 9 (TLR9) activation. This stems from the “trapping” of TLR7 in the late endosome and lysosomal compartments of pDCs, and is strongly associated with an SLE phenotype in humans (118).
In particular, TLR7 directly associates with LAMP1 and Rab7 in the lysosomal and late endosomal compartments, respectively. This study also found that SLE disease activity index (SLEDAI) positively correlated with IFNα levels generated by pDCs. These results imply that deficient trafficking and trapping of TLR7 in the late endosome elicited greater IFNα levels and increased disease activity, as detected by SLEDAI (118). The coordination of intercellular and intracellular signaling is tightly regulated. One mechanism of such regulation is through redox signaling within the cell. Oxidative stress occurs because of an overproduction of oxidative species and an underproduction of reducing species, generating ROS.
ROS are formed through normal physiological and metabolic processes. Five classical ROS are hydrogen peroxide (H2O2), superoxide anion (O2 −), hypochlorous acid (HOCl), singlet oxygen (1O2), and hydroxyl radical (•OH) (22). The single valence electrons present on superoxide, 1O2, and hydroxyl radicals can compromise cellular stability, nucleic acid integrity, and organelle function.
In a normally functioning, healthy cellular environment, these damaging ROS are neutralized by enzymatic reactions, which prevent further cellular damage, specifically superoxide dismutase (SOD), catalase, and glutathione peroxidase (62). In addition, a basal amount of ROS maintains cellular structure and function. For example, ROS are necessary signaling requirements for adaptive T cell responses (48).
ROS signaling modifies integrin and CAM expression, and, by extension, organ-specific margination of circulating cells. In the case of α2bβ3 integrin, ROS can change integrin shape through redox-sensitive cysteine residues, which ultimately alter binding affinity (56). ROS are critical signaling intermediates in promoting intracellular signaling. External stimuli lead to upregulation of enzyme systems such as Cytochrome P450 and myeloperoxidase. Combining this with the activity of the mitochondrial respiratory chain, ROS generation upregulates phosphatases, Ras/Rho GTPases, and other enzyme systems responsible for altering cell shape and migration/attachment. This ultimately leads to changes in cell adhesion, migration, proliferation, differentiation, and survival (51).
Indeed, we see a classic overlap between liver pathologies that overwhelm redox reactions with the invasion of inflammatory cells, such as dendritic cells and eosinophils (98). Migration of leukocytes through VAP-1 is dependent on ROS generation. Inhibition of the catalytic activity of VAP-1 or removal of the H2O2 from its environment eliminated the enzymatic role of sVAP-1, the soluble form of VAP-1. This reduced the migrating cells to the liver by ∼50%, suggesting that the outside-in signaling of ROS through VAP-1 enzymatic activity plays an important role in leukocyte margination in the liver (184).
The enzymatically active VAP-1 localizes to early endosomes and is involved in lysosomal trafficking (183). Whether the recycling of VAP-1 occurs by GTPases, enzymes involved in membrane and endosomal trafficking, is poorly understood. However, since it is localized to early endosomes, it is reasonable to hypothesize that Rab GTPases associated with the early endosome, such as Rab4 (180) and Rab5 (54, 162), may mediate this recycling. While VAP-1 is implicated in T cell, B cell, monocyte, and granulocyte binding in HEVs, its high upregulation in liver sinusoids makes it a critical target of hepatitis investigation (53).
ROS itself can modulate integrin expression and modification through the regulation of phosphatases, as all protein tyrosine phosphatases also contain a redox-sensitive cysteine residue—when oxidized, the phosphatase is inactive. Protein tyrosine phosphatase (PTP) with PEST (proline-, glutamic-acid-, serine-, and threonine-rich) domains (PTP-PEST), SH2 domain-containing protein tyrosine phosphatase-2 (SHP-2), and low molecular weight protein tyrosine phosphatase are all phosphatases that regulate integrin expression, which are inactivated by oxidation (170).
Catalase directly binds to the SHP-2 phosphatase and the adapter protein growth factor receptor-bound protein 2 (Grb2) upon integrin binding to protect these proteins from ROS, specifically H2O2-mediated oxidation (192, 193). Hydrogen peroxide-inducible clone-5 (Hic-5), a protein involved in the binding of integrin with the actin cytoskeleton, has a ROS-sensing domain on its N-terminus (170). Under conditions of profound oxidative stress, Hic-5's ROS-sensing domain forces release of Hic-5 and signals its movement into the nucleus (158).
The GTPases responsible for intracellular trafficking are also sensitive to ROS. Clustal Omega sequence alignment of 12 members of the murine Rab GTPase family shows the existence of either the GXXXGK(S/T)

Abnormal CAM and Integrin Expression Lead to Immune Dysfunction
The transition from innate to adaptive immune response is complex and tightly regulated. Defects, either in this transition or in general immunity, can lead to autoimmunity, which affects 3%–5% of people globally (107). One such example is SLE, an autoimmune disease of unknown etiology; its prevalence ranges from 20 to 150 per 100,000 people worldwide. Our current knowledge of SLE pathogenesis involves the dynamic interplay of ultraviolet light exposure, genetic predisposition, hormonal dysregulation, and other factors, which lead to disease manifestation in susceptible individuals. This ultimately leads to immune cell dysregulation and the generation of immune complexes, cytokines, antibodies, and autoreactive immune cells (176).
SLE affects the liver (98), kidney, lung, heart, brain, and skin (176). The spectrum of disease varies among patients, with different patients having different organ involvement. For example, one of the most common causes of death in SLE is kidney failure, secondary to immune complex deposition in kidney glomeruli (172). Yet other patients suffer from diffuse alveolar hemorrhage, a rare complication that affects <10% of SLE patients but is fatal in up to 50% of such cases (81). In addition, up to 20% of patients suffer from liver inflammation marked by elevated liver function tests (LFTs) and pathologic liver histology (98).
Genome-wide association studies have found that variation in the ITGAM gene, which codes for CD11b (a β2 integrin family member), is one of the strongest risk factors for SLE (43). A proposed mechanism for its pathogenesis is through defects in clearing apoptotic debris and self-reactive immune cells (141). As a whole, CD11b variants can reduce ligand binding and phagocytosis, increase cytokine production, elevate anti-dsDNA (double-stranded DNA) antibodies, and promote the autoreactivity of B cells (83).
ITGAM gene mutations correlate with increased skin, kidney, and neuropsychiatric manifestations of SLE. However, in mouse models of lupus, the absence of CD11b (downstream of ITGAM) has a protective effect. Using a pristane-induced mouse model of lupus, Zhuang et al. found that CD18−/− mice had little to no evidence of lung injury (200). Whether this observation holds true in humans is unknown.
Integrins and CAMs are also implicated in neuropsychiatric manifestations of SLE. Anti-SLE and anti-RA (rheumatoid arthritis) medications impact circulating CAMs, which correlate with neuropsychiatric manifestations of SLE (30). SLE patients generally have higher CAM levels than healthy controls, and circulating CAM levels correlate with disease severity (31).
Specifically, prednisolone and cyclophosphamide significantly decreased P-selectin and VCAM-1 expression in patients presenting with neuropsychiatric lupus (104). Functionally, P-selectin serves to bind and capture a circulating lymphocyte (100), while VCAM-1, recently proposed as a biomarker for lupus nephritis (112), acts as an adhesion molecule, helping to traffic lymphocytes and inflammatory cells (28). These CAMs may represent specific targets for treating neuropsychiatric lupus, potentially through preventing leukocyte migration into the central nervous system.
Liver disease in autoimmunity is also linked to biomarkers of SLE, such as autoantibodies and SLEDAI. Conventional treatment of SLE includes treatment with anti-inflammatory drugs, such as corticosteroids (176). Liu et al. found that treatment with the corticosteroid prednisone decreases both SLEDAI and LFTs (98). This suggests that liver dysfunction in the setting of SLE may originate autoimmunity and downstream inflammation. The liver is a complex organ, and its role in SLE further complicates its study. SLE-associated liver diseases can be classified as either immunological comorbidities (overlap syndromes), nonimmunological comorbidities related to SLE, and SLE-mediated direct hepatic injury (9).
Rabs, CAMs, and Integrins Modulate Liver Inflammation
The liver contains a mix of different cells, each of which integrates signals through Rabs and cell surface proteins (Fig. 1). While hepatocytes promote detoxification, antioxidation, and protein synthesis, immune cells are primarily anti-inflammatory and immunomodulatory. Collagen, integrins, and CAMs exist to anchor immune cells together. In states of pathology, dysfunctional liver cells lead to ROS toxicity, lipid peroxidation, autoimmunity, fibrosis, and cirrhosis. The heterogeneous population of liver cells use integrins, CAMs, and endosomal trafficking to accomplish their physiological tasks; in states of pathology, or inflammation, these pathways are dysfunctional and lead to hepatic inflammation.
Responsible for drug and lipid metabolism, hepatocytes are the primary functional units of the liver. The antioxidant pathways outlined above take place in hepatocytes and are subject to integrin-mediated sequestration of immune cells. Rab GTPases are implicated in a variety of hepatocellular functions outside of immunity. For example, Rab10 interacts with Dynamin 2 to promote the maturation of lipophagic organelles (96). Rab7 has also been established as a “central regulator” of lipophagy in the hepatocytes, promoting lipid droplet breakdown (153). Rab9A mediates carcinogenesis in Hep3b and HepG2 human liver carcinoma cell lines, potentially through the AKT signaling pathway (168). Male Wistar rats fed a Lieber-DeCarli diet, which is a model of experimental alcoholic liver disease, show decreases in Rab3d and Rab18 in whole liver sections, while also showing a decrease in Rab2, Rab5, Rab7, and Rab18 in isolated lipid droplet fractions (139).
Hepatic stellate cells (HSCs; Ito cells) are the primary mediators of hepatic fibrosis in response to liver injury and are important in their storage of vitamin A. These cells differentiate into cells that resemble myofibroblasts in response to liver injury to promote fibrosis downstream of transforming growth factor beta (TGFβ) signaling (60). Rab18 is implicated as a driver of autophagy and fibrosis in human HSC cell lines, and has been hypothesized as potential target of therapies against liver fibrosis (6). Rab18 is a “retinoid responsive mediator” required for the activation of HSCs in mouse models of liver fibrosis (124).
Kupffer cells (KCs) are resident tissue macrophages that are unique to the liver and are critical to the liver's response to toxicity. They are important in autoimmunity for their ability to regulate their neighboring cells. They can directly impact immune tolerance by secreting interleukin 10 (IL-10) to activate regulatory T cells. They can also activate HSCs to promote fibrosis, and have been shown to respond to hepatitis B virus (HBV) and hepatitis C virus (HCV) infections by secreting interleukin 18 (IL-18) and interleukin 1 (IL-1) to stimulate natural killer (NK) cells in liver pathologies (179).
Natural killer T (NKT) cells are lymphocytes with innate-like properties. In the liver, they exist to recognize both self and pathogenic lipid antigens, and promote immune tolerance. KC signaling can prime NKT cells to activate HSCs, kill hepatocytes, and activate neutrophils under pathologic states. However, NKT Type II can serve tolerance roles through conventional dendritic cell (cDC) signaling to inhibit Type I NKT cells. NKT Type I-mediated hepatic destruction is mediated by IFN-γ signaling, (5) which is regulated by Rab3D and Rab19 (74). Cholangiocytes are a rare population in the liver, but have recently come to the forefront of immunobiology. While the mechanism defining their ability to present antigens to T cells is unclear, cholangiocytes can present lipid antigens and derivatives of riboflavin to NKT cells, specifically through a CD1-iTCR (invariant T cell receptor) interaction (4).
NK cells comprise up to 40% of intrahepatic lymphocytes in humans and up to 20% of intrahepatic lymphocytes in mouse. Here, they facilitate tolerance to the diverse repertoire of pathogens that flow through the general circulation from the gut. Like NKT cells, NK cells integrate responses from other liver cells to mediate hepatic injury. HSCs can inhibit NK cell activity through TGF-b, and regulatory T cells can inhibit NK cell activity through IL-10 and TGF-b. NK cells can be activated by IFN-γ from NKT cells and dendritic cells, or through interleukin 12 (IL-12) and IL-18 from KCs to directly damage hepatocytes to promote liver injury and inhibit both liver regeneration and liver fibrosis through HSC inhibition (174).
Hepatocytes and HSCs both express α1β1 (very late antigen-1; VLA-1) and α2β1 (very late antigen-2; VLA-2) (67). α1β1 retains monocytes and circulating T cells under physiological conditions (49), while α2β1, which is upregulated during inflammatory states (58), mediates monocyte adhesion and NK cell proliferation in the liver (166). HSCs also express α5β1 (very late antigen-5; VLA-5), also known as the fibronectin receptor, in times of inflammation (67). VLA-5 has been shown to promote monocyte localization to infarcted myocardial tissue (175). Whether this interaction takes place in the liver is unknown.
While integrins are composed of heterodimers, the individual components are also trafficked through the activities of Rab GTPases. α1 is regulated by Rab21 and functions in cell migration. β1 is not only trafficked by Rab21, but also by Rab5, myosin X (Myo10), disabled homolog 2 (Dab2), and Numb, and functions in cell migration and cancer cell invasion. α2β1 (VLA-1) is trafficked by Rab11, and α5β1 (VLA-5) is trafficked by Rab11, Rab25, receptor component protein (RCP), chloride intracellular channel protein 3 (CLIC3), GIPC PDZ domain-containing family, member 1 (GIPC1), protein kinase D1 (PRKD1), syntaxin-6 (STX6), and vesicle-associated membrane protein 3 (VAMP3) (33).
Rab11 mediates the surface translocation of the TGFβ receptor in HSCs, suggesting a role for Rab11 in mediating hepatic fibrosis, as that is driven by TGFβ signaling (177). In addition, Rab25 regulates autophagy and lipid droplet degradation in HSCs in a ROS-dependent manner (198), suggesting that α5β1 (VLA-5) may also be involved in ROS-dependent surface expression. α1β1 is endocytosed by Dab2 in a clathrin-dependent manner, but the involvement of Rab GTPases in this regulation is unknown (11) (Fig. 1).
Mechanistic Target of Rapamycin Acts as a Sensor of Oxidative Stress and Effector of Inflammation in the Liver
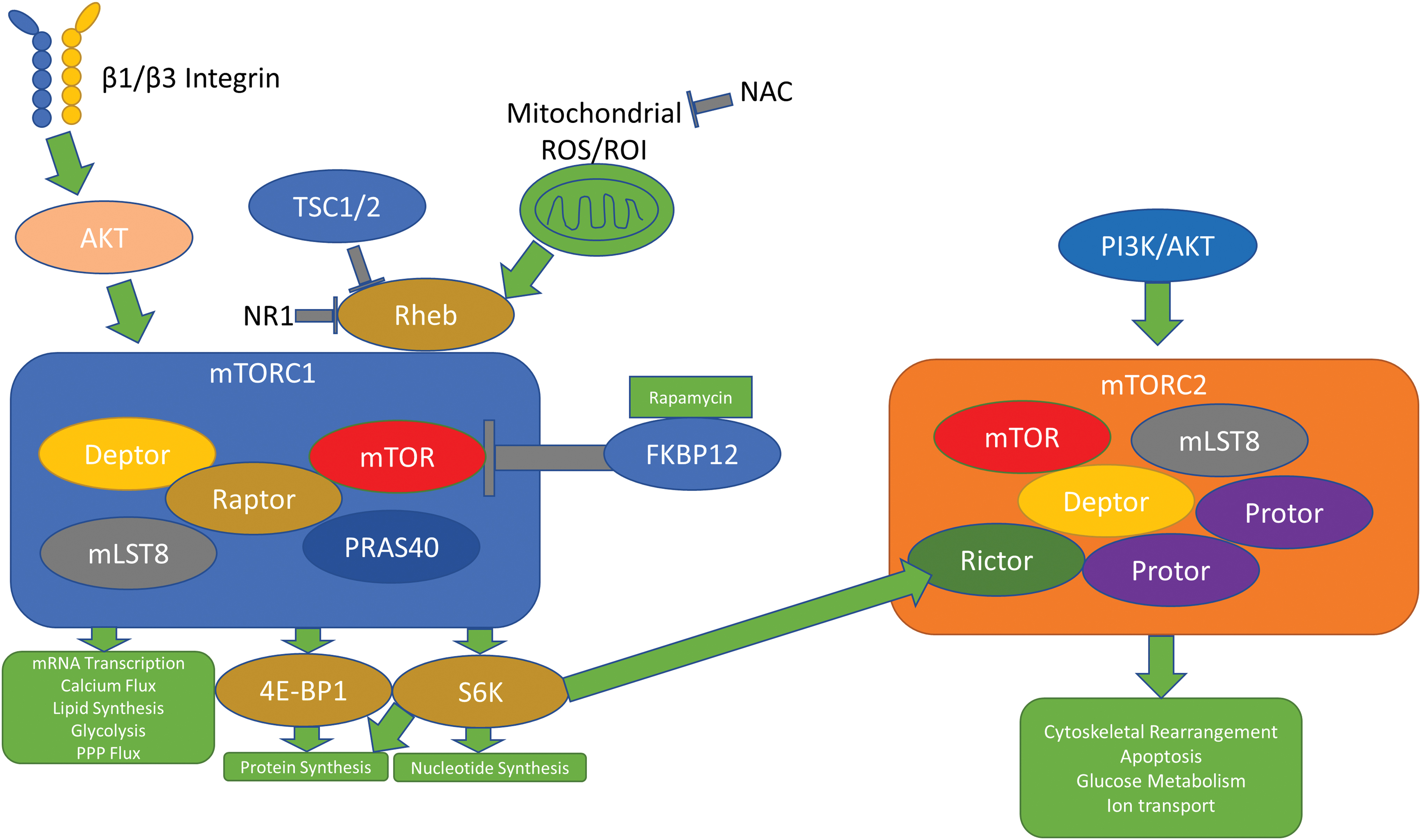
The mechanistic target of rapamycin (mTOR) is a target of immunomodulation in autoimmune diseases, including SLE (188). mTORC1 and mTORC2, two different mTOR complexes, control distinct regulatory pathways in the cell. When acted upon by rapamycin, a macrolide synthesized by soil microbes first discovered on Easter Island, the mTOR/AKT/phosphatidylinositol 3-kinase (PI3K) pathway is inhibited, ultimately leading to decreased cell growth, proliferation, and survival. mTORC1 acts downstream of amino acids, glucose, immune cytokines, hormones, and mitochondrial ROS to promote messenger RNA (mRNA) transcription, calcium flux, nucleotide synthesis, protein synthesis, lipid synthesis, glycolysis, and flux through the pentose phosphate pathway (PPP).
mTORC1's ability to phosphorylate and activate eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and S6 kinase (S6K) leads to increases in protein synthesis and nucleotide synthesis. mTORC2 acts downstream of the PI3K/AKT pathway to promote cytoskeleton rearrangement, apoptosis, glucose metabolism, and ion transport (152).
mTORC1 reacts to oxidative stress through its interaction with the Rheb protein, a member of the Ras superfamily of G proteins (127). The small molecule inhibitor NR1 preferentially binds the switch II domain of Rheb, halting mTORC1 downstream activities, but leaving mTORC2 signaling unaffected (106). HEK293T cells treated with phenylarsine oxide (PAO), a cell-permeable reagent that causes crosslinkage of vicinal thiol groups and oxidizes cysteine residues, result in activation of mTORC1 and not mTORC2, suggesting that ROS promote mTORC1 activity through the activation of the tuberous sclerosis complex (TSC) 2/Rheb pathway (197) (Fig. 4).
In times of increased stress, this pathway is responsible for generating antioxidant molecules. Cancer cells experience immense oxidative stress, and signaling through PI3K/AKT pathways mediates the upregulation of the classical antioxidant signaling molecule nuclear factor erythroid 2-related factor 2 (Nrf2) in addition to the metabolism of glutathione and the production of nicotinamide adenine dinucleotide phosphate (NADPH). Nrf2, when activated, acts as a transcription factor, upregulating the genes responsible for generating glutathione-S-transferase, SOD, and PPP enzymes, which generate NADPH (85).
Studies in mouse breast cancer models show crosstalk between the AKT/mTORC1 pathway and integrin expression/ECM remodeling. Mammary tissue-specific deletion of the β1 and β3 integrin subunits decreases mTORC1 activity (13). Since β1 and β3 integrins facilitate outside-in signaling, it is reasonable to conclude that integrin signaling modulates ROS responses in an mTOR-dependent manner. β1 integrins are critical to liver regeneration (163), and it is worth noting that mTOR signaling mediates physiological autophagy in the liver, which, when dysfunctional, can lead to liver fibrosis, predisposition to HBV infection, nonalcoholic fatty liver disease, liver ischemia and reperfusion injury, and liver cancer (181). Dysfunctional β1 integrin signaling has also been shown to promote hepatocellular carcinoma through the activation of AKT (2).
Paradoxically, long-term usage of rapamycin leads to increased ROS. Rapamycin administration in mouse models of liver dysfunction shows that while, in the short term, rapamycin can decrease ROS generation and lipid droplet formation (thus decreasing the incidence of nonalcoholic steatohepatitis and hepatocellular carcinoma), chronic, long-term administration can increase interleukin 6 (IL-6) and signal transducer and activator of transcription 3 (STAT3) signaling, which ultimately overcomes the beneficial effects of rapamycin (178). Case reports have even suggested that long-term rapamycin usage can reactivate latent HBV, and in vitro studies found that this may be linked to inducing hepatocellular autophagy (72).
Antioxidant Mitigation of Liver Dysfunction
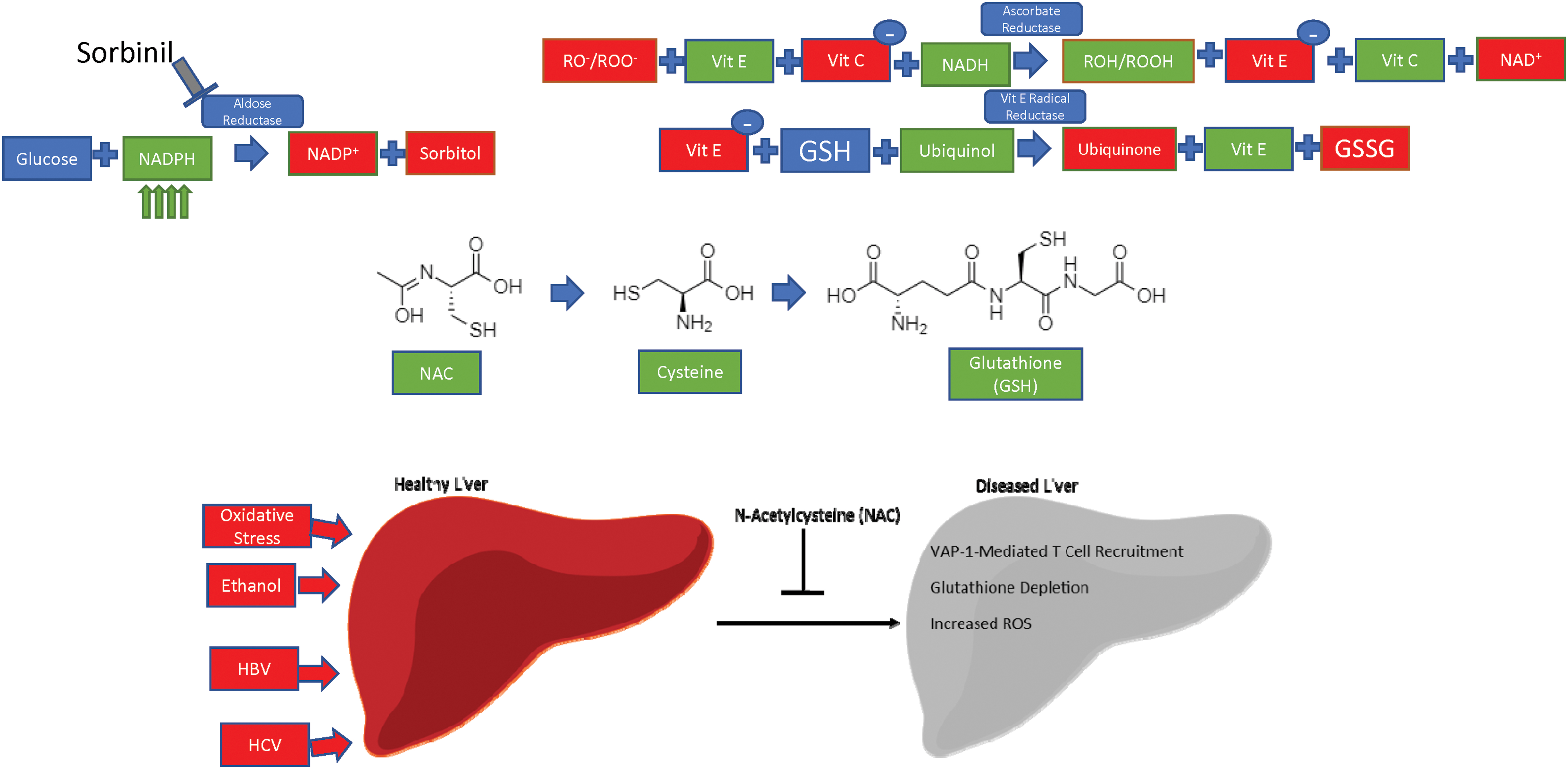
While ROS play a key role in regulating integrin and CAM expression, both dependently and independently of endosomal trafficking, ROS are also harmful to cellular components. The liver has more phagocytic cells than anywhere else in the body, and is uniquely positioned to mount an anti-inflammatory and redox response due to its continuous encounter with foreign food antigens (87). For example, liver damage caused by increased oxidative stress from chronic alcohol exposure predisposes to liver inflammation and hepatitis (116). Oxidative stress is implicated in the pathology and treatment of SLE—mitigation of oxidative stress by N-acetylcysteine (NAC) improves disease activity in mouse models of SLE (133).
In addition, ethanol-mediated ROS generation exacerbates the liver-damaging effects of HCV infection, which can cause liver fibrosis, hepatocellular carcinomas, and chronic, long-standing hepatitis (136). Mammalian redox systems exist to neutralize the damaging effects of ROS. The key players in these systems are NADPH and reduced glutathione (GSH), which act as electron carriers—most of the following pathways, in one way or another, exploit either NADPH [and its nonphosphorylated counterpart nicotinamide adenine dinucleotide (NADH) (196)] or glutathione to reduce the damaging effects of ROS on the cell (Table 2).
Implication of Antioxidant Enzymes in Human Disease Pathogenesis
Enzymes responsible for catalyzing antioxidant reactions in the human body are also implicated in human disease.
ALS, amyotrophic lateral sclerosis; AML, acute myeloid leukemia; AR, aldose reductase; ARDS, acute respiratory distress syndrome; G6PD, glucose-6-phosphate dehydrogenase; HCC, hepatocellular carcinoma; IDH2, isocitrate dehydrogenase 2; NNT, nicotinamide nucleotide transhydrogenase; SLE, systemic lupus erythematosus; SOD, superoxide dismutase; TAL, transaldolase.
The PPP, also known as the hexose monophosphate shunt, generates intermediates for protein synthesis and nucleotide synthesis. The PPP is responsible for 5-carbon sugar metabolism and the generation of NADPH, crucial for recharging the antioxidant systems in response to oxidative stress (64). The PPP contains two arms: a nonoxidative branch and an oxidative branch, so named after their effects on the cell. The nonoxidative branch is regulated by the rate-limiting, reversible enzyme transaldolase (TAL), which catalyzes glyceraldehyde-3-phosphate and sedoheptulose-7-phosphate to erythrose 4-phosphate and fructose-6-phosphate (135). Haploinsufficiency of TAL has been reported in 4 of 27 patients with acetaminophen-induced liver failure, who were then responsive to NAC therapy (125). As of 2019, there have been 34 patients reported with complete TAL deficiency (187).
Glucose-6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme of the PPP, and is the most common enzyme deficiency present in ∼400 million people worldwide (15). In physiological states, G6PD converts glucose-6-phosphate into 6-phosphogluconate, reducing NADP+ into NADPH in the process. Deficiencies in this enzyme lead to a reduced capacity of erythrocytes to clear oxidative stress, which can lead to hemolysis and favism in response to oxidative drugs such as primaquine (malaria drug) and sulfamethoxazole (sulfa drug/antibiotic) (47).
Glutathione exists in two forms: oxidized (GSSG) and GSH. Glutathione acts as an electron carrier, preventing free electrons from binding to oxygen (O2) and wreaking havoc on the cell. In this case, NADPH harnesses the electrons from GSSG and is converted into NADP+. GSSG is then converted to GSH, along with a •OH (103).
Superoxide is generated as a by-product of O2 metabolism through natural processes and is a potent free radical (61). SOD acts to convert the highly active superoxide radical into molecular O2 and H2O2 (171). The remaining H2O2 is acted upon by the enzyme catalase to produce pure O2 and water (H2O) (21).
Aldose reductase (AR) is an enzyme located in the cytosol, which preferentially uses NADPH to promote its enzymatic activity. Using NADPH, AR converts glucose into sorbitol (88), a compound that is known to cause cataracts and visual disturbances through changes in osmotic pressure. Sorbinil is a pharmacological compound being investigated for use as a therapeutic against these diseases by acting as an inhibitor of AR (Fig. 5) (71).
Thioredoxin (TRX) represents a family of proteins that are important in reducing oxidized proteins into reduced proteins by acting as molecular intermediates between oxidized proteins and NADPH (23).
Isocitrate dehydrogenase 2 (IDH2) is a reversible mitochondrial enzyme. IDH2 either goes in the forward direction to generate α-ketoglutarate and NADPH from isocitrate, or goes in the reverse direction, generating isocitrate and NADP+ from α-ketoglutarate. IDH2 can also convert 2-oxoglutarate into D-threo-isocitrate (101). In parallel, CO2 and NADPH are used up and generate NADP+ in the process. Preferentially targeting IDH2 can serve to dramatically decrease NADPH utilization, thus increasing antioxidant stores in the cell. IDH2 mutations are implicated in the progression of liquid and solid tumor cancers. Enasidinib, an FDA-approved glioblastoma multiforme therapy, preferentially inhibits the mutant R140Q IDH2 enzyme to kill cancer cells in mouse and human models (194). The incidence of IDH2 mutations in leukemia is up to 19% depending on the subtype of leukemia (109).
Nicotinamide nucleotide transhydrogenase (NNT) is a nuclear-encoded gene located in the inner mitochondrial membrane (70). Hoek and Rydström describe NNT as a physiological buffer system, serving to transfer electrons to NADPH from NADH and vice versa (70). The forward and reverse reactions are catalyzed by the proton motive force in the mitochondria, which is hypothesized to be due, in part, to a conformational change in the enzyme itself (145).
In the forward reaction, the buildup of the proton motive force from the cytoplasm into the mitochondria pushes the reaction forward, reducing NADP+ into NADPH. Conversely, the reverse reaction takes place under pathologic conditions (69), and is catalyzed by protons moving across the inner membrane from the mitochondrial compartment into the cytoplasmic compartment (70). The incidence of NNT mutations is ∼10% among patients with familial glucocorticoid deficiency, although prevalence information is unavailable at this time (143) (Fig. 6).
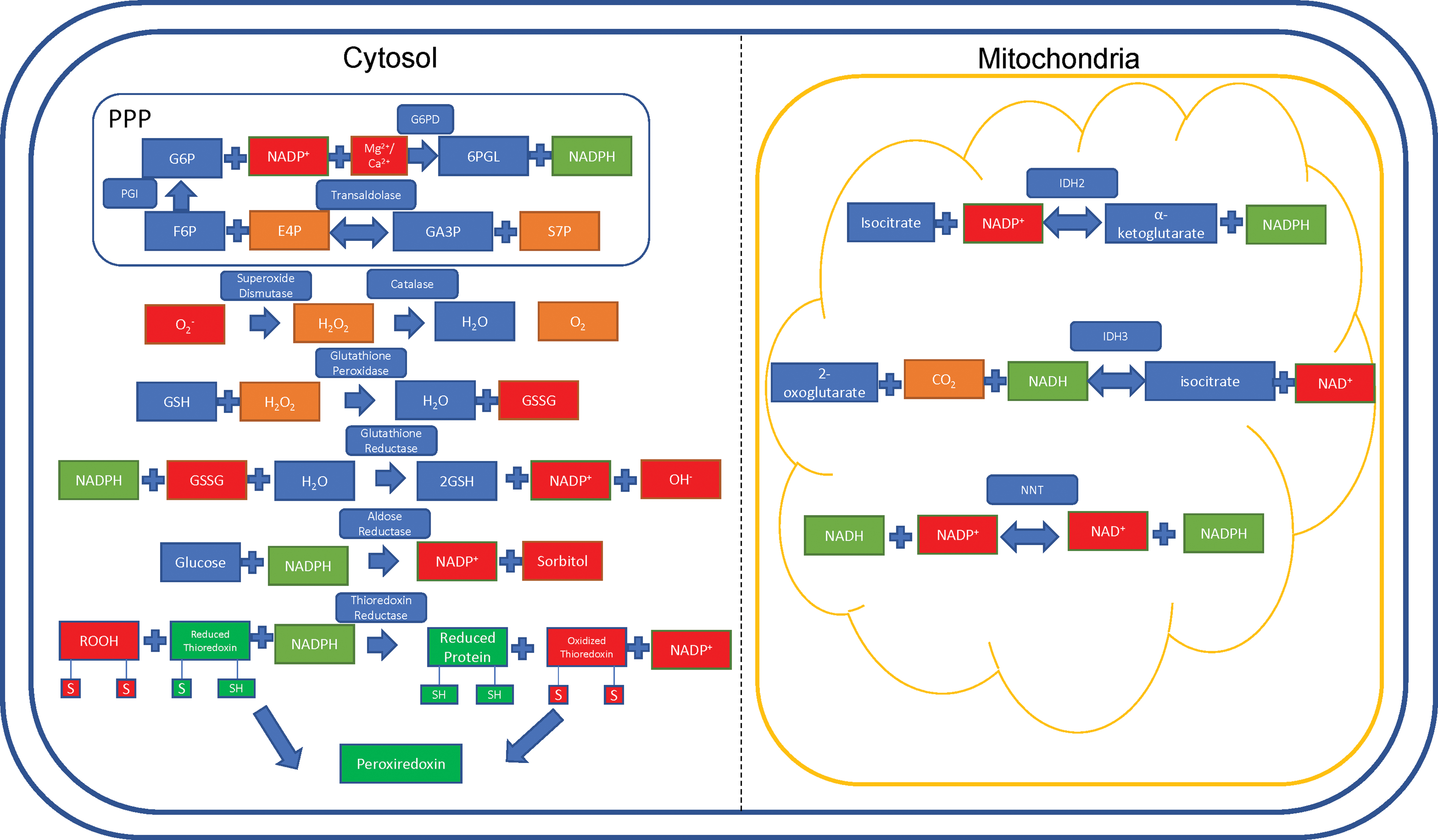

Peroxiredoxins (Prxs) are thiol-dependent peroxidases, which act to remove ROS and come in three subtypes. In typical 2-Cys Prxs, the main peroxidative cysteine residue (Cp) reacts with the resolving cysteine residue (Cr) that is contained in the second subunit of the Prx dimer. In atypical 2-Cys Prxs, the oxidized Cp reacts with the Cr residue on the same subunit molecule. In 1-Cys Prxs, the Cp residue generates sulfenic acid that is then regenerated directly through the transfer of an electron to the thiol group when ascorbate is present (122).
In states of acute liver damage, Prxs have been shown to translocate to the mitochondria and become hyperoxidized, eliminating their antioxidant function in the cell. Interestingly, Prxs can protect against ROS by reducing the expression of adhesion molecules in vascular endothelial cells and inhibit inflammatory response (102), two processes that, when dysfunctional, can directly lead to liver dysfunction. Autoantibodies against Prxs have also been shown to be upregulated in patients with autoimmune diseases, such as SLE and RA (80) (Fig. 7).
Targeting mTOR, ROS, and Integrins Has Therapeutic Potential in Autoimmune Liver Disease
We are only just starting to understand the importance of hepatic injury in the context of SLE. About 20% of SLE patients suffer from liver dysfunction (98). Lifestyle factors and interactions with infectious agents also predispose to inflammatory states in the liver, which involves central lymphocyte migration into the hepatic parenchyma (116).
While the liver is an organ of immense immune activity, pathologic inflammation almost always involves migration of peripherally circulating leukocytes and lymphocytes into the liver. For example, AIH, an autoimmune condition in which the immune system attacks its own hepatocytes, is characterized by CD4+ and CD8+ effector T cell responses (182). Primary biliary cirrhosis, a disease marked by inflammatory destruction of biliary epithelial cells, is also predominated by inflammatory infiltrates of CD4+ and CD8+ T cells, B cells, macrophages, eosinophils, and NK cells (77).
That mTOR inhibitors, such as rapamycin (sirolimus), everolimus, and mTOR kinase, are used to prevent immune rejection of transplanted livers and are used as therapies for hepatocellular carcinoma also suggest mTOR's important role in mediating hepatic inflammation, and that targeting mTOR has therapeutic benefits against liver inflammation (46). Indeed, rapamycin is used in the treatment of SLE, and considering liver dysfunction is present alongside SLE and can be reduced with rapamycin (either alone or with other immunosuppressants) (98) generates interesting questions in its role as an inhibitor of autoimmune-mediated hepatic dysfunction. Rapamycin has also been shown to treat refractory and post-transplant AIH (20, 82).
Reducing oxidative stress through NAC administration has beneficial effects outside of its classical role as a reversal agent of acetaminophen overdose (Table 3). Due to the impact of oxidative stress on integrin signaling, the effect of antioxidant therapy on integrins and abrogating hepatic dysfunction remains an outstanding question. Aberrant ROS production is hypothesized to be a driver of T cell dysfunction, which then leads to autoimmunity. We have reported that NAC blocks ROS production by complex I of the mitochondrial electron transport chain in lupus T cells (37).
Therapeutic Benefit of N-Acetylcysteine in Patients with Liver Failure
NAC and other antioxidant therapies may improve liver disease and inflammation in other organs of patients with systemic autoimmunity.
IV, intravenous; NAC, N-acetylcysteine; NAI-ALF, nonacetaminophen-induced acute liver failure.
While hepatic dysfunction in SLE and autoimmunity occurs as a result of KC, NK cell, and NKT cell dysfunction (Fig. 1), we posit that targeting the T cells that mediate systemic autoimmunity may also inhibit liver dysfunction, which may lead to a decrease of autoreactive intrahepatic inflammation in the setting of SLE. The mechanisms that define the interactions between T cells and hepatocytes are poorly understood, and this is an open area for investigation.
Our laboratory has defined hepatic manifestations of SLE in mouse models (128). This squares well with clinical manifestations of SLE, which show dysfunctional LFTs correlating with SLEDAI scores. Treatment of SLE patients with corticosteroids not only reduced SLEDAI but also decreased liver disease severity as shown by decreased LFTs (98).
In a study of 85 patients with liver disease (49 patients with autoimmune cholestatic disease and 36 patients with AIH), patients with either disease condition had increased lipid and protein oxidative injury products, suggesting that either of these autoimmune hepatic conditions is associated with increased oxidative stress. Whether the oxidative stress leads to the disease or the disease leads to increased oxidative stress is unknown, but the study concluded that the increase of oxidative stress corresponded to “liver inflammatory activity and fibrosis” in both diseases (79).
In the treatment of inflammatory bowel disease (IBD), anti-integrin therapies, such as Natalizumab (α4 integrin), vedolizumab (α4β7 integrin), and ertolizumab (β7-integrin) (126), decrease disease flares by preventing the interactions between circulating immune cells and endothelial CAMs. While these therapies are effective for treating IBD, anti-integrin therapies have been shown to have adverse side effects, including hepatocellular injury and liver failure in <5% of patients receiving therapy (though this is still less than the ∼29% of IBD patients who develop abnormal liver tests without therapy) (68). Mouse studies have found that loss of αvβ5 promotes liver regeneration through inhibition of TGFβ signaling, though no studies have confirmed this in humans (55).
Targeting integrin expression via endosomal trafficking remains an open area of investigation. As of now, no therapeutics exist to reduce endosome-mediated disease burden in liver dysfunction. However, a completed clinical trial sought to exploit the endosomes of cancer cells using fimaporfin, a light-activated compound that incorporates into tumor cell endosomes (14).
Since Rab GTPases can regulate endosomal trafficking and sorting, Rabs have been an active area of research regarding what diseases they can cause. Overexpression of a variety of Rab isoforms is implicated in cancers, neurodegenerative diseases, and immune disorders (57). While nonspecific Rab inhibitors exist, such as 3-PEHPC (which inhibits many different Rab isoforms) (16), few, if any, inhibitors exist to directly target a single Rab GTPase, perhaps due to the high homology among members of the Rab family.
Antioxidants have also been used as disease therapeutics. Vitamins E and C work through vitamin E radical reductase and ascorbate reductase, respectively, to reduce redox agent burden (17). Metadoxine is an antioxidant that has been shown to improve survival in patients with alcoholic hepatitis when combined with the standard treatment regimen of prednisone and pentoxifylline due to its ability to synthesize glutathione (66). Alcoholic hepatitis involves immune cell margination into the liver, which may be dependent on ROS-mediated trafficking of cell surface receptors. Silymarin/Silybin, active extract derivatives from milk thistle, act to enhance both glutathione peroxidase activity and SOD, and have been found to improve clinical outcomes in patients with inflammatory liver diseases (25, 161).
Two dietary supplements, Resveratrol and Quercetin, have been under extensive study as antioxidant molecules. Resveratrol is a natural polyphenol, which has antioxidant, anti-inflammatory, and anticancer properties. It stimulates expression of sirtuins, which then promote antioxidant enzyme production (144). Quercetin is a flavonoid found in fruits and vegetables that has been shown to increase catalase, SOD, and glutathione peroxidase activities to reduce ROS in the cell (189). Relevant for our studies of immunity, quercetin can block TNFα-mediated NF-κB signaling in vitro and ultimately decreases cytokine release in vivo (95). NAC regenerates glutathione stores, which are depleted in acute acetaminophen toxicity (63).
In the context of autoimmunity, our group found that NAC provides a therapeutic effect when administered to SLE patients by blocking the mTOR pathway (90). Our group is currently expanding on this finding in a phase 2 clinical trial (134) (Fig. 5).
Conclusions
The tightly controlled immune response to a pathogen, when dysfunctional or retargeted to a self-antigen, can lead to systemic autoimmunity. SLE, a prototypical systemic autoimmune disease, is characterized by potentially life-threatening involvement of all organs of the body, underlain by ROS generation, dysregulated endosomal trafficking, and abnormal CAM and integrin expression. Changes in CAMs and integrins facilitate liver invasion by immune cells—the redox control of integrin expression is crucial to controlling hepatic dysfunction in the setting of systemic autoimmunity. Through the direct modification of integrins or the endosomal trafficking of integrins via redox-sensitive Rab GTPases, ROS can modulate immune attack in states of autoimmunity. Targeting ROS-mediated signaling and pathways that exploit ROS generation and neutralization can serve as potentially powerful therapeutics to help alter immune-mediated liver injury.
Footnotes
Acknowledgment
The authors thank Brandon Wyman for his careful reading and editing of the article.
Authors' Contributions
All the authors conceived and wrote this review in its entirety.
Author Disclosure Statement
The authors do not have any competing financial interests to disclose.
Funding Information
This work was supported by grants R01-AI072648 and R01-AI122176 from the National Institutes of Health and the Central New York Community Foundation.