
Abstract
Significance:
Reactive oxygen species (ROS) are well known to promote innate immune responses during and in the absence of microbial infections. However, excessive or prolonged exposure to ROS provokes innate immune signaling dysfunction and contributes to the pathogenesis of many autoimmune diseases. The relatively high basal expression of pattern recognition receptors (PRRs) in innate immune cells renders them prone to activation in response to minor intrinsic or extrinsic ROS misbalances in the absence of pathogens.
Critical Issues:
A prominent source of ROS are mitochondria, which are also major inter-organelle hubs for innate immunity activation, since most PRRs and downstream receptor molecules are directly located either at mitochondria or at mitochondria-associated membranes. Due to their ancestral bacterial origin, mitochondria can also act as quasi-intrinsic self-microbes that mimic a pathogen invasion and become a source of danger-associated molecular patterns (DAMPs) that triggers innate immunity from within.
Recent Advances:
The release of mitochondrial DAMPs correlates with mitochondrial metabolism changes and increased generation of ROS, which can lead to the oxidative modification of DAMPs. Recent studies suggest that ROS-modified mitochondrial DAMPs possess increased, persistent immunogenicity.
Future Directions:
Herein, we discuss how mitochondrial DAMP release and oxidation activates PRRs, changes cellular metabolism, and causes innate immune response dysfunction by promoting systemic inflammation, thereby contributing to the onset or progression of autoimmune diseases. The future goal is to understand what the tipping point for DAMPs is to become oxidized, and whether this is a road without return. Antioxid. Redox Signal. 36, 441–461.
Introduction
Essentially all living organisms, from simple prokaryotes to multicellular eukaryotes, rely for their entire life on their innate immune system for protection against microbial infections (46, 80). In the past, innate immunity was deemed simple and unsophisticated compared with the specific and long-lasting responses of the adaptive immune system. However, it has since become clear that innate immunity depends on vastly diverse, immunologically active preexisting proteins, not just immune cells, to trigger an effective host response to a pathogen and to prime adaptive immunity (76). Further, it is now recognized that innate immune responses are not only accountable for inducing chronic inflammation and autoimmunity, but they are as well the direct cause of cell, tissue, and organ damage that is generally associated with autoimmunity (153).
Pattern recognition receptors (PRRs) are the key players for the induction of innate immunity and can be divided into several classes. Starting with the extracellular space, plasma membrane-positioned type 1 transmembrane proteins, known as Toll-like receptors (TLRs), recognize hydrophobic lipids and proteins exposed on the surface of pathogens (91). Within the cell, endosomal TLRs have the distinct ability to recognize the genetic material of pathogens (e.g., DNA, RNA). Alike is another set of cytoplasmic PRRs, known as retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs), which are able to recognize specific patterns within the genomic material of pathogens, such as conserved motifs from different RNA-viruses or RNA-intermediates from DNA-viral replication (114).
Pathogen-associated DNA is recognized by other distinct cytosolic sensors localized freely in the cytoplasm, such as cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS) (170, 197); interferon-gamma-inducible protein 16 (IFI16), a member of the pyrin and HIN domain-containing (PHYIN) protein family (181); Z-DNA-binding protein 1 (ZBP1), also known as DNA-dependent activator of interferon-regulatory factors (DAI) (172); or the absent in melanoma 2 (AIM2) protein, which is not only a DNA sensor, but also reportedly an inflammasome activator and inducer of pyroptosis, an inflammatory form of cell death (53, 71).
In addition to inducing proinflammatory cytokines postengagement, PRRs can mobilize adaptive immune responses by increasing major histocompatibility complex class II expression and by inducing the expression of costimulatory molecules, such as cluster of differentiation (CD) −40, −80, and −86 on antigen-presenting cells (177). Further, PRRs may recognize endogenous motifs that are known as danger-associated molecular patterns (DAMPs) of “self”-derived material origin, which comprise cellular or organelle components rather than a pathogen-associated “nonself” structure of motif (Table 1). During cellular stress or injury, otherwise spatially confined components can relocate between the sub-cellular compartments and/or be chemically modified to become mimics of “nonself” motifs that typically induce immunity (111).
Intra- and Extracellular Pattern Recognition Receptors for Mitochondria-Derived Danger-Associated Molecular Patterns
CD, cluster of differentiation; cGAMP, cyclic guanosine-adenosine-monophosphate
Reactive oxygen species (ROS) are generated in all cell types, and under normal physiologic conditions limited ROS production is known to be overall beneficial and necessary to maintain biological functions of the cell (165). This includes gene expression, molecular signaling between organelles, control of the balance between the cell's proliferation and death, and response to microbial infections (126). Increased and/or chronic, potentially harmful levels of ROS occur during tissue injury, microbial infection, or exposure to environmental factors, such as ionizing radiation, ultraviolet (UV) irradiation associated with sun exposure, or heavy metal ions present in drinking water or overall increased air pollution (184) (Fig. 1).

Chronically increased ROS may lead to the accumulation and release of DAMPs, which can continuously activate the innate immune system (74). Studies of innate immunity activation in the absence of pathogens suggest that the various forms of ROS can be considered as DAMPs on their own, leading to the increased secretion of proinflammatory cytokines, changes in cellular metabolism, and induction of inflammation that could contribute to the development of autoimmune diseases (23, 148).
The ROS still posit a conundrum, because their increased presence in one cell type might not correspond to the effect it would have in a different cell type, or even in the same cell under analogous physiological conditions, as such an effect depends on the cell localization in the tissue, cellular density, molecular interactions, and timespan of the events (6). The activity of ROS, although when it was determined to be at the same concentration, was shown to depend not only on a level of multitude of relating cellular antioxidants, but it was also suggested to depend on the density of the cytoplasmic milieu, which might control the rate of ROS or antioxidant diffusion, respectively (192, 198).
Further, latest findings suggest that the mobility of different types of ROS within one cell can be important in distributing oxidative stress at the tissue or even entire organ level (116). The role of ROS in becoming a DAMP and the role of ROS-mediated modifications of DAMPs in the onset or potential progressive acceleration of autoimmune disease development are just at the beginning of being evaluated.
The term ROS comprises a heterogenous group of various oxygen-containing molecules (Fig. 1), produced from molecular oxygen, which can participate in electron transfer processes (56). The reactivity of different ROS molecules differs by at least 11 orders of magnitude respective to their kinetic interaction rate with a given substrate (125). However, the type of ROS and the level of relative concentration that is necessary to promote specific DAMP accumulation and oxidation, which in turn leads to changes in cellular metabolism to promote chronic inflammation and development of autoimmunity, is not known.
Eighty five percent to 90% of cellular oxygen is metabolized in mitochondria (Fig. 1), where superoxide anion (O2˙−) is produced after the interaction of a single electron with molecular oxygen at the electron transport chain (ETC) (56). The O2˙− can also be produced in the cytoplasm and at the plasma membrane (Fig. 1), where the catalytic action of many cytoplasmic enzymes has been shown to involve O2˙− (165). The capacity of O2˙− to become an efficient DAMP modifier is limited by its very short lifetime, caused by spontaneous or enzymatic dismutation to less reactive hydrogen peroxide (H2O2) (56). The rate of spontaneous O2˙− dismutation is fast and the enzymatic reaction catalyzed by cytoplasmic, mitochondrial, and extracellular compartmental-specific superoxide dismutases (SODs) is at least 103 times faster and close to the rate of free diffusion (56).
Hence, O2˙− is not able to travel far from where it is produced, or passage across cellular membranes, therefore unlikely being directly responsible for DAMP modification (56). The immediate targets of the negatively charged O2˙− in the mitochondria are positively charged iron/sulfur (Fe-S) centers in proteins, such as tricarboxylic acid (TCA) cycle enzyme aconitase or succinate dehydrogenase (SDH), both of which were shown to be more sensitive to oxidation by O2˙− than by H2O2 (27). The interaction of O2˙− with aconitase occurs in the mitochondrial matrix and was indicated to lead to the release of hydroxyl radical (HO˙) (182) (Fig. 1).
HO· is one of the strongest oxidants known, and because of its very low activation energy and quick diffusion has been suggested to be responsible for mitochondrial DNA (mtDNA) oxidations (137, 190). The protonated form of O2˙−, perhydroxyl radical (HOO˙), on the other hand, was suggested to be responsible for modification of mitochondrial phospholipids, for example cardiolipin (20). Another strong ROS, which can modify proteins and is responsible for lipid peroxidation, is peroxynitrite (ONOO−) (167). ONOO− is a product of near-diffusion-controlled reaction of O2˙− with nitric oxide (NO), which is on its own capable of oxidizing biomolecules (12, 167).
H2O2, although it is not a free radical like O2˙− or HO· and is less redox active because of its high activation energy, is relatively longer lived and was suggested to disseminate through membranes by passive diffusion (165) (Fig. 1). H2O2 has augmented reactivity toward cysteine residues in certain proteins, depending on the structure and environment, providing a basis for selectivity and specificity of H2O2 under physiological conditions (191, 195). Under oxidative distress conditions, when the H2O2 concentration increases in the cell from 1–10 to ∼100 nM and higher, nonspecific oxidation of proteins and damage to DNA and other biomolecules was suggested to occur (165) (Fig. 2).

It should be noted that the overall H2O2 concentration in the mitochondria was estimated to be at least two to five times higher than that detected in the cytoplasm, but lower than this was observed in the lumen of the endoplasmic reticulum (ER; ∼700 nM), where the formation of each disulfide bridge during the physiological folding and maturation of proteins leads to the generation of one molecule of H2O2 (165). The exact concentration of ROS in mitochondria to induce DAMP formation in immune cells, which in turn has a higher capacity to induce DAMP-dependent signaling, is not known and will most probably fluctuate with every organ (Fig. 2).
Future studies will need to focus on evaluating how innate immune cells respond to different levels of extracellular molecular oxygen in a tissue-specific context, simply because of their highly migratory nature. Only under this consideration we will be able to better understand the necessary conditions for the generation and release of DAMP and its associated pathologies.
The aim of this review is to present a comprehensive summary of how ROS modify mitochondrial components and become DAMPs on release, and how these intrinsic “self”-targets influence metabolic reprogramming, which ultimately contributes to the onset or promotes the progression of various autoimmune diseases.
Genomic Material: mtDNA and mtRNA
Mitochondrial DNA
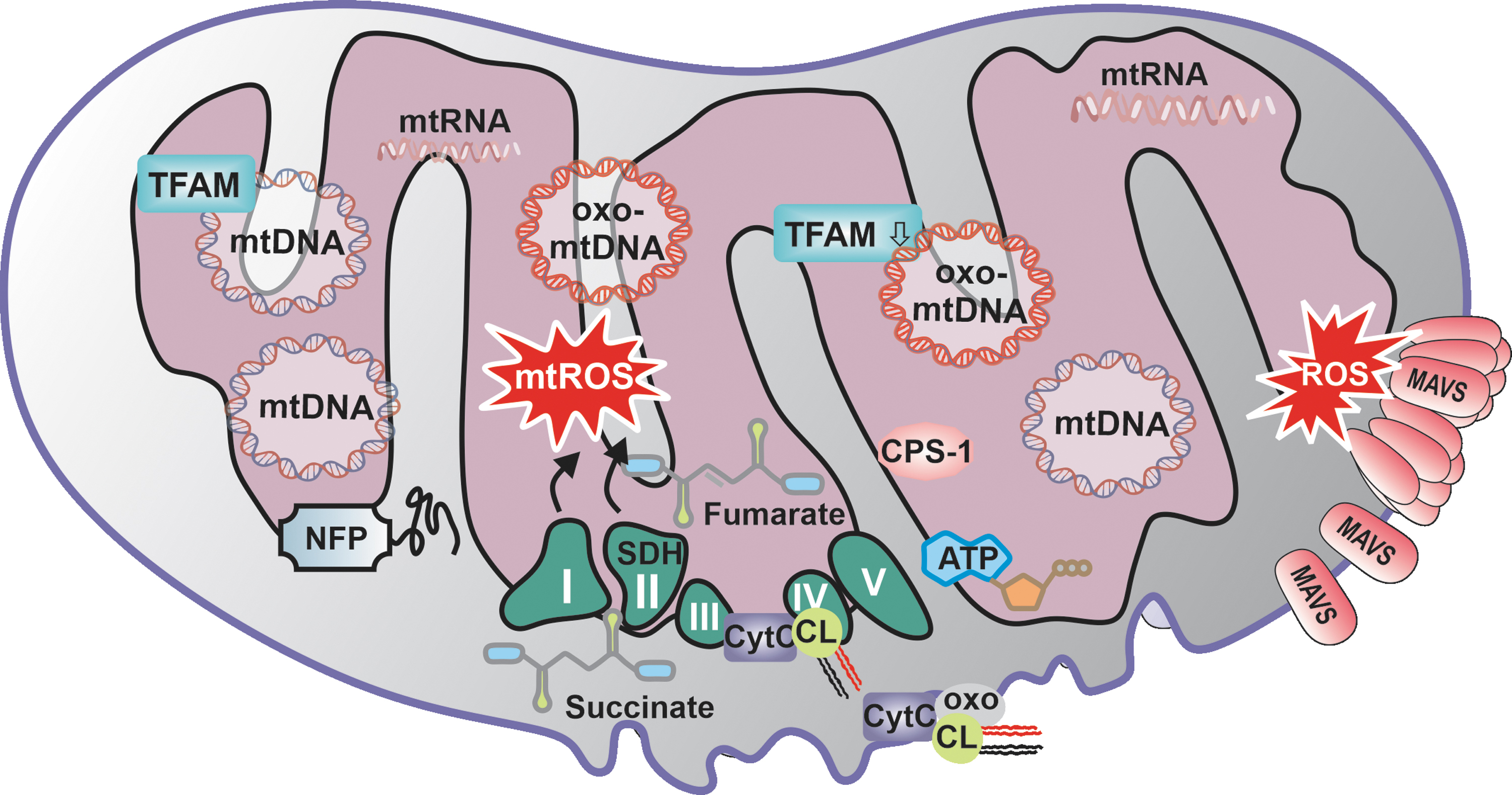
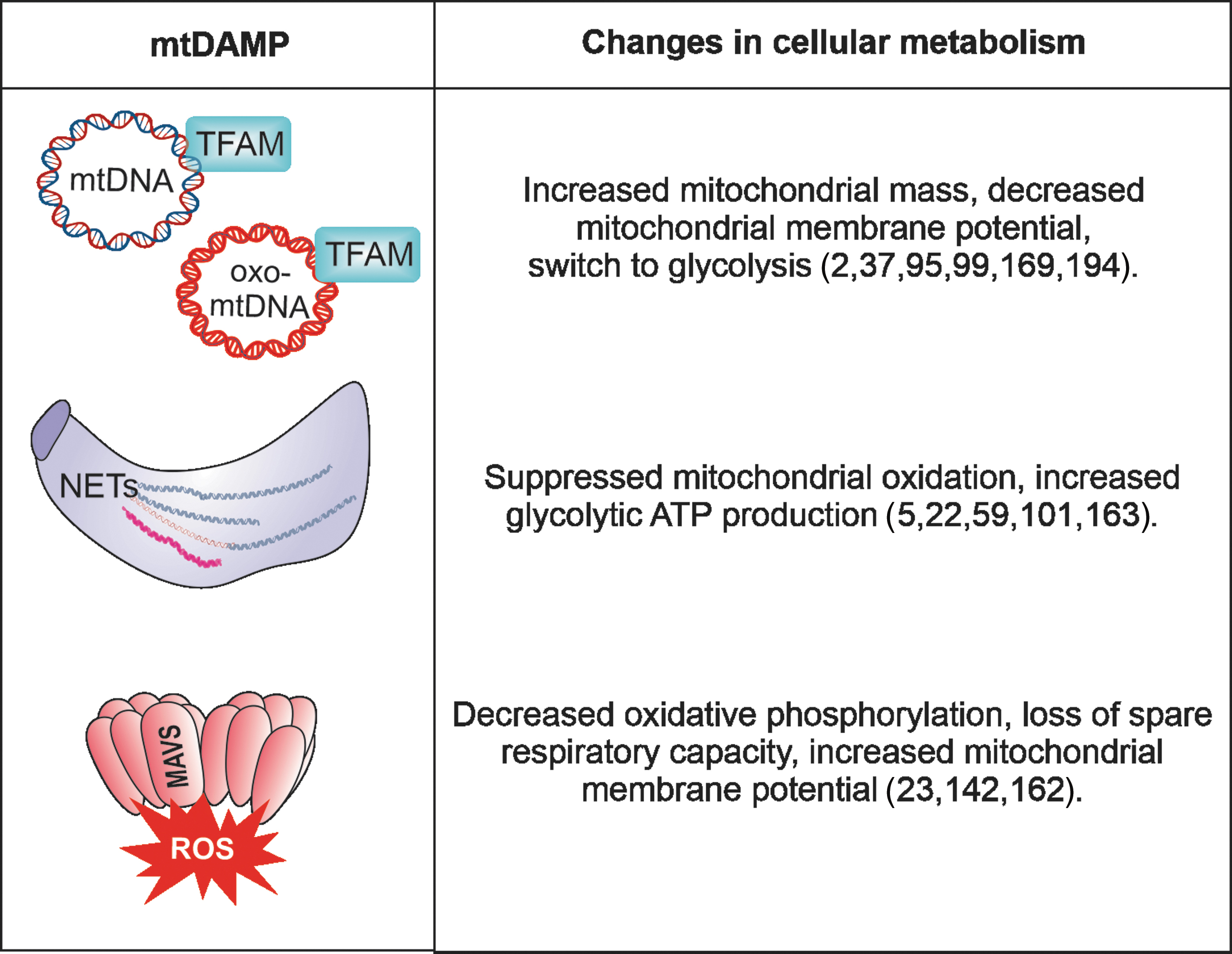
As bacterial descendants, mitochondria possess plenty of DNA motifs that resemble pathogen-associated molecular patterns (PAMPs); hence, mtDNA is often considered to be the prototypical mitochondrial DAMP. Similar to bacterial DNA, mtDNA is circular in structure and arranged in nucleoids, which, on average, contain one copy apiece of mtDNA and mitochondrial transcription factor A (TFAM) (Fig. 3). TFAM is responsible for inducing mtDNA transcription and is also known to regulate mtDNA metabolism by directing mtDNA to lysosomes for degradation (95). Due to its localization in the mitochondrial matrix and thus in close vicinity to the ETC—a major source of ROS—mtDNA is known to be vulnerable to oxidation (194) (Fig. 3).
Murine studies have shown that intra-articular injection of mtDNA purified from cell extracts, but not DNA isolated from the nucleus, causes increased localized inflammation, suggesting a major difference in the capacity of the two DNA to induce inflammation (37). This observation was confirmed with the injection of a synthetic, sequence-analogue DNA with and without oxidized nucleotides, which when oxidized lead to the secretion of proinflammatory cytokines or inflammation (37) (Figs. 3 and 4). mtDNA can simulate several PRRs, including cytosolic cGAS, which promotes type I interferon (IFN) responses, and AIM2, which leads to the formation of inflammasome and endosomal TLR9 (150, 189, 196).

The release of mtDNA into the cytoplasm acts as a danger signal and can occur during infection, cell death, or in response to the exposure to diverse environmental factors (Figs. 4 and 5). In fact, it has been suggested that the release of mtDNA into the cytoplasm during infection strengthens a cell's antiviral response by activating the cGAS pathway and prompting the secretion of antiviral type I IFNs and proinflammatory cytokines, which could prevent the spread of the virus to neighboring cells.

For example, several herpes viruses have acquired the ability to diminish mtDNA release as a defense mechanism by directly degrading mtDNA, which results in the complete loss of mtDNA from infected cells (39). Interestingly, it has been reported that mtDNA depletion does not affect viral replication; however, to date, it has yet to be investigated as to what impact virus-mitigated loss of mtDNA has on the metabolism of a cell affected by infection (49). mtDNA release is associated not only with DNA virus infection, but also with small RNA viruses that lack DNA or DNA intermediates in their genome, such as dengue virus (DENV) (159).
During DENV infection, it has been shown that cytosolic mtDNA interacts directly with cGAS in an oxidized state (oxo-mtDNA), and is further released from the cell, where it can interact with TLR9 and promote antiviral immunity (2, 99, 169). Interestingly, inflammatory stimulation of bystander cells with interleukin (IL)-1β, which is secreted in response to viral infection, also leads to the release of mtDNA into the cytoplasm and cGAS activation (Fig. 5). However, this mtDNA-cGAS interaction occurs without apparent loss of mitochondrial mass and increase in mitochondrial membrane potential (Δψm), and absence of cytochrome C release, which indicates lack of cell death induction (1).
It is currently not known whether there is a difference in the efficiency or kinetics of cGAS or TLR9 stimulation between mtDNA and oxo-mtDNA and whether there are differences between mtDNA and oxo-mtDNA in bystander cell activation, nor how it influences bystander cells' metabolism or inflammatory responses (Fig. 2). mtDNA-TLR9 interaction is known to occur not only after viral infection but it is also implicated in the progression of autoimmune diseases such as rheumatoid arthritis (RA), atherosclerosis, diseases associated with sterile inflammation, steatohepatitis, or acute liver injury (57, 188, 196).
Although identified under many different clinical conditions, the exact molecular mechanism of mtDNA interaction with TLR9 is still unclear due to the controversy that surrounds how to determine the precise degree of methylation of CpG motifs in mtDNA (70, 138). mtDNA is also being discussed to play a role in regulating hyperglycemia during type 2 diabetes, where circulating and cell-free mtDNA was suggested to contribute to AIM2 inflammasome activation (7).
The rate and level of mtDNA release is now being evaluated in many other than autoimmune diseases and it is becoming clear that mtDNA has a longer lifetime in circulation, even in healthy individuals, as compared with DNA originating from the nucleus (112, 179). To date, most studies have focused on mtDNA's extracellular secretion from neutrophils, which extrude fibrous networks known as neutrophil extracellular traps (NETs)—easily detectable by fluorescence microscopy—that contain mtDNA in response to microbial activation (22). In addition, it was more recently reported that not only neutrophils but also macrophages and monocytes can release extracellular traps with nuclear DNA; however, the presence of mtDNA was not investigated (163).
The NET formation can also occur spontaneously in the absence of microbial infection and was associated with a variety of autoimmune diseases, such as atherosclerosis, psoriasis, RA, gout, anti-neutrophil cytoplasmic antibodies-associated vasculitis, or systemic lupus erythematosus (SLE) (101). It has been suggested that NET formation and release induces changes in cellular metabolism, associated with an increase in glycolytic adenosine triphosphate (ATP) production and ROS-regulated dynamics of cytoskeleton (5) (Figs. 5 and 6).
Recent evidence indicates that activated eosinophils can also release extracellular traps (EET), however the exact molecular mechanism of EET formation remains unknown (72, 166). It is also unclear whether EETs are performed in eosinophils and subsequently released, as recent studies suggest that the kinetics of mtDNA release from eosinophils differs from EET formation (60). Whether extracellular EET signaling can affect cellular metabolism of bystander cells is not known.
Dissemination of mtDNA may occur through the direct fusion of mitochondrial membranes with the cell membrane; when this occurs, the levels of oxo-mtDNA have been shown to be high (24, 106). oxo-mtDNA released from neutrophils in SLE patients was reported to interact with TLR9 on plasmacytoid dendritic cells (pDCs), drive type I IFN production, and have increased immunostimulatory potency compared with nonoxidized mtDNA (24, 139). The exact mechanism responsible for the increased interferogenicity of oxo-mtDNA is not known. One study suggests that oxidized DNA, regardless of mitochondrial or nuclear origin, is less susceptible to 3′ repair exonuclease 1 (TREX1)-mediated degradation (59).
Other studies were unable to show differences between oxo-mtDNA and nonoxidized mtDNA regarding susceptibility to degradation by extracellular deoxyribonucleases or differences in the quantitative uptake, intracellular compartmentalization, or stability within pDCs (139). This supports the idea that there may be a unique TLR9-oxo-mtDNA interaction, or a recruitment of an as-of-yet-unidentified co-receptor that facilitates the interaction and determines the specificity (139).
Mitochondrial RNA
During mitochondrial gene expression, both strands of mtDNA are simultaneously transcribed in reverse, resulting in the formation of lengthy polycistronic mitochondrial RNA (mtRNA) transcripts (171) (Fig. 3). These are then processed by endoribonucleases, yielding mature messenger RNAs (mRNAs), ribosomal RNAs (rRNAs), transfer RNAs (tRNAs), and large amounts of complementary, noncoding RNA from the opposite strand of mRNA and rRNA (4). The untranslated light strand is under physiological conditions quickly degraded in mitochondria by polynucleotide phosphorylase (PNPase) and further un-winded in the cytoplasm by the helicase HSuv3 to become a substrate for cytoplasmic RNases (17, 96).
PNPase localization at the inter-mitochondrial membrane space ensures that any mtRNA of pathogen-resembling structure, such as long double stranded (ds) RNA, are detected before escaping into the cytoplasm to become a DAMP. In fact, antisense mtRNA, if not degraded but accumulated in mitochondria, was shown to form long dsRNA helices analogous to viral genomes, which can specifically be recognized by the antiviral RLR-family member melanoma differentiation gene 5 (MDA5) helicase, causing type I IFNs and proinflammatory cytokines secretion (45, 132).
Notably, patients carrying mutations in the PNPase-encoding PNPT1 gene—whose depletion is embryonically lethal—display mitochondrial dsRNA buildup linked with upregulation of type I IFN-stimulated genes and other proinflammatory markers frequently found in SLE patients (45). ds-mtRNA can also be recognized by the protein kinase R (PKR) (88): A PKR ds-mtRNA complex was shown to induce type I IFN secretion, and to influence eukaryotic initiation factor 2 alpha's (eIF2α) phosphorylation, respectively suggesting that indirectly may impact translation and cell signaling (88). Interestingly, mtRNA can be interferogenic not only because of the formation of dsRNA stretches but can also be mistaken for viral RNA because of the specific structure of the 5′-end untranslated regions.
Since mtRNA is not capped at the 5′-end like other cellular mRNAs found in the cytoplasm, it can bind to RLR-helicases such as RIG-I, which then induces type I IFN secretion (10). Finally, similar to bacterial RNA, mtRNA contains less of naturally occurring chemically modified nucleotides, which were shown to affect primary, secondary, and tertiary structures of cytoplasmic RNA and suggested to render them inefficient to interact, for example with TLR 3, 7, and 8 (55, 86).
Emerging studies suggest that RNA oxidation plays a significant role in the development of various diseases (30). Whether mtRNA exists in an oxidized form, and whether it could reach the cytoplasm to become a PRR substrate, remains unknown; to-date, no peer-reviewed reports address a role for oxidized mtRNA. It is also not known whether the escape of mtRNA, or potentially its oxidized form, into the cytoplasm could lead to changes in cellular metabolism, such as those that occur through translational or cell-cycle interference (193), which could be expected because of the interaction with PKR and its ribosomal localization.
Mitochondrial Proteins: TFAM, N-Formyl Peptides, Carbamoyl Phosphate Synthetase-1, Cytochrome C
Mitochondrial transcription factor A
TFAM is an indispensable element of the mtDNA transcription machinery (51). TFAM has nonspecific DNA-binding properties that are similar to family members of the high mobility group (HMG) proteins, which interact with nuclear DNA and are known as nucleus-derived DAMPs (134) (Fig. 2). However, the similarity between TFAM and HMGs is only structural, as the amino acid sequences differ substantially (134). Similar to HMGs, TFAM can be released from a variety of immune and nonimmune cells during cellular stress, alone or in complex with mtDNA, which were shown to stimulate dendritic cells and macrophages, causing a dose-dependent increase in tumor necrosis factor (TNF)-α secretion (81, 84, 157).
Interestingly, exogenous HMGs are stronger immunogenic factors when associated with nuclear DNA (151). Whether the same signal amplification applies as well to TFAM and mtDNA has yet to be investigated in detail. Injection of purified glutathione S-transferase-tagged TFAM, but not the tag alone, was shown to upregulate circulating levels of proinflammatory cytokines, increase neutrophil infiltration of the lungs, and cause organ injury in healthy animals, all of which suggest that extracellular TFAM can trigger inflammation in the absence of mtDNA (31).
Further, TFAM augmented TNF-α secretion in response to cytosine and guanine with phosphodiester backbone (CpGA) DNA, which were then in complex recognized extracellularly by the receptor for advanced glycation endproducts (RAGE) (Fig. 5), an immunoglobulin superfamily transmembrane receptor, and TLR9 (82). Interestingly, TFAM was also shown to be recognized by brain microglia and proposed in the human brain to have intercellular signaling function, which causes pro-inflammatory and cytotoxic responses (104).
Recent data suggest that even though TFAM transcript levels increase in the nucleus and cytoplasm during sepsis, the levels of intra-mitochondrial TFAM protein and its level of interaction with mitochondrial transcription factor 2B (a key for mtDNA transcription initiation) do not (145). The exact mechanism underlying spatial misplacement of TFAM during sepsis is unknown. One possible explanation is that TFAM may either be post-translationally modified, and thus unable to enter mitochondria, or it may nonspecifically interact with other proteins or nuclear DNA, and subsequently be extruded from the cell as a DAMP.
In fact, it has been shown that ubiquitination of TFAM blocks its ability to enter mitochondria, resulting in dysfunction of retinal cells, and a subsequent loss of sight (156). In addition to ubiquitination, TFAM can also be acetylated and phosphorylated (89). Phosphorylation of human TFAM in mitochondria is vital for the maintenance and expression of the mitochondrial genome, and it has been also shown to occur within other HMGs by cAMP-dependent protein kinase (PKA) and result in the rapid and selective degradation of TFAM (107).
Interestingly, the PKA-dependent phosphorylation of Saccharomyces cerevisiae's TFAM analogue appears to be tied in to metabolic processes, since TFAM's affinity to mtDNA under conditions of glucose repression was shown to be decreased (33). Moreover, TFAM can be phosphorylated by extracellular signal-regulated protein kinases (ERKs), which translocate to the mitochondria (44, 185). Although this phosphorylation reduces TFAM binding to transcription initiation sites, it does not decrease binding to nonspecific-mtDNA sequences (44, 185). Although phosphorylation and other modifications have been well described, it has not yet been established whether human TFAM can be modified by ROS at the two cysteines that are encoded in its sequence, either in mitochondria, in the cytoplasm, or in extracellular space (120).
In contrast, ROS modification was observed in HMG homologues, where three conserved cysteines were found in an oxidized state on translocation from the nucleus into the cytoplasm or extracellular space (109). One set of cysteines in the N-terminus was shown to form intramolecular bridges, thus altering the structure of the protein and causing at least a 10-fold decrease in its affinity for DNA. On the other hand, the oxidation of cysteine positioned in the middle of the protein was shown to be responsible for its translocation (109, 136). Nuclear HMGs have been also shown to become immune-silent after caspase-dependent oxidation (87).
Based on TFAM's localization in the middle of mitochondria, it is unlikely that oxidation could be selective enough to regulate TFAM, like it was observed for HMGs, to promote the immune function associated with tissue regeneration, chemotaxis, and inflammation (174). Therefore, it is not surprising that TFAM levels are responsive to more selective redox modifiers such as hydrogen sulfide, which was shown to not directly modify TFAM per se, but to sulfhydrate interferon regulatory factor 1 (IRF-1), which inhibits the methylation of the TFAMs gene promoter and represses its expression (103).
Somewhat surprisingly, TFAM expression levels are not solely contributors to the development of danger in the cell; increased expression of TFAM can also facilitate cardioprotection, prevent skeletal muscle atrophy, or positively impact mtDNA copy number, as seen in a rat model of diabetic neuropathy (29, 97). In conclusion, TFAM can have a pleiotropic effect on cellular function, and simply associating its increased qualitative expression with cellular stress in a given organism should be approached with caution. This is especially true, considering that the impact of TFAM modification associated with oxidative stress and danger signals secondary to posttranslational phosphorylation or other modification that originates from mitochondria had not yet been carefully addressed in any autoimmune disease.
N-formyl peptides
N-formyl peptides (NFPs) are derivatives of mitochondrial and/or bacterial translation (158). Mitochondria, similar to bacteria, start protein synthesis with an N-formylmethionine residue; after the release of mitochondrial proteins into extracellular space, this residue can function as a chemoattractant that is sensed by high-affinity formyl peptide receptors (FPRs) (26, 115). Interestingly, these FPRs are not only found to be expressed on immune cells such as neutrophils, monocytes, and dendritic cells, but as well on cells of the nervous system, endothelial cells, and hepatocytes (94). Mitochondrial NFPs attract and activate various immune cells that result, for example, in the secretion of proinflammatory signals such as matrix metalloproteinase-8 (MMP-8) and IL-8, but also attract thrombocytes, thereby potentially triggering unwanted blood coagulation (43, 146).
The oxidation of NFPs is so far not widely studied; however, early studies have shown that human neutrophils can induce oxidation of the synthetic chemotactic peptide N-formyl methionyl-leucyl-phenylalanine by the myeloperoxidase system (180) (Fig. 4). Oxidation was proposed to diminish NFPs' chemotactic activity, which may be one of the mechanisms by which neutrophils modulate the inflammatory process. The ratio of oxidized to nonoxidized mitochondrial NFPs released from the cell under different stress conditions has not yet been investigated, but it would be interesting to determine whether possible differences in NFP oxidation under different types of cell stress and death exist. Overall, this implies that oxidation and thereby inactivation of humoral mediators plays a significant pathophysiologic role as a negative feedback inflammatory control mechanism (34).
Carbamoyl phosphate synthetase-1
Carbamoyl phosphate synthetase-1 (CPS-1) is a 165-kDa protein that is translated as a proenzyme in the cytoplasm and, after transfer into the mitochondrial matrix, is cleaved to yield the mature protein, which is responsible for generating the precursor of citrulline (80a) (Figs. 3 and 4). The expression of CPS-1 is mostly restricted to the liver, where it constitutes up to 15%–20% of total hepatic mitochondrial protein; it is characterized by a long half-life of ∼8 days (35). More recent work described CPS-1 expression also in intestinal enterocytes and intestinal mucosa, where it appears to contribute to pyrimidine biosynthesis and citrulline/arginine production (61).
A possible role for CPS-1 as an extracellular DAMP molecule triggering innate immune responses has only recently been considered. Early findings showed that CPS-1 can be released by the liver into circulation after lipopolysaccharide (LPS) stimulation, although pro- or anti-inflammatory consequences of this process were not evaluated (168). The release of CPS-1 after LPS stimulation shows, however, interesting kinetics, as compared with the release of acute inflammation cytokines such as TNF-α (168). Unlike TNF-α, which can be detected several minutes after LPS stimulation in circulation and can return to basal level within a few hours, extracellular CPS-1 levels have been shown to increase with a delay of several hours and to remain sustained for extended periods of time (168).
The release of CPS-1 into circulation has also been observed after induction of cecal ligation and puncture sepsis, where it is proposed to be associated with increased mitochondria depletion due to increased lysosomal clearance rather than cell death (3). Interestingly, increased circulating CPS-1 has also been associated with mitochondrial morphology abnormalities in the liver (42). A later study showed that the liver also releases CPS-1 into circulation during Fas-ligand-induced apoptosis; CPS-1 has under these conditions a rather shorter half-life of merely 2 h, suggesting that its lifetime in circulation may directly be dependent on necrotic, apoptotic, or cell-death-independent release from mitochondria and the cell (187).
However, besides the mere observation of the highly variable stability of CPS-1, the underlying molecular mechanism is not known. Of note, recent in vivo studies suggest a CPS-1 release during acute liver injury into the blood, followed by a rapid uptake by monocytes, which become as a result anti-inflammatory, and homing to the liver (135). CPS-1's function in monocytes may be independent of its enzymatic ability to produce carbamoyl phosphate (135). This would suggest that CPS-1, instead of acting as a DAMP, would have protective and anti-inflammatory properties, although it is not clear what precise mechanism would underlie this monocyte/macrophage activation. At present, no information is available about CPS-1 stress-related modifications or what impact released CPS-1 may have on cell metabolism, especially in monocytes.
Cytochrome C and cardiolipin
Cytochrome C and cardiolipin release into circulation is fairly well documented in patients who have SLE, liver injury, suffered a myocardial infarction, or are undergoing chemotherapy (11, 78, 110, 117, 144). Cytochrome C's role as an intra- and extracellular danger signal is not as well understood as its canonical function in the ETC at mitochondria, where it interchanges from a reduced to an oxidized form by transferring electrons from complex III to complex IV (64) (Fig. 3). Besides its functional role in mitochondria, cytochrome C is structurally essential for the proper association of the entire respiratory supercomplexes (140). In addition, cytochrome C is also responsible for the import of cysteine-rich proteins into the mitochondrial intermembrane space via sulfhydryl oxidase-mediated redox reactions (15).
Cytochrome C escapes from mitochondria on detection of proapoptotic stimuli such as DNA damage, metabolic stress, accumulation of unfolded proteins, chemical triggers such as H2O2, etoposide, actinomycin D, or exposure to UV light (62). The mechanisms of cytochrome C release from mitochondria are still controversial, as it seems it can occur either via nonspecific pores in the outer mitochondrial membrane (OMM), via “apoptotic pores” formed by the B cell lymphoma protein-2 (Bcl-2) family of proteins, or via mitochondrial porins, which are formed by the voltage-dependent anion channel (VDAC) protein (62).
Altogether, this suggests that cytochrome C escape from mitochondria is cell-type specific and depends not on one but diverse apoptotic triggers. Several studies now suggest that cytochrome C release from mitochondria is a gradual process that may also take place under nonapoptotic conditions, without mitochondrial membrane permeabilization (47).
Interestingly, under homeostatic conditions and at maximal mitochondrial respiration, not all cytochrome C is bound to the inner mitochondrial membrane (IMM), but at least 20% can be also found firmly attached via electrostatic and hydrophobic interactions to the cytosolic interface (58). Some studies suggest that after release into the cytoplasm, cytochrome C migrates into the nucleus, where it interacts with a series of chromatin-binding factors, thereby changing its function from a “killer” to a “healer” by regulating the dynamics of damaged chromatin (64).
The best described nonapoptotic cytoplasmic receptor for cytochrome C is inositol-1,4,5-trisphosphate receptor (IP3R), a member of ion channels, which facilitate the release of calcium from the ER into the cytosol by the binding of IP3 (14). Cytochrome C was shown to bind to IP3R, before apoptosis induction and independently of caspase activation, and to promote calcium release, which was suggested in a feedback loop to promote mitochondrial depolarization and augment cytochrome C release (131). Interestingly, the role of cytochrome C binding to IP3R and increased calcium release from the ER was not studied in the context of NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) activation, which is well documented to occur after calcium influx in the cytoplasm (123).
As it is not known which form of cytochrome C binds to IP3R, whether it being oxidized or nonoxidized, post-translationally modified, or in a complex with another compound, differences in cytochrome C's redox state may offer an explanation that yet has to be answered. Cytochrome C in its reduced form in neurons is not able to interact with proapoptotic proteins to form the apoptosome (183). Early studies suggested that cytochrome C, when exogenously added to cytoplasmic extracts, is rapidly reduced—within less than 5 min—and that only oxidized species can activate caspases (67).
In line with this observation, later studies that were performed in a cell-free system suggested that the presence of cytochrome C reductases, such as glutathione, L-cysteine, ascorbate, or N-acetylcysteine, prevents the formation of the apoptosome (133).
In contrast, more recent studies suggest that the presence of the cytochrome C reductant tetramethyl-p-phenylenediamine (TMPD) does not entirely eliminate cytochrome C activity in the induction of apoptosome and activation of caspase 3, as it was studied in cell-free extracts isolated from either murine macrophage J774 cells or human HeLa cells (18). Also, recent studies using an in vitro purified system and cytochrome C point mutation variants, which can accommodate different oxidation states, suggest that the redox state of cytochrome C is irrelevant for the induction of the apoptosome complex (113).
Cytochrome C's role is, therefore, complex and should not be examined in an isolated state. Physiologically, cytochrome C is noncovalently attached to the mitochondrial membrane via interaction with acidic phospholipids primarily found in the IMM, such as cardiolipin (75). Under physiological conditions, cardiolipin is equally distributed between the inner and outer leaflet of the IMM and not present at the OMM (83). Cytochrome C interaction with cardiolipin leads to peroxidation and subsequent relocation of cardiolipin to the OMM, which is associated with the OMM permeabilization and release of pro-apoptotic factors (83, 130).
Although the role of oxidized cardiolipin as a DAMP signal has not been widely investigated, it is known that anti-cardiolipin antibodies are present in SLE patients and individuals with antiphospholipid syndrome (141). Anticardiolipin antibodies after the interaction with their target antigen beta-2-glycoprotein-I, an anionic phospholipid-binding serum protein, can bind to TLRs on monocytes, macrophages, and platelets, which subsequently leads to their activation and pro-inflammatory responses (48, 122, 155). Another example of the pathologic role of cardiolipin was shown in lung pathology associated with pneumonias, where increased extracellular cardiolipin was associated with lung malfunction (147).
In this study, cardiolipin was shown to interact with the P-type ATPase transmembrane lipid pump (ATP8b1) and internalized into lung epithelium; however, the fate of the cardiolipin after uptake or proinflammatory signaling was not investigated (147). In vitro, experiments suggest that lack of cardiolipin impairs NLRP3 inflammasome activation and cannot be rescued by other phospholipids, as it was tested in cell-free extracts by measuring caspase-1 activity (77). A contradictory, beneficial function was identified for extracellular cardiolipin, when bound to mitochondria and possibly cytochrome C.
Extracellular mitochondria coated with both oxidized and nonoxidized forms of cardiolipin have been shown to be efficiently engulfed by professional phagocytes through the receptor CD36, causing an attenuation of innate immunity responses (9). Further, extracellular cytochrome C has been shown to interact with serum leucine-rich α-2-glycoprotein-1 (LPG), and this interaction was shown, instead of being pathologic, to protect lymphocytes from cell death (36).
The role of cardiolipin in the assumption of distinct metabolic programs and mitochondrial function in immune cells was only just recently investigated. Cardiolipin was shown to be necessarily present in the cell before stimulation, which leads to immune memory development (41). In conclusion, DAMP properties of cytochrome C and its binding cofactor cardiolipin suggest that a cytoplasmic localization favors the development of a proinflammatory phenotype (Figs. 3 and 4). In contrast, cytochrome C and cardiolipin in the extracellular space were thus far shown to be recognized by sensors that trigger an anti-inflammatory response. Future research should also focus on potential differences of oxidized versus nonoxidized forms of cytochrome C and cardiolipin in innate immune cells to precisely dissect their pro-apoptotic and/or pro-inflammatory functions.
Mitochondrial antiviral signaling protein
Mitochondrial antiviral signaling (MAVS) protein is located downstream of PRRs of the RIG-I-like helicases family, which are activated by either viral RNA recognition during infection, or self-RNA interaction that was, for example, released by mitochondria (161) (Figs. 3 and 4). MAVS has been demonstrated by our group to be activated by oxidative stress alone, independent of viral infection and upstream RLR interaction (23) (Fig. 6). In fact, chemically generated oxidative stress in the presence or absence of pan- or mitochondria-specific ROS scavengers demonstrated that specifically mitochondrial ROS (mtROS) lead to the formation of a disulfide bond between a highly conserved cysteine at position 79 (C79) in neighboring MAVS molecules.
This disulfide-dependent dimer formation triggers further MAVS oligomerization, functional formation of the MAVS signaling complex with its downstream proteins, and secretion of proinflammatory cytokines such as type I IFN and IL-6 (23). In contrast, inhibition of mtROS by the mitochondria-targeted antioxidant MitoQ inhibited MAVS aggregation and type I IFN secretion. Interestingly, these stable MAVS oligomers could be detected in immune cells and in the circulation of SLE patients (23, 162). The observation that overall disease intensity in SLE patients carrying an MAVS-mutation of cysteine 79 to phenylalanine (C79F) is significantly reduced substantiates the notion that mtROS-dependent MAVS oligomerization drives type I IFN production (23, 142).
Of note, this C79F variation is found naturally in 30% of the healthy sub-Saharan population; at this geographical location, the C79F variant appears to be a natural adaptation that offers some protection from ROS-driven MAVS-oligomerization due to continuous exposure to UV-irradiation (142). Taken together, these findings suggest that mtROS modified MAVS in SLE patients serves as a DAMP molecule and contributes to the type I IFN signature characteristic of the disease (23). Of particular interest, mtROS-dependent MAVS oligomers have been shown to not only affect the proinflammatory state of the cell, but also to cause mitochondrial hyperpolarization, reduced spare respiratory capacity, and decreased ATP production (23).
Mitochondria-associated membrane PRRs: cGAS and stimulator of interferon genes
Another example of direct ROS modulation of a PRR has been observed for the cytosolic DNA sensor cGAS and its interacting partner stimulator of interferon genes (STING) (176). Exposure of murine bone marrow-derived macrophages (BMDMs) to vitamin K3 (menadione) can lead to the oxidation of STING at cysteine 147 (176). Contrary to MAVS, oxidation of STING did not lead to accelerated signaling and secretion of proinflammatory cytokines; in fact, there was an opposite effect, as STING signaling was suppressed. STING is not the only example for a PRR to be directly and/or specifically modified by mtROS, especially during microbial infections. For example, during infection with Listeria monocytogenes, specifically mtROS, but not cytoplasmic ROS, was shown to induce disulfide bridges in the NF-κB essential modulator (NEMO) (69).
It was further demonstrated that mtROS-dependent dimerization of NEMO led to the secretion of proinflammatory cytokines such as IL-1β, TNF α, and IL-6, and induced macrophage polarization (69). mtROS was also implicated in the activation of the inflammasome, a supramolecular signaling complex that converts inactive pro-IL-1β into its active pro-inflammatory form (IL-1β) (199). The exact role of mtROS versus cytoplasmic ROS in activation of inflammasome is still quite controversial. Interestingly, NRLP3, one of the key proteins forming a DAMP- but not PAMP-specific inflammasome, has been shown to contain a highly conserved and redox-sensitive disulfide bond (8).
It would be valuable to evaluate the role of this disulfide bridge formation in the presence of MitoPQ, a mitochondria-specific redox cycler, which increases superoxide production within the mitochondrial matrix (149). Additional research will be needed to determine how this spatiotemporal action of mtROS occurs and whether mtROS plays a more direct role in the activation of nonmitochondrial PRR rather than via the ancillary proteins in the downstream complex.
Metabolites: ATP, succinate, mtROS
Adenosine triphosphate
ATP is a vital mitochondrial metabolite that fulfills multiple roles as an energy molecule, a phosphate donor, and a signaling molecule inside the cells. Intracellularly generated ATP is under oxidizing conditions largely produced in mitochondria, whereas cytoplasmic glycolysis is the most important extra-mitochondrial source of ATP. ATP that is present in the extracellular space is now recognized as a compound that not only can change different cellular functions associated with cellular metabolism but also can play a key role in promoting an innate immunity response during bacterial infections, in addition to becoming a DAMP signal by itself (65).
The release of ATP is now known not to be a random process that is associated with gross leakage of other cellular content after cell rupture, but rather being specifically controlled by pannexin 1 plasma membrane channels and recognized by purinergic P2 receptors as a “find me” signal on phagocytes (32, 50).
Despite ATP's essential part in regulating a proper immune response during microbial infection or cancer, extracellular ATP has been shown be a sensor in its own right that induces the release of pro-allergic mediators and contributes to chronic inflammation in lungs under certain conditions (73, 93). Extracellular ATP can elicit NLRP3 inflammasome activation in the presence or absence of secondary PAMPs or DAMPs (63). Extracellular ATP that interacts with P2 receptor on DCs promotes T cell priming, possibly setting a chronic inflammatory state, as it was, for example, described in inflammatory bowel disease (98).
Whether stress-released ATP is oxidized is not known. Very early studies indicate that oxidized ATP is an inhibitor of purinergic macrophage receptors and rather attenuates proinflammatory signaling (13, 124). Follow-up studies on the enzymes responsible for nucleotide pool recycling and DNA repair uncovered further that the influence of free nucleotides' oxidation is surprisingly larger than that of direct oxidation of DNA (128). Interestingly, a so-called sanitizing enzyme for the oxidized nucleotide pool, mutator T homolog 1 (MTH1), also known as nudix hydrolase 1 (NUDT1), was shown to efficiently hydrolyze not only free not-incorporated-in-DNA deoxy-triphosphate purines, but also purine ribonucleoside triphosphates such as 2-OH-ATP, 8-oxo-ATP, and 8-oxo-GTP (129, 154).
This suggests that oxidized ATP could exist in the pool of free nucleotides released from the cell and disturb signal transduction or metabolic function, where ATP is an essential mediator (Fig. 4). Twelve modified variants of endogenous NTPs were recently discovered in various eukaryotic cells, and NUDT1 was shown to colocalize to mitochondria (79, 85). Whether oxidized ATP would pose as a relatively stronger or weaker DAMP is presently unknown; interestingly, a polymorphism of the NUDT1 gene that results in a less stable variant of the protein may be associated with the development of type 1 diabetes in the female population (119).
Succinate
Succinate appears in the TCA cycle as a metabolic intermediate. It is oxidized by SDH into fumarate in complex II of the ETC in mitochondria. Recent studies suggests that succinate, besides being a metabolite, can act as a DAMP in the cytoplasm and in the extracellular space (118, 127). The multifaceted function for succinate as a DAMP, aside from its classical role as mitochondrial “fuel,” can only be understood when the role of succinate in mtROS homeostasis under different conditions is considered. Under saturating levels of succinate, when the function of either complex I or III is not pharmacologically inhibited, the flow of electrons is slow, and all adenosine diphosphate (ADP) is converted to ATP (so-called state 4 respiration).
In this case, succinate can reverse electron transfer (RET) through complex I when Δψm increases (90). RET then leads to the generation of mtROS at a very high rate, increased Δψm, and a slow flow of electrons (28, 92). Under RET conditions, succinate in the cytoplasm was demonstrated to inhibit the activity of prolyl hydroxylase domain (PHD) enzyme by direct interaction. In turn, the succinate-mediated inhibition of PHD leads to stabilization of the hypoxia-inducible factor 1α (HIF-1α), which further interacts with HIF response elements (HREs) of proinflammatory genes such as IL-1β and the HREs of genes responsible for the induction of glycolysis (160).
Of note, succinate-dependent HIF-1α stabilization can occur both under hypoxia, when oxygen levels are low and hence the activity of SDH dampened, and during normoxia, when SDH is unable to convert succinate to fumarate, for example due to genetic aberrations (21, 175) (Fig. 4). Stabilization of HIF-1α can also occur independently from succinate's interaction with PHD, as HIF-1α levels were shown to be increased by direct action of mtROS (66, 143). It has been suggested that mtROS-mediated stabilization of HIF-1α may be the dominant mechanism in most cells, whereas succinate-mediated activation of HIF-1α occurs specifically in inflammatory macrophages (118, 127).
It is noteworthy that, in addition to a reduction in the function of SDH or RET, there are several other ways in which accumulation of succinate can occur in a cell (118). For example, succinate levels can rise as a result of increased glutamine metabolism through anaplerosis of α-ketoglutarate through the TCA cycle and γ-aminobutyric acid (GABA) and its transporters, which, in turn, generates succinate by way of the so called “GABA-shunt” or “glyoxylate shunt,” which leads to the conversion of isocitrate to succinate by isocitrate lyase (175, 186).
Although the impact of succinate's accumulation under different conditions and its function as a DAMP are still being precisely determined, the general consensus is that the pathological outcome of succinate buildup will depend on the accumulation mechanism, the level of mtROS released into the cytoplasm, and the cell's oxygen supply. A consequence of cytoplasmic succinate accumulation, aside from the possibility that it will engage with mitochondria, mtROS, SDH, or other processes, is its release into the extracellular space. Once there, it interacts with G protein-coupled receptor 91 (GPR91), likewise known as succinate receptor 1 (SUCNR1) (68).
GPR91 is strongly expressed on dendritic cells and was shown to play an important role in adipose, hepatic, renal, splenic, and retinal tissues, and associated tissue-specific diseases (105, 152, 178). It has also been suggested that extracellular succinate activates adaptive immunity in the gut via Tuft cells, which express high levels of GPR91 (102). In SLE patients, succinate has been shown to support the expansion and metabolism of a specific subtype of T-follicular helper cells (Tfh cells), which provide B cell help (25).
Interestingly, similar T cells can be also generated from naive CD4+ T cells that have been exposed to pDC activated by oxo-mt DNA (24). Although here the discussion posits succinate as a DAMP, it is also important to mention that itaconate, another mitochondrial metabolite that acts as an endogenous SDH inhibitor, has recently been shown to regulate succinate levels and function and mitochondrial respiration, as well as reduce inflammatory cytokine production (40, 100). In SLE patients, it has been observed that 4-octyl itaconate has inhibited proinflammatory cytokine production in peripheral blood mononuclear cells (173). Lastly, it has been proposed that succinate and its dehydrogenase may not only promote mtROS production but also control mitochondrial lipid peroxidation, a DAMP signal in its own right as discussed for cytochrome C—cardiolipin interaction (16).
Mitochondrial ROS
mtROS has the ability to directly activate PRRs, thus bypassing any interaction with other DAMPs to induce a downstream signaling cascade. Therefore, mtROS per se qualifies as a DAMP. Under normal physiological conditions, mitochondria contribute at least one-third of total cellular H2O2 production (52) through at least 11 distinct sites within the mitochondrial ETC that generate mtROS in the form of H2O2 or superoxide anions O2
The sites with the utmost capacity to generate ROS are associated with respiratory chain's complex I, which releases mtROS into the matrix, and complex III, which is able to disseminate superoxide toward either side of the inner mitochondrial membrane (19). Theoretically, superoxide in the intermembrane space has easier access to cytosol than matrix-localized O2
Although less reactive than the superoxide anion, H2O2 can oxidize any given PRRs thiol group of cysteines to form sulfenic acid, thereby acting as a quasi-cytoplasmic PRR ligand. This cysteine-oxidation can then lead to glutathionylation, the formation of intra- or intercellular crosslinking disulfide bonds with an adjacent thiol, or a reaction with an amide group to form sulfenyl amide (54).
In summary, in this review, we discussed how mitochondria-derived DAMPs, which undergo oxidation, start forming an independent, strongly interferogenic family of ligands for PRR, which are associated with chronic changes in cell metabolism and are associated with the development of autoimmunity (Fig. 5).
Author's Contributions
All the authors contributed equally to this review.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
The authors were supported by grants NIH/NIAMS R01AR073282, NIH/NIAID R21AI128510, and NIH/NIAMS R21AR069830 (I.A.B.-K.) and the Harold S. Geneen Charitable Trust Awards Program for Coronary Heart Disease Research (A.K.).