
Abstract
Significance:
Aging is a natural process that affects most living organisms, resulting in increased mortality. As the world population ages, the prevalence of age-associated diseases, and their associated health care costs, has increased sharply. A better understanding of the molecular mechanisms that lead to cellular dysfunction may provide important targets for interventions to prevent or treat these diseases.
Recent Advances:
Although the mitochondrial theory of aging had been proposed more than 40 years ago, recent new data have given stronger support for a central role for mitochondrial dysfunction in several pathways that are deregulated during normal aging and age-associated disease.
Critical Issues:
Several of the experimental evidence linking mitochondrial alterations to age-associated loss of function are correlative and mechanistic insights are still elusive. Here, we review how mitochondrial dysfunction may be involved in many of the known hallmarks of aging, and how these pathways interact in an intricate net of molecular relationships.
Future Directions:
As it has become clear that mitochondrial dysfunction plays causative roles in normal aging and age-associated diseases, it is necessary to better define the molecular interactions and the temporal and causal relationship between these changes and the relevant phenotypes seen during the aging process. Antioxid. Redox Signal. 36, 824–843.
Aging and the Main Aging Theories
Aging is the natural process of gradual loss of function in biological systems that affects living organisms, resulting in an increase in mortality rate with time. This process, which affects most species, with a few notable exceptions (57, 211), has been subject to scientists and philosophers for millennia, with the first speculations on the nature of the aging process dating as far back as Aristotle (241). In fact, the first work of literature ever written, The Epic of Gilgamesh, narrates Gilgamesh's quest to find eternal youth (68).
The field of biology of aging has been rapidly developing, with significant discoveries made in the past few decades. One of the earliest aging theories was Denham Harman's Free Radical Theory of Aging (74), which suggested that oxidative damage by reactive oxygen species, mainly produced in mitochondria, causes age-associated dysfunction. Although the relationship between aging and oxidants turned out to be not so straightforward, with many researchers adapting or abandoning the theory [reviewed in Liu et al. (126)], mitochondria remained a focal point in aging research, with mitochondrial dysfunction being considered one of the hallmarks of aging (130), as shown in Figure 1, which presents a schematic view of the proposed mechanistic links between mitochondria and the hallmarks.
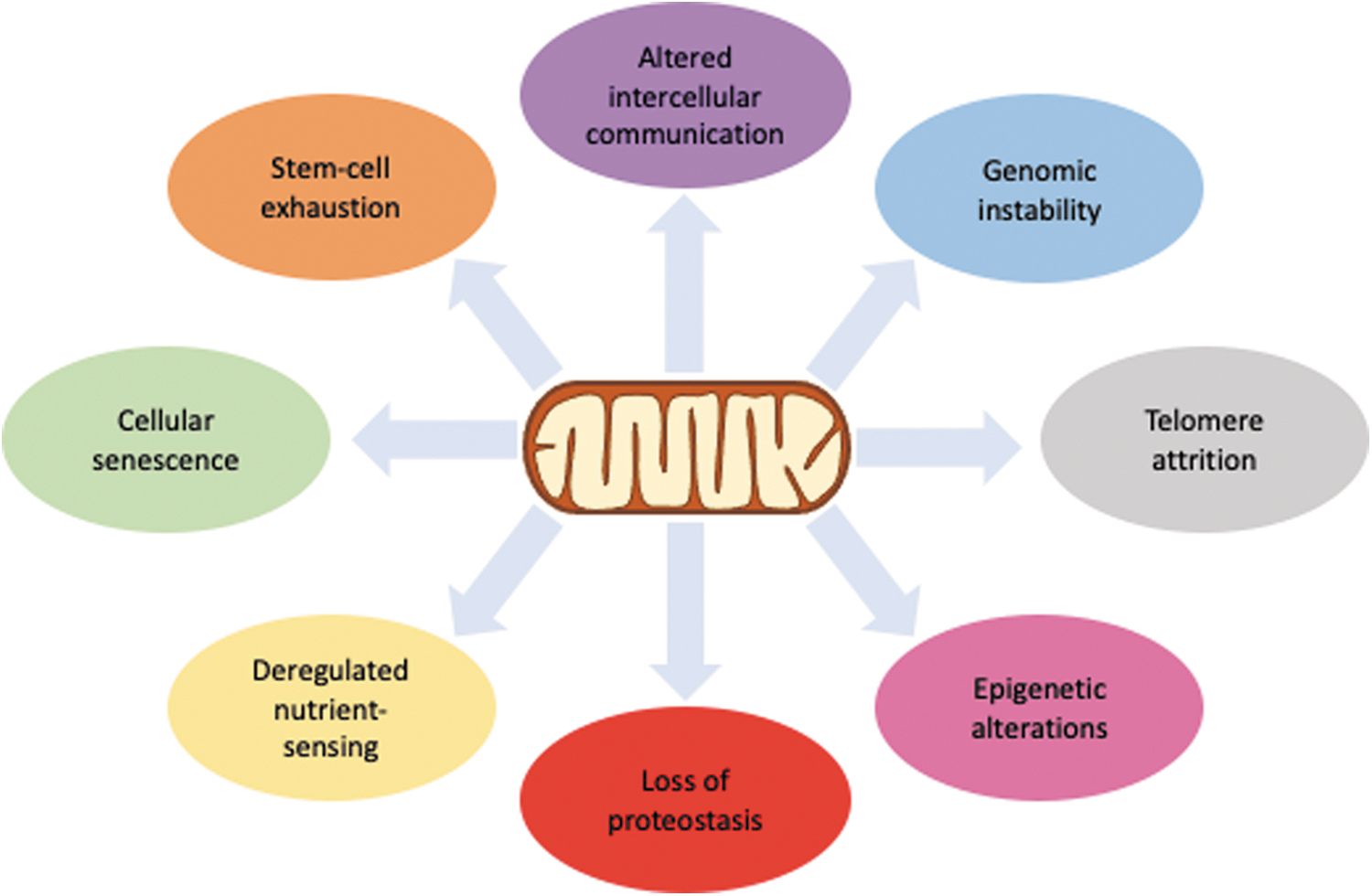
Causal relationships among the different molecular hallmarks of aging are still unclear. Although it is still premature to say that mitochondria have a more prominent role in relation to other hallmarks, it is certain that many of the alterations observed during aging parallel changes in mitochondrial function. This review will focus on the many roles played by mitochondria in this process. However, we consider it useful to briefly review other molecular theories of aging, as well as some of the evolutionary aspects, to provide a better context of where mitochondria lie in this puzzle.
Mitochondria have always had a central place in aging research. Even when not explicitly mentioned, most theories of aging have a mitochondrial component. The paradigm in modern evolutionary aging theory includes the antagonistic pleiotropy theory, which posits that aging is caused by the effects of several alleles that have late life detrimental, but early life beneficial effects (183).
These genes with antagonistic pleiotropy are not selected out of the gene pool as natural selection is weaker with advancing age because, as time passes, the probability of survival, and, therefore, reproduction, decreases due to diseases or exogenous factors, making effects that manifest early in life more relevant to the organismal fitness. Thus, due to early life beneficial effects, their net effect remains positive, such that natural selection favors them. Different rates of fecundity with age also create a similar effect on the force of natural selection with time.
In this theoretical framework, aging has no evolutionary purpose, but it is only a side effect of the declining force of natural selection not being able to clear genes with detrimental late life effects when the same genes are beneficial in early life. However, some dispute this idea and posit that aging does have an evolutionary role, and an evolutionarily conserved aging program could exist (69, 128).
Another theory that composes the modern paradigm is the disposable soma theory (104), based on August Weismann's early work, which postulates that due to a limitation in the amount of energy, organisms are not capable of having optimal investments in both maintenance and reproduction, making that the organism must choose where to allocate its limited resources. This leads to suboptimal investment in repair mechanisms, eventually resulting in accumulation of damage with time and aging.
A major challenge to this theory is the observation that single-gene interventions can increase lifespan without affecting reproduction (43, 138, 200). On the other hand, it is interesting how both these paradigmatic theories provide a framework to understand some of the phenomena observed in mitochondria during aging. As the main ATP producers in cells, variations in their capacity to produce energy would impact repair mechanisms in a way aligned with the disposable soma theory.
On the other hand, many cellular processes that are regulated by mitochondria, such as oxidants production and cellular senescence, have early life beneficial effects and late life detrimental effects, when their regulation becomes defective.
The Free Radical Theory of aging has long been associated with mitochondria, both as a main source of oxidants in cells and as a primary target to their damaging effects, with all mitochondrial biomolecules, including mitochondrial DNA (mtDNA), accumulating oxidation damage. The idea that DNA alterations are involved in aging was first proposed by Szilard (216) and Failla (52), from the simple idea that damage to the main repository of information could be the cause of aging.
Today, a major role for DNA damage in aging continues to be one of the leading theories in the field, with genomic instability being considered one of the hallmarks of aging (Fig. 1) (130). In fact, DNA damage accumulates with time in several models, including mammals such as mice (140) and humans (49). A meta-analysis of 36 studies on the theme has revealed a strong positive correlation between DNA damage and aging (206).
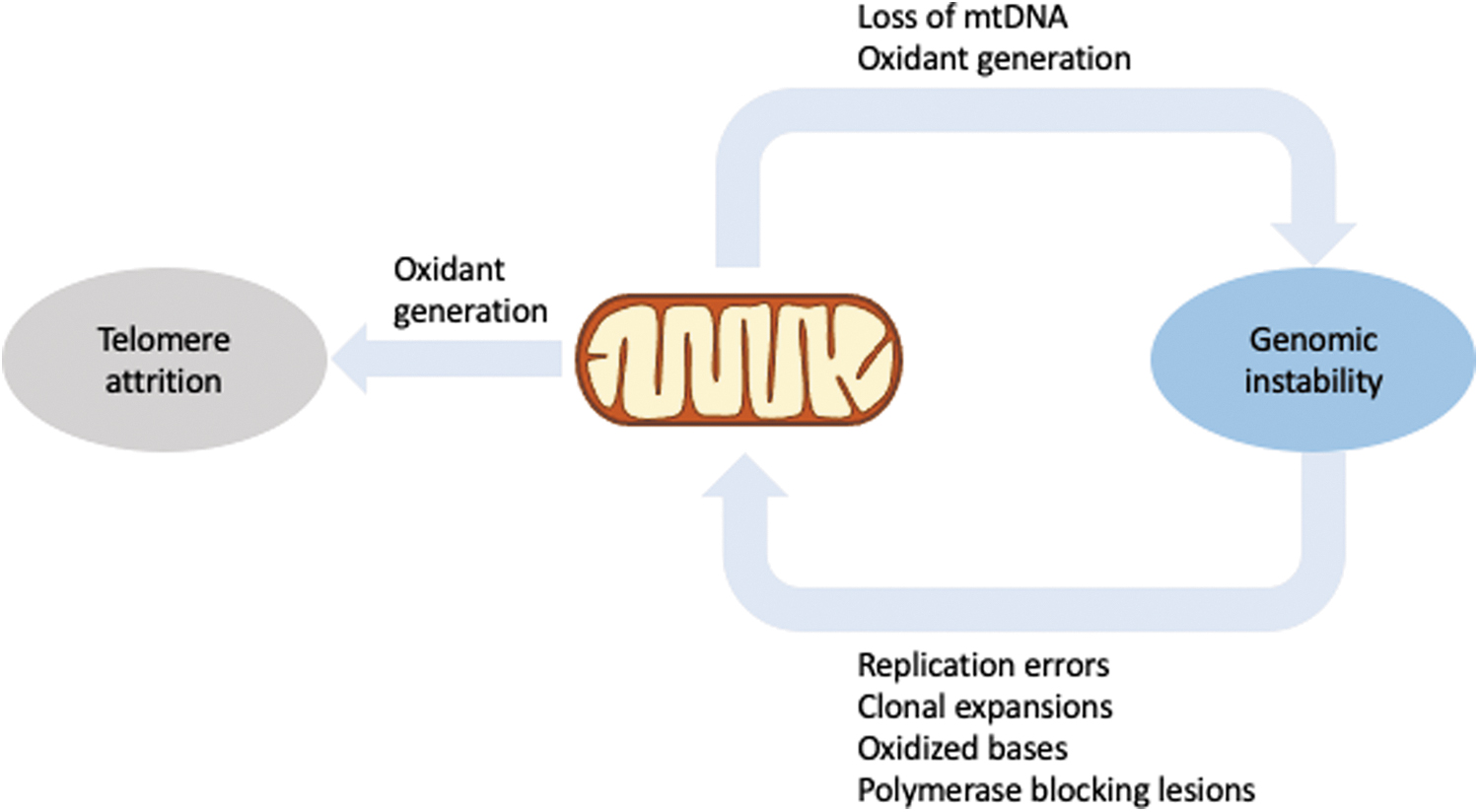
Moreover, two interventions that increase longevity in mice, calorie restriction and suppression of the somatotrophic axis, cause a reduction in mutation frequency (64). In addition, age-associated changes in DNA repair pathways have also been observed by several groups [reviewed in Rao (180)]. The fact that the mtDNA accumulates more damage with age (85) and mtDNA repair is impaired with age as well [reviewed in Muftuoglu et al. (155)] directly links mitochondrial dysfunction and genomic instability.
Telomere attrition is another well-established hallmark of aging (Fig. 1). Telomeres are DNA–protein macromolecular complexes that are found at the ends of chromosomes and function mostly to protect chromosomes from nucleolytic degradation [reviewed in Chakravarti et al. (28)]. Due to the intrinsic DNA end-replication problem, telomeres are shortened with cell division, and critically shortened telomeres induce cell senescence.
Cellular senescence is a state of growth arrest and altered pattern of gene expression and secretion of molecular factor, such as cytokines. Senescent cells accumulate with age, and their clearance in model organisms has been shown to extend lifespan (243). Moreover, due to their high guanine content and some sequence effects, telomeres are also particularly prone to oxidative DNA damage, which has been implicated in telomere attrition (10), also linking this hallmark with mitochondrial dysfunction.
Several other molecular alterations have been observed in aging, including: (i) accumulation of misfolded and damaged proteins (189); (ii) low-grade, sterile, chronic inflammation, often called “inflammaging” [reviewed in Franceschi et al. (62)]; (iii) epigenetic alterations (16); (iv) deregulated nutrient-sensing (93); and (v) altered intercellular communication (Fig. 1) (204). Although these do not refer to specific types or targets of damage, these events are possibly a consequence of the types of damage related to the hallmarks described earlier. Nonetheless, mitochondrial dysfunction can be mechanistically linked to all the alterations observed in aging, as it will be further developed in this article.
Mitochondrial Function and Redox Balance
Mitochondria are present in variable quantities in most aerobic cells and despite being initially considered to be static and isolated organelles only responsible for oxidative phosphorylation (101), it is now clear that mitochondria are the site for many essential biosynthetic pathways and play several other important cellular functions, such that mitochondrial integrity is essential even in conditions where cells obtain most of their energy through glycolysis.
The Chemiosmotic Theory, developed by Mitchell (147, 148), provided a framework for understanding mitochondrial function. According to this theory, the thermodynamically favorable downhill flow of electrons through the respiratory chain is conserved in the form of an electrochemical proton potential (ΔP) via proton-pumping outward the inner membrane, at the level of respiratory complexes I, III, and IV. This generates a negative inside electrical potential (ΔΨ) and consequently an alkaline inside ΔpH (ΔP = ΔpH + ΔΨ).
The mitochondrial ATP synthase, an integral inner membrane protein, operates as a coupling device allowing for the extruded H+ to return through the enzyme's proton channel (Fo), thus providing a mechanism for ΔP transduction into chemical energy in the form of ATP.
This theory has changed the field of mitochondrial physiology, placing mitochondria at the center stage of many cellular physiological and pathological processes, including cell death (8, 184). The coupling of respiration to phosphorylation requires inner mitochondrial membrane properties that were neglected by the previous proposed hypotheses for oxidative phosphorylation (148, 147, 203), such as the inner membrane being asymmetrically organized and highly impermeable to H+ and other ions. It should, therefore, be emphasized that any agent or process that alters the proton gradient will certainly compromises ATP synthesis and other energy-dependent processes (147).
The new concepts developed considering the Chemiosmotic Theory increased the general interest in mitochondria, and this knowledge was key in understanding the mechanisms of aging and accidental or programmed cell death (8, 184, 231). It is now amply accepted that mitochondria control cellular life/death decisions through highly regulated processes such as mitochondrial oxidant production (110), Ca2+ transport (231), and formation of the mitochondrial permeability transition pore (mPTP) (257).
The proton-motive force used by the ATP synthase to phosphorylate ADP is also involved in the regulation of other mitochondrial processes such as oxidant generation by respiration, in addition to being the driving force for: (i) ion (including Ca2+) transport across the inner membrane (89, 231); (ii) nonshivering thermogenesis promoted by UCP1 (158); (iii) ADP/ATP exchange via the adenine nucleotide translocator (105); (iv) import of cytosolic proteins and substrates for matrix metabolic pathways (133); (v) K+ influx through the ATP-sensitive channel (mitoKATP) (67); (vi) the reduction of NADP+ by NADH catalyzed by the nicotinamide nucleotide transhydrogenase (162, 186); and (vii) transport of metabolites across the inner mitochondrial membrane, such as pyruvate and phosphate.
It should be noted that NADPH is the main source of electrons to the enzymatic systems that detoxify peroxides and reduce mitochondrial dithiols (186). Therefore, the continuous energy-dependent re-reduction of NADP links the ΔP to the maintenance of mitochondrial redox balance. This is a critical mechanism to protect mitochondria against oxidant stress and is discussed in more detail in this issue (Francisco et al., 2021) (258).
Mitochondrial oxidant generation
Mitochondrial respiration generates superoxide radical (O2 •−) via one-electron transfer to molecular oxygen during electron transport through the respiratory complexes. Subsequently, O2 •− is converted to H2O2, hydroxyl radical (•OH), and peroxynitrite (ONOO−) (56, 176, 240). The chemical properties of O2 •− favor its reaction with •NO (•NO + O2 •− → ONOO−) over its dismutation into H2O2 by superoxide dismutase (SOD) under in situ conditions, thus producing higher amounts of ONOO− than H2O2 (176). About 20%–39% of this anion is protonated (ONOOH) in physiological pH (7.4–7.0), rendering it diffusible through cellular membranes. Peroxynitrite's high reactivity can cause oxidative damage to biomolecules and it has been shown to cause mitochondrial dysfunction by induction of the mPTP via direct oxidation of the mitochondrial thiol pool (63, 176).
In the respiratory chain, oxidants are mainly generated in Complex I (including forward and reverse electron transfer), Complex II, and the ubiquinone cycle within Complex III (240), although other mitochondrial enzymes also produce O2 •− or H2O2 as a byproduct. These include glycerol-3-phosphate, pyruvate, alfa-ketoglutarate, dihydroorotate, and very long chain acyl-CoA dehydrogenases and the electron transfer flavoprotein [reviewed in Mailloux (134)]. Under some conditions, mitochondrial oxidant production by these enzymes can be equivalent or even larger than that of the respiratory chain (23, 209).
Although in the past mitochondrially generated oxidants were considered always detrimental to cellular homeostasis, it is now clear that, under controlled situations, mitochondrial oxidants have important roles in redox signaling of multiple mitochondrial and cellular processes (56, 73, 196). Nonetheless, mitochondria have antioxidant defense systems that both scavenge mitochondrial oxidants and are regulated by them (73). The SOD is found in the matrix and in the intermembrane space, since O2 •− is produced in both compartments (56, 81).
H2O2 is removed by catalase in liver and heart mitochondria, or thioredoxin and glutathione peroxidases in the mitochondrial matrix (56, 159, 177, 188), using redox equivalents from thioredoxin or glutathione, which are then re-reduced by mitochondrial thioredoxin reductase or glutathione reductase, using NADPH as the electron donor (239). In addition, mitochondrial radicals are also scavenged by compounds such as vitamin C, E, CoQ10, and others (55).
The rate of O2 •− generation is strongly regulated by the proton motive force, such that O2 •− is higher at high membrane potentials than at lower potentials. High membrane potential decreases the rate of respiration, thus increasing the amount of partially reduced electron carriers that are capable of one-electron transfer to O2 (73, 110). In addition, lower respiration rates increase the availability of O2 in the microenvironment where one-electron donors are located, favoring the one-electron transfer reaction [reviewed in Turrens (225)]. In contrast, lower membrane potential inhibits reverse electron transfer from Complex II to Complex I, a main mechanism of oxidant formation under some metabolic conditions (56).
Mitochondria developed mechanisms to modulate oxidant generation via fine-tuning the proton motive force through slow return of external protons to the matrix, known as mild uncoupling (202), which increases respiration rate and, consequently, decreases the availability of respiratory intermediates for one-electron transfer to O2. Inner membrane integral proteins such as the uncoupling proteins (UCPs) (106, 202); the mitoKATP channel (67); and possibly the H+ transport activity of the ANT translocator (13) have been shown to, directly or indirectly, catalyze slow return of H+ to the matrix. For a comprehensive review of mitochondrial oxidant production, we refer the reader to (56).
The mPTP
The mPTP is a proteinaceous mega-channel in the inner mitochondrial membrane (257) whose opening is promoted by redox imbalance coupled with matrix Ca2+ overload (108, 228). The molecular structure of the mPTP remains controversial (25, 89, 228), but evidence suggests that both matrix and membrane proteins participate in its structure via thiol-mediated interactions (89). The opening of this mega-channel leads to ΔΨ collapse, which results in an additional increase in oxidant production, creating an uncontrolled cascade of oxidative stress, mitochondrial damage, and cell death (248).
The mPTP also occurs in the form of a pore of lower conducting state that may display physiological functions such as fast mitochondrial Ca2+ efflux and possible regulation of mitochondrial volume (12, 86, 229). The transition from low to high conductance state of the mPTP seems to depend on the mitochondrial redox balance (247), as the mPTP opening is stimulated by mitochondrial NADPH depletion (119, 147, 230), oxidants (51, 72), and exogenous (26, 110) or endogenous oxidant-generating systems (22, 27).
The redox nature of mPTP is further supported by its blockage by antioxidants (108, 231), anoxia (26), catalase (26, 109), peroxiredoxin (111), or o-phenanthroline (26). The involvement of H2O2 in mPTP assembly is strongly supported by its ability to produce protein disulfides (51, 108). Indeed, evidence has been provided that redox signals generated by cysteine oxidation via sulfenylation, S-glutathionylation, and S-nitrosylation regulate mPTP opening (135), suggesting that the mPTP is not a molecularly defined channel but rather a permeability transition pore originated by protein–thiol crosslinking (231).
The role of mPTP in aging and age-associated diseases has also been extensively demonstrated, and for a comprehensive review we refer the reader to Rottenberg and Hoek (184).
Mitochondrial Dysfunction and Genomic Instability
Genomic instability and DNA damage accumulation have been some of the earlier recognized hallmarks of aging in several model organisms, including humans. Accumulation of nucleotide base damage and DNA single- and double-strand breaks with age have been shown by several groups [for review, see Maynard et al. (143)]. In addition, accumulation of DNA damage markers such as phosphorylated histone H2A (H2AX) and p53-binding protein 1 foci has also been demonstrated.
Support for a direct role for DNA damage in aging also comes from human segmental premature aging syndromes caused by mutations in genes that code for proteins with varied roles in genomic maintenance, such as the Werner syndrome, caused by mutations in the WRN helicase, Cockayne syndrome, caused by mutations in the repair factors CSA and CSB, and progeria, caused by mutations in the nuclear lamin A (LMNA) [reviewed in Kipling et al. (102)].
Several mouse models lacking DNA repair genes also display a segmental premature aging phenotype. Interestingly, the mouse model with the phenotype that closest resembles normal aging is the mitochondrial mutator mouse, expressing a proof-reading deficient DNA polymerase γ, the mtDNA replicative polymerase (115, 224). These mice accumulate somatic point mutations and deletions in the mtDNA and show significantly decreased lifespan accompanied by hair graying, hearing loss, lipodystrophy, and cardiac disease. In fact, it was later demonstrated that DNA deletions and clonal mutations drive the aging phenotype in these mice (232), establishing a causal relationship between mtDNA instability and aging.
Owing to its location, associated with the inner mitochondrial membrane and close to the sites of mitochondrial oxidant generations, it was proposed that the mtDNA accumulated more oxidative damage than the nuclear DNA (nDNA). Despite differences in the analytical methods used, and some variation in the absolute values obtained, several groups, including ours, have shown that steady-state levels of 8-hydroxyguanine (8-oxoG) are around 10 times higher in mitochondrial than in nDNA (85). Levels of other oxidized bases are also higher in mtDNA, at least in the few tissues in which it has been analyzed (139). Levels of polymerase-blocking lesions are also higher in mtDNA than in nDNA (244).
As it was initially believed that mitochondria lacked efficient repair mechanisms, these differences were attributed to the damage not being removed from the mtDNA. However, it is now clear that mitochondria are proficient in most DNA repair pathways, except for the nucleotide excision repair [reviewed in Alencar et al. (4)], meaning that the higher steady-state levels of DNA damage must be related to higher formation. This would not be unexpected as, in addition to being the main cellular site of O2 •− and H2O2 formation, mitochondria also accumulate lipophilic cations such as several genotoxins (185). For more details on mtDNA maintenance mechanisms see Carvalho et al., 2021 (259), in this issue.
The mammalian mtDNA codes for 13 polypeptides, all integral components of 4 of the 5 oxidative phosphorylation complexes (5). Complex II is the only oxidative phosphorylation system (OXPHOS) complex with no subunit coded in the mtDNA. As mutations in the mitochondrially encoded subunits lead to respiratory dysfunction, and mtDNA accumulates mutagenic lesions, such as 8-oxoG, with age, it was proposed that mtDNA may act as a “biological clock” (75). According to this hypothesis, oxidative damage to mtDNA would result in mutations in the OXPHOS subunits, leading to a vicious cycle where increased oxidant production would fuel accumulation of damage and mutation (80).
However, two recent studies in Drosophila (87) and mice (100) have challenged this hypothesis, showing that the predominant mutations found in mtDNA from old individuals are not due to oxidative base damage. Moreover, knockout of repair enzymes for oxidized bases does not change the age-associated mutation signature. Although the exact origin of these mutations is not yet clear, the types of mutations suggest that they result from replication errors and clonal expansion (107). Moreover, it should be noted that most individual mtDNA mutations require high heteroplasmy to cause significant respiratory deficiency, although the effect of a high load of rare mutations is unclear (115, 213).
A direct relationship between genomic instability and mitochondrial dysfunction was demonstrated by using mice defective in the repair protein XPA, where hyperactivation of the DNA damage sensor poly-ADP-ribose polymerase 1 (PARP1) leads to decreased mitophagy and decreased clearing of damaged mitochondria. This effect seems to be mediated by lower NAD+ levels, consumed by PARP1, and decreased activity of the PGC-1α/SIRT1 axis (54).
In addition, several of the clinical phenotypes seen in mitochondrial diseases, caused by either mutation in the mtDNA or mutations in nuclear-encoded mitochondrial proteins, overlap those of DNA repair/maintenance diseases (192).
Conversely, mitochondrial dysfunction has been causally implicated in genomic instability. In yeast, loss of mtDNA leads to nuclear genomic instability not only through impaired iron–sulfur cluster metabolism (228), but also through mitochondrially generated oxidants (181). Moreover, mouse embryos (proficient in telomerase activity) treated with the mitochondrial uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrozone (FCCP) showed disrupted intracellular calcium homeostasis, increased oxidant generation and telomere loss, and chromosome fusion and fragmentation.
These effects were completely prevented by the antioxidant N-acetylcysteine, suggesting that mitochondrial dysfunction caused telomere attrition (125). Similar results were obtained in other studies [reviewed in Zheng et al. (255)], highlighting the intricate relationship between three major hallmarks of aging, mitochondrial dysfunction, genomic instability, and telomere attrition.
For comprehensive reviews on mitochondria and genomic instability, we refer the reader to Refs. (53, 97). The interplay between mitochondrial function and genomic stability is also depicted in Figure 2.
Mitochondria and Protein Quality Control
Loss of proteostasis has been recently recognized as a hallmark of aging (218). Given that most mitochondrial proteins are nuclear encoded and must be transported and properly assembled in the organelles, it is essential for mitochondria to have mechanisms to repair, refold, and eliminate damaged or misfolded proteins. Ultimately, severely damaged mitochondria might be recycled through mitophagy; however, before mitochondria reach a critical level where mitophagy is deemed necessary, other mechanisms will try to ensure proteostasis.
Misfolded proteins can be generated by several mechanisms, including as errors during translation, either in the mitochondria or in the cytosol, oxidative damage to proteins themselves, or mutations in the coding sequence of the respective genes. Estimates point to an ∼10% rate of mistranslation in newly synthesized proteins, even under optimal conditions (103), and to a 20%–30% rate of degradation of newly synthesized polypeptides due to folding errors (173, 194).
Therefore, proper functioning of these quality control systems is fundamental to prevent accumulation of defective proteins and to maintain the proper functioning of mitochondria and cells.
Proper mitochondrial proteostasis begins with protein import. Proteins are imported into the mitochondria through two multi-subunit complexes, the transporter of the outer membrane, and the transporter of the inner membrane. The available evidence suggests that mitochondrial protein import is not significantly affected with age in mice skeletal muscle (84, 217). However, the increased expression of Tim23, Tim17, mtHsp70, and Hsp60 was observed after exercise in young animals was severely affected in old animals, whereas the expression of cytosolic chaperones was increased (84), suggesting that aging may impair mitochondrial biogenesis itself.
During import, proteins are unfolded, and they are subsequently refolded inside the mitochondria. An indirect factor that may affect mitochondrial protein quality is that errors in protein translation in the cytosol may introduce proteins with erroneously incorporated amino acids inside the mitochondria, putting pressure in the mitochondrial protein folding machinery (103, 173, 194). Moreover, the activity of the matrix protease Lon has been shown to be reduced with aging (7, 14), whereas its overexpression in postmitotic cells of the fungus Podospora anserina increases lifespan by more than 50% (131).
Several proteins accumulate in insoluble protein aggregates during Caenorhabditis elegans aging, including in the mitochondria, and a significant overlap was found between these proteins and proteins that regulate lifespan (39). The mitochondrial unfolded protein response (UPRmt) is a stress response pathway that regulates protein folding, metabolism, antioxidant defense, immune response, and regulation of iron sulfur cluster assembly (195). In addition, UPRmt has a role in retrograde signaling to the nucleus, to coordinate the stress response to reestablish proteostasis of mitochondrial proteins.
UPRmt can be activated in response to stresses, including increased mitochondrial protein synthesis or import (153, 254), as well as impaired protein import, in both C. elegans and mammals (178). Knockdown of mitochondrial proteases and chaperones in C. elegans also serves as a trigger, as well as treating cells with the redox cycling compound paraquat (245). UPRmt seems to mediate lifespan extension in C. elegans with cytochrome C oxidase subunit 1 knockdown (44). Similar results were observed in Drosophila and mice (83, 90), even though both models display lower mitochondrial function.
In C. elegans, UPRmt is mediated by the activating transcription factor associated with stress-1 (ATFS-1), which localizes to both the nucleus and the mitochondria. ATFS-1 is predominantly imported into the mitochondrial matrix in healthy conditions, where it is degraded. When mitochondrial import is defective, or when mitochondrial chaperones and proteases are inhibited, ATFS-1 cannot be imported into mitochondria and, instead, accumulates in the nucleus, where it induces expression of stress response genes (3, 44).
In mammals, ATF5 (the ATFS-1 homologue), ATF4, and CHOP are involved in the regulation of UPRmt (59, 145, 175, 226), with evidence suggesting that ATF5 promotes oxidative phosphorylation and cell growth during mitochondrial dysfunction (59).
Mitochondrial Dynamics and Mitophagy in Aging
Early electron microscopy observations by Palade (166) and Sjöstrand (201) revealed that mitochondria were, on average, ∼1.0 μm in length and 0.5 μm in diameter, although at that time they already noted great variations in shape, size, number, and arrangement of substructures. Later studies demonstrated impressive configurational changes during meiosis in yeast cells (20, 149). This ability of mitochondria to fuse and divide was later confirmed in mammalian cells and tissues, suggesting that mitochondrial morphology play an important role in the mitochondrial life cycle (20).
It is now clear that mitochondria fuse and divide through highly regulated events, in a process called mitochondrial dynamics, which is relevant to physiological and pathophysiological noncanonical mitochondrial functions (112, 197). Liesa and Shirihai (121) demonstrated that the physical association between mitochondria allows for exchange of their components, thus enabling the selective maintenance of functional mitochondria. Results showing that ablation of fission-related proteins leads to the formation of abnormally long and dysfunctional mitochondria that fail to divide and remove the damaged parts also support a critical role for dynamics in mitochondrial function.
It has, since, become clear that mitochondrial dynamics, in addition to serving as a quality control mechanism, permits the adaptation of mitochondrial bioenergetics to cellular needs (121). Indeed, dynamic changes in mitochondrial morphology and location have been recognized as critical regulators of metabolism and cellular resistance to stresses in such a way that defects in this complex array of signals lead to mitochondrial dysfunction and diseases (120, 121, 146). Several groups have shown that impaired mitochondrial dynamics leads to accumulation of dysfunctional mitochondria and contributes to age-related disorders, such as diabetes and neurodegeneration (112, 197).
Mitochondrial fusion and fission are controlled and executed by a series of highly conserved proteins, including mitofusins 1 and 2, OPA1, DRP1, FIS1, and DNM2 [reviewed in Tilokani et al. (221)], and their balance controls the shape of the mitochondrial network. Although the mechanisms are not yet fully understood, several observations support that mitochondrial dynamics affects lifespan, likely through mitochondrial bioenergetics. Changes in mitochondrial network with age have been reported in several models, with the types of changes varying greatly [reviewed in Liu et al. (127)].
It does seem, however, that the alterations in the balance between fusion and fission may mediate life extension in some organisms. In yeast, reducing mitochondrial fission results in increased lifespan (191). In line with that, increased mitochondrial fusion promotes survival of older C. elegans (30). On the other hand, promoting fission in midlife improves health and prolongs lifespan in Drosophila, through a mechanism dependent on autophagy (179). Lifespan extension induced by calorie restriction (CR) in C. elegans is also mediated, at least in part, by changes in mitochondrial dynamics (235), whereas in mice CR also induces mitochondrial network remodeling, through the PGC-1α coactivator (58).
It is, nonetheless, clear that mitochondrial dynamics is essential for cellular stress response (48). Mitochondrial fission is essential not only for organellar proliferation and distribution during cell division but also for removal of damaged mitochondria through mitophagy (164). Mitophagy is a specialized type of autophagy targeting dysfunctional mitochondria, thus contributing to maintaining a “healthy” mitochondrial network. Thus, it is not surprising that it has a prominent role in aging, as it has been reported in several model organisms, including mammals. Mitophagy is activated by a series of mitochondrial stressors and depends on signaling pathways that allow for the identification and targeting of the damage mitochondrion to autophagosomes, by two major pathways, a receptor-mediated and a ubiquitin-mediate mitophagy [reviewed in Onishi et al. (163)].
Defective mitophagy has been demonstrated in age-associated diseases, notably Alzheimer's [reviewed in Tran and Reddy (223)] and Parkinson's disease [reviewed in Clark et al. (35)]. In fact, mutations in the genes enconding for PINK-1 and Parkin, which are key proteins in ubiquitin-mediated mitophagy, cause early onset PD. Moreover, several studies in C. elegans have directly linked mitophagy efficiency to lifespan, showing that upregulated mitophagy is required for lifespan extension in the daf-2 long-lived mutants (167). Mitophagy also mediates the lifespan extension effects of tomatidine, a tomato-derived alkaloid (55), and iron starvation (193).
Conversely, defective mitophagy is implicated in premature aging, as described earlier. Figure 3 presents a schematic representation of the relationship between mitochondrial function, dynamics, and mitophagy in the aging process.

The Role of Mitochondria in Inflammaging
With aging, the immune response profile changes, leading to a state of low-grade, sterile, chronic inflammation, referred to as “inflammaging.” A pathological inflammatory profile can also be found in several diseases of aging (242). The immune system responds to pathogen infection through recognition of damage-associated molecular patterns (DAMPs) by pattern recognition receptors, which are present both on the cell surface and in the intracellular compartments. Families of receptors involved in this process include cytosolic DNA sensors (cGAS and STING), nucleotide oligomerization domain-like receptors (NLRs), retinoic acid-inducible gene I-like receptors, and Toll-like receptors (TLRs) (219).
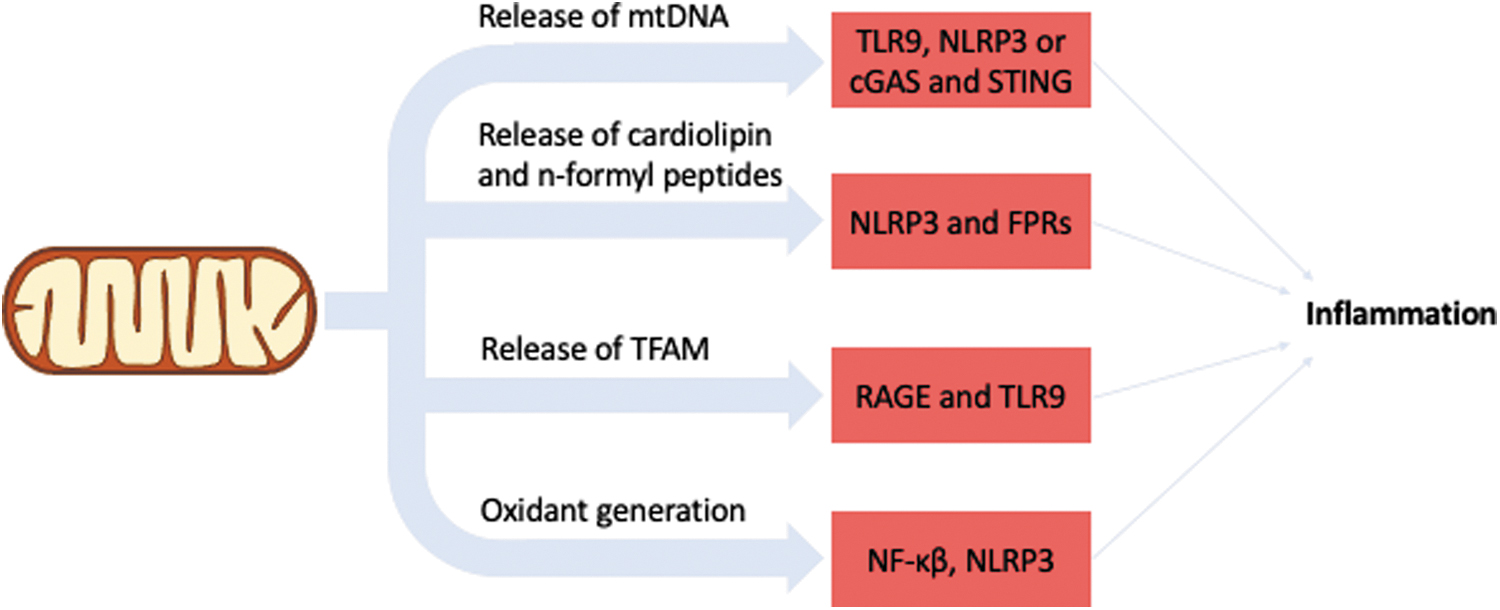
Inflammation can be triggered by several types of stresses, both endogenous and exogenous. In the case of endogenous types of stresses, which would be characteristic of aging, some of them have a mitochondrial origin. DAMPs are molecular products released because of damage, and several mitochondrial components when found outside the mitochondrial compartment elicit an immune response. MtDNA is a potent DAMP as, in contrast to nDNA, it lacks methylation, likely due to its bacterial origin. This prompts the immune system, through the activity of TLRs, NLRs, or cGAS and STING, to recognize the mtDNA and activate the immune response (252).
cGAS/STING activation by mtDNA induces a type 1 interferon (IFN) response. Interestingly, during Bax/Bak-induced apoptosis there is a suppression of IFN production by preventing mtDNA-mediated activation of cGAS/STING, ensuring that apoptosis does not elicit an immune response (182, 237). On the other hand, some experimental evidence suggests that circulating mtDNA does, indeed, increase with age and correlates with increased levels of pro-inflammatory cytokines (171).
Cardiolipin and n-formyl peptides also elicit an immune response when found outside the mitochondrial compartment (76, 88). Similar to mtDNA, these two components also have a bacterial origin, pointing to an evolutionary pattern to sequestering components of prokaryotic origin to signal inflammation. Another mitochondrial component that has been found to elicit an immune response when found outside mitochondria is the mitochondrial transcription factor A (TFAM) (31). TFAM is a transcription factor and it packs the mtDNA in a manner analogous to the packing of nDNA by histones. When found outside the mitochondrial compartment, TFAM triggers immune cells via RAGE and TLR9.
Oxidants also have a role in inflammation and the release of pro-inflammatory cytokines (18). H2O2 induces translocation of NF-κβ to the nucleus independent of IκBα degradation, which is the canonical NF-κβ activation via TNF-α (21). The exact influence of this signaling pathway, however, may depend on the location where the oxidation takes place (95), as mitochondrial H2O2 contributes to endothelial NF-κβ activation in arteries of aged rats (227), but the inhibition of mitochondrial H2O2 production in endothelial cells prevents NF-κβ activation and interleukin (IL)-6 secretion induced by hypoxia (249).
Overexpression of SOD2 also inhibits the oxidants-NF-κβ signaling (32). Further, mitochondrial oxidants also seem to have a role in NLRP3 activation, and induction of mitochondrial oxidant production with rotenone and antimycin (complex I and III inhibitors, respectively) increases NLRP3 activation (1). In addition, NLRP3 has also been shown to relocate to the mitochondria on activation, through a mechanism mediated by mitochondrial anti-viral signaling protein (1).
Considering that mitochondrial oxidant production and mtDNA damage increase with age, these mechanisms point to a central role for mitochondrial dysfunction in inflammaging, as schematized in Figure 4.
Mitochondria and Cellular Senescence
Cellular senescence is another hallmark of aging. It is induced in response to stresses, including DNA damage, telomere dysfunction, oncogene activation, and organelle stress [reviewed in Di Micco et al. (42)], and it limits proliferation of the damaged cells. Senescent cells are characterized by cessation of cell division despite the availability of nutrients and mitogenic stimuli, an altered pattern of gene expression, and secretion of varied factors referred to as senescence-associated secretory phenotype (SASP).
They are also highly resistant to apoptosis due to an upregulation of anti-apoptotic pathways, including BCL-2 proteins and related pathways (34). In fact, the signaling pathways regulating cellular senescence and apoptosis often overlap, suggesting that there might be some mechanism that regulates whether defective cells will be eliminated through apoptosis or arrested by senescence (34). On the other hand, senescent cells also have a tumor suppressor role, by arresting replication of damaged cells that could potentially become cancerous (198, 199), as well as roles in embryonic development (156) and wound healing (94).
Several groups have suggested that senescent cells accumulate with age in different tissues and species (78, 91, 114), and recent studies showed that selective clearance of senescent cells in mice is capable of alleviating symptoms of aging (243). The impact of senescent cells in aging is usually believed to be related to the pro-inflammatory factors secreted through SASP, which contribute to the chronic inflammation observed during the aging process. The role of SASP in young organisms seems to be to serve as a signal to the immune system so that senescent cells are eliminated. With aging, the immune system weakens and loses the capacity to clear senescent cells, but the pro-inflammatory signal remains, creating chronic inflammation.
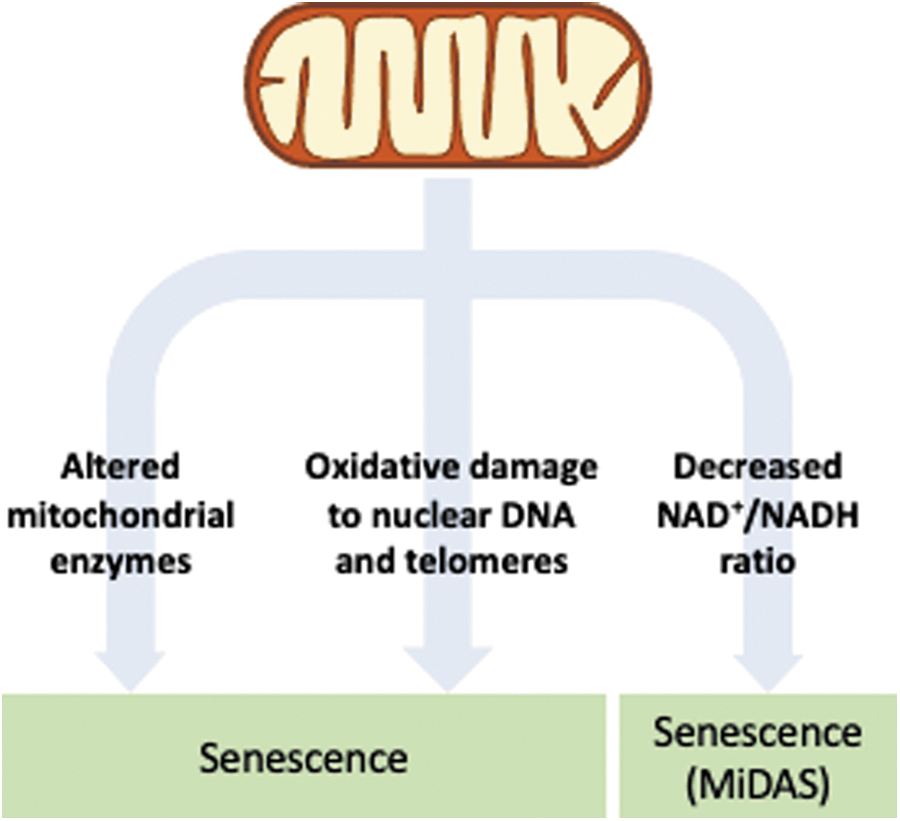
Evidence has accumulated, demonstrating a link between cellular senescence and mitochondrial dysfunction. Several studies have demonstrated that mitochondrial dysfunction can induce cellular senescence (38, 151, 233), most likely through the production of oxidants. The observation of a relationship between oxidants and cellular senescence was first proposed by Packer and Fuehr (1977), when they found that culturing human cells in a low-oxygen environment resulted in an extension in their replicative lifespan (165).
Oxidant production is higher in senescent cells (98, 117, 169), accompanied by a decrease in antioxidant defenses (169, 190). Oxidants may also induce senescence by causing damage to DNA (33), including the telomeres (79). Further, the reduction of oxidant levels through several interventions prevents telomere attrition, leading to an increase in replicative lifespan in cultured cells (169, 190).
Not surprisingly, regulators of cell cycle arrest are upregulated in senescent cells, including p16, p21, p53, and ARF, all of which will be upregulated depending on the stimuli that induced senescence. For instance, p21 regulates cellular senescence in response to oxidants (132, 168), whereas both p21 and p53 are upregulated by the DNA damage response (DDR) pathway, which can lead to senescence. P53-binding protein-1 foci and phosphorylated histone 2A (H2AX), markers of DDR, are abundant in senescent cells (19). p16 and ARF, on the other hand, are upregulated by mitogenic stress (122).
Cellular senescence may also be directly linked to mitochondrial metabolism, likely in a tissue-specific fashion. The expression of mitochondrial malic enzyme (ME2), which converts malate to pyruvate, is repressed by p53 activation. Further, knockdown of ME2 induces cellular senescence, and its overexpression reverts the phenotype (92). Also, the overexpression of malate dehydrogenase in yeast results in lifespan extension (45). Notably, low NAD+/NADH ratio induced by malate dehydrogenase depletion has been shown to induce cellular senescence (118).
In oncogene-induced senescence, overexpression of the BRAF oncogene caused an alteration in the activity of the pyruvate dehydrogenase complex, leading to more pyruvate oxidation and increased generation of oxidants, inducing a senescent state (98).
The NAD+ levels seem to be involved in central pathways that regulate aging, either through their redox role or through a signaling role as substrate for PARP and SIRTs. NAD+ levels decline with age in several tissues (15, 70, 246), and a decreased NAD+/NADH ratio activates AMPK and subsequently p53, inducing a specific type of cellular senescence called mitochondrial dysfunction-associated senescence, which lacks the IL-1 mediated arm of the SASP (238).
The same study also demonstrated that a mitochondrial mutator progeroid mouse model displays lower NAD+/NADH ratio and accumulation of senescent cells without IL-1-dependent SASP factors, linking genomic instability to NAD+ levels. Mechanistically, this link may be mediated by PARP1, which when activated on DNA damage uses NAD+ to ADP-ribosylate itself and various other proteins. This accumulation of poly-ADP ribose at damaged sites causes the recruitment of chromatin modifiers and helps dictate the choice of DNA repair pathway.
Inhibition of PARP can induce cellular senescence due to unrepaired DNA damage (46), indicating that cells with low NAD+/NADH ratio are more susceptible to cellular senescence. In addition, the mitochondrial sirtuins SIRT3 and SIRT5, which display an NAD-dependent deacetylase function, have been shown to regulate the pyruvate dehydrogenase complex (141) and cellular senescence (238). Alternatively, a low NAD+/NADH ratio may be leading to more ROS formation due to more electrons being available to react with molecular oxygen. It can also serve as an indicator of impairment of respiratory chain activity, since NADH is not being consumed by it.
The main mitochondria-related senescence pathways are shown in Figure 5.
The Role of Mitochondria in Preserving Stem Cell Function
Several molecular mechanisms have been implicated in deteriorating cellular function with time. However, in theory, given the infinite capacity of stem cells to maintain their function and to replace some types of cells in the tissues where they are present, damage to these cell types into which stem cells can differentiate would not result in deleterious effects to the organism because stem cells would be able to replace them. In effect, however, stem cell function does decline with aging, and replacement of malfunctioning cells does not occur in a perfect manner.
Decline in stem cell function is also considered a hallmark of aging, and much evidence points to an important role of mitochondria in maintaining stem cell function. Of note, it has been shown that human mammary stemlike cells display asymmetric apportioning of mitochondria during division, with one daughter cell getting preferentially younger mitochondria, and the other getting old ones (99). Daughter cells receiving younger mitochondria maintain stem cell traits, highlighting the role that mitochondria play in maintaining stem cell function.
In the same study, it was shown that inhibition of mitochondrial fission eliminated the asymmetric apportioning of mitochondria and caused daughter cells to lose stem cell properties. However, despite this apparent importance of mitochondria in maintaining stem cell function, adult stem cells rely mostly on glycolytic metabolism, with mitochondria contributing little to the energy generation of most stem cell types [reviewed in Zhang et al. (250)]. In fact, during reprogramming of cells in induced pluripotent stem cells, there is a shift from oxidative phosphorylation to glycolysis (61). On the other hand, oxidative phosphorylation seems to be required in several adult stem cell lines for differentiation (250).
This reliance on anaerobic metabolism has been proposed as an adaptation to the hypoxic nature of some stem cell niches, the capacity of glycolytic metabolism to provide intermediates to support anabolic pathways that are essential to stem cell function, and to the fact that avoiding aerobic metabolism minimizes the generation of oxidants produced by mitochondria, preventing oxidative damage during aging (214).
Several mitochondrial metabolic intermediates have been shown to regulate stem cell fate decisions [reviewed in Mattson et al. (142)]. A decline in NAD+ levels with age in mouse muscle stem cells leads to cellular senescence, whereas supplementation with nicotinamide riboside (NR), an NAD+ precursor, decreases muscle stem cell senescence and increases lifespan (251). In neural stem cells, a similar influence of NAD+ levels in the aged stem cell phenotype could be observed, and, likewise, NAD+ supplementation renewed old neural stem cells (210).
In hematopoietic stem cells, a decline in SIRT3 and SIRT7, two NAD+-dependent deacetylases, leads to hematopoietic stem cell dysfunction, and their supplementation restores cellular function (150).
Another relevant link between mitochondria and stem cell function may be mediated by the control of gene expression through DNA or histone methylation. Both methyltransferases and demethylases are dependent on mitochondrial metabolites since they use as substrates S-adenosyl-methionine (SAM) and α-ketoglutarate. Although SAM is not produced directly in the mitochondria, its production depends on the folate cycle, which depends on the one-carbon metabolism inside the mitochondria.
In muscle stem cells, the histone trimethylation marker H3K4me3, which promotes an increase in gene expression, decreases during aging, whereas the marker H3K27me3, which decreases gene expression, increases. In hematopoietic stem cells, on the other hand, H3K4me3 levels increase in genes related to self-renewal and identity during aging. This is in line with the observation that the number of hematopoietic stem cells increases with age in mice (215).
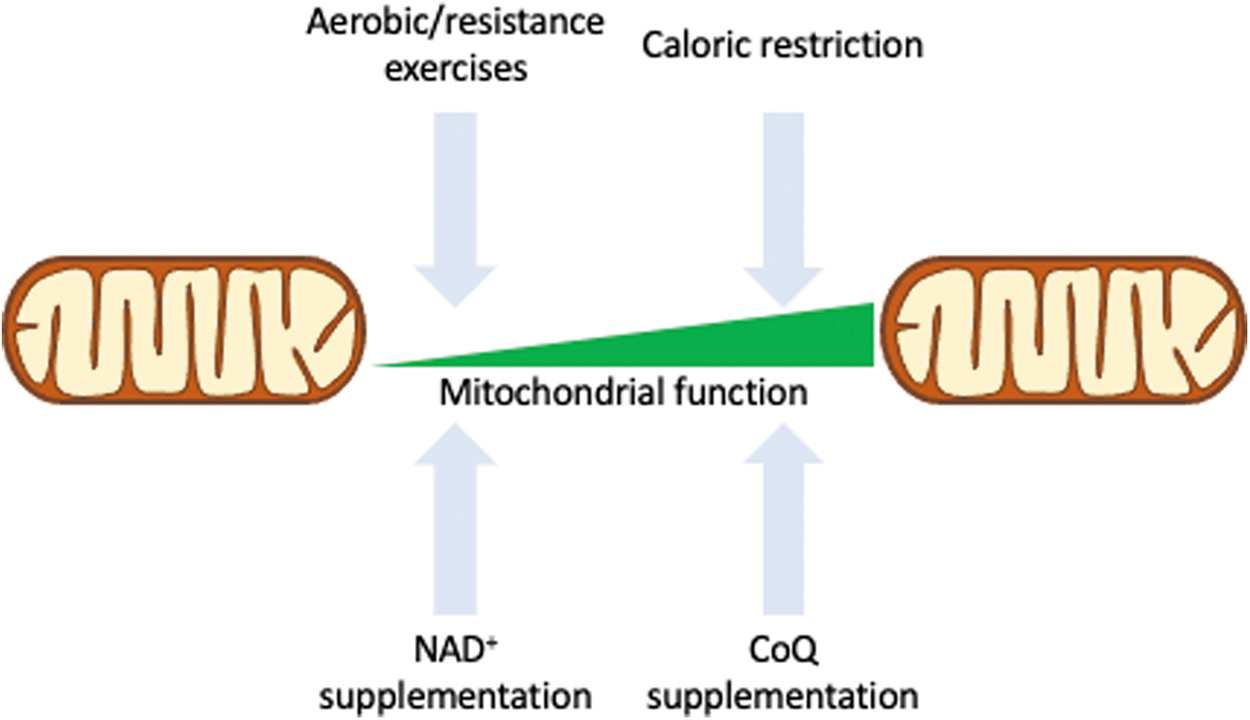
Strategies to Preserve Mitochondrial Function During Aging
Given the known age-associated changes in mitochondrial function and the potential effects they may have in the proper functioning of organisms, it is of interest to review possible interventions that might help to preserve mitochondrial function and to postpone the decline in fitness characteristic of aging, enabling for a longer health and, possibly, lifespan. One of the simplest and most effective ways to preserve mitochondrial function with aging is through aerobic and resistance training. Exercising is a widely accepted method of preventing age-related disabilities and diseases (157), and it has numerous benefits in relation to mitochondrial function.
Aerobic exercise enhances the capacity of oxidative enzymes in rat mitochondria (82), and aerobic and resistance exercise increase protein synthesis and mitochondrial biogenesis in human skeletal muscle (137). Endurance exercises also improve insulin sensitivity, mitochondrial oxidative capacity, and SIRT3 levels, as well as they stimulate mitochondrial biogenesis in several tissues (47).
Muscle strength declines at an average of 1% per year in older adults, with that rate increasing 2- to 4-fold in 70 or older individuals (71), in a process known as sarcopenia. Regular physical activity and endurance exercises demonstrate improvements in several age-related pathologies, including sarcopenia (29, 212). In murine muscle, increased PGC-1α expression, a master regulator of mitochondrial biogenesis, protects from sarcopenia and metabolic disease during aging (236). PGC-1α is also a major regulator of exercise-induced adaptation.
In the mtDNA mutator mice, endurance exercise rescues the progeroid phenotype and induces systemic mitochondrial rejuvenation (187). However, it is important to note that exhaustive exercise can lead to ROS-mediated negative effects (77).
Calorie restriction (CR) is the most robust environmental intervention known to slow aging and extend lifespan in several model organisms, including mammals (36, 136). Applications of CR typically involve a reduction in up to 40% in caloric intake, without malnutrition. The molecular mechanisms responsible for regulating the effects of CR are not completely understood, but many of its components have been characterized, demonstrating that these mechanisms are largely conserved between.
CR promotes lower cell proliferation, oxidant production, and body temperature (17, 24, 205), and its effect is related to the insulin-like growth factor-1 receptor pathway and the target of rapamycin (TOR) pathway (170). CR also promotes a reduction in NF-κβ and decreases inflammaging (170), and it upregulates Forkhead-box transcription factors, which, in turn, activate mitophagy and expression of antioxidant enzymes (129). The expression of antioxidant enzymes is also increased by CR through the activation of the nuclear factor erythroid 2-related factor 2 (129). Mitochondrial activity is induced by CR through the activation of AMPK and sirtuins. Higher levels of mitophagy were also found in mouse liver and rat kidney after CR (37, 172), and an upregulation of PINK1 or Drp1 in mouse cardiac tissue was observed on physical exercise or CR, respectively (253).
In yeast, CR has been found to increase replicative lifespan by increasing mitochondrial respiration (124) and maintain cells phenotypically younger during chronological lifespan (41). Yeast grown in 0.5% glucose exhibits an increase in chronological lifespan through a mechanism that involves the Ras/PKA and the TOR pathways (234). Deletions in the proteins of these pathways, such as tor1, sch9, and ras2, mimic some of the CR effects (50, 96), and they display increases in respiration and redox signaling, as well as increases in H2O2 and SOD activity (144).
It is important to note that the TOR pathway regulates autophagy, including mitophagy, and is a TOR, a drug with proven potential to extend lifespan in multiple organisms (208). Therefore, the modulation of mitophagy through this pathway could be essential for maintaining mitochondrial quality with aging. Extension of chronological lifespan in yeast by calorie restriction also required mitochondrial respiration levels above a certain threshold during exponential growth (160).
CR has been shown to increase the NAD+/NADH ratio in yeast, leading to activation of Sir2 (123), although the importance of Sir2 in mediating the CR-related lifespan extension has been recently questioned (96). Since NAD+ levels decline with age in several species (122, 256), interventions aiming at restoring its levels have been promoted recently as a possible antiaging strategy. Upregulation of NAD+, through genetic or pharmacological means, has been shown to protect against obesity and type 2 diabetes in rodents and to improve glucose homeostasis and mitochondrial dysfunction (113, 246 ).
Methods used to increase NAD+ levels include inhibition of enzymes that consume NAD+ (6, 9, 66), inhibition of nicotinamide N-methyltransferase (113), and supplementation of NAD+ precursors (246). Supplementation with NR and nicotinamide mononucleotide (NMN), two NAD+ precursors, in yeast and C. elegans results in increased lifespan (11, 154). In skeletal muscle, supplementation with NMN increases mitochondrial function and ATP production (70). The NR supplementation in old mice fed a high-fat diet improves muscle oxidative metabolism, thermogenic capacity, and increased endurance. Another study found that NR supplementation improves mitochondrial function in muscle stem cells, increasing the number and quality of muscle stem cells in old mice, as well as increasing lifespan (251).
In humans, some NAD+ precursors are already used for certain treatments. Niacin has been widely studied and is used to treat hypercholesterolemia (65). Several clinical trials investigating the effects of NMN and NR in humans are being conducted.
Coenzyme Q (CoQ) is a central component of the electron transport chain, where it serves as an electron carrier from complex I and complex II to complex III. It also serves as an antioxidant in redox reactions with some mitochondrial proteins. CoQ synthesis is reduced with age (2), potentially compromising its functions. In humans, a high CoQ10H2/CoQ10 ratio seems to be a risk factor for sarcopenia, as it was shown to be negatively associated with upper body muscle strength, peak flow, and muscle mass in aged individuals (60).
An analysis of the levels of CoQ10 in plasma of young and old individuals showed that older individuals have higher levels of plasma CoQ10, and that when each cohort was stratified in terms of level of physical activity, the response of the two cohorts was different, with younger individuals displaying a correlation between higher activity and lower CoQ10 plasma levels, and older individuals displaying a correlation between higher activity and higher CoQ10 plasma levels, which were related to lower lipoperoxidation and oxidized LDL levels (40).
CLK-1, the C. elegans homologue of CoQ7, mediates the retrograde signaling of UPRmt in the nucleus in response to mitochondrial oxidants (152). And mCLK1+/− (CoQ7 homologue) mice display an increased lifespan, despite several signs of mitochondrial dysfunction (116). Supplementation with ubiquinone-10, the oxidized form of CoQ10, has been shown to suppress nuclear and mtDNA damage in rats (161, 174), whereas supplementation with ubiquinol-10, the reduced form of CoQ10, produces beneficial effects in age-related diseases (207, 222).
Ubiquinol-10 supplementation also enhanced mitochondrial activity and decreased levels of oxidative damage in a mouse model of accelerated aging (220). All these point to CoQ being an interesting target for further studies to better establish its potential as a dietary supplement for preserving mitochondrial function. Figure 6 shows a schematic representation of the possible innervations to preserve mitochondrial function or revert dysfunction.
Concluding Remarks
Aging is a multifactorial complex biological process involving several distinct cellular targets and signaling pathways. Nonetheless, given the many roles that mitochondria play in cellular homeostasis, it has become clear that mitochondrial dysfunction is central in the intricate net of events that result in loss of cellular function and organismal fitness. As we begin to understand the changes that mitochondria undergo with age, at a mechanistic level, we move toward gaining a better view of these causal relationships and of the possible intervention targets to prevent some of the deleterious health effects of the aging process.
Footnotes
Authors' Contributions
C.M.P.F.B., A.E.V., and N.C.S-P. designed, wrote, and approved the final article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Fundação de Apoio à Pesquisa do Estado de São Paulo (FAPESP) grants 2017/04372-0 to NCSP and 2017/17728-8 to AEV. Caio M.P.F. Batalha is supported by Conselho Nacional de Pesquisa e Desenvolvimento (CNPq).