
Abstract
Aims:
Although prebiotics, probiotics, and fecal transplantation can alter the sensation of hunger and/or feeding behavior, the role of the constitutive gut microbiota in the short-term regulation of food intake during normal physiology is still unclear.
Results:
An antibiotic-induced microbiota depletion study was designed to compare feeding behavior in conventional and microbiota-depleted mice. Tissues were sampled to characterize the time profile of microbiota-derived signals in mice during consumption of either standard or high-fat food for 1 h. Pharmacological and genetic tools were used to evaluate the contribution of postprandial endotoxemia and inflammatory responses in the short-term regulation of food intake. We observed constitutive microbial and macronutrient-dependent control of food intake at the time scale of a meal; that is, within 1 h of food introduction. Specifically, microbiota depletion increased food intake, and the microbiota-derived anorectic effect became significant during the consumption of high-fat but not standard food. This anorectic effect correlated with a specific postprandial microbial metabolic signature, and did not require postprandial endotoxemia or an NOD-, LRR-, and Pyrin domain-containing protein 3-inflammasome-mediated inflammatory response.
Innovation and Conclusion:
These findings show that the gut microbiota controls host appetite at the time scale of a meal under normal physiology. Interestingly, a microbiota-derived anorectic effect develops specifically with a high-fat meal, indicating that gut microbiota activity is involved in the satietogenic properties of foods. Antioxid. Redox Signal. 37, 349–369.
Introduction
The gut is inhabited by trillions of microbes, which directly influence host metabolism and health (88). It has recently been proposed that the gut microbiota influences food intake (2, 34, 52, 89). Actually, dietary prebiotic supplementation can reduce food intake (5, 6, 18, 47, 53). Moreover, some probiotics can alter appetite in humans and change feeding behavior in laboratory animals (7, 33, 50, 60, 82). Fecal microbiota transplantation can also modify food intake (86).
Several nonexclusive mechanisms have been proposed to explain the regulatory effect of the gut microbiota on feeding behavior. Strikingly, dietary prebiotic and probiotic supplementation studies show a strong correlation between gut microbiota activity and the release of gastrointestinal peptides involved in appetite regulation (17, 29, 39). Furthermore, specific microbiota-derived molecules are known to be gut hormone-releasing factors (58, 87). In addition, the gut microbiota produces neuroactive peptides that modulate the activity of the enteric nervous system, which is notably involved in the control of food intake (55). Moreover, bacterial toxins, such as lipopolysaccharide (LPS), the plasma concentration of which is closely associated with energy intake, can activate vagal afferent terminals and alter their sensitivity to leptin (3, 28, 37).
Circulating bacterial molecules might also directly activate brain areas that control appetite. For instance, it has been suggested that gut microbiota-derived acetate reduces appetite after reaching central homeostatic feeding centers (36). Finally, recent studies revealed the existence of microbiota molecular mimetics that might act on receptors expressed in the brain and thus impact behavior (34, 77). Indeed, the caseinolytic protease B is a mimetic of the host α-melanocyte-stimulating hormone (α-MSH), and is able to activate anorectic pro-opiomelanocortin neurons within the hypothalamus (11).
Experimental and clinical manipulations of gut microbiota by prebiotics, probiotics, fecal transplantations, or nutritional interventions, and dysbioses found during obesity, diabetes, and anorexia nervosa clearly support a relationship between microbes and host feeding behavior. However, the role of the constitutive gut microbiota in food intake under normal physiology, that is, without preliminary microbiota manipulation and in a nondisease state, remains to be defined. Moreover, the exact role of the constitutive gut microbiota in appetite and satiety, if any, has not been elucidated.
Innovation
Role of the constitutive gut microbiota is not well understood. In an antibiotic-induced microbiota depletion study, we show the role of the constitutive gut microbiota in the short-term regulation of food intake. In addition, we describe the time profile of microbiota-derived signals in mice, over the course of a meal, during consumption of either a standard diet (SD) or a high-fat diet (HFD). We show that the gut microbiota can modulate food intake according to food composition. Specifically, the microbiota-derived anorectic effect of the gut microbiota is apparent during consumption of HFD during 1 h but not SD. Moreover, this anorectic effect correlates with a specific postprandial microbial metabolic signature. Conversely, molecular, pharmacological and genetic studies do not support a role for postprandial NLRP3-dependent inflammation, gut oxidative stress, TLR4 signaling, and GLP-1 release in the ABX-sensitive behavioral effects that develop within 1 h (Fig. 1).

In addition, mechanistic studies have determined diverse neuroendocrine routes in the gut–brain axis that microbiota-derived signals probably use to affect food intake. Notably, this includes gut inflammatory/oxidative stress, systemic endotoxemia, and glucagon-like peptide-1 (GLP-1). Nevertheless, it is still unclear whether these relays that are involved in the host response to a modified microbiota are also engaged by the constitutive microbiota to trigger a behavioral response under normal conditions.
Results
The microbiota is involved in the short-term regulation of food intake when consuming high-fat food
To assess the role of the microbiota in the short-term regulation of food intake, we examined feeding behavior in microbiota-depleted and control mice. To deplete microbes before behavioral analyses, a mix of ABX was given orally in drinking water for 2 weeks, while control mice received no treatment (Fig. 2A). The ABX treatment dramatically increased the size of the cecum, the content of which became dark, and strongly reduced DNA content and bacterial 16S rRNA gene copy number in feces (Fig. 2B–E), indicating effective gut microbiota depletion in ABX-treated mice.
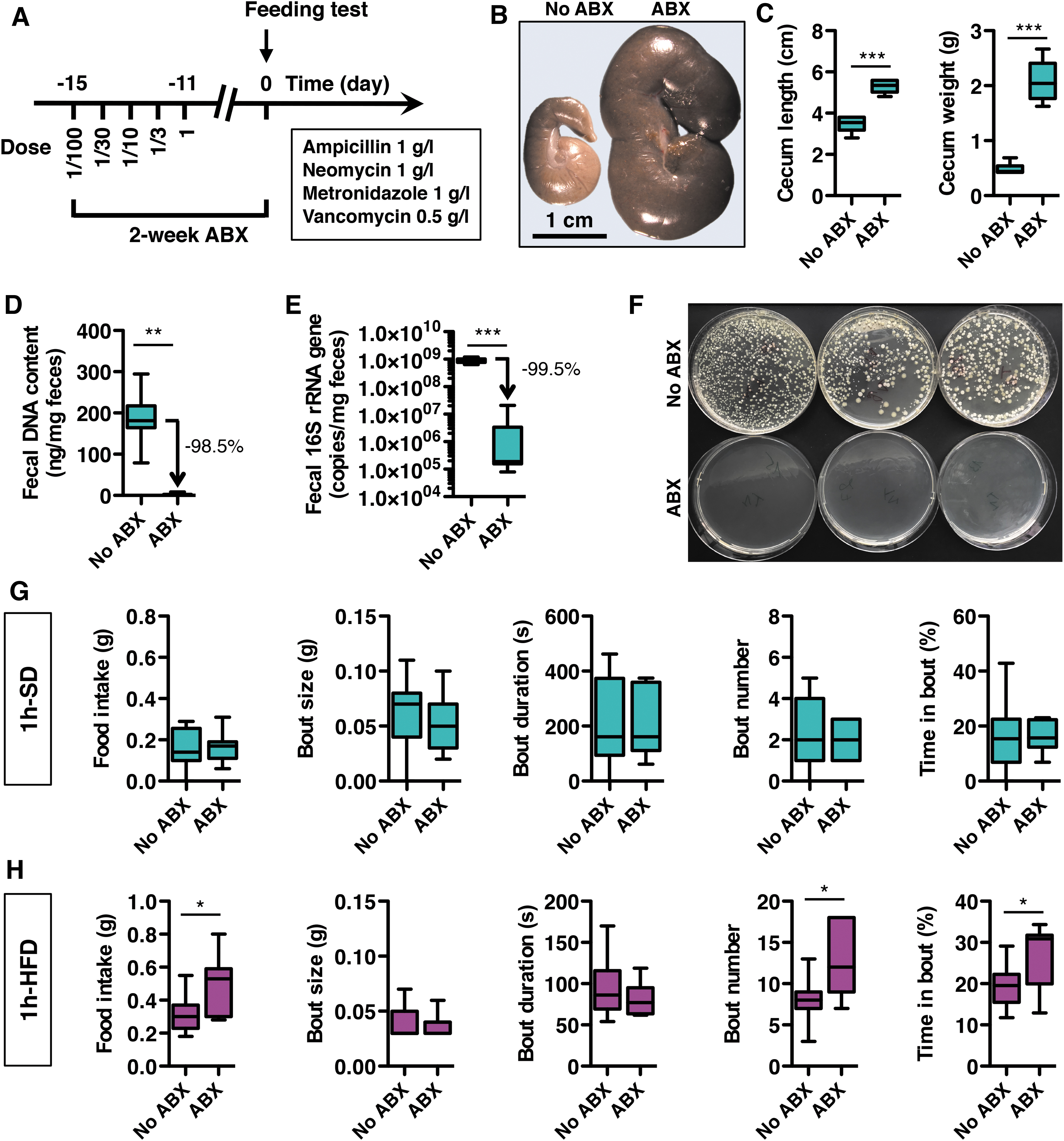
Moreover, fecal cultures prepared from control and ABX-treated mice revealed no colony-forming units of viable aerobic bacteria in the ABX group, and confirmed that ABX resulted in microbiota depletion in the gut. Importantly, the ABX treatment did not alter food intake and body weight as measured under SD over the 24 h prior acute feeding tests (Supplementary Fig. S1A) and did not cause discomfort, clinical symptoms, or mortality. A few episodic decreases in daily water intake were observed during ABX treatment, but these were normalized before the analyses (Supplementary Fig. S1A).
In addition, ABX treatment did not alter mRNA expression of interleukin 1 beta (IL-1β) and cyclooxygenase-2 (COX-2), two proinflammatory markers, while it markedly downregulated expression of tumor necrosis factor alpha (TNFα; Supplementary Fig. S1B). Notably, ABX treatment did not change the mRNA expression of a set of redox markers in the gut, including the anti-inflammatory and antioxidant transcription factor nuclear factor erythroid 2-related factor 2 (NRF2), and the two antioxidant enzymes: catalase (CAT) and superoxide dismutase-1 (SOD-1). Thus, the ABX treatment per se did not initiate gut inflammation or oxidative stress.
Mice were then fed either a SD or HFD, and feeding behavior was monitored for 1 h during the active period of the mice, at the onset of the night period (Supplementary Fig. S1C, D). ABX had no effect on feeding behavior in mice fed a SD for 1 h (Fig. 2G). In contrast, ABX increased HFD food intake over the 1-h period (Fig. 2H). This increase in food consumption for the microbiota-depleted mice fed HFD was explained by a higher number of feeding bouts without any change in the size or duration of these bouts. As a result, the time spent feeding was markedly increased in ABX-treated mice on HFD compared with controls, with consequently reduced interbout intervals. These results reveal a constitutive ABX-sensitive short-term control of food intake that develops during a high-fat meal, supporting the existence of a macronutrient-dependent release of microbiota-derived satietogenic signals.
HFD changes the cecal microbial metabolic signature at the time scale of a meal
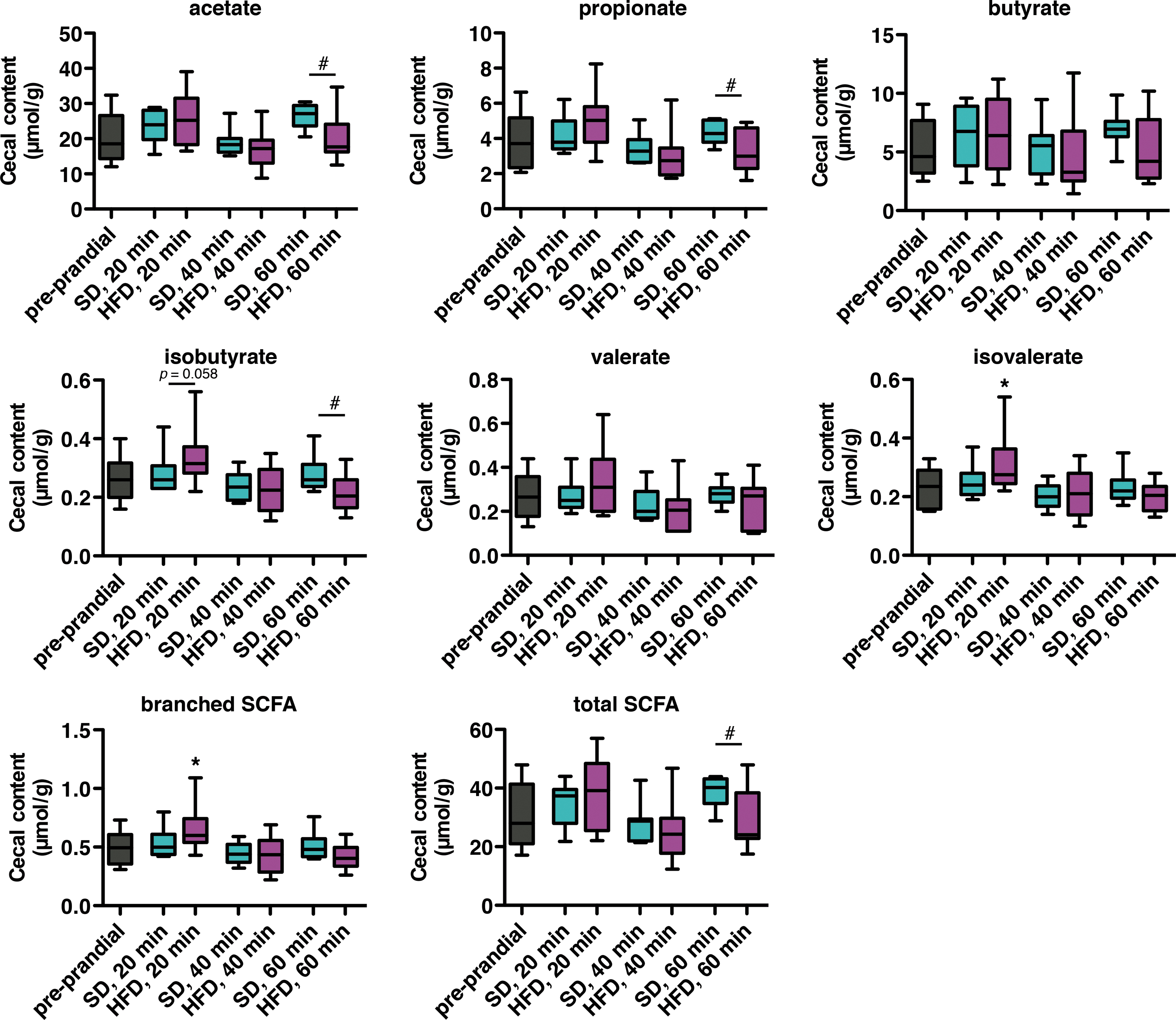
Feeding behavior in microbiota-depleted mice varied depending on the type of food ingested (SD or HFD). To determine the source of this differentiation, we first investigated postprandial changes in microbial metabolites produced in the cecum during SD and HFD consumption in non-ABX–treated mice. Levels of cecal short-chain fatty acids (SCFAs) were analyzed by gas chromatography. While consumption of SD did not significantly change SCFA concentration in the cecum compared with the preprandial state, HFD briefly increased cecal branched SCFA, and isovalerate in particular, after 20 min (Fig. 3). In addition, the level of total SCFA was reduced after 1 h of HFD feeding compared with SD. This is largely explained by the reduction in acetate and propionate levels. These data show that the postprandial microbial metabolic signature varies according to the type of ingested diet.
Postprandial GLP-1 response varies according to food composition
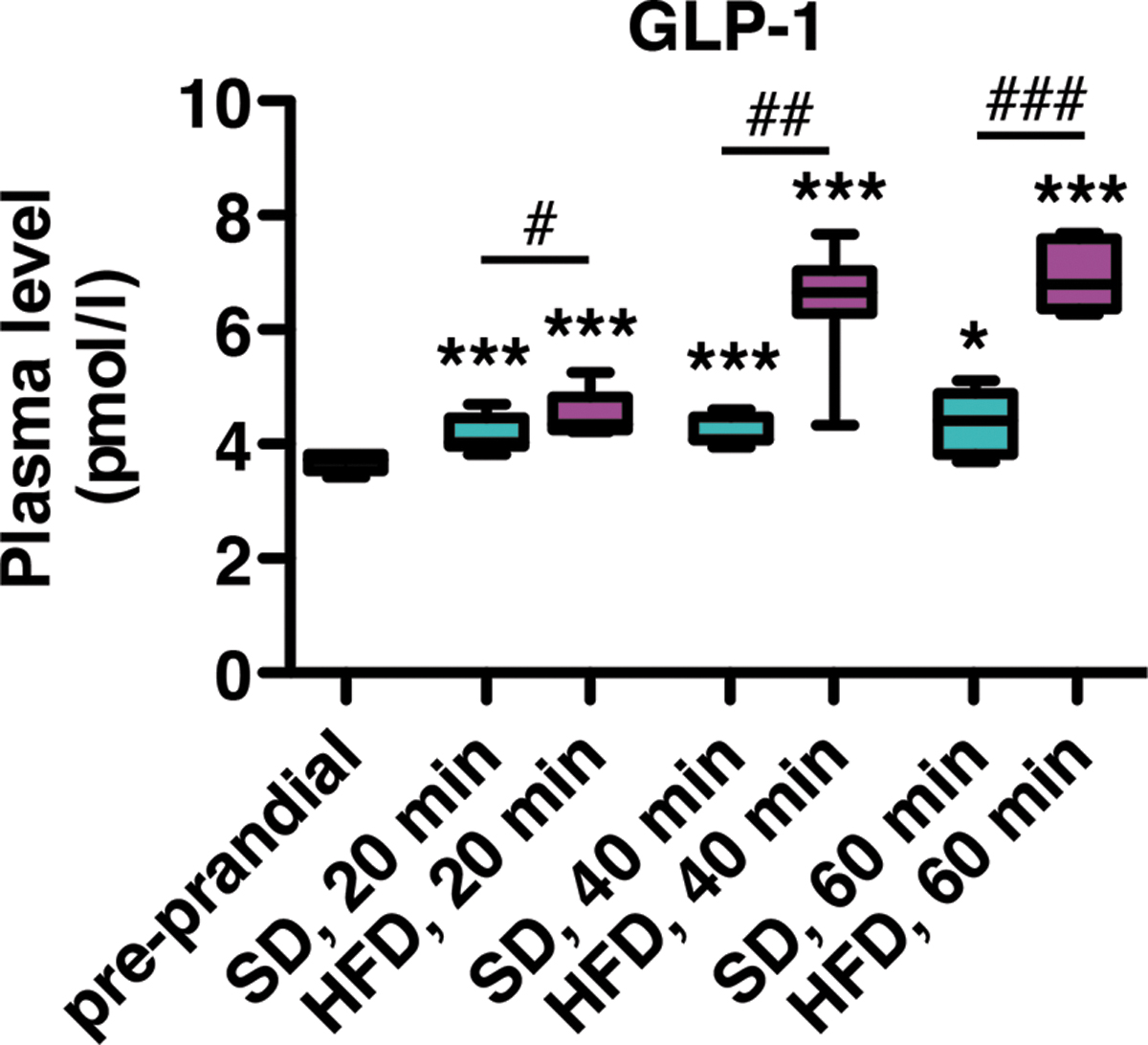
During food consumption, the endocrine gut releases several hormones that stimulate meal termination and satiety. The gut microbiota has been shown to modulate the release of endogenous stores of gut hormones into the blood (40). Before examining the effect of ABX treatment on the postprandial release of gut hormones in our model, we first monitored the postprandial GLP-1 response as an indicator of gut hormone pathway activation. As expected, plasma levels of total GLP-1 increased in mice after consumption of SD (Fig. 4). This release was further increased after HFD consumption, indicating differences in hormonal signaling that are dependent on food composition.
HFD induces a proinflammatory response in the gut at the time scale of a meal
Gut microbiota is a critical inducer of immune responses, and the immune system constitutes one of the routes of communication for the gut–brain axis (9, 26, 44). Gut-associated immune cells are components of this system and contribute to the maintenance of tissue homeostasis. Thus, we assessed the abundance of cluster of differentiation 4 (CD4)-positive and CD8-positive T lymphocytes and F4/80-positive macrophages in histological tissue sections of intestine from mice fed SD or HFD for 1 h (Supplementary Fig. S2).
We found that SD consumption increased CD4 immunostaining in the small intestine, suggesting the recruitment of CD4-positive thymus-derived lymphocytes (T cells) in the gut after a balanced meal (Fig. 5A). A slight increase in CD8 and F4/80 staining was also observed in some intestinal segments after 1 h-SD, but it remained below the significance threshold in most cases, suggesting that immune cells were not recruited in the gut for HFD for 1 h (1 h-HFD). Instead, samples from HFD-fed mice had reduced F4/80 staining in the jejunum and the colon, suggesting withdrawal of gut macrophages after HFD ingestion. More generally, these observations suggest reduced immune defense in the gut after HFD consumption.

Inflammatory molecules that are released in the intestinal microenvironment dictate immunological reactions and propagate inflammatory responses (72). We investigated the postprandial regulation of key inflammatory markers in intestinal tissues of mice fed SD or HFD for 1 h. Expression levels of IL-1β, TNFα, and COX-2 mRNA were significantly increased in both the jejunum and duodenum after HFD consumption (Fig. 5B). No changes were detected in distal parts of the gut. Taken together, these data indicate specific postprandial immune responses in the gut as a function of the type of diet ingested, and support the idea that HFD consumption induces local proinflammatory responses.
We also assessed the postprandial regulation of the key redox markers in the intestines of mice fed SD or HFD for 1 h, including NRF2, CAT, and SOD. Analyses revealed some changes in the duodenum but not in distal parts of the gut (Fig. 5C). NRF2 was upregulated after 1 h-HFD, but not after SD, and conversely, CAT was upregulated in response to SD but not HFD. These data suggest rapid postprandial changes in redox homeostasis in the upper parts of the gut.
HFD induces a systemic inflammatory response at the time scale of a meal
Next, we compared the systemic postprandial inflammatory response in mice fed SD or HFD for 1 h by measuring plasma level of several inflammatory factors. Biochemical analyses revealed a rapid and sustained elevation of the chemokine C-C motif chemokine ligand-5 (CCL5) (alias regulated upon activation, normal T-cell expressed and secreted [RANTES]) in the plasma, 20 min after the introduction of HFD food, but not after SD feeding (Fig. 6). HFD, but not SD, consumption also increased plasma levels of IL-1β, IL-6, and TNFα within 1 h. Plasma levels of other factors such as CCL2 (alias monocyte chemoattractant protein-1 [MCP1]), IL-10, and interferon γ were not affected after feeding, regardless of the diet. No change in blood cell counts was found after either diet (Supplementary Fig. S3).

These results show the induction of a rapid systemic inflammatory response after HFD but not after SD consumption.
HFD induces a postprandial endotoxemic response
Nutritional obesity is associated with increased blood levels of LPS, a constituent of Gram-negative bacteria (78). This bacterial component acts on peripheral metabolism and might also affect food intake (16, 28). Thus, we examined blood levels of lipopolysaccharide (LPS) in SD- and HFD-fed mice. We found that consumption of SD for 1 h did not alter levels of circulating LPS (Fig. 7A). However, consumption of HFD significantly increased levels of LPS in the blood as soon as 40 min after food introduction.
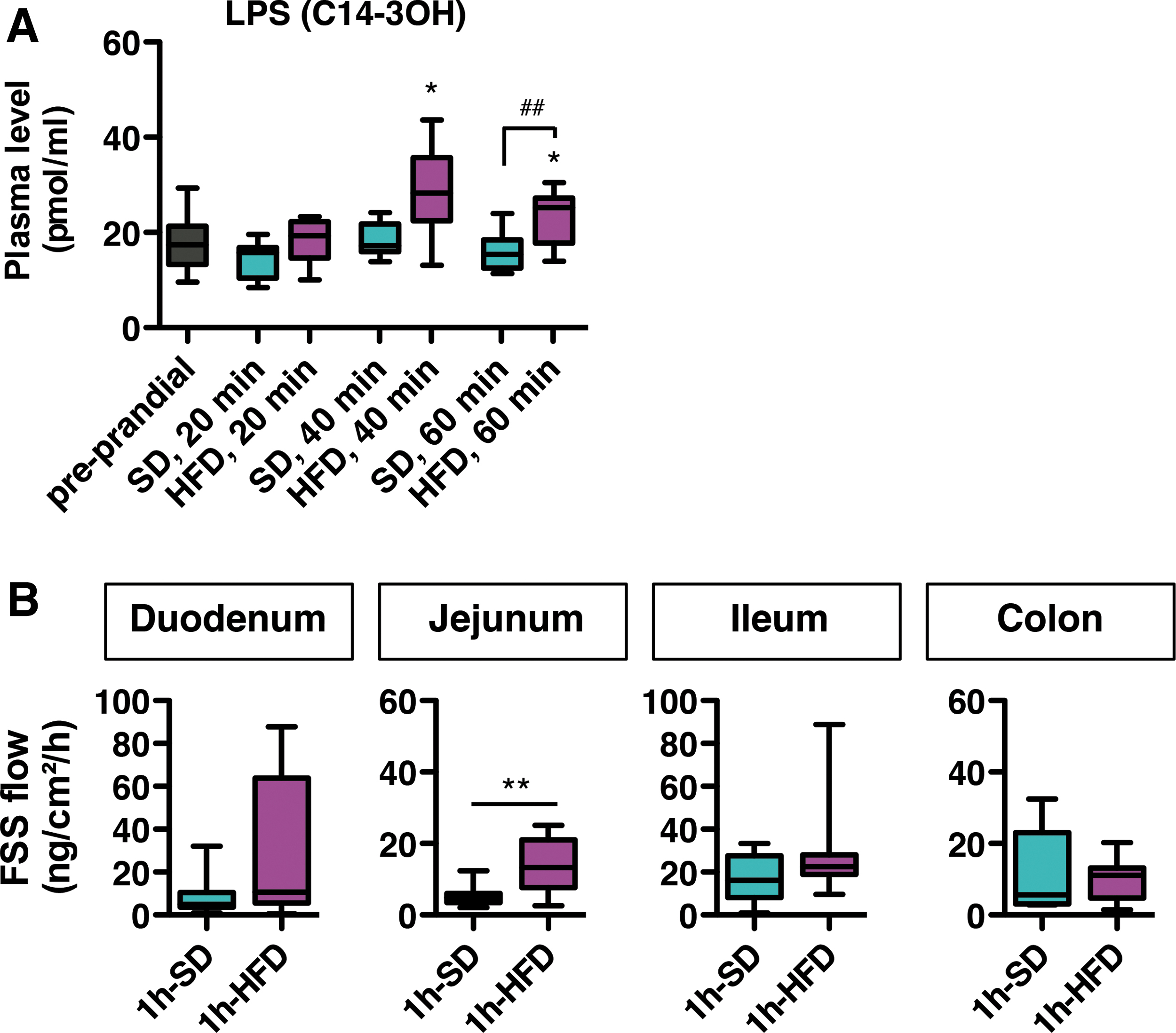
LPS enters systemic circulation via a transcellular pathway during fat absorption and can also translocate from the gut lumen to blood vessels via a paracellular pathway once the gut barrier is altered. We evaluated intestinal paracellular permeability in mice after 1 h-SD and 1 h-HFD by measuring fluorescein sodium salt (FSS) diffusion through gut biopsies mounted in Ussing chambers. We found higher permeability in the jejunum from 1 h-HFD mice compared with 1 h-SD (Fig. 7B). These results indicate impaired gut permeability and endotoxemia at the time scale of a meal after HFD but not SD consumption.
ABX-induced microbiota depletion affects postprandial metabolic responses but does not inhibit postprandial GLP-1, inflammatory, or endotoxemic responses
ABX treatment affected feeding behavior only under HFD. Therefore, we evaluated the effects of ABX treatment on different biological outcome parameters affected by 1 h-HFD. We compared 1 h-HFD and preprandial (control) mice that received or did not receive ABX treatment. We first examined the effect of ABX treatment on levels of cecal SCFA, which were analyzed by gas chromatography. Although the amount of cecal SCFA was substantial in ceca from untreated mice, these contents became undetectable in 1 h-HFD ABX-treated mice (Fig. 8A), indicating that microbiota metabolism was dramatically reduced by the ABX treatment.
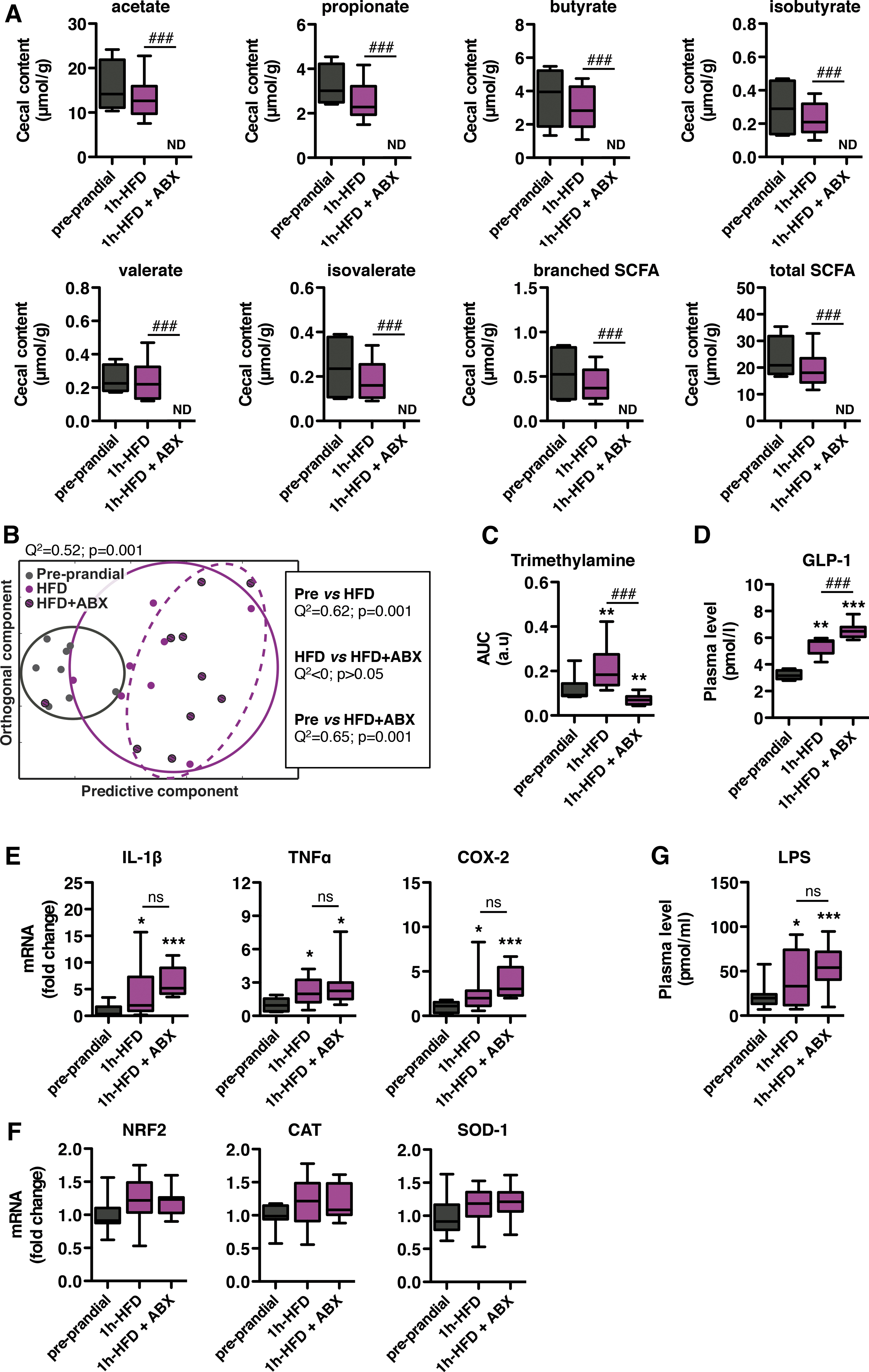
Cecal biochemical assays were completed by 1H NMR-based metabolomic analysis of plasma samples from the same mice. Statistical orthogonal projection on latent structure-discriminant analysis revealed a specific metabolic profile in plasma from 1 h-HFD mice as compared with preprandial mice (Fig. 8B). Conversely, the global analysis indicated no significant difference between samples from untreated and ABX-treated 1 h-HFD mice. Interestingly, of all the identified NMR spectra, trimethylamine, a molecule produced by the gut microbiota, was the only circulating metabolite found to be affected by ABX. In fact, postprandial plasma levels of trimethylamine were significantly increased in 1 h-HFD–fed mice, but ABX treatment completely prevented this HFD-induced postprandial rise (Fig. 8C).
We subsequently examined the effect of ABX treatment on the postprandial GLP-1 response. Again, plasma levels of total GLP-1 were elevated in 1 h-HFD mice compared with those of preprandial mice (Fig. 8D). Remarkably, plasma levels of total GLP-1 were further increased in samples from ABX-treated mice under HFD for 1 h.
To investigate the influence of the microbiota on the HFD-induced postprandial inflammatory response, we analyzed expression of proinflammatory markers in jejunum tissues. As observed before, expression levels of IL-1β, TNFα, and COX-2 mRNAs were increased in 1 h-HFD mice relative to preprandial mice (Fig. 8E). Interestingly, ABX treatment did not alter postprandial induction. In addition, expression levels of NRF2, CAT, and SOD in the jejunum remained unchanged in 1 h-HFD samples and were unaffected by the ABX treatment (Fig. 8F).
Finally, we analyzed the HFD-induced postprandial endotoxemic response in microbiota-depleted mice. The level of circulating LPS was increased in plasma samples from 1 h-HFD mice, and this HFD-induced endotoxemic response was not affected by the ABX treatment (Fig. 8G).
The NLRP3-inflammasome is not involved in the short-term regulation of food intake
The postprandial release of IL-1β regulates metabolism and behavior (30, 61). Maturation of pro-IL-1β into its active and secreted form usually requires the proteolytic activity of caspase-1, which is under the control of a multiprotein complex, the inflammasome (79). Apoptosis-associated Speck-like protein containing a CARD (ASC) and NLRP3, two components of inflammasome, are both involved in the outcome of metabolic disorders (80).
To determine the role of ASC and NLRP3 in the short-term regulation of food intake, we examined the feeding behavior of genetically modified mice lacking ASC or NRLP3 (ASC-knockout [KO] and NLRP3-KO mice, respectively). Deficient mice were exposed to either SD or HFD for 1 h, and compared with wild-type (WT) mice. The feeding behavior of ASC-KO mice was similar to that of WT mice, regardless of the diet (Fig. 9A, B). Similar results were found in NLRP3-KO mice (Fig. 9C, D). At the time of testing, ASC-KO mice had significantly lower body weight compared with controls, and there were no significant differences in body weight for NLRP3-KO mice versus controls (Supplementary Fig. S4).
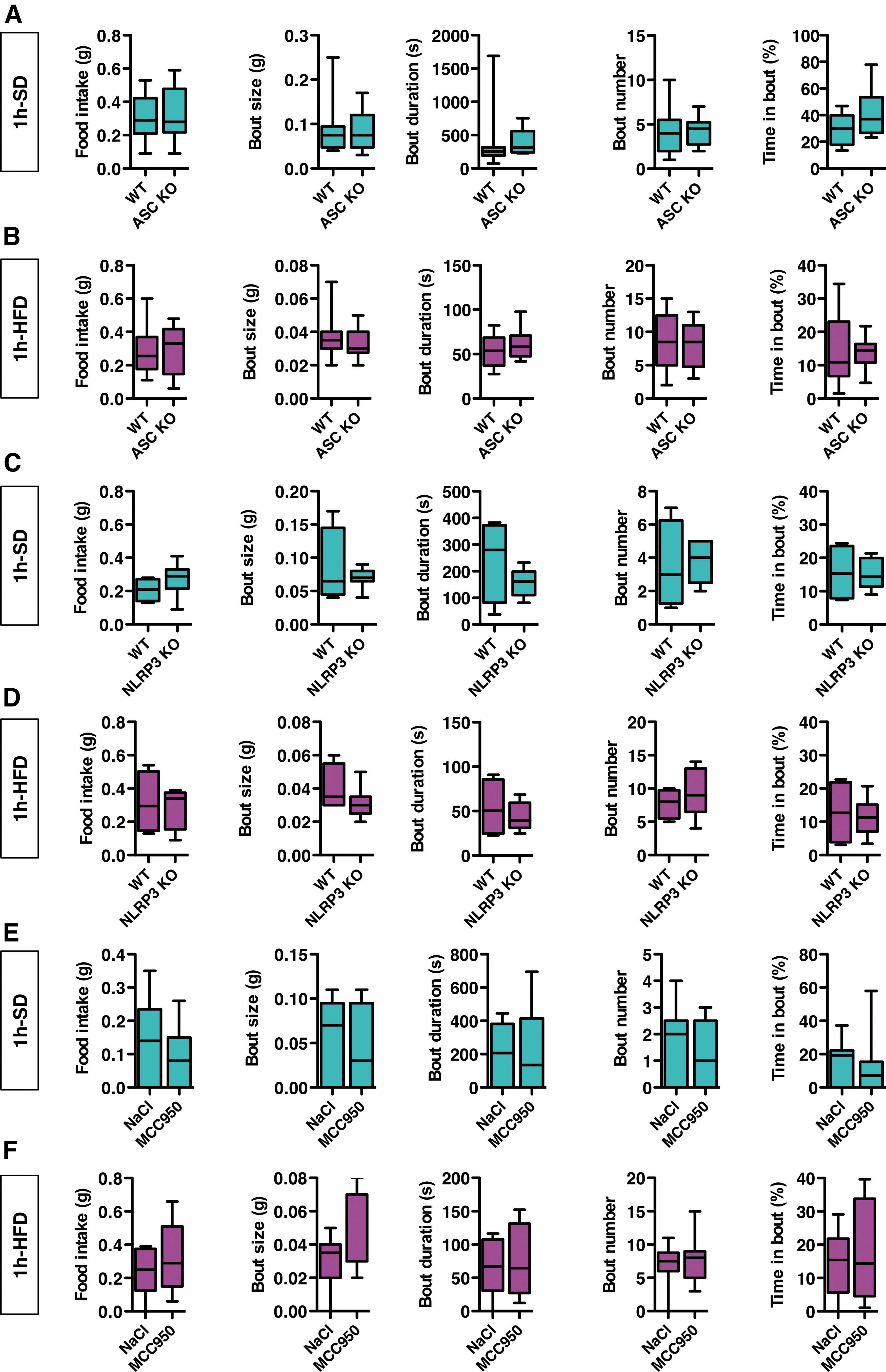

Since compensatory mechanisms that are inherent to genetic models might have masked the role of ASC and NLRP3 in the short-term regulation of feeding behavior, we performed a pharmacological experiment in adult WT mice using MCC950, a small-molecule inhibitor of the NLRP3-inflammasome (25). Treated mice received an intraperitoneal injection of MCC950 (20 mg/kg) 2 h before the test. Control mice received vehicle (saline). Per the experiments described above, mice were fed either a SD or a HFD for 1 h. MCC950 did not alter feeding behavior in mice under SD or HFD (Fig. 9E, F). Taken together, these experiments suggest that the NLRP3-inflammasome is not involved in the short-term regulation of food intake.
LPS-binding TLR4 receptor is not involved in the short-term regulation of food intake
In the host, LPS is sensed by the pattern-recognition TLR4 receptor (45). To address the role of TLR4 in the short-term control of food intake, we used mice deficient in TLR4 (TLR4-KO mice). We compared feeding behavior in TLR4-KO and WT mice. As described above, behavior was analyzed with either SD or HFD for 1 h. The feeding behavior of TLR4-KO mice was not different from that of WT mice, regardless of the diet (Fig. 9G, H).
We performed an additional pharmacological experiment in adult WT mice using TAK-242, a TLR4 antagonist (83). Mice received either an intraperitoneal injection of TAK-242 (10 mg/kg) or vehicle (saline), 2 h before the behavioral test. TAK-242 did not alter feeding behavior in mice in response to SD or HFD for 1 h (Fig. 9I, J). Taken together, these results suggest that TLR4 is not involved in the short-term regulation of food intake.
Discussion
In this study, we provide evidence for the constitutive microbial control of food intake at the time scale of a meal in mice. We found that food intake was abnormally increased in microbiota-depleted mice after a high-fat meal, showing that the microbiota is able to regulate food intake under normal physiology, that is, in healthy mice, with no need of additional microbiota boosters. This result is consistent with current theories supporting a beneficial effect of the microbiota on appetite regulation (2, 34, 54, 89).
Importantly, the behavioral effect of the microbiota appeared after a high-fat meal only and was not observed after a balanced meal, showing that an interaction between food and the microbiota elicits the anorectic effect. Similar results have been found in overfed mice kept on HFD for 3 days, for which excessive calorie consumption was aggravated by microbiota depletion (56). Likewise, no microbiota-dependent anorectic effect has been established in control mice maintained on SD (56, 92). Thus, these data reveal the constitutive and macronutrient-dependent anorectic effect of the gut microbiota.
Characterization of the constitutive microbial effect on feeding behavior
We found that the number of feeding bouts increased and the time between bouts decreased in microbiota-depleted mice fed HFD, suggesting that microbial activity under the HFD increases interbout intervals, and that microbiota-derived factors are satietogenic. The positive effect of the microbiota on satiety has been already proposed, but most studies (i) found that the microbiota altered host satiety-related signaling and brain pathways without evidence for a functional outcome, and/or (ii) reported an effect on cumulative food intake but not on the structure of feeding behavior (12, 33, 74, 85).
Other preclinical studies have addressed the anorectic role of microbiota specifically during obesity or diabetes (4, 20, 36). In the few behavioral studies showing an effect of the microbiota on satiety, the microbiota was stimulated experimentally with pre- or probiotics before analyses, or isolated bacterial molecules were given before a feeding test (5, 11, 17, 18, 21, 76). In this study, we demonstrate that constitutive microbes can contribute to satiety in normal physiological conditions after consumption of a high-fat meal.
Seeking the satietogenic pathway that mediates the anorectic effect of microbiota during HFD consumption
How could the gut microbiota influence satiety under physiological conditions? Details of this relationship are not completely understood. In this study, we explore the contribution of four well-known microbiota-associated routes in the gut–brain axis, including metabolic, hormonal, immune, and endotoxemic pathways.
We found that the satiety effect of the microbiota occurred after a high-fat meal only, with the postprandial inflammatory response. In particular, our data indicate reduced immune cell infiltration in intestinal tissues under HFD, which could lower local immunity and increase susceptibility to pathogens. A shift in gut immune cell populations has been already reported in response to HFD for a few days, likely reflecting cell migration toward the peritoneal cavity, adipose tissues, and liver (30, 38, 49, 62).
Here, we show that gut immune cell remodeling is induced very rapidly after HFD consumption. Thus, this process might contribute to the postprandial inflammatory response. Although induction of postprandial inflammatory reactions by a high-fat meal has been already reported (22, 42, 43, 65), the physiological role of this response remains unclear. Recently, the postprandial release of IL-1β was found to be required for the accurate homeostatic control of peripheral glucose metabolism (30). Moreover, IL-1β signaling is involved in the initiation of postprandial fatigue, suggesting that the postprandial surge of this cytokine might act centrally (61).
Nevertheless, behavioral studies from our genetic and pharmacological models, including ASC-KO, NLRP3-KO, and MCC950-treated mice, show that inhibition of the NLRP3-inflammasome, a master regulator of inflammatory responses, does not alter HFD feeding during the first hour, suggesting that postprandial inflammation does not contribute to the regulation of food intake within 1 h of HFD. Interestingly, the commensal gut microbiota composition remains unchanged in inflammasome-deficient mice, which minimizes confounding factors (66).
In addition, molecular studies show that HFD-induced intestinal inflammatory markers were not suppressed by the ABX treatment. This is in line with a previous study showing that the postprandial rise in circulating IL-1β is not affected by microbiota depletion (30). Thus, ABX-insensitive postprandial inflammatory responses are probably not involved in the HFD-dependent anorectic effect of microbiota. Although this study does not support a role for the postprandial NLRP3-mediated inflammatory response in feeding behavior within 1 h, possible roles for other inflammatory pathways in feeding behavior should not be totally excluded. More delayed effects of the gradual inflammatory response should be also examined. Indeed, disruption of the nuclear factor-kappa B signaling in the brain impairs the homeostatic feeding response to dietary fat (13).
We also found that ABX did not change basal expression of key redox markers in the gut. Moreover, levels of redox markers in the jejunum were not affected by either HFD or ABX. These data suggest that the postprandial microbiota-dependent regulation of food intake during HFD does not engage marked changes in the intracellular redox state in the gut.
Consistent with previous rodent and human studies, we described a specific postprandial endotoxemic response to HFD (22, 32, 59, 90). Interestingly, the level of circulating LPS is positively correlated with energy intake, and sustained endotoxemia might deregulate food intake (3, 28). However, in our study, HFD-induced postprandial endotoxemia was not reduced by the ABX treatment while food intake increased. The remaining systemic LPS in microbiota-depleted mice fed with a HFD for 1 h might be derived from ingested foods, as was previously proposed to explain detection of basal levels of blood LPS in germ-free (GF) animals (15). LPS might reach the blood compartment more easily when dietary fats are consumed and/or when gut permeability is altered.
Thus, short-term control of food intake after a high-fat meal does not seem to depend on circulating LPS, which has a well-established anorectic effect. Indeed, genetic and pharmacological interventions targeting TLR4, the mammalian endotoxin sensor, failed to inhibit food intake under HFD, indicating that TLR4 signaling is not involved in the control of food intake at the time scale of a meal. Interestingly, the postprandial inflammatory response does not require TLR4 activation either (43). Although LPS alters peripheral metabolism during sustained HFD exposure and might influence long-term regulation of food intake, the functional effect of the early postprandial rise of blood LPS remains to be elucidated, and the role of additional LPS sensors should be considered in this case (68).
To investigate whether the hormonal route could mediate the microbiota-derived satietogenic effect in our model, we examined the postprandial GLP-1 response as a relevant marker. We observed an elevated level of total GLP-1 in plasma of mice fed a HFD for 1 h. Remarkably, ABX did not reduce this hormonal response in 1 h-HFD mice but further increased it.
This result is consistent with previous studies, suggesting that the microbial metabolism negatively regulates GLP-1 levels (91). At the same time, abnormal elevated food intake was found in HFD-fed ABX-treated mice. Thus, the elevated plasma levels of total GLP-1 in ABX-treated mice corroborate the ABX-induced hyperphagia under HFD and can be viewed as a physiological consequence of the excessive feeding without an immediate behavioral effect. Since GLP-1 is anorectic (14, 84), these data show that during the 1 h-HFD test, the microbiota does not modulate feeding behavior via the GLP-1 signaling pathway. If GLP-1 sensitivity is maintained in ABX-treated mice, delayed GLP-1 action would probably take place after the 1-h test period.
Finally, the roles of hormonal, immune, and endotoxemic pathways in the mediation of the microbiota-derived anorectic effect during a high-fat meal appear unlikely. More studies are needed to identify pathways and mediators underlying this effect. Nevertheless, we observed that the microbiota-derived anorectic effect happens under HFD only, a nutritional condition characterized by a specific microbial metabolic signature detectable both in the gut and in the blood.
Indeed, we found a brief elevation of cecal branched SCFA after 20 min of HFD consumption. Cecal acetate, propionate, butyrate, and valerate also increased under HFD at this time point, but did not reach statistical significance. Branched SCFAs, such as isovalerate and isobutyrate, are end-products of bacterial proteolytic fermentation (64). The production of branched SCFAs increases during acute consumption of animal-based food (27). Although some studies reported an effect of branched SCFAs on gut motility, colonocyte functions, and liver metabolism, the impact of these molecules on the host feeding behavior is still unknown (10, 41, 48).
The proton NMR-based metabolomic analysis failed to discriminate a global ABX-sensitive postprandial metabolic response in the blood after HFD consumption; however, it is clear that the HFD-induced postprandial increase in blood trimethylamine was lessened by the ABX treatment. This result shows that some microbial molecules can reach the systemic compartment in response to the type of ingested food.
Thus, microbial metabolites produced in the gut lumen could have initiated a HFD-specific behavioral response either locally in the gut or directly in brain centers via the humoral route. Further analyses in the proximal intestine and over shorter postprandial periods will help better delineate all these metabolic changes. Likewise, more precise assays, for instance using liquid chromatography/mass spectrometry to measure microbial molecules in arterial and portal blood, are required to obtain a comprehensive view of postprandial changes in these metabolites. It would also be interesting to investigate the role of trimethylamine in the control of feeding behavior.
Other bacterial enterosynes, including microbiota-derived neurotransmitters, gaseous active molecules, aromatic compounds, and mimetics, which are known to modulate host metabolism and behavior, should be also investigated in the future (1, 34, 55). The gut endocannabinoid system could be involved as well since related bioactive lipids can mediate the effects of the gut microbiota on host metabolism (70). Most of these molecular actors can be sensed locally by the enteric nervous system to modulate the brain postprandial response (19).
Methodological considerations
One limitation of our study is that we did not include prandial drinking in the meal definition although “drinking-explicit” models are proposed to be the most appropriate methods for analyzing meal patterns (81, 93). Thus, the measurement of feeding bouts alone does not fully reflect the entire ingestive behavior and might provide an approximation of meal structure. Second, we analyzed raw feeding bouts without any mathematical corrections. Reanalyzing data in meals using a standard 5-min intermeal interval provided similar results (Supplementary Fig. S5).
The size and duration of meals tended to increase in HFD-fed mice treated with ABX, suggesting that microbiota could be also involved in satiation. Indeed, we found increased cumulative food intake and increased time of feeding bouts and meals within the first hour of HFD in microbiota-depleted mice. Thus, regardless of the model used for assessing feeding behavior, these results indicate that the microbiota modulates the short-term regulation of food intake under physiological conditions.
Based on the ABX-induced microbiota depletion approach, we found evidence for the contribution of the microbiota in the feeding behavior in adult mice. In contrast, we failed to reproduce a similar phenotype in GF mice. Indeed, GF mice did not eat more than conventional (conv) mice during 1 h-HFD (Supplementary Fig. S6), despite careful matching in age and weight of GF and conv mice during the experiment. For the ABX depletion, we used a typical mix of drugs, including ampicillin, neomycin sulfate, metronidazole, and vancomycin.
Anatomical, molecular, biochemical, and bacteriological analyses of cecum content and feces indicated that our protocol strongly depleted gut bacteria. Despite this, the presence of some ABX-resistant bacteria living after the pharmacological intervention was still probable. However, unlike in GF mice in which the absence of microbes is complete and constant over the life span, the use of ABX avoids critical defects in immune and homeostatic systems observed in adult GF mice (51). Thus, one can speculate that defects in GF mice might have altered the microbiota-derived satietogenic effect that happens in conventional mice.
In our study, the ABX dosage was increased progressively to acclimate the mice to the bitter taste and to ensure normal hydration. Treatment was not supplemented with sweeteners or antifungals. It was designed first and foremost to maintain normal metabolism and behavior before the feeding test. Indeed, the body weight and feeding behavior of ABX-treated mice were unchanged under SD using this protocol. Nevertheless, we found that the treatment increased food intake when fatty foods were consumed, highlighting the role of microbiota in the control of food intake. Since the gut microbiota influences various satiety-related pathways, the effect of the treatment was likely derived from gut microbiota depletion.
On the contrary, commensal bacteria reside in various parts of the body, including the mouth, and recent studies suggest a link between the oral microbiota and food preference (23, 31, 67). Similar studies established a link between olfactory microbiota and olfactory epithelium functioning (35, 71). Accordingly, a change in taste and smell perception might account for the increase in food intake in HFD-fed mice under ABX treatment. However, we found that the size and duration of feeding bouts were not changed in microbiota-depleted mice, which is not in favor of an alteration in olfactogustatory sensing, in food preference and satiation.
Conclusion
This work provides evidence that commensal bacteria promote satiety in mice under healthy conditions. We found that the microbial anorectic effect develops according to the composition of the meal. Strikingly, the microbiota limits excessive calorie consumption in mice eating high-fat food, highlighting the homeostatic role of the microbiota over the course of normal physiology. This finding strengthens the concept that appetite is shaped by complex interactions between diet and commensal bacteria.
Materials and Methods
Electronic laboratory notebooks were not used.
Animals
All protocols involving animals were reviewed and approved by the local ethics committee, and were in strict accordance with the European Community guidelines. All protocols were agreed by the French Ministry of Higher Education, Research and Innovation (accreditation no. APAFIS no. 18117 and no. 1285-2015102618593445v3).
Most experiments were carried out on 7/9-week-old male C57Bl/6J mice from Charles River Laboratory (France). We also examined transgenic homozygous Asc−/− , Nlrp3−/−, and Tlr4−/− mice (named hereinafter ASC-KO, NLRP3-KO, and TLR4-KO mice, respectively), which were bred in a C57Bl/6J genetic background. Transgenic animals were compared with WT mice bred in the same environment. All animals were housed at 22.5°C ± 1°C on a 12–12 h reversed light–dark cycle (light off at 10:30 a.m.; light on at 10:30 p.m.), in a specific pathogen-free facility. Before experiments, mice had free access to food and water.
For the study on GF and conventional (conv) mice, GF mice were obtained from Anaxem GF animal facilities of the Micalis Institute, INRAE (France), and conv mice were purchased from Charles River Laboratory (France). Mice were 8-week-old C57Bl/6J males. GF and conv mice were transferred to two separated sterile isolators (Eurobioconcept, France) allowing the continuous maintenance of sterility as described previously (69). Inside the isolators, the animals were housed in individual cages containing sterile bedding, and had free access to autoclaved tap water and γ-irradiated (45 kGy) food. The animal room was maintained at 22°C ± 2°C and kept on a 12 h light/dark cycle (lights on at 7:30 a.m.).
ABX treatment
At the beginning of the treatment, animals were housed individually. ABX were provided in drinking water for 15 days. The mix contained ampicillin (1 g/L; cat. no. A-9518; Sigma-Aldrich), neomycin sulfate (1 g/L; cat. no. N1876; Sigma-Aldrich), metronidazole (1 g/L; cat. no. M3761; Sigma-Aldrich), and vancomycin (0.5 g/L; cat. no. 00315; Chem Impex International). Drugs were diluted in a final volume of 15 mL. The dosage was increased gradually (1/100, 1/30, 1/10, 1/3, 1) to reach final concentration after 4 days (Fig. 1A). ABX were changed every day for the first 4 days, and then renewed every second day.
Experimental design
For experiments, mice were single caged, and food was removed for 2 h before lights off as previously described (73) (Supplementary Fig. S1C). Mice were trained to the food removal three times. Preprandial mice were sacrificed at the end of the food restriction period, at 10:30 a.m. Postprandial mice received food at the beginning of the dark period and were sacrificed at different time points (i.e., 20, 40, and 60 min) after food introduction. During the tests, food was either the SD (cat. no. A04; SAFE) or a semisynthesized HFD (cat. no. U8954 v7; SAFE) (Supplementary Fig. S1D). To prevent food neophobia, mice received one HFD pellet for 10 min 1 day before trial. For axenic experiments, food was γ-irradiated (45 kGy) SD or HFD. Before sacrifice and dissection, mice were anesthetized by intraperitoneal injection of a ketamine–xylazine mix (120–24 mg/kg).
Drug administration
TAK-242, a TLR4 antagonist (cat. no. R-3152492-1; Merck), was prepared in 0.9% NaCl containing 1% DMSO. MCC950, a NLRP3-inflammasome inhibitor (cat. no. HY-12815A; CliniSciences), was prepared in phosphate-buffered saline (PBS). Dosage was 10 mg/kg TAK-242 and 20 mg/kg MCC950, injected intraperitoneally (100 μL), 2 h before lights off, according to previous pharmacokinetic/pharmacodynamic studies (24, 25, 46). Controls received saline. Before experiments, mice were trained 3 times with ip injections of saline once a day before lights off. For experiments, mice were randomly assigned to receive TAK-242, MCC950, or vehicle.
Feeding behavior analysis
Except in axenic experiments, food intake was recorded using the BioDAQ System (Research Diets). Mice were acclimatized for 3 days to single housing and feeding through the food hopper. Cages contained enrichment and bedding material. Water was given ad libitum from regular bottles placed on the top of the cage. During habituation and experiments, food hoppers were closed for 2 h before lights off. Data were collected continuously and were analyzed using the BioDAQ DataViewer software (Research Diets). For the analysis, the minimum food amount was filtered at 0.02 g, and minimum interbout interval was set at 5 s. For the axenic experiment, food intake was measured manually.
Blood biochemistry
Mice were anesthetized, and blood was collected by cardiac puncture in ethylenediaminetetraacetic acid-coated tubes. Aliquots were sent to the CerbaVet laboratory for complete blood count (Wissous, France). Plasma samples were obtained by centrifugation at 2650 g for 10 min at 7°C.
Plasma levels of total GLP1 were measured by enzyme-linked immunosorbent assay (ELISA) using the Total GLP-1 NL-ELISA kit (cat. no. 10-1278-01; Mercodia). Plasma levels of cytokines were measured using a V-Plex multiplex assay (Meso Scale Discovery), according to the manufacturer's protocol. The plasma LPS concentration was determined by direct quantitation of 3-hydroxymyristate (or 3HM) by high-performance liquid chromatography coupled with tandem mass spectrometry as previously described (75). An initial HCl hydrolysis step was conducted to distinguish between unesterified 3HM and esterified 3HM; that is, true LPS. Thus, the LPS concentration was defined as total 3HM (as assessed after HCl hydrolysis) minus unesterified 3HM concentration (as assessed without HCl hydrolysis).
Quantitative assay of the bacterial 16S rRNA gene
The analysis was performed on fresh feces, which were first snap-frozen and stored at −80°C. The DNA extraction and assay were performed by the Biomnigene company. Samples were lysed and extracted using the E.Z.N.A DNA Stool kit (Omega Bio-tek). Extracted DNA was quantified using a Qubit 4 Fluorometer (Thermofisher).
Quantitative polymerase chain reaction (qPCR) targeting the 16S rRNA gene was performed using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems), with the following primers: forward 5′ CCT ACG GGA GGC AGC AG 3′, reverse, 5′ ATT ACC GCG GCT GCT GGC A 3′. A dilution series (from 0.0064 to 100 ng) of a sample derived-plasmid construct containing the target amplicon was run in parallel for absolute quantification. Reactions were performed on a QuantStudio 3 thermocycler (Applied Biosystems), with the following PCR cycling conditions: 50°C for 2 min, 95°C for 15 min followed by 35 cycles, including 95°C for 15 s, 60°C for 30 s, and 72°C for 30 s, followed by a step at 80°C for 20 s and a final step for melt curve analysis.
Fecal cultures
Fresh feces were collected in sterile tubes and mixed with PBS (1 g/mL feces/PBS). Feces were emulsified by vigorous vortex agitation for 5 min; 500 mL of surnageant was spread on plates containing solid sterile lysogeny broth agar. Plates were incubated under aerobic conditions for 3 days at 37°C.
Cecal SCFA assay
Cecal samples were water extracted, and proteins were precipitated with phosphotungstic acid as described previously (57). For the assay, 0.3 μL of the supernatant together with 2-ethylbutyrate as an internal standard were injected into a gas–liquid chromatograph (Agilent 6890, France), which was equipped with a split-splitless injector 7850, a flame ionization detector, and a capillary polyethylene glycol column (15 m, 0.53 mm, 0.5 μm). The flow rate of the carrier gas (He) non H2 was 10 mL/min. Inlet, column, and detector temperatures were 200, 100, and 240°C, respectively. The column temperature was first set at 100°C, held for 15 min, then programmed from 100 to 180°C at a rate of 20°C/min and held 2 min. 2-Ethylbutyrate was used as an internal standard.
Data were collected, and peaks integrated with openLab software, CDS GC ChemStation (Agilent, les Ulis, France).
Proton nuclear magnetic resonance-based metabolomics
Plasma samples were prepared for NMR analysis as described previously (8). All proton nuclear magnetic resonance (1H-NMR) spectra were obtained on a Bruker DRX-600-Avance NMR spectrometer (Bruker) using the AXIOM metabolomics platform (MetaToul) operating at 600:13MHz for 1H resonance frequency. Data acquisition, pretreatment, and statistical analysis were performed as described previously (63). Areas under the curve of several signals of interest were integrated, and significance tested with a Mann–Whitney test.
Gene expression assay in gut biopsies
Mice were anesthetized, and fragments of duodenum, jejunum, ileum, and colon were collected, snap-frozen, and stored at −80°C until use. Total RNAs were extracted using RNeasy Plus Mini Kit (Qiagen), according to the manufacturer's protocol. In brief, tissues were disrupted using lysis buffer and a TissueLyser (Qiagen). Lysates were first spun through gDNA Eliminator spin columns to remove genomic DNA. Total RNA was then purified using RNeasy Mini spin columns. The concentration and integrity of extracts were assessed by spectrophotometry (Nanodrop 2000; ThermoFischer Scientific) and with the Experion automated electrophoresis system (Bio-Rad). Reverse transcription was performed on 500 ng of total RNA using SuperScript IV VILO Master Mix (ThermoFischer).
PCRs were carried out in a 10 μL reaction volume containing 2 μL of 1:5 diluted cDNA, 2 μL of nuclease-free water, 5 μL of Taqman fast advanced Master Mix (Applied Biosystems), and 1 μL of appropriate Taqman probes (Applied Biosystems) for polymerase II subunit A (POLR2A) (Mm00839502), IL-1β (Mm00434228), TNFα (Mm00443258), COX-2 (Mm00478374), NRF2 (Mm00 477784_m1), CAT (Mm00 437992_m1), and SOD (Mm0 1344233_g1). Real-time PCRs were run on a StepOnePlus thermocycler (Applied Biosystems). The comparative cycle threshold method was used for relative quantification of target genes, using POLR2A as a reference gene and the preprandial condition as a control.
Analysis of gut permeability
Mice were anesthetized, and intestinal tissues were removed and opened longitudinally. Desired intestinal sections were cut and mounted on an Ussing chamber system (Physiologic Instrument, San Diego). The chamber opening exposed 0.5 cm2 of tissue surface area to oxygenated buffered solutions at 37°C. The serosal chamber was filled with 10 mM glucose-Krebs solution to provide fuel to the tissue, and the mucosal chamber was filled with 10 mM mannitol-Krebs solution to maintain osmotic balance. Paracellular permeability was determined using FSS (0.38 kDa), which was added to the mucosal chamber at t0. Medium from serosal chambers was sampled every 30 min for 120 min. The concentration of fluorescent sodium salt was measured by fluorimetry at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Immunostaining in histological gut sections
Mice were anesthetized, intestines were dissected, and intestinal segments were selected. Collected tissues were fixed overnight in 4% paraformaldehyde before embedding in paraffin. Paraffin-embedded tissues were cut, and sections were deparaffinized and rehydrated. Endogenous peroxidases were quenched with 3% hydrogen peroxide. Nonspecific binding of antibodies was prevented with 3% bovine serum albumin (BSA). Sections were incubated with primary antibodies diluted with 1% BSA in PBS, overnight at 4°C.
After washing, sections were incubated with a horseradish peroxidase enzyme-conjugated secondary antibody for 30 min at room temperature. Immune complexes were detected with NovaRed peroxidase substrate (cat. no. SK4800; Vector Labs). Sections were counterstained with Harris hematoxilin, washed, and mounted. Immunolabeling was analyzed using ImageJ software, using the Color Deconvolution and Statistical Region Merging parameters.
Primary antibodies were as follows: rat antimouse F4/80 for detection of macrophages (dilution: 1/100, cat. no. MCA497; AbD Serotec); rabbit anti-CD4 for detection of T-helper lymphocytes (dilution: 1/1000; cat. no. ab1836858; Abcam); and rabbit anti-CD8 for detection of cytotoxic T lymphocytes (dilution: 1/1000, cat. no. ab217344; Abcam). Secondary antibodies were as follows: antirat immunoglobulin G (IgG) (cat. no. MP-7404; Vector Labs) for F4/80-positive cells; and antirabbit IgG (cat. no. MP-7401; Vector Labs) for CD4-positive and CD8-positive cells.
Statistics
Statistical analyses were performed using Prism 5.0 software (GraphPad Software, Inc.). No statistical method was used to predefine sample size. Results on graphs were expressed as boxplot with individual data from min and max. Multiple comparisons of groups were carried out using one-way analysis of variance (ANOVA) followed by a post hoc Newman-Keuls test. Equality of variances and normality of distribution were checked before each analysis. The Mann–Whitney test was used for two group comparison and for nonparametric multiple comparisons. No correction was applied on data for analysis. A p value <0.05 was considered statistically significant.
Footnotes
Acknowledgments
We thank the animal facility of the Centre des Sciences du Goût et de l'Alimentation (CSGA), especially Anne Lefranc, Martin Bertomeu, and Virginie Cadiou for animal care, the CellImaP imaging facility of the Université de Bourgogne, especially André Bouchot, and Audrey Geissler for histological studies, and the Lipidomic facility of the Université de Bourgogne, especially Jean-Paul Pais de Barros for LPS assay.
We also thank the Biomnigene Company, especially Alexandre Douablin for 16S RNA gene analyses; students who assisted researchers, especially Alice Crétin-Anciaux, Sophie Lahmar, Marie Janod, and Laura Guénot; Vishva Dixit (Genentech) for providing ASC −/− mice and the ANIRA-PBES animal facility of SFR Biosciences (UAR3444/CNRS, US8/Inserm, ENS de Lyon, UCBL), for breeding these mice; and Abby Cuttriss (Office of International Scientific visibility) for editing this article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the French “Investissements d'Avenir” program, project ISITE-BFC (contract National Research Agency (ANR)-15-IDEX-0003), by the ANR (contract ANR-21-CE14-0033), by the Région Bourgogne and FEDER program (contract 2019-6200FEO047S00409) to A.B.; by the INRAE grant “Action Prioritaire du Département Alimentation Humaine” to A.B., G.B., and V.D.; by the Medisite Foundation and the French government, managed by the ANR, through the UCAJEDI Investments in the Future project (contract ANR-15-IDEX-01) to C.C., C.S., J.L.N., and C.R.; by the LABEX SIGNALIFE program (contract ANR-11-LABX-0028-01) to J.L.N. and C.R.; and by the Fondation pour la Recherche Médicale (contract DEQ20170336744) to V.P.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.