
Abstract
Significance:
Hydrogen sulfide (H2S) is reported to be an important mediator involved in numerous physiological processes. H2S and hydropersulfides (RSSH) species are intimately linked biochemically. Therefore, interest in the mechanisms of the biological activity of H2S has led to investigations of the chemical biology of RSSH since they are likely to coexist in a biological system. Currently it is hypothesized that RSSH may be responsible for a least part of the observed H2S-mediated biology/physiology.
Recent Advances:
It has been recently touted that thiols (RSH) and RSSH have some important differences in terms of their chemical biology and that the generation of RSSH from RSH is purposeful to exploit these chemical differences as a response to a physiological or biological stress. This transformation may represent an unappreciated/unrecognized biological mechanism for dealing with cellular stresses.
Critical Issues:
Although recent studies indicate a diverse and potentially important chemical biology associated with RSSH species, these ideas have their foundations in early studies (some over 60 years old). It is vital to recognize the nature of this early work to fully appreciate the current ideas regarding RSSH biology. Importantly, these early studies were performed before the realization of purposeful H2S biosynthesis (before 1996).
Future Directions:
Taking clues from the past studies of RSSH chemistry and biology, progress in delineating the chemical biology of RSSH will continue. Determination of the possible relevance of RSSH chemical biology to signaling and cellular physiology will be a primary focus of many future studies. Antioxid. Redox Signal. 36, 244–255.
Introduction
Small di- and triatomic molecules such as nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) are endogenously generated biological effectors/mediators and, along with dioxygen (O2), are important to the regulation and function of a variety of physiological signaling pathways (7, 36, 50). Among these small-molecule signaling agents (i.e., NO, CO, H2S, and O2), NO is the most defined and understood in terms of the primary biological target and the biochemical and physiological mechanisms of action. The predominant biological target responsible for the physiological actions of NO is the enzyme soluble guanylyl cyclase (sGC). NO can bind to the regulatory iron heme of sGC resulting in activation that leads to a dramatic increase in the rate of biosynthesis of the second messenger cyclic guanosine monophosphate (cGMP) with subsequent effects on the downstream physiology (49). There are also other possible mechanisms independent of cGMP formation by which NO can affect physiological function such as the generation of S-nitrosothiols on proteins and free radical trapping, among others (90).
Unlike NO, H2S is much less understood regarding its biological targets and mechanisms of action. Indeed, based on chemical arguments, it has been hypothesized that H2S may not be the sole effector but that H2S-related or derived species may be equally, if not more, important (38). For example, it is important to note that H2S and hydropersulfides (RSSH) are intimately linked via Reaction 1 and thus can be formed in proteins or small molecules via reaction of H2S with disulfides (RSSR) or other oxidized sulfur species (22, 81). Moreover, RSSH are intermediates in several H2S biosynthetic pathways (since H2S can be formed from RSSH via the reverse of Reaction 1) (21).
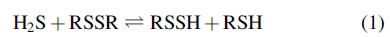
Importantly, cysteine hydropersulfide (Cys-SSH, also referred to as thiocysteine) has been reported to be generated by cysteinyl transfer RNA (tRNA) synthetase and can actually be translationally incorporated into proteins (4), making Cys-SSH a candidate as the 22nd amino acid in eukaryotes. Thus, the chemical biology and physiology of RSSH species are topics of recent and significant interest since their generation may account for at least some of the biological activity attributed to H2S and, indeed, in many cases H2S and RSSH can be considered markers for the presence of each other. Due to the intimate association between H2S and RSSH, it is unwise and likely naive to assume that H2S is the sole biologically important effector species.
The −SSH functional group of RSSH contains a type of sulfur atom not present in most other, more studied sulfur species (e.g., thiols [RSH], thiyl radicals [RS·], RSSR, sulfenic acids, sulfinic acids, and sulfonic acids). RSSH contains a “sulfane sulfur,” which refers to the sulfur atoms in molecules that are bound only to other sulfur atoms (100). For example, the bolded sulfur atoms in the following molecules are considered to be sulfane sulfurs—RS

Thus, assays for sulfane sulfur in molecules based on Reaction 2 have been used for many years (and important to the discussion of several studies below).
Numerous reviews of the relevant chemical biology of RSSH species and the potential physiological utility of their chemical attributes attest to the recent interest in this novel biological functional group (8, 28, 29, 39, 57, 76). Due to the availability of the many recent publications reviewing RSSH biochemistry and chemical biology, a comprehensive and detailed discussion of these points is not be given here (and will only be summarized later). Instead, the primary focus herein is on the early studies that portend the relatively recent discoveries regarding the physiological importance of RSSH and related species. That is, work done decades before the establishment of H2S as a biological signaling molecule foretold the discoveries being made today in this area of research. However, before reviewing and discussing these past reports, it is worthwhile to first review the work that led to the current interest in RSSH and related polysulfur species.
H2S As an Endogenous Biological Signal/Effector Species
Due, in part, to the numerous reports in the late 1980s and early 1990s of the endogenous generation and associated physiology of NO (69), many began to consider other endogenously generated small-molecule species as potential signaling agents. H2S can be biosynthesized via several biochemical pathways involving sulfur-containing amino acids such as cysteine (Cys-SH) or methionine (21, 89). Although H2S had long been recognized and studied as a toxin (82), in 1996 it was reported that endogenous H2S can act as a neuromodulator that enhances NMDA receptor-mediated responses leading to long-term potentiation in the brain (2). A year later, a role for H2S as an endogenous smooth muscle relaxant that exhibited synergy with NO was reported (47). Since these early publications, numerous reports have appeared describing a variety of physiological functions associated with H2S (for reviews, see, 53, 58, 59, 64, 97). For example, in addition to the abovementioned roles as a neuromodulator and vasorelaxant, H2S has also been reported to inhibit smooth muscle cell proliferation (24), activate KATP channels (61), and protect the brain from ischemic damage (60), just to name a few. In a particularly fascinating article, Blackstone et al. reported that exposure of mice to sublethal levels of gaseous H2S induced a suspended animation or hibernative state (low metabolic rate, low O2 consumption, low core body temperature) that was readily reversible with no apparent deficit when H2S exposure was ceased (12). Despite the apparent and ubiquitous examples of H2S signaling and biological effects, the chemical/biochemical mechanisms of many of these actions remain obscure. That is, H2S (either physiologically generated or pharmacological administered) clearly has effects on numerous tissues and cell systems, but the mechanism by which these occur is largely unestablished.
To begin to try to understand the mechanisms by which H2S might act physiologically and/or pharmacologically, the biochemical reactivity and chemical properties of H2S became an important topic. As already discussed, one of the most obvious possible reactions of H2S in a biological system is with other sulfur species such as RSSR (Reaction 1). To be sure, Reaction 1 has been known for over 60 years (81) but became of increasing recent interest due to the realization that endogenously generated H2S has important physiological functions and that H2S generation can lead to RSSH formation. Thus, much of the recent interest in RSSH species stems primarily from a desire to delineate the mechanisms of H2S bioactivity. Importantly, before this recent attention, many others had discovered unique and rich biochemistry associated with RSSH species and many of the currently considered ideas associated with the biological utility of RSSH are derivative of these early proposals/discoveries. To be sure, there is much more to be learned regarding RSSH biochemistry/chemistry as we are still in the very early stages of understanding the biochemical mechanisms and physiological roles of this unique functional group. Regardless, the discussion below focuses on some of the early, seminal work in this field that serves as the basis of many of the current hypotheses and ideas regarding H2S/RSSH biology.
Early Studies on the Fundamental Chemistry of RSSH
During the 1960s, a series of studies by Tsurugi and coworkers regarding the general chemical reactivity of RSSH species were published. Using aralkyl hydrodisulfides [i.e., benzyl (C6H5-CH2-SSH) and benzhydryl ((C6H5)2-CH-SSH) hydrodisulfides] as model compounds (structures shown in Fig. 1), numerous fundamental chemical properties associated with the −SSH functional group were elucidated. For example, RSSH was shown to be a far superior and “cleaner” reductant when reacting with radical species (e.g., 2,2-diphenyl-1-picrylhydrazyl [DPPH]; Fig. 1) compared with RSH, giving near quantitative amounts of the reduced 2,2-diphenyl-1-picrylhydrazine (DPPH2) and the corresponding tetrasulfide (RSSSSR) product. This reaction was proposed to occur via a stepwise process, through which RSSH reduces DPPH to DPPH2 (possibly by simple hydrogen atom donation), followed by dimerization of the perthiyl radical (RSS·) intermediates to the RSSSSR (Reactions 3 and 4) (73).
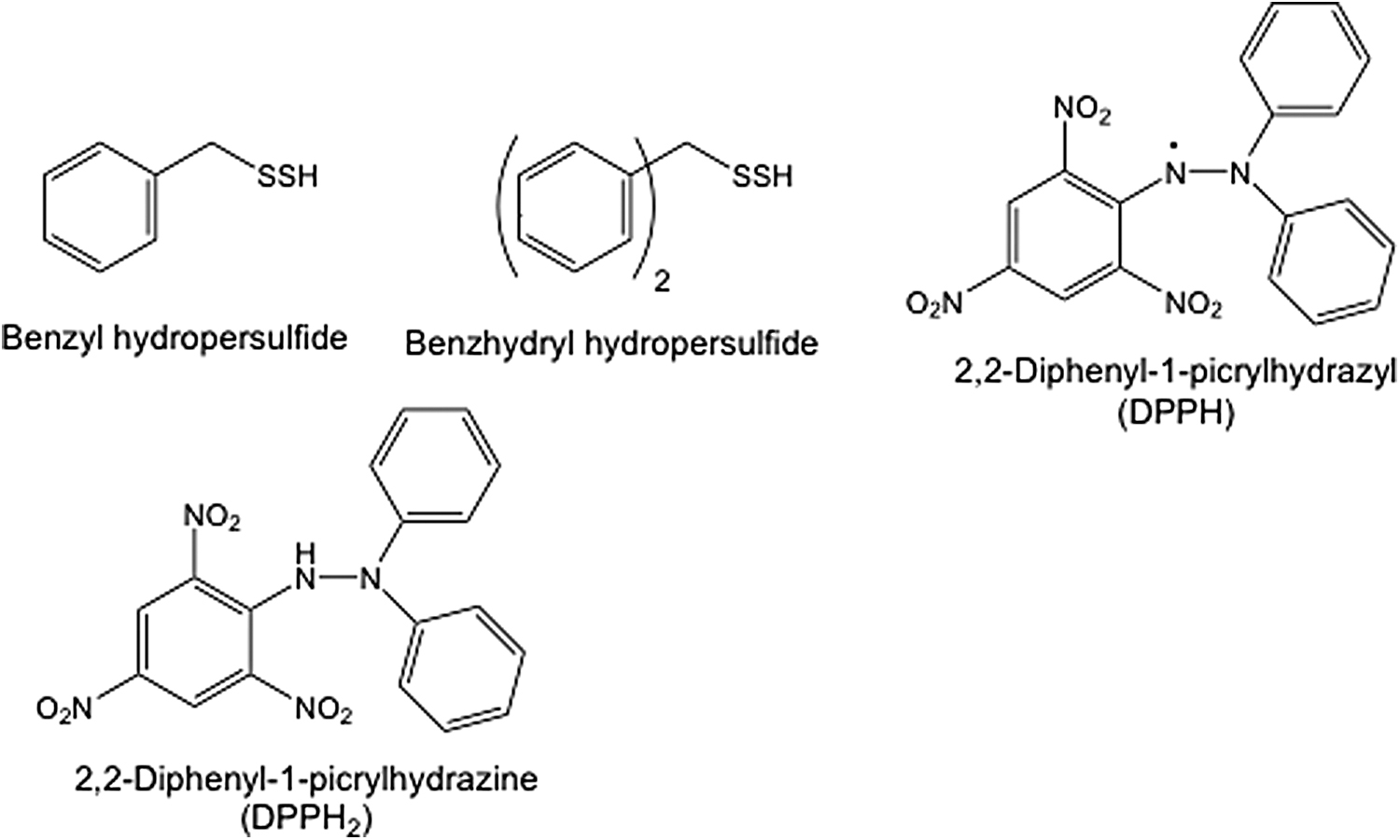

The reaction of DPPH with RSH is far slower and follows a slightly different pathway. Similar to what happens with RSSH, oxidation of RSH gives the corresponding RS· and DPPH2. However, RS· then reacts with a second equivalent of DPPH to give an adduct DPPH-SR, presumably the result of covalent bond formation between the two radicals RS· and DPPH (Reactions 5 and 6).

This observation alludes to the relative lack of reactivity associated with RSS· versus RS· since RSS· does not appear to have the propensity to react with a second equivalent of DPPH and instead dimerizes to RSSSSR.
The reaction of the oxidant/electrophile benzoquinone with RSSH also gave the corresponding RSSSSR and the fully reduced hydroquinone (Fig. 2). Interestingly, no addition product was observed, unlike RSH, which add to quinones via Michael addition (Fig. 2). The mechanism of reaction of RSSH with benzoquinone is not established, although it is telling and interesting that the products from RSSH versus RSH are distinct, further establishing a fundamental difference between their chemistries. It is possible that RSS− reacts via electron-transfer mechanisms with RSS· intermediates, while RSH reacts via typical 1,4-Michael addition chemistry, although this will need to be further examined.

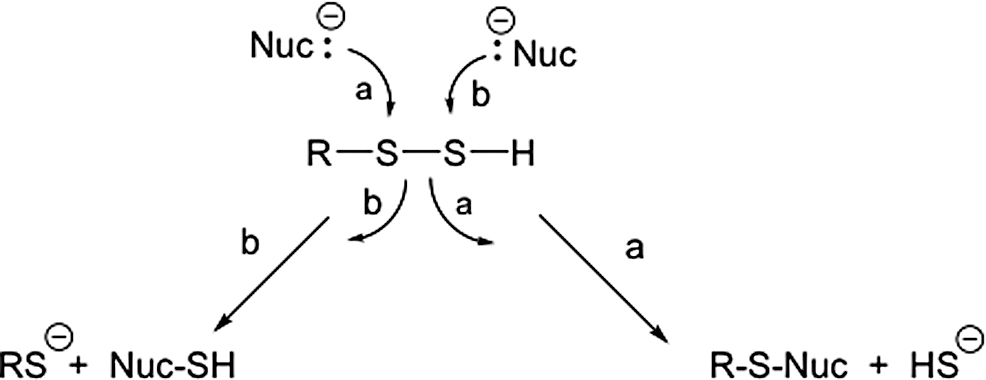
This group also examined the electrophilic properties of RSSH by studying the reaction of both benzyl and benzhydryl hydrodisulfides with a series of different nucleophiles (e.g., phosphines, phosphites, RSH, cyanide, hydroxide, arsines, sulfite) in organic solvents (52, 72, 74, 93, 94). As is established for RSSR, they found that either sulfur atom of RSSH can be attacked by nucleophiles. For example, phosphine nucleophiles can react at either the sulfenyl sulfur (the sulfur atom attached to carbon) or the sulfane sulfur (the sulfur atom attached to the sulfenyl sulfur) depending on the sterics of the electrophilic reaction center and the strength of the nucleophile (Fig. 3). Weak nucleophiles, in the absence of steric restrictions, appear to prefer the sulfenyl sulfur atom for attack, while stronger nucleophiles such as cyanide or thiolate are capable of attacking either sulfur atom of RSSH. To be sure, these reactions were examined in organic solvents, and the general trends/reactivity observed may be different in aqueous solution.
Finally, this group examined the reaction of their model RSSH compounds with ferric ion (Fe3+) in dioxane and found that RSSH readily reduces Fe3+ to ferrous ion (Fe2+) via Reaction 7 (51). Importantly, they determined that RSSH was stable in the presence of Fe2+, noting that the RSSH species are unlike analogous hydroperoxides (ROOH) in this regard, which can oxidize Fe2+ to generate potent and potentially deleterious oxidants (e.g., HO· generated via the Fenton reaction) (Reaction 8).

Thus, these early reports clearly established RSSH as good reductants and electrophiles, as well as established the relative lack of reactivity associated with the RSSH oxidized species RSS·—all properties that have been recently touted as important with regard to the possible biological utility of RSSH. Also, the finding that RSSH chemical reactivity is distinct from ROOH (e.g., RSSH does appear to participate in Fenton-like chemistry) is an important observation that may have significant biological implications.
Early Reports of Biological Per- and Polysulfide Prevalence/Activity
Although the idea that RSSH species can be important biochemical effectors and mediators seems relatively new, it must be emphasized that previous literature, some published over 50 years ago, reported the existence, activity, and likely importance of the −SSH functional group along with related polysulfur species in eukaryotic systems. Below is a brief account of some of these early previous studies (i.e., reports published before the general idea that endogenously synthesized H2S and associated persulfides possess important physiological functions).
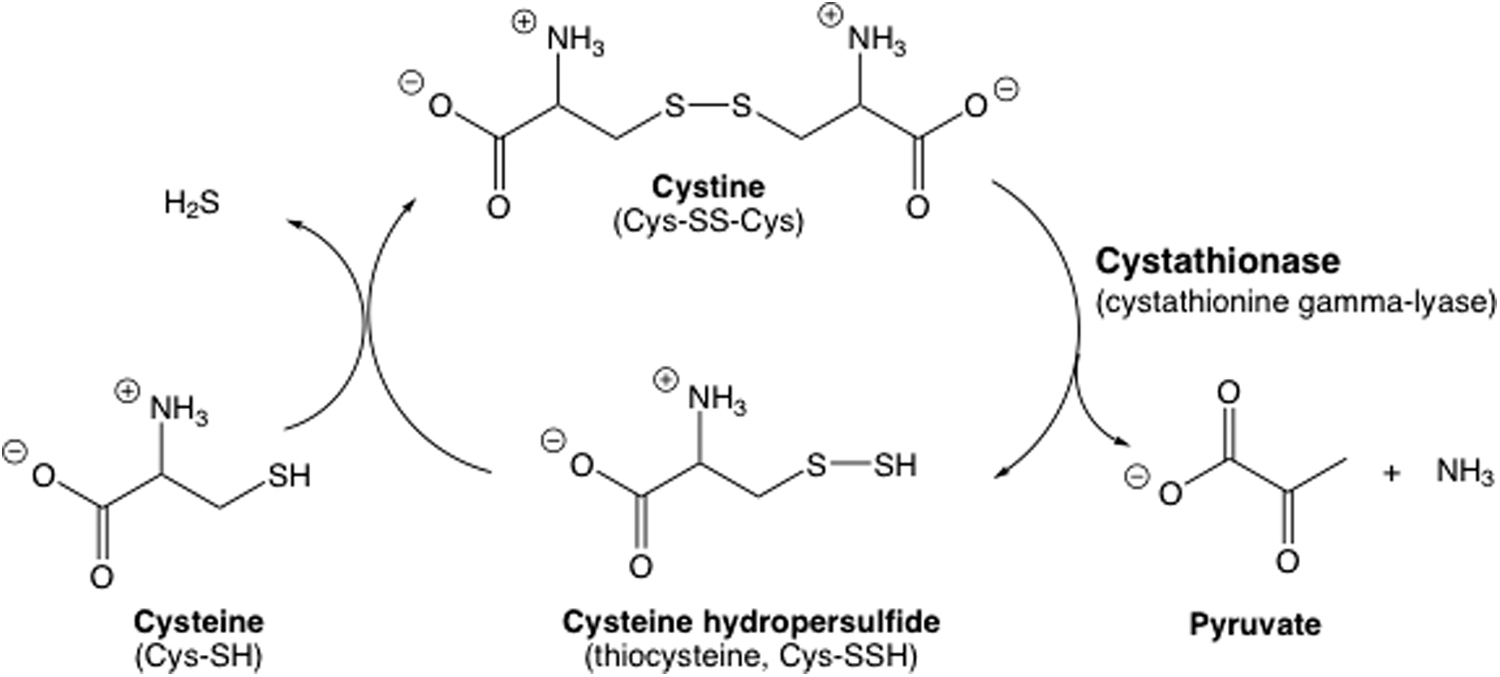
Although RSSH species can be generated via Reaction 1 from H2S, it was demonstrated long ago that the presence of RSSH in biological systems is not entirely dependent on initial H2S formation. The generation of Cys-SSH from cystine (Cys-SS-Cys) via a pyridoxal phosphate-dependent enzyme from rat liver (16) and in Neurospora (30) has been known for almost 60 years, a reaction that was observed previously in the presence of pyridoxal phosphate even in the absence of enzyme (15). The reaction scheme proposed by Cavallini et al. (16) accurately described the ability of cystathionase (a.k.a., cystathionine gamma-lyase) to generate Cys-SSH from Cys-SS-Cys and for subsequent generation of H2S from Cys-SSH via reaction with a thiol (Fig. 4). This work established the possible link between H2S biosynthesis and RSSH generation. That is, Cys-SSH can be a precursor to H2S generation, and along with Reaction 1, indicates an intimate biochemical link between RSSH and H2S, an idea that is almost 60 years old. Thus, this work made clear that the examination of H2S biological activity must consider the possibility of RSSH generation and its biological activity as well (and vice versa). Importantly, in all of these early studies, the metastability of RSSH was discussed and is clearly fundamental to the study of these species. Therefore, the possible generation of RSSH in biological systems via either biochemical or enzymatic processes, the intimate biochemical link between RSSH and H2S, as well as the potentially fleeting nature of RSSH have all been known for a long time.
One of the most important aspects of RSSH biological reactivity was established by the Massey laboratory in 1971 (66) while investigating a curious finding reported in the following previous study. Froede and Hunter (34) were examining the effect of oxidized glutathione (GSSG) on the glutathione (GSH)-mediated reduction of ferric cytochrome c (FeIIICyt c) and found that the slow rate of reduction of ferric to ferrous cytochrome c (FeIICyt c) by GSH was markedly increased when GSSG was added. This effect was metal independent and also possible with other RSH (i.e., GSSG addition enhanced the rate of FeIIICyt c reduction by RSH such as 2-mercaptoethanol or Cys-SH). Moreover, the slow reduction of FeIIICyt c by Cys-SH could be enhanced by the addition of Cys-SS-Cys as well, indicating that this effect is not specific to GSH/GSSG. This is a very curious observation since it is not immediately evident how an oxidized species, GSSG, can enhance the reducing capability of a weakly reducing species, GSH. Moreover, considering the observations above, GSH-mediated reduction of FeIIICyt c should be autocatalytic since GSH reduction of FeIIICyt c should generate GSSG, which would be expected to increase the overall rate. Interestingly, this was not seen (i.e., without the addition of exogenous GSSG, no autocatalytic effect was observed). That is, the GSSG generated in situ did not appear to have the same effect as exogenously added GSSG. Surprisingly, it was also reported that this system, GSH/GSSG, could lead to the generation of adenosine diphosphate (ADP) to adenosine monophosphate (AMP) and inorganic phosphate (Pi) and adenosone triphosphate (ATP) from AMP and pyrophosphate, leading investigators to propose a possible role for GSH/GSSG in oxidative phosphorylation (77 –80). These provocative results prompted the proposal of a mechanism, through which GS− and GSSG form an electron rich, three-membered, substituted sulfur ring that can reduce FeIIICyt c and facilitate the synthesis of a phosphoanhydride bond from Pi and AMP (to make ADP) (80). To be sure, based on subsequent work by Massey (discussed immediately below), the validity of the existence and reactivity of this unusual intermediate is highly questionable.
In examining the above phenomenon, Massey et al. (66) found that the rate enhancement of GSH-mediated reduction of FeIIICyt c by GSSG was highly dependent on the source and batch of GSSG used, indicating the possibility that it was a contaminant that was responsible for the effect. They also reported that the FeIIICyt c reducing capabilities of the GSSG samples correlated with sulfane sulfur content, as determined by incubation with cyanide (CN−) and generation of SCN− (vide supra). Further experimentation using Cys-SH-based analogues revealed that the dialkyltrisulfide, GSSSG, was responsible for the observed rate enhancement, not GSSG. Massey et al. proposed that GSH reacted with the contaminant GSSSG to make the glutathione hydropersulfide (GSSH) (Reaction 9), which served as the ultimate reductant in this system (Reaction 10). The GSS· formed from GSS− oxidation could then simply dimerize to give GSSSSG (Reaction 11).
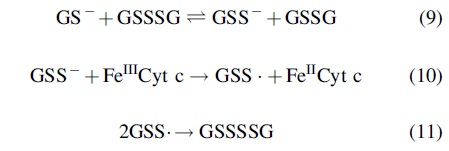
This study revealed several important aspects of RSSH and polysulfide chemistry/biochemistry. First, this work helped establish the superior one-electron reducing capability of RSS−/RSSH versus RS−/RSH. Second, it indicates that dialkylpolysulfides such as GSSSG are relatively stable in that they can persist as contaminants in, for example, GSSG preparations. Finally, it highlighted the possible relevance and prevalence of dialkylpolysulfides such as GSSSG. Although the origins of GSSSG contaminants in the commercial GSSG samples were not addressed in this study (and may have been a result of the commercial preparation of GSSG from GSH), it is worth noting that more recent work has shown that polysulfides such as GSSSG are present in animal tissues and plasma (48) and could have also been present in preparations derived from natural sources. It is worth noting that recent work in prokaryotic systems has discovered that structural tetrasulfide (RSSSSR) formation in proteins can lead to functional changes. For example, the binding of the transcriptional repressor SqrR in Rhodobacter capsulatus to the DNA operator is inhibited by formation of an intramolecular RSSSSR leading to gene expression (86).
Further work from the Massey laboratory postulated protein RSSH species as important entities in the catalytic activity of two molybdenum enzymes. Xanthine oxidase (XO) had long been known to be inhibited by CN− (33). While examining this phenomenon, Massey and Edmondson discovered that CN− inhibition coincided with the generation of SCN− and postulated that CN−-mediated inhibition of XO occurred via destruction of a crucial active-site persulfide (via Reaction 2) (65). Subsequent work then led to a proposal that XO activity relies on a nucleophilic attack by an active-site RSS−/RSSH species (presumably a protein Cys-SSH residue) on the substrate xanthine, which results in electron donation (via hydride) to other redox components of the enzyme (25). Embedded in this mechanism is the implication that RSS− is a superior nucleophile capable of reacting with a weakly electrophilic xanthine molecule. It was also shown that aldehyde oxidase behaves similarly to XO and could contain an active-site RSSH species (13). It needs to be stressed, however, that the idea of an active-site persulfide in XO has been disputed. Indeed, numerous reports provide strong evidence that supports the idea that the cyanolyzable sulfur does not come from an active-site Cys-SS− but rather from a terminal molybdenum sulfide (MoS) (42, 75, 96). However, it needs to be mentioned that it has been proposed in more recent work that other molybdenum-containing enzymes of the DMSO reductase family (e.g., nitrate reductase) may be capable of forming a transient RSS-Mo complex from an MoS and a nearby Cys-SH residue via a sulfur-shift mechanism (Fig. 5), although it is unclear that this is possible with XO (20, 46, 71). The catalytic function of the sulfur-shift mechanism proposed for the Mo-containing nitrate reductases involves the opening up of an open coordination site for substrate binding (e.g., NO2 −), as well as a change in the formal oxidation state of the Mo center (17, 18, 87). Again, the relevance of this mechanism has only been demonstrated and/or hypothesized for certain members of the DMSO reductase family of Mo enzymes and may not be relevant for other Mo-containing enzymes such as XO. It is also important to note that nitrate reductase can be inhibited by CN− in a noncompetitive manner (nitrate and CN− do not bind at the same site) with generation of SCN− and with no evidence of CN− coordination to the molybdenum – all consistent with the idea that CN− is destroying an important persulfide species in this enzyme (14). To be sure, these experimental observations are also consistent with CN− destruction of an MoS species (as proposed for XO) as well.

Implicit in the proposals by Massey and coworkers (mentioned above) are several important aspects of RSS−/RSSH biochemistry: (i) RSS−/RSSH are superior reductants compared with RS−/RSH capable of, for example, rapidly reducing FeIIICyt c to FeIICyt c, (ii) oxidized RSS· species are not reactive oxidants and simply dimerize (consistent with the strong reducing capabilities of RSS−/RSSH), and (iii) RSS− are likely strong nucleophiles, proposed to react with, for example, xanthine leading to hydride release and oxidation (although this proposal seems unlikely in light of more recent work, vide supra). Thus, these provocative initial studies, along with subsequent work by many others, provide a basis for current speculation regarding the biological/physiological utility of the −SSH functional group.
Further work by others reinforced the idea that RSSH species (and other sulfane sulfur-containing molecules) are biologically accessible and possess unique biochemical properties not seen with other sulfur species. For example, Wood (100) reported a protective effect associated with the ingestion of the sulfane-sulfur donor thiocystine (Cys-SSS-Cys) by mice against CN− poisoning and X-ray irradiation toxicity (although the protection against X-ray toxicity was moderate). This finding may not be surprising since Cys-SSS-Cys can react with another thiol to give Cys-SSH (Reaction 12) (1, 10), which can detoxify CN− via Reaction 2 and act as an antioxidant against damaging X-ray-derived radical species (see paragraph below). Alternatively, it is also possible that Cys-SSS-Cys reacts directly with CN− leading to detoxification since, as discussed earlier, CN− can generally react with sulfane sulfur-containing species.
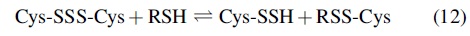
Just before the explosion of interest regarding the biological signaling and utility of endogenous H2S production (around 1996), the potential antioxidant properties (e.g., the reducing capabilities) of RSSH were further established by Everett et al. (26) and Everett and Wardman (27). They showed that RSS−/RSSH were capable of reacting very rapidly (k > 109 M−1/s) with oxidizing radicals either by electron donation (from RSS−) or hydrogen atom donation (from RSSH) (Reactions 13 and 14). Moreover, it was also shown that RSS· are far less oxidizing compared with RS· and therefore less apt to initiate pro-oxidative events (e.g., lipid peroxidation). Thus, the idea that the −SSH functional group can serve as a reducing antioxidant became firmly established.
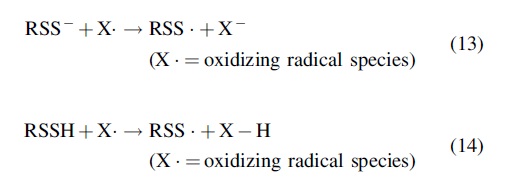
This work also led to the finding that RSSH species are significantly more acidic compared with the corresponding RSH (by 1–2 pKa units), indicating a higher proportion of the more nucleophilic and reducing anionic species with RSSH compared with RSH.
Several other early reports proposed that persulfide formation in proteins can be regulatory. For example, Agro et al. (3) proposed that the generation of a persulfide (made via rhodanese and thiosulfate) in the enzyme malate dehydrogenase increases activity, and Valentine et al. reported that the activities of numerous enzymes in erythrocytes can be dramatically inhibited via persulfidation (95). In a more specific study, an RSSH generating system was shown to inhibit tyrosine amino transferase, presumably via transfer of sulfane sulfur to the enzyme (although the physiological relevance of this process was questioned) (44). In 1982, it was reported that a non-Cys-SSH may also be present and regulatory. Williamson and coworkers (including the Massey laboratory) (99) were examining a green, inhibited form of butyryl-coenzyme A (CoA) dehydrogenase and found that the green color of this inhibited enzyme was due to a coenzyme A persulfide (CoA-SSH) that formed a charge transfer interaction with a protein-bound FAD (which exhibits a green color). Thus, it was reported that CoA-SSH strongly binds to this enzyme, blocking access of the natural substrate and therefore inhibiting the enzyme. Interestingly, butyryl-CoA-dehydrogenase from most mammalian sources is isolated as a green-colored protein (indicating the prevalence of the CoA-SSH inhibited form). Moreover, other analogous dehydrogenases are also isolated as green proteins, which are presumably CoA-SSH inhibited (41, 83, 84). Therefore, a regulatory role of CoA-SSH for enzymes, which utilize CoA-S-esters, seems clear. Further importance of this work is the demonstration that the −SSH functional group is not solely associated with Cys-SH-containing peptides and proteins.
In 1986, Toohey reported that systems capable of slowly generating low levels of persulfides greatly enhance the growth of L1210 murine lymphoma cells, which are known to be defective in sulfur metabolism (91). Interestingly, the growth enhancing effects of the persulfide generating systems occurred within a very narrow concentration range as only slightly higher levels of persulfide precursors did not enhance growth. The reason for the lack of effect for these persulfide generating systems at higher levels is unknown but could be due to either the levels of precursor or to the persulfide itself. Regardless, this study established a potentially vital physiological role for low-level persulfides (or sulfane sulfur) in cell health/growth. The above studies, along with many others, prompted Toohey to prophetically predict an important regulatory role for sulfane sulfur (proposed long before interest in biological H2S-mediated signaling in eukaryotes) (92). In this review, Toohey presents a comprehensive account of studies that describe the chemical and biological generation of sulfane sulfur as well as allude to the ability of persulfides and other sulfane sulfur species to regulate the activity of enzymes, as well as have effects on general cell health, the immune system, in the etiology of cancer and protein translation, among others. Thus, a general and important role, as opposed to isolated and specific functions, for persulfides in overall cellular signaling and health was put forth. Importantly, many of these effects have received further recent support and it is becoming increasingly clear that these early thoughts and ideas regarding the possible physiological importance of sulfane sulfur were well-conceived.
Thus, the recent interest in the −SSH functional group (as well as other sulfane sulfur species) and many of the prevalent ideas/hypotheses regarding RSSH biological/physiological activity have their roots in some of this older, seminal literature. The progress in this area made since 1996 (the time of the initial reports associated with the physiological actions of endogenous H2S biosynthesis) has, to be sure, been significant and the following section generally and briefly describes some of these more recent advances that have built upon this early work.
More Recent Work on the Prevalence and Utility of RSSH Generation
Although past studies identified and characterized numerous ways in which RSSH species can be formed biochemically (vide infra), more recent reports indicate the high prevalence of both small-molecule- and protein-associated RSSH. One of the first studies that proposed significant hydropersulfide content in proteins (PSSH) was by Mustafa et al. (70), who reported that numerous proteins from mouse liver contain Cys-SSH residues (especially after H2S treatment). However, it needs to be mentioned that the assay used in this report is highly questionable and likely not specific to PSSH (102). Regardless, more rigorous subsequent studies report the prevalence and general ubiquitous nature of PSSH (23, 35, 48, 63, 102). Ida et al. (48) also report significant levels of small-molecule RSSH species such as GSSH and Cys-SSH in the plasma and tissues of mammals (with levels >100 μM for GSSH reported in some tissues). Moreover, recent work on CoA-SSH further establishes it as a regulatory agent for some enzymes utilizing CoA thioesters as substrates (68). Furthermore, mechanisms of CoA-SSH biosynthesis (56) and degradation (85) have been reported. One potentially monumental finding (briefly mentioned earlier) is the report that cysteinyl tRNA synthetase is capable of converting Cys-SH to Cys-SSH, which is either released or translationally incorporated into proteins (4). This novel biosynthetic pathway is reminiscent of the biosynthesis of selenocysteine, an amino acid with similar chemical properties as Cys-SSH (39). Thus, one of the major advances made since the pre-1996 work is the realization of the ubiquitous and prevalent nature of the RSSH (and polysulfide) functional group in numerous proteins and small molecules and that the biological actions of H2S can be the result of RSSH/PSSH generation.
A greater understanding of some of the chemistry originally proposed in early reports has occurred in more recent times. It is clear that conversion of RSH to RSSH results in an RSSH functional group that has similar, although enhanced, chemical reactivity compared with RSH. For example, RSS−/RSSH are more nucleophilic (9, 22), and, as discussed previously, more reducing (11, 32, 40) and a superior hydrogen atom donor (11, 19) compared with the corresponding RS−/RSH. Moreover, the idea that RSSH is more acidic compared with RSH has been further established with pKa differences as much as 4 pKa units reported (9, 22), indicating that an even greater percentage of the nucleophilic and reducing anionic species can be present than originally proposed (26, 75). Thus, it can be said that RSSH has analogous general chemical reactivity to that of RSH except that RSSH is significantly more reactive. RSSH also possesses chemical properties that RSH does not have. As an oxidized sulfur species, RSSH can be an electrophilic oxidizing agent, akin to the reactivity of RSSR (as demonstrated previously by Tsurugi and coworkers, vide infra). RSH, on the contrary, is fully reduced and cannot act as an oxidant/electrophile. One important aspect of the conversion of RSH to RSSH is that this transformation represents an oxidation and therefore will be more apt to occur under oxidizing conditions (e.g., cellular oxidative stress). What this means is that RSSH, a superior reductant, will more likely be generated under oxidizing conditions. The abovementioned enhanced chemical properties of RSSH versus RSH, as well as the redox relationship between RSH and RSSH, led to the idea that conversion of RSH to RSSH could be a cellular response to oxidative and/or electrophilic stress (5, 67, 76). That is, cellular oxidative stress promotes the conversion of RSH to RSSH generating a potentially protective reductant (to protect from oxidative stress) and nucleophile (to protect from electrophilic stress). When an RSSH is generated on a protein (PSH → PSSH), this too may be a protective mechanism since PSH can be irreversibly modified by oxidants and electrophiles, while PSSH modification by oxidants/electrophiles generates intermediates that are readily reduced back to the initial PSH (67). Moreover, the fact that RSSH can also act as an oxidant may allow it to activate transcription factors responsible for cellular protection against oxidative stress (37). For example, sulfane sulfur species have been shown to react with KEAP1 thiols, presumably via oxidation to polysulfides, leading to the activation of the protective transcription factor Nrf2 (54, 55). Although the general idea that per- and polysulfide generation is a cellular response to oxidative/electrophilic stress remains somewhat speculative, it is worth mentioning that numerous recent reports appear consistent with this general hypothesis (10, 28, 43, 45, 48, 98).
Despite the recent support for the idea of RSSH protection against oxidative and/or electrophilic stress, it needs to be noted that RSSH is an oxidized species with respect to RSH and thus may itself pose a danger via oxidative stress at improper concentrations. That is, although RSS−/RSSH is a good nucleophile/reductant, it is an oxidant [when in the protonated RSSH form (37)] akin to Cys-SS-Cys or GSSG and can therefore also elicit an oxidative stress. Toohey may have observed this when noting that the growth enhancing effects of RSSH generating systems on cells had a very narrow concentration window (91), with levels above the optimum range exhibiting an inhibitory or toxic effect. In possible support of this idea, Lin et al. found that some cells export Cys-SSH when overall intracellular RSSH levels get too high (62). Although there may be numerous reasons for this export, one possibility may be to avoid an oxidative stress caused by high levels of the oxidized RSSH species (akin to why, for example, GSSG is transported out of a cell). Thus, as Toohey (91) observed, a bell-shaped curve associated with effects of RSSH species on cell viability or cell protection may not be unexpected.
Summary
The more recent interest in RSSH stems from the realization that H2S is an endogenously generated signaling species with numerous physiological effects. The mechanisms of H2S biological/physiological activity remain unestablished and the fact that H2S is biochemically linked to RSSH species prompted many to begin to examine the possible chemical biology of RSSH as a means of trying to understand H2S physiology/biology.
It is becoming increasingly clear that the −SSH functional group is an important member of the many biologically relevant/active sulfur species. It is somewhat unique among these species in that it can be a superior reductant and nucleophile compared with other sulfur congeners and yet can also be electrophilic and oxidizing (37). The chemical properties of RSSH (i.e., reductive, nucleophilic) predict that it can be an important response to cellular oxidative and/or electrophilic stress and indeed the fact that it is an oxidized species itself (relative to a thiol) makes its biosynthesis more reasonable under stress conditions. However, the electrophilic and oxidizing properties of RSSH also indicate that it can participate in this type of chemistry under the proper conditions, and therefore, care must be taken when interpreting the biological effects associated with this unique functional group. Figure 6 schematically summarizes some of the possible roles discussed herein for RSSH species.
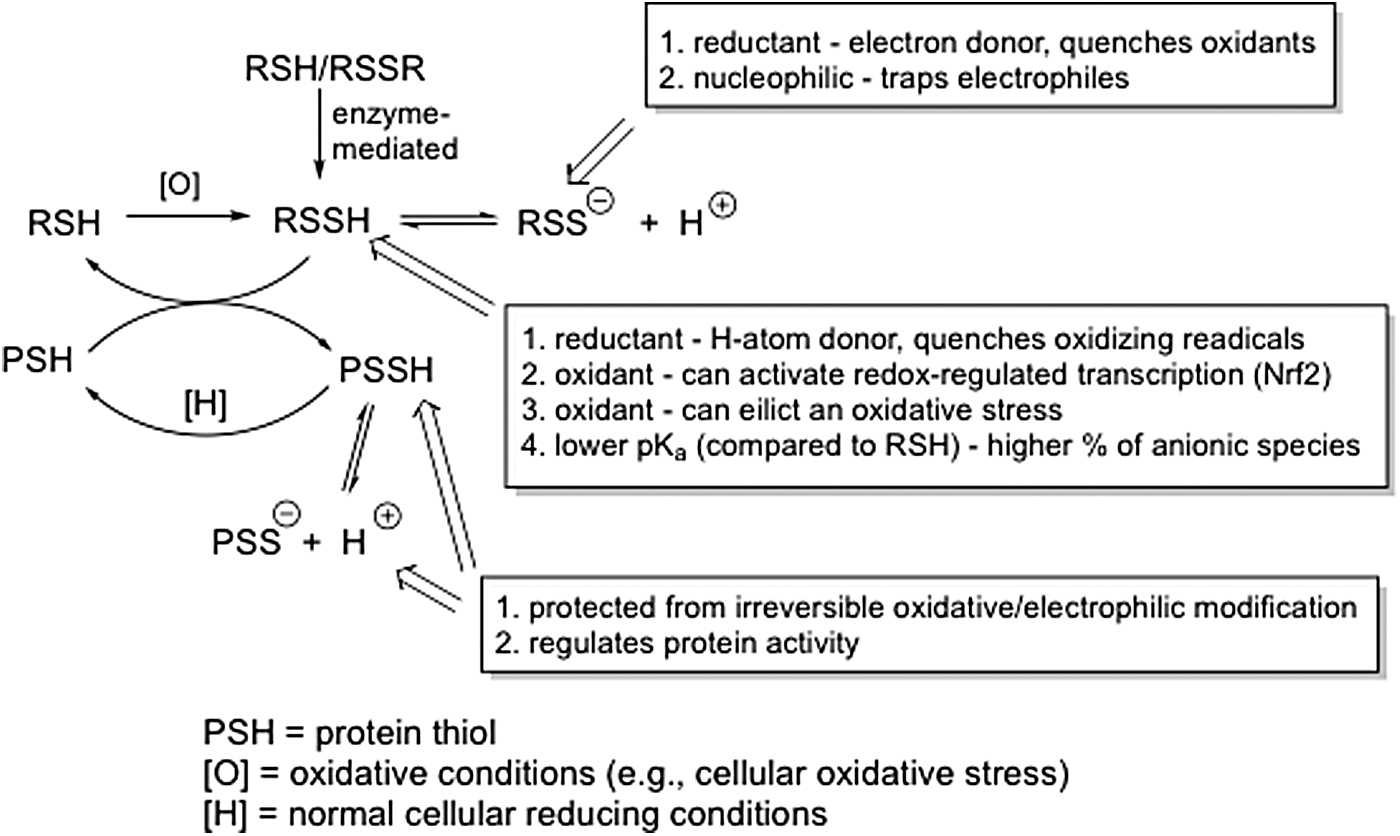
As discussed above, many of the fundamental chemical and biochemical properties of RSSH were initially elucidated many years ago before the realization of purposeful H2S biosynthesis. The superior reducing capabilities, the electrophilicity, and the possible regulatory properties of the −SSH functional group were established long ago. More recent work has built upon these ideas and definitely has expanded the general scope of possible RSSH relevance. For example, based on the chemical properties of RSSH, the idea of RSSH-mediated cellular protection has received recent attention and may be an important role for this functional group. However, the mechanisms of H2S biology/physiology and the utility of RSSH in this regard are still a matter of significant speculation and are not firmly established. That is, whether RSSH generation is fundamental to H2S biology remains to be determined. Although this field (the study of RSSH and polysulfur species) is over 60 years old, it is still in its infancy with regard to establishing whether the ideas, discussed herein, associated with RSSH biological utility are valid. Clearly, even after all of these years, much more work will be required to determine the possible importance or relevance of biological RSSH species.
Footnotes
Acknowledgments
The author would like to thank Adrian Hobbs, Taka Akaike, Yoshito Kumagai, Masahiro Akiyama, Peter Nagy, John Toscano, and Joseph Lin for helpful discussions and input.
Author Disclosure Statement
The author has no conflicts of interest.
Funding Information
No funding was provided for this article.