
Abstract
Aims:
Peripheral artery disease (PAD) is a severe complication of diabetes, characterized by defective traffic of hematopoietic stem/progenitor cells (HSPCs). We examined the hematopoietic versus nonhematopoietic role of p66Shc in regulating HSPC traffic and blood flow recovery after ischemia in diabetic mice.
Results:
Using streptozotocin-induced diabetes, chimeric mice with green fluorescent protein (GFP)+ bone marrow (BM), and the hind limb ischemia model, we found that the physiologic mobilization and homing of HSPCs were abolished by diabetes, along with impaired vascular recovery. Hematopoietic deletion of p66Shc, obtained by transplanting p66Shc −/− BM cells into wild-type (Wt) recipients, but not nonhematopoietic deletion, constrained hyperglycemia-induced myelopoiesis, rescued postischemic HSPC mobilization, and improved blood flow recovery in diabetic mice. In Wt diabetic mice transplanted with BM cells from GFP + p66Shc −/− mice, the amount of HSPCs homed to ischemic muscles was greater than in mice transplanted with GFP + p66Shc +/+ cells, with recruited cells displaying higher expression of adhesion molecules and Vegf. In 40 patients with diabetes, p66Shc gene expression in mononuclear cells was correlated with myelopoiesis and elevated in the presence of PAD. In 13 patients with diabetes and PAD, p66Shc expression in HSPC-mobilized peripheral blood cells was inversely correlated with VEGF expression.
Innovation:
For the first time, we dissect the role of hematopoietic versus nonhematopoietic p66Shc in regulating HSPC traffic and ischemic responses.
Conclusion:
Hematopoietic deletion of p66Shc was sufficient to rescue HSPC mobilization and homing in diabetes after ischemia and improved blood flow recovery. Inhibiting p66Shc in blood cells may be a novel strategy to counter PAD in diabetes. Antioxid. Redox Signal. 36, 593–607.
Clinical Trial No.: NCT02790957.
(Color images are available online).

Innovation
Peripheral artery disease (PAD) is a severe complication of diabetes, characterized by defective traffic of hematopoietic stem/progenitor cells (HSPCs). We found that hematopoietic deletion of the redox enzyme and adaptor protein p66Shc was sufficient to rescue the defective HSPC mobilization and homing observed in diabetes after ischemia and improved blood flow recovery. While the molecular mechanisms through which p66Shc governs myelopoiesis and vascular endothelial growth factor signaling have been explored before, our new data provide salience to the role of p66shc as a molecular hub and to HSPCs as cellular messengers of the response to ischemia. In view of the consistent results obtained on p66Shc expression in humans, we propose that inhibiting p66Shc in blood cells may be a novel strategy to counter PAD in diabetes.
Introduction
P
Remote tissue ischemia triggers mobilization of HSPCs, which, in turn, home to ischemic sites, where they contribute to tissue repair. Ischemia-activated hypoxia-inducible factor-1α (HIF-1α) induces expression and releases of chemokines (e.g., C-X-C- motif ligand-12 [CXCL12]) and proangiogenic growth factors (e.g., vascular endothelial growth factor [VEGF]) that signal to the BM eliciting HSPC release (14, 57). Blunted activation of HIF-1α in ischemic diabetic tissues limits the HSPC mobilization (15, 23). Of note, diabetes also impairs the recruitment of BM-derived progenitors to sites of tissue damage (2). Such defective HSPC traffic from the BM to peripheral tissues is thought to compromise tissue repair.
In murine and human diabetes, BM responsiveness to the HSPC mobilizing effect of granulocyte-colony stimulating factor (G-CSF) is abrogated (20, 25), while hematopoiesis is skewed toward an exacerbated generation of myeloid cells (40, 41). These alterations amplify systemic inflammation and promote the rapid development of CVD (41). Mechanistically, we have shown that an excess accumulation of BM macrophages due to myelopoiesis results in overproduction of oncostatin M (Osm) that signals through p66Shc in stromal cells to induce the expression of CXCL12, thereby retaining HSPC within the BM (3). Deleting either Osm or p66Shc within the BM restored G-CSF-induced mobilization and constrained myelopoiesis in diabetic animals (1).
p66Shc is the 66 kDa isoform of SHC (Src homology 2 domain containing) transforming protein 1, encoded by the mammalian ShcA gene. Besides its adaptor role in the Osm signaling pathway (1), p66Shc acts as a mitochondrial redox protein that produces reactive oxygen species (ROS) by oxidizing cytochrome C and reducing O2 to H2O2 (27). This propagates oxidative stress, but can also exert physiological effects, such as in regulating insulin signaling (6). As an adaptor protein, p66Shc transduces the signaling pathways of several growth factor receptors involved, for example, in the cell proliferation or apoptosis, also in hematopoietic cells (7, 46). p66Shc has been proposed as a longevity gene, since its genetic deletion decreased levels of ROS in vitro and prolonged the life span in a controlled environment (38). p66Shc −/− mice are also protected against tissue damage in murine diseases characterized by oxidative stress (11, 28, 43) and in models of diabetic complications (21, 37). Hyperglycemia induces activation of p66Shc via Ser-36 phosphorylation by protein kinase C (PKC)-ßII, thus inducing oxidative damage (49). This pathway is responsible for pericyte damage in patients with PAD (60). Activation of the p66Shc pathway in diabetes is also linked to hyperglycemic memory, through which high glucose induces epigenetic fingerprints, such as increased histone-3 acetylation and decreased promoter methylation, lasting beyond restoration of euglycemia, and developing a vicious cycle of p66Shc activation and oxidative damage (10, 48). Furthermore, p66Shc expression is associated with epigenetic changes in human CD34+ cells exposed to high glucose, which impaired CXCR4 expression and responses toward CXCL12 (59). Importantly, p66Shc modulates the response of skeletal muscle to ischemic damage (61), and postischemic perfusion was improved in diabetic p66Shc −/− animals, possibly by limiting hyperglycemia-induced oxidative stress (21).
Based on these observations and having demonstrated that p66Shc cell intrinsically stimulates myelopoiesis and blunts the mobilizing response to G-CSF, we herein hypothesize that hematopoietic p66Shc regulates the traffic of HSPCs and the responses to ischemia.
Results
Diabetes impairs postischemic HSPC mobilization and homing in mice
Three days after hind limb ischemia, the levels of PB-HSPCs (defined as Lin−c-Kit+Sca-1+ [LKS], cells) peaked at approximately sixfold in nondiabetic control mice (Supplementary Fig. S1A), but did not change in streptozotocin (STZ)-induced diabetic mice (Supplementary Fig. S1B), a difference that was highly statistically significant and robust (Supplementary Fig. S1C, D). This finding is in line with previous studies performed in mice and rats with STZ-induced diabetes (1, 23). We then evaluated whether the traffic of BM-derived cells to the ischemic tissue was impaired in STZ diabetes by using mice transplanted with BM green fluorescent protein (GFP)+ cells (Fig. 1A). After reconstitution, these chimeric mice harbored >95% of GFP+ hematopoietic cells (Supplementary Fig. S2) and can be used to track the fate of BM-derived cells in peripheral tissues. Two weeks after inducing hind limb ischemia in BM GFP+ chimeric mice, the microvasculature of adductor muscles in STZ diabetic mice displayed a significant 25% lower capillary/fiber ratio compared with that in nondiabetic control mice (p = 0.03; Fig. 1B, C), indicating poor angiogenic response. This was consistent with the significant reduction in hind limb perfusion in STZ diabetic versus nondiabetic mice, evidenced by a lower ischemic/nonischemic perfusion ratio upon laser Doppler imaging (p < 0.01, Fig. 1E, F). While about 2.0% of muscle capillaries in nondiabetic mice harbored GFP+ cells, GFP-bearing capillaries were reduced to 0.2% in STZ diabetic mice (p = 0.01; Fig. 1D), suggesting that diabetes compromised the traffic of BM-derived cells to the ischemic tissue.

Ubiquitous p66Shc deletion does not rescue postischemic HSPC mobilization
Since we have previously found that deletion of p66Shc rescued G-CSF-induced HSPC mobilization, we tested whether and how p66Shc is involved in diabetes-associated defective postischemic HSPC mobilization. No difference was observed in the degree of hyperglycemia achieved 4 weeks after STZ administration in wild-type (Wt) and p66Shc −/− mice (Fig. 2A). Unexpectedly, while nondiabetic p66Shc −/− mice showed a normal approximately sixfold increase in LKS cell counts after ischemia, p66Shc deletion did not rescue the defective postischemic HSPC mobilization observed in diabetic mice (Fig. 2B). Differently from HSPC mobilization induced directly by stimulation with G-CSF, ischemia-induced mobilization requires signals from ischemic tissues to the BM via growth factors (e.g., VEGF) and chemokines (e.g., CXCL12). We previously reported that both arms of this response were affected by STZ diabetes (23). As intrinsic BM responsiveness to G-CSF in diabetes was improved by p66Shc deletion, we hypothesized that lack of rescue of ischemia-induced mobilization in diabetic p66Shc −/− mice was attributable to an impaired signaling from ischemic muscles to the BM, which relies on hypoxia-sensing and HIF-1α activation (14). Indeed, induction of HIF-1α target genes in the hypoxic muscles at day 3 after ischemia was impaired by diabetes (Vegf) and worsened by p66Shc deletion (involving also Cxcl12, Hk1, and Glut1) (Fig. 2C). This finding is consistent with prior studies showing an interplay between the mitochondrial activities of p66Shc and HIF-1α in inducing the proangiogenic response (42).
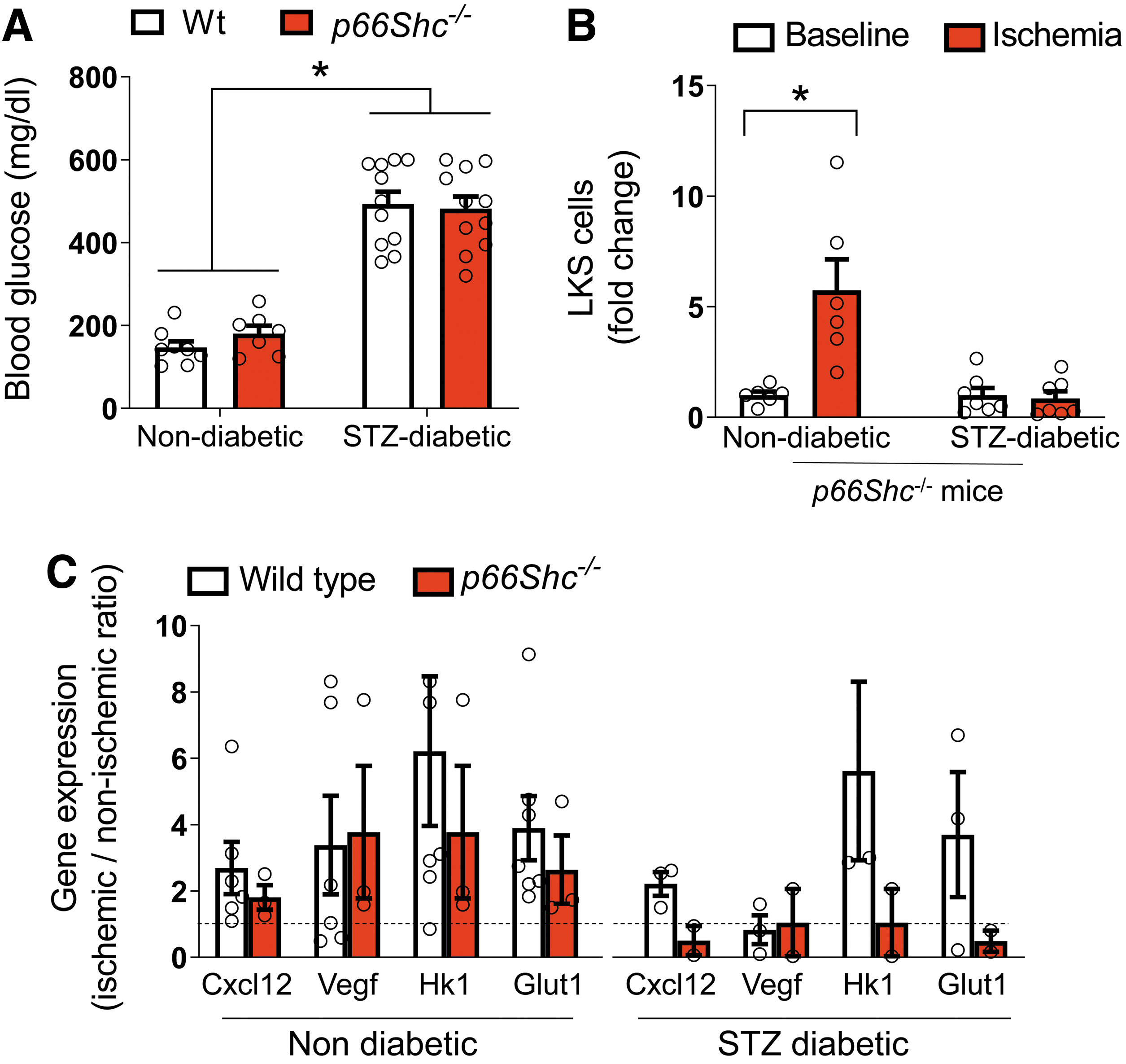
Hematopoietic p66Shc deletion rescues postischemic responses
Our prior observation that, in diabetic mice, p66Shc deletion in the BM rescued G-CSF-induced HSPC mobilization along with the new finding that ubiquitous p66Shc deletion did not rescue postischemic HSPC mobilization led us to hypothesize that, in skeletal muscle, p66Shc may switch off the signals required for eliciting HSPC mobilization. We thus assessed the contribution of hematopoietic versus nonhematopoietic p66Shc to postischemic HSPC mobilization using cross bone marrow transplantation (BMT) experiments (Fig. 3A). In all BMT experiments, after 4 weeks of reconstitution, we achieved a robust degree of chimerism, with >95% of circulating leukocytes being derived from the grafted BM, tracked by the CD45.1/CD45.2 ratio (Supplementary Figs. S2 and S3). Since we aimed to assess the contribution of selective hematopoietic versus nonhematopoietic p66Shc, we first confirmed that the host BM stromal compartment was preserved after BMT. We analyzed the BM of GFP+ chimera, confirming that more than 98% of CD45−CD90+ putative stromal cells were GFP− (Supplementary Fig. S4A). Furthermore, BM-derived stromal cells in vitro emerged only from the GFP− fraction (Supplementary Fig. S4B), while GFP+ cells formed scattered nonadherent cells (a feature of hematopoietic cells). Therefore, cross BMT involved only the hematopoietic component and spared the stromal compartment.
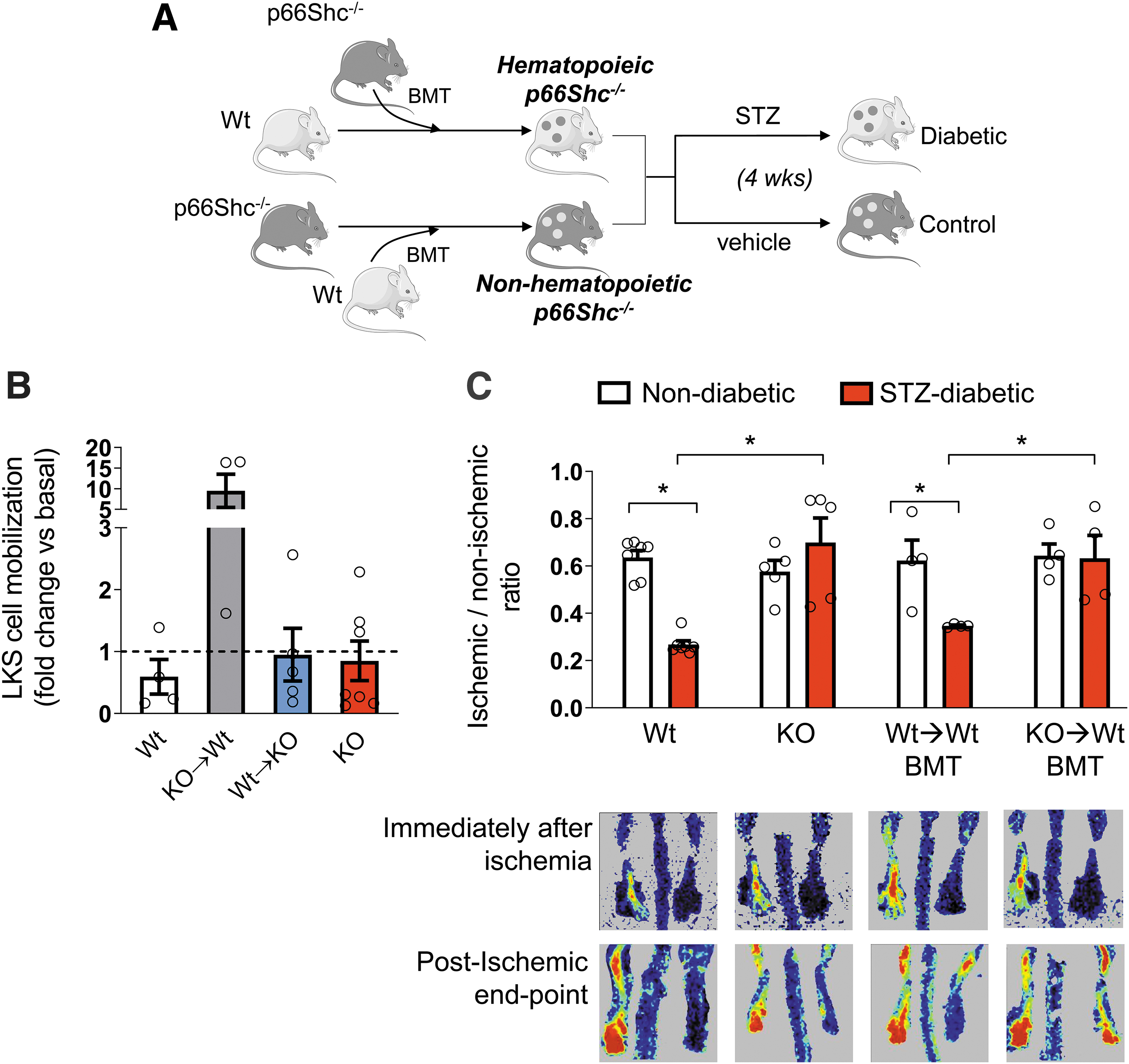
Remarkably, only p66Shc −/−→Wt BMT diabetic mice showed a recovery of postischemic HSPC mobilization, while Wt→p66Shc −/− BMT diabetic mice showed a postischemic LKS cell mobilization response similar to that of ubiquitous p66Shc −/− diabetic mice and Wt→Wt BMT diabetic mice, which served as controls (Fig. 3B). This finding is consistent with the hypothesis that absence of p66Shc in ischemic muscles prevents signaling to the BM via factors elicited by HIF-1α, such as VEGF and CXCL12. We then evaluated whether the recovery of postischemic HSPC mobilization by hematopoietic p66Shc deletion was sufficient to rescue reperfusion of the ischemic tissue in diabetic mice. We found that while in Wt→Wt BMT mice, diabetes still compromised blood flow recovery (ruling out an unspecific effect of BMT), in Wt→p66Shc −/− BMT mice, the adverse effect of diabetes on blood flow recovery disappeared (Fig. 3C). Thus, hematopoietic p66Shc deletion was sufficient to rescue postischemic blood flow, implying that diabetes-associated HSPC mobilopathy can affect the responses of distant tissues under pathological conditions.
To assess the contribution of mobilized HSPCs to the recovery of ischemic tissue, we transplanted Wt or p66Shc −/− GFP+ BM cells into Wt mice and analyzed gastrocnemius muscles 2 weeks days after hind limb ischemia in STZ diabetic mice (Fig. 4A and Supplementary Fig. S5A). This enabled us to compare hematopoietic p66Shc deletion (p66Shc −/− → Wt BMT) versus a control condition (Wt → Wt BMT). We confirmed that hematopoietic p66Shc deletion limited diabetes-induced myelopoiesis (1), because of lower granulocyte levels, despite a similar degree of hyperglycemia (Fig. 4B, C), while lymphocyte counts were not affected (Supplementary Fig. S5B). Deletion of p66Shc did not modify the polarization of BM macrophages in vitro (Supplementary Fig. S5C).

Flow cytometry analysis showed that total GFP+ cells in the ischemic gastrocnemius did not differ in hematopoietic p66Shc −/− versus control animals, but GFP+ LKS cells were more abundant in the gastrocnemius of animals transplanted with p66Shc −/− BM than in those transplanted with Wt BM (Fig. 4D). By analyzing sections of ischemic compared with nonischemic contralateral gastrocnemius muscles, we found that hematopoietic p66Shc deletion increased the capillary/fiber ratio by twofold (0.43 ± 0.07 vs. 0.84 ± 0.08, p < 0.01), with more GFP+ cells in close proximity of capillaries (Fig. 4E).
We then obtained cell suspensions from ischemic muscles and sorted GFP+ BM-derived cells. Immune cell subpopulations were detected with similar proportions within the ischemic muscles of mice transplanted with Wt or p66Shc −/− BM (Supplementary Fig. S6), suggesting that hematopoietic p66Shc deletion allowed a selective LKS traffic to ischemic sites. We also analyzed gene expression of GFP+ cells sorted from ischemic muscles. Adhesion molecules CD164 and CD62L (Sell) and VEGF were all upregulated in lineage+ (mature) or lineage− (immature) fractions of p66Shc −/− cells compared with p66Shc+/+ cells (p < 0.05 for p66Shc −/−) (Fig. 4F).
Hematopoietic p66Shc gene expression in humans
In a cross-sectional cohort of 40 patients with diabetes, we examined expression of p66Shc in relation to features of myelopoiesis, such as the white blood cell (WBC) count and the neutrophil/lymphocyte (N/L) ratio. Clinical characteristics of these patients are summarized in Supplementary Table S1. Briefly, they were 62.8-year olds, 70% males, with an average 8.7 years of diabetes duration. They had an HbA1c of 7.7% and most were affected by microvascular (82.5%) or macrovascular (77.5%) complications. We found that p66Shc gene expression in peripheral blood mononuclear cell (PBMC) was directly correlated with WBC (r = 0.33; p = 0.038; Fig. 5A) and with the N/L ratio (r = 0.44; p = 0.004; Fig. 5B). Furthermore, patients with PAD, defined as an ankle-brachial index below 0.9, had significantly higher p66Shc gene expression in PBMC (Fig. 5C), supporting the findings in mice that hematopoietic p66Shc deletion improved responses to ischemia.
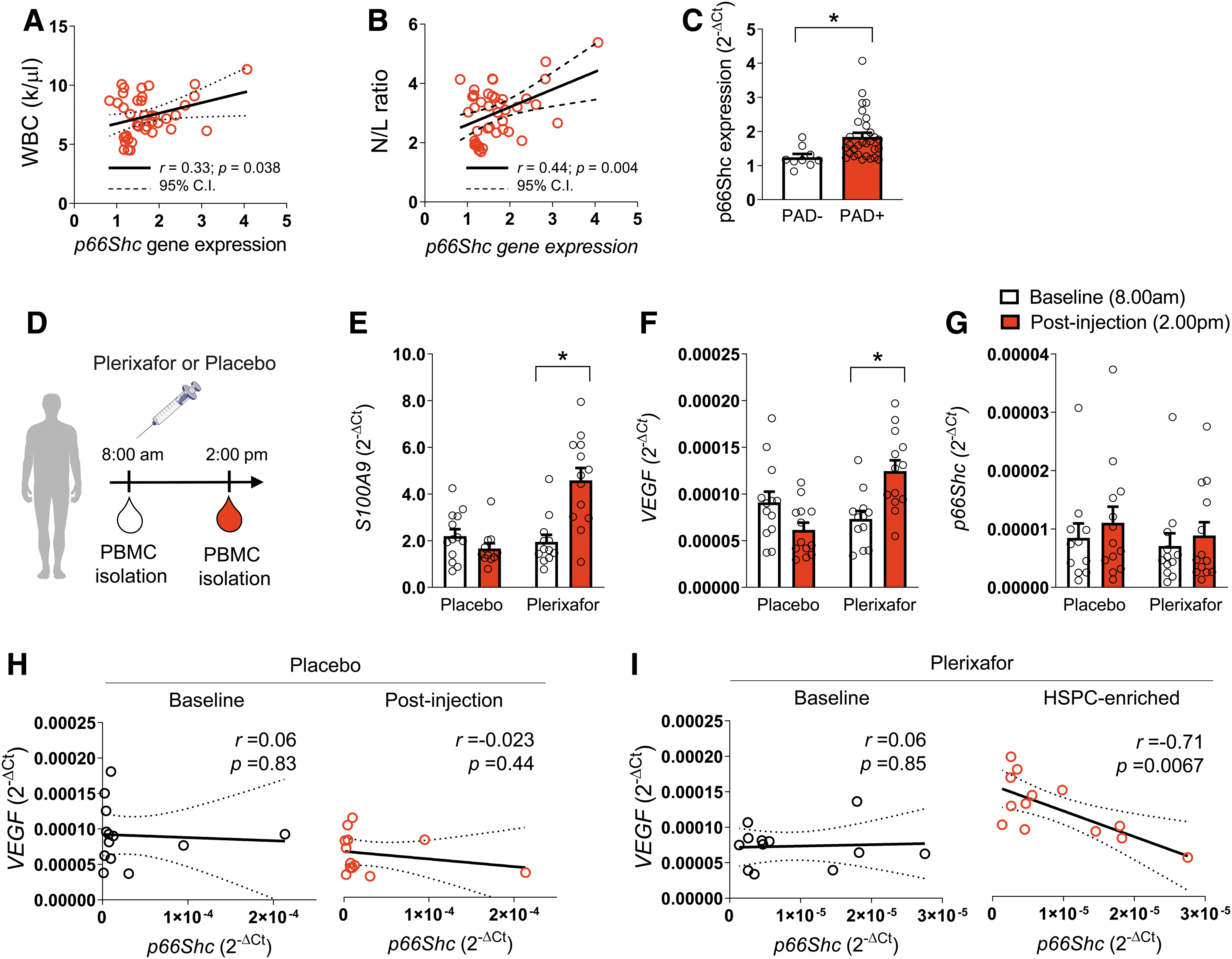
We then analyzed gene expression in PBMCs obtained during a previous clinical trial in which 26 patients with diabetes and ischemic wounds were randomized to receive a single subcutaneous injection of plerixafor or placebo (saline; Fig. 5D) to evaluate the effects of HSPC mobilization on wound healing (8). Clinical characteristics of these patients have been described in detail elsewhere (8). Briefly, they were on average 69-year olds, 85% males, with a diabetes duration of about 15 years, the vast majority also being affected by hypertension (100%) and dyslipidemia (81%). Glucose control was overall poor (average HbA1c 9.2%) and several additional chronic complications were present. Patients were receiving state-of-the-art therapies for diabetes and concomitant risk factors. Plerixafor-induced mobilization of HSPCs was robust (fold increase 7.7 ± 4.4) compared with the placebo group (fold increase 1.1 ± 0.3; p < 0.01 vs. plerixafor). Therefore, PBMCs isolated from patients who received plerixafor were markedly enriched in CD34+ HSPCs. VEGF gene expression was upregulated in PBMCs collected after treatment with plerixafor compared with placebo (fold increase 2.1 ± 0.5 vs. 0.8 ± 0.2; p < 0.01, Fig. 5E). Similarly, alarmin S100A9 (also known as calgranulin B) was increased in PBMC after treatment with plerixafor (fold increase 2.5 ± 0.4 vs. 1.0 ± 0.2; p < 0.01, Fig. 5F). S100A9 is highly expressed in neutrophils and granulocytes and drives myelopoiesis via p66Shc (1). p66Shc gene expression was not affected by treatment and did not differ between the two groups (p = 0.56, Fig. 5G). However, within CD34+ HSPC-enriched PBMCs isolated from patients after treatment with plerixafor, we found a significant negative correlation between VEGF and p66Shc expression (r = −0.70; p = 0.007; Fig. 5H, I). These data are in line with findings obtained in the STZ diabetes mouse model and support the concept that hematopoietic cell-intrinsic p66Shc modulates myelopoiesis and proangiogenic signals.
Discussion
We show that the redox enzyme and adaptor protein p66Shc hematopoietic cell intrinsically govern the traffic of HSPCs to peripheral ischemia, ultimately affecting blood flow recovery. We used the STZ-induced diabetes mouse model, which features an impaired HSPC mobilization from the BM and homing to the ischemic skeletal muscles, along with blunted vascular recovery and tissue reperfusion. This model parallels human disease, because diabetes compromises HSPC mobilization in response to G-CSF and tissue ischemia, which is associated with poor outcomes (20, 23, 35). The inability to adequately form collaterals and repair the damage is mechanistically linked to the severity of PAD in diabetes (18), which can lead to major amputations, worsening quality of life, and shortening survival. The molecular bases of such condition are manifold and incompletely understood, making the therapeutic armamentarium limited. Cell therapies based on HSPC infusion or injection have shown beneficial effects in patients with PAD (54), but they can hardly be applied on a large scale. Thus, we aimed to identify molecular pathways governing HSPC traffic that could be targeted to improve PAD outcomes in diabetes.
Since conditional knockout of the p66Shc isoform is unavailable (39), we performed BMT to obtain selective deletion of p66Shc in the BM hematopoietic compartment. The BM stem cell niche is composed by hematopoietic cells (e.g., HSPCs and macrophages) and stromal cells (e.g., endothelial cells, osteoblasts, and mesenchymal stem cells [MSCs]), but only the hematopoietic component is substituted by BMT. We have previously shown that, within stromal cells, p66Shc participates in Osm signal transduction, which culminates in the release of Cxcl12 that keeps HSPCs attached to the niche, preventing them from reaching periphery blood (1). Yet, the Osm-p66Shc pathway is also active within hematopoietic cells and stimulates myelopoiesis in an autocrine/paracrine loop (1). With cross-BMT experiments, we untangled the contribution of p66Shc in the stromal versus hematopoietic compartment of the BM because the stroma was not radioablated and remained of host origin. Herein, we confirmed that selective deletion of p66Shc in the hematopoietic compartment limited diabetes-induced myelopoiesis (i.e., the surge in neutrophil count) and was sufficient to rescue ischemia-induced HSPC mobilization and homing to the ischemic tissue, as well as blood flow recovery in mice with diabetes. These results were not recapitulated in ubiquitous p66Shc −/− mice, which displayed no postischemic HSPC mobilization. This is in sharp contrast with the efficient mobilization achievable in diabetic p66Shc −/− mice after G-CSF (1). While the mobilizing response to G-CSF only involves the BM compartment, mobilization induced by ischemia requires activation of hypoxia-sensing systems in the ischemic tissue, followed by release of soluble mediators that stimulate the BM to mobilize HSPCs, which, in turn, are recruited to the ischemic site via chemokine receptors and adhesion molecules. This complex sort of reflex can be interrupted by either defective sensing of hypoxia or BM dysfunction. Diabetes disrupts the physiologic HIF-1α regulation required for HSPC traffic (14, 26, 58) and p66Shc has been shown to interact with the HIF-1α pathway to activate responses to hypoxia (12, 42). In agreement with the notion that p66Shc is required for HIF-1α accumulation and mounting of the hypoxic response, we found that whole-body p66Shc deletion prevented induction of the typical hypoxic signature of HIF-response genes in the ischemic muscle, especially in the presence of diabetes. Arguably due to impaired release of soluble factors (e.g., Cxcl12 and Vegf) (23) from the ischemic site, diabetic p66Shc −/− mice were unable to mobilize HSPC after hind limb ischemia. In addition, we previously demonstrated that deletion of p66Shc diminished ischemic tissue damage (21), possibly below the threshold needed to elicit a BM response.
Transplanting p66Shc −/− BM in Wt diabetic mice rescued cell-intrinsic HSPC mobilization (1), while the ischemic skeletal muscle, still bearing p66Shc, could elicit a sufficient response to trigger mobilization. Hematopoietic deletion of p66Shc restrained myelopoiesis induced by hyperglycemia without affecting PB lymphocyte count. Polarization of BM macrophages in vitro was not affected by p66Shc deletion, suggesting an early role of p66Shc in modulating myelopoiesis by committing HSPCs. An escape from myelopoiesis might contribute to rescue the responsiveness of BM upon ischemia, similarly to what occurs upon G-CSF stimulation (1). Others have shown that p66Shc modulated the survival of progenitor cells when exposed to high glucose in vitro and rescued their angiogenic properties in hyperglycemic mice in vivo (56). All these clues suggest that hematopoietic deletion of p66Shc improves the ability of HSPCs to egress the BM and provide vascular support for angiogenesis.
By generating GFP-expressing p66Shc −/− mice and transplanting their BM into Wt mice, we developed a new tool to verify whether the restored HSPC mobilization in hematopoietic p66Shc-deleted diabetic mice improved recruitment of HSPCs to the ischemic site. Defective recruitment of BM-derived cells at sites of vascular damage has been reported consistently in diabetes (2, 9). Gene expression analysis of GFP+ cells homed to the ischemic muscles revealed that p66Shc −/− cells had higher expression of adhesion molecules that are critical for homing, such as CD62L and CD164. Intriguingly, an increased expression of CD62L in diabetic HSPCs isolated from the BM can be reverted by the deletion of p66Shc and has been associated with improved mobilization after G-CSF (4, 25). This highlights how HSPC traffic is differentially regulated in time and space, as mobilized HSPCs require upregulation of adhesion molecules such as CD62L to reach target tissues. By analyzing sections of ischemic skeletal muscles, we found a higher number of GFP+ vessels in mice receiving p66Shc −/− versus p66Shc +/+ BM. Yet, it should be carefully noted that, in absolute terms, the number of GFP+ cells in close proximity of blood vessels was small. Indeed, the extent to which BM-derived cells physically contribute to neovessel formation is debated (29), as lineage-tracing experiments failed to confirm morphological studies on endothelial transdifferentiation (24). Rather, it is supposed that BM-derived cells recruited to ischemic sites stimulate angiogenesis in cooperation with resident cells mainly by producing paracrine factors (13). Intriguingly, p66Shc −/− BM-derived GFP+ cells homed to ischemic muscles also showed higher Vefg expression, in both Lin+ (mature) and Lin− (immature, including HSPCs) populations, which is consistent with the paracrine activity of these cells in promoting angiogenesis. The role of soluble factors produced locally by blood cells (macrophages, neutrophils, and T cells) has been demonstrated also in the setting of solid cancer angiogenesis (44). Altogether, these data support a cell-intrinsic role for p66Shc in negatively modulating the proangiogenic responses in diabetes (48).
Therefore, dissecting the contribution of p66Shc in the hematopoietic system and in the target ischemic tissue, we herein show that cell-autonomous p66Shc in the BM fosters myelopoiesis and acts against HSPC mobilization in response to ischemia, blocking HSPC homing to the target tissue, ultimately impairing blood flow recovery and arguably leading to a worse course of PAD.
In patients with diabetes, impaired HSPC mobilization, called “diabetic stem cell mobilopathy” (16), is followed by a stable reduction of circulating HSPCs, which is linked to the progression of diabetic complications (53). In the presence of PAD, pauperization of BM-derived stem/progenitor cells (17) predicts future PAD-related adverse events (30). It is important to remind that p66Shc gene expression is upregulated in hematopoietic cells (PBMCs) of patients with diabetes, is directly correlated with plasma 8-isoprostanes, a validated marker of oxidative stress (47), and predicts new onset of complications, especially macroangiopathy (19). We now show a direct correlation between p66Shc gene expression in PBMC and indexes of myelopoiesis (WBC count and the N/L ratio), as well as an upregulation of p66Shc in patients with PAD. Furthermore, in plerixafor-mobilized diabetic patients with PAD, enrichment in CD34+ HSPCs might explain the surge in VEGF expression observed in PBMCs. Intriguingly, we found an inverse correlation between p66Shc and VEGF gene expression, confirming that p66Shc can regulate the hematopoietic proangiogenic response and lending further support to the pathophysiological importance of hematopoietic p66Shc in diabetic vascular disease. While p66Shc is involved in VEGF signaling in endothelial cells (45), regulation of VEGF expression and production by p66Shc appear to be tissue and stimulus specific since our data support the finding that in skeletal muscle (61), the deletion of p66Shc did not rescue ischemic Vegf upregulation as it does in T cells (42).
Conclusion
Our findings highlight a novel differential role of p66Shc in modulating the cross talk between ischemic tissues and the BM. While p66Shc might be necessary to elicit a proper response to ischemia in peripheral tissues, its deletion in hematopoietic cells represents a putative strategy to improve HSPC traffic and vascular recovery after ischemia. While no specific chemical inhibitor of p66Shc is available, directly delivering p66Shc siRNA to hematopoietic cells may be feasible using nanoencapsulation (33). Alternatively, epigenetic manipulation may uncover new treatment opportunities in view of the signature that hyperglycemia imprints on CD34+ HSPCs and endothelial cells (48, 59). For example, apabetalone (RVX-208), an FDA-approved drug, has shown promising results in removing detrimental epigenetic traits in the contest of CVDs (50, 51).
Materials and Methods
Mice
C57BL/6J Wt and C57BL/6-Tg(UBC-GFP) mice were purchased from The Jackson Laboratory and established as a colony since 2001 and 2017, respectively. Mice were randomly assigned to treatments or experimental groups. p66Shc−/− mice were obtained from Pier Giuseppe Pelicci's laboratory (European Institute of Oncology, Milan, Italy); a colony was established in 2010 and backcrossed on the C57BL/6J background for 10 generations. p66Shc−/−UBC-GFP mice were generated by crossing p66Shc−/− mice and C57BL/6-Tg(UBC-GFP) mice. Ly5.1 CD45.1 C57BL/6 mice (
Induction of diabetes
For all the experiments, 3- to 4-month-old male mice were used. Type 1-like diabetes was induced by a single intraperitoneal injection of 175 mg/kg STZ (AdipoGen Corp) in 100 mM pH 4 Na-citrate buffer (23). Blood glucose levels were measured using a point-of-care glucometer (FreeStyle; Abbott, Abbott Park, IL). Diabetes onset was confirmed for blood glucose ≥300 mg/dL 48 h after STZ injection, but only mice with persistent hyperglycemia in the subsequent 2 weeks were used.
BM transplantation
Recipient mice (3 months old) were treated with a myeloablative dose of total body irradiation with 10 Gy, split in two doses of 5 Gy 3h apart, and followed by an intravenous injection of unfractioned BM cells from donor p66Shc−/− mice, C57BL/6-Tg(UBC-GFP), p66Shc−/−UBC-GFP, or Ly5.1 CD45.1 C57BL/6 mice (4 × 107/each) isolated by flushing both femurs and tibias with sterile ice-cold phosphate-buffered saline (PBS). Animals were housed with sterile cages, water, food, and bedding for 4 weeks to allow BM reconstitution. Engraftment was assessed on PB by performing blood cell count using the CELL-DYN Emerald hematology analyzer (Abbott). Complete engraftment and development of the BM-GFP chimera were confirmed when ≥95% of WBCs were GFP+ or CD45.1+ using by flow cytometry.
Hind limb ischemia
Animals were sedated with inhaled isoflurane (Iso-Vet, Piramal Healthcare, United Kingdom). The femoral artery and the vein were surgically dissected from the femoral nerve, then cauterized with low-temperature cautery, and excised between inguinal ligament and hackle. Stem/progenitor cell mobilization was assessed after 3 days, whereas we measured hind limb microvascular perfusion with Perimed Periscan Pim II Laser Doppler System (Perimed AB, Sweden) and collected the limb muscles for analysis 14 days after surgery. Skeletal muscle from GFP+ transplanted animals were fixed with 4% paraformaldehyde and dehydrated with ascending concentrations of sucrose before freezing in liquid nitrogen-cooled isopentane. In separate experiments, ischemic muscles from GFP+ transplanted animals were minced and processed with 2 mg/mL of collagenase II (Worthington, United Kingdom) for 1h at 37°C to obtain a cell suspension for flow cytometry analysis and cell sorting.
Flow cytometry
Flow cytometry was performed on BM, ethylenediaminetetraacetic acid-treated PB, or tissue lysates. BM cells were isolated by flushing femurs and tibias with ice-cold MACS Separation Buffer (Miltenyi Biotec, Gladbach, Germany) through a 40 mM cell strainer. Cell suspensions were incubated with antibodies for 15 min in the dark at room temperature. After red blood cell lysis, samples were resuspended in 200 mL PBS, and data were acquired with an FACSCanto (BD Biosciences) cytometer or sorted with an FACSAria IIIu cytometer followed by analysis using FlowJo software (Tree Star, Inc.). The antibodies used are listed in Table 1.
List of Antibodies
Patients
Patients with diabetes were recruited at the University Hospital of Padova. The trial of stem cell mobilization with plerixafor for the treatment of diabetic ischemic wounds due to PAD was approved by the local ethics committee (NCT02790957) and was conducted in accordance with the Declaration of Helsinki as revised in 2000. It was a pilot, phase IIa, single-center, randomized, double-blind, placebo-controlled trial to test whether HSPC mobilization with the CXCR4 antagonist plerixafor improved healing of ischemic diabetic wounds (8). PB was collected at baseline (8:00 am) and 6 h after (2:00 pm) patients had received a subcutaneous injection of plerixafor 0.24 mg/kg or matching volume of 0.9% saline (placebo). PBMCs were isolated with Histopaque®-1077 (Merck) according to the manufacturer's protocol and kept in QIAzol (Qiagen) at −80°C.
In addition, we reanalyzed data collected during a previous cohort study (17) designed to evaluate the relationship between p66Shc gene expression in PBMCs and diabetic complications. We selected patients for whom the following information was available: p66Shc gene expression, total and differential WBC count, presence/absence of PAD, according to the standard definition of an ankle-brachial index (the ratio of systolic blood pressure measured at the ankle over systolic blood pressure at the arm) <0.9. Informed consent was obtained from all human participants.
Immunofluorescence
Gastrocnemius muscles were frozen in isopentane-cooled liquid nitrogen and stored at −80°C. Muscle 10-um-thick cryosections were incubated with a DyLight™594-conjugated Isolectin B4 (Bandeiraea simplicifolia, 1:100; Vector Laboratories) and costained with the rabbit anti-GFP antibody (1:100, Catalog # A-11122; Invitrogen) followed by an incubation with the goat anti-rabbit AlexaFluor®488 (1:200, Catalog # 111-545-046; Jackson ImmunoResearch, Cambridge, United Kingdom). Nuclei were counterstained with Hoechst 33258 (Sigma-Aldrich).
Cell culture
Murine BM-derived mesenchymal stem cells (BM-MSCs) were isolated by flushing the BM and cultivating the cells with minimum essential media-α containing 10% fetal bovine serum (FBS), glutamine (2 mM, all from Corning), and penicillin/streptomycin. Passage 3–6 was used in all experiments.
BM-derived macrophages
BM cells were obtained by flushing with sterile ice-cold PBS both the femurs and tibia of 3-month-old mice. RBC were lysed with ammonium-chloride-potassium lysing buffer. To obtain resting (M0) macrophages, 150,000 cells/cm2 were plated on six-well plates (BD Falcon, NY) with RPMI-1640 medium (Corning) supplemented with glutamine 2mM, penicillin/streptomycin, and 10% FBS +10 nM murine macrophage-colony stimulating factor (ImmunoTools) for 7 days without medium change. For M1 macrophages, cells were incubated 48 h with lipopolysaccharide (1 μg/mL) and mouse recombinant IFN-γ (10 ng/mL). For M2 polarization, resting macrophages were incubated with IL-4 (20 ng/mL) and IL-13 (5 ng/mL).
Gene expression
RNA was isolated from cells or tissues by use of QIAzol or with a Total RNA Purification Micro Kit (Norgen Biotek, Canada) and quantified with a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized from 500 ng RNA using a SensiFAST cDNA Synthesis Kit (Bioline, London, United Kingdom). Quantitative PCR was performed using the SensiFAST SYBR Lo-ROX Kit (Bioline) via a QuantStudio 5 Real-Time PCR System (Thermo Fisher Scientific). The primers used are listed in Table 2.
List of Primers
Statistics analysis
Continuous data are expressed as mean ± SE unless otherwise specified. Normality was checked using the Kolmogorov–Smirnov test, and non-normal data were log-transformed before analysis. Comparison between two or more groups was performed using the Student t test and analysis of variance (ANOVA) for normal variables or the Mann–Whitney U test and Kruskal–Wallis test for non-normal variables that could not be log-transformed (e.g., because of frequent zero values). To analyze data from experiments with two groups and two treatments, two-way ANOVA was used, and the effect of group and treatment was analyzed. All tests were two-tailed. Bonferroni adjustment was used to account for multiple testing. Biological replicates (individual mice) are shown as individual data points superimposed to bar charts. Significance was conventionally accepted at p < 0.05.
Footnotes
Authors' Contributions
M.A. performed the experiments, researched and analyzed data, and wrote the article. M.D.A. performed the experiments, researched and analyzed data, and revised the article. B.M.B. performed the experiments, researched and analyzed data, and revised the article. G.Z. performed the experiments, researched and analyzed data, and revised the article. A.R. designed the experiments, contributed to discussion, and revised the article. E.I. performed the experiments, contributed to discussion, and revised the article. A.A. provided supervision and funding, contributed to discussion, and revised the article. G.P.F. researched and analyzed the data, provided supervision and funding, and wrote the article. M.A. and G.P.F. are guarantors of this work and take full responsibility for its content.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from the Italian Ministry of Education, University and Research (PRIN2015ZTT5KB and PRIN201793XZ5A).
Data Availability
Original data obtained during this study are available from the corresponding author upon reasonable request.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.