
Abstract
Aims:
Trimethylamine-N-oxide (TMAO) is a metabolite generated from dietary choline, betaine, and
Results:
This study was conducted using a murine model of acutely disturbed flow-induced atherosclerosis induced by partial carotid artery ligation. 3,3-Dimethyl-1-butanol (DMB) was used to reduce TMAO concentrations. Wild-type mice were divided into four groups [regular diet, high-TMAO diet, high-choline diet, and high-choline diet+DMB] to investigate the effects of TMAO elevation and its inhibition by DMB. Mice fed high-TMAO and high-choline diets had significantly enhanced neointimal hyperplasia and advanced plaques, elevated arterial elastin fragmentation, increased macrophage infiltration and inflammatory cytokine secretion, and enhanced activation of nuclear factor (NF)-κB, the NLRP3 inflammasome, and endoplasmic reticulum (ER) stress relative to the control group. Mice fed high-choline diets with DMB treatment exhibited attenuated flow-induced atherosclerosis, inflammasome expression, ER stress, and reactive oxygen species expression. Human aortic smooth muscle cells (HASMCs) were used to investigate the mechanism of TMAO-induced injury. The HASMCs were treated with TMAO with or without an ER stress inhibitor to determine whether inhibition of ER stress modulates the TMAO-induced inflammatory response.
Innovation:
This study demonstrates that TMAO regulates vascular remodeling via ER stress.
Conclusion:
Our findings demonstrate that TMAO elevation promotes disturbed flow-induced atherosclerosis and that DMB administration mitigates vascular remodeling, suggesting a rationale for a TMAO-targeted strategy for the treatment of atherosclerosis. Antioxid. Redox Signal. 38, 215–233.
Innovation
We discovered that the inhibition of TMAO attenuates neointimal formation. TMAO increases vascular inflammation, mitochondrial dysfunction, and oxidative stress through activation of ER stress. DMB has therapeutic potential for the treatment of vascular inflammation by competing for the absorption of choline. In addition, inhibition of the ER stress signaling pathway could be exploited as a molecular target for therapeutic applications.
Introduction
Cardiovascular disease (CVD) is the leading cause of death worldwide, especially in developed countries, and atherosclerosis is the most common pathological cause of CVD. Vascular remodeling of the carotid artery, clinically defined as intima-media thickening, is an essential predictive phenotype for CVD (Ravani et al, 2015). Although multiple risk factors for atherosclerosis are well known, this pathological condition preferentially occurs in particular areas of disturbed flow in branched and curved arteries (Ku et al, 1985).
Disturbed flow-induced atherosclerosis with intimal injury affects several cell types, including smooth muscle cells, endothelial cells, platelets, and inflammatory cells, causing the release of mediators, including growth factors and proinflammatory cytokines (Bentzon et al, 2014; Libby et al, 2011). Excessive neointimal hyperplasia frequently occurs after percutaneous coronary intervention (PCI) with stent implantation or in arterial locations with disturbed blood flow, often necessitating a secondary revascularization procedure (VanderLaan et al, 2004).
Moreover, disturbed flow-induced vascular injury stimulates inflammatory cytokine and chemokine secretion and oxidative stress (Koka et al, 2017). Despite advances in PCI technology, neointimal hyperplasia and luminal restenosis continue to be the most frequent causes of target lesion failure after revascularization.
Trimethylamine N-oxide (TMAO), an important gut microbe-dependent metabolite, is generated from dietary choline, betaine, and
The TMAO increases reactive oxygen species (ROS) production and activates the NLRP3 inflammasome in the endothelium via inhibition of the SIRT3/SOD2/mtROS pathway (Chen et al, 2017) and activation of the ROS/TXNIP/NLRP3 pathway (Sun et al, 2016). In addition, TMAO activates the nuclear factor (NF)-κB pathway and increases the expression of inflammation-related genes, including interleukin (IL)-6, tumor necrosis factor-α (TNF-α), COX-2, intercellular adhesion molecule-1 (ICAM-1), and E-selectin, which leads to leukocyte recruitment and vascular inflammation (Seldin et al, 2016).
Treatment with a TMAO inhibitor increased the anti-inflammatory cytokine IL-10 in a TMAO-supplemented murine model (Chen et al, 2019a). Moreover, TMAO promoted the transformation of macrophages into foam cells by activating the CD36/MAPK/JNK pathway and inducing abnormal macrophage activation by increasing the expression of endoplasmic reticulum (ER) stress-related proteins, such as GRP78, GRP94, and HSP70 (Mohammadi et al, 2018; Mohammadi et al, 2015). Cholesterol metabolism is crucial in the progression of atherosclerotic lesions (Ross and Glomset, 1973).
The TMAO decreases the mRNA levels of the bile acid synthetic enzymes Cyp7a1 and Cyp27a1 and the bile acid transporters Oatp1, Oatp4, Mrp2, and Ntcp in the liver. The TMAO also decreases the mRNA levels of the intestinal cholesterol transporters Npc1L1 and Abcg5-Abcg8 in the gut (Koeth et al, 2013). In addition, TMAO promotes platelet hyperreactivity and thrombosis risk by enhancing the stimulus-dependent release of Ca2+ from intracellular Ca2+ stores (Zhu et al, 2016). Emerging evidence indicates that TMAO is a novel risk factor for CVD (Koeth et al, 2013; Mente et al, 2015; Tang et al, 2013; Wang et al, 2014; Wang et al, 2011), as it is associated with vascular inflammation and reduced circulating endothelial progenitor cell levels (Chou et al, 2019), leading to atherosclerosis (Seldin et al, 2016).
These findings suggest a rationale for the use of TMAO inhibition as a therapeutic target for atherosclerosis. However, some studies have provided evidence suggesting that TMAO may protect against CVD (Nowinski and Ufnal, 2018). Supplementation with
Vegans and vegetarians produce less TMAO from
In contrast, the use of probiotics or their functional products might be an effective and safer method. In animal studies, administration of a probiotic markedly decreased choline-induced cecal TMA and serum TMAO levels by changing the microbiota (Qiu et al, 2018) and decreasing cholesterol levels (Huang et al, 2018b). Although probiotics have been shown to decrease TMAO levels, there were no dramatic effects in human studies. Treatment with the probiotic strain Lactobacillus casei Shirota in patients with metabolic syndrome did not alter plasma TMAO levels in response to a high-fat diet (Tripolt et al, 2015).
However, some small molecules protect against atherosclerosis by decreasing TMAO levels and changing the microbiota composition, such as resveratrol (Chen et al, 2016) and berberine (Shi et al, 2018). FMO3 is a critical enzyme in the conversion of TMA to TMAO. Although knockdown or silencing of hepatic FMO3 in different mouse strains reduced plasma TMAO levels and attenuated atherosclerosis, FMO3 knockdown promoted hepatic inflammation and ER stress by dampening LXR activation (Warrier et al, 2015) and changing the composition of cholesterol. Moreover, the inhibition of FMO3 expression led to the accumulation of TMA, resulting in trimethylaminuria (Messenger et al, 2013).
In previous studies, treatment with TMAO increased proinflammatory cytokines but decreased anti-inflammatory cytokine and protein expression. Zhu et al (2017) reported that TMAO-induced platelet hyperresponsiveness was attenuated by a low dose of aspirin. In addition to TMAO in CVD, high levels of TMAO contribute to renal dysfunction and mortality risk in CKD (Gupta et al, 2020). However, there were some limitations and adverse effects of non-steroidal anti-inflammatory drug treatments in CKD (Baker and Perazella, 2020). The effects of TMAO were directly abrogated by anti-inflammatory therapeutics, and the side effects should be further investigated.
The 3,3-Dimethyl-1-butanol (DMB), a structural analog of choline, reduced plasma TMAO levels in mice fed high-choline and high-carnitine diets by inhibiting microbial TMA formation. Reducing TMAO concentrations through the nonlethal targeting of gut microbial pathways is an attractive approach to inhibit atherosclerosis (Wang et al, 2015). However, the effects of TMAO and its inhibition by DMB on vascular remodeling in response to disturbed flow-induced atherosclerosis remain unclear.
We hypothesized that TMAO activates inflammation by promoting ER stress and that treatment with DMB inhibits the effects of TMAO on vascular remodeling by inhibiting inflammation. In this study, we investigated the effects of high-TMAO and high-choline diets on vascular remodeling in a partial carotid artery ligation model and examined the TMAO-lowering effects of DMB in vivo. In addition, treatment with TMAO promoted inflammation through ER stress induction in vitro. Tauroursodeoxycholic acid (TUDCA), an ER stress inhibitor, was used to reverse the TMAO-induced activation of ER stress.
Results
Effect of high-TMAO and high-choline diets on plasma TMAO levels
To investigate the effect of TMAO, mice were fed diets containing choline or TMAO daily for 6 weeks. Plasma TMAO levels were significantly elevated in the high-TMAO and high-choline groups compared with the control group (control vs. high-TMAO vs. high-choline group, 3.2 ± 1.3 μM vs. 171.3 ± 30.8 μM vs. 76.5 ± 9.5 μM, p < 0.01 vs. control; Fig. 1B) and were higher in the high-TMAO group than in the high-choline group.

The administration of DMB significantly reduced plasma TMAO concentrations in mice fed the high-choline diet (high-choline group vs. high-choline+DMB group, 76.5 ± 9.5 μM vs. 43.5 ± 15.3 μM, p < 0.01). There were no significant differences in body weight changes, liver or renal function, fasting blood glucose levels, or total cholesterol levels among the four groups (data not shown).
Effect of high-TMAO and high-choline diets on vascular remodeling after carotid artery ligation
In this study, all mice survived after carotid artery ligation. Representative images of the ligated and contralateral carotid artery cross sections are shown in Figure 1. No morphological differences in the contralateral carotid arteries were observed among the groups. Four weeks after carotid artery ligation, neointimal hyperplasia and increased intima/media ratios were identified in wild-type (WT) mice, even with a regular diet; however, these effects were substantially greater in the high-TMAO and high-choline groups than in the control group (Fig. 1C).
The high-TMAO and high-choline diets also significantly promoted ligation-induced arterial elastin fragmentation (Fig. 1D). Of note, reduced neointimal hyperplasia, a lower intima/media ratio, and less arterial elastin fragmentation were observed in the DMB group than in the high-choline group (Fig. 1E–G).
Effects of TMAO elevation on vascular inflammation after carotid artery ligation
To further investigate the impact of increased TMAO levels on vascular inflammation, macrophage infiltration in arterial tissues was assessed by immunofluorescence staining using an F4/80 antibody. The fluorescence intensity value was calculated from microscopic images using Metamorph software. The relative intensity refers to the fluorescence intensity value/high power field in each group compared with the sham group.
Increased infiltration of F4/80-positive cells in the intimal area was observed in the high-TMAO and high-choline groups and was ameliorated in the DMB group (Fig. 2A, B). Vascular injury triggers NLRP3 signaling, resulting in the formation and activation of the NLRP3 inflammasome. NLRP3 and ASC expression was upregulated and colocalized in the vascular neointimal area in the high-TMAO and high-choline groups, with lower NLRP3 and ASC expression in the DMB group (Fig. 2C, D).
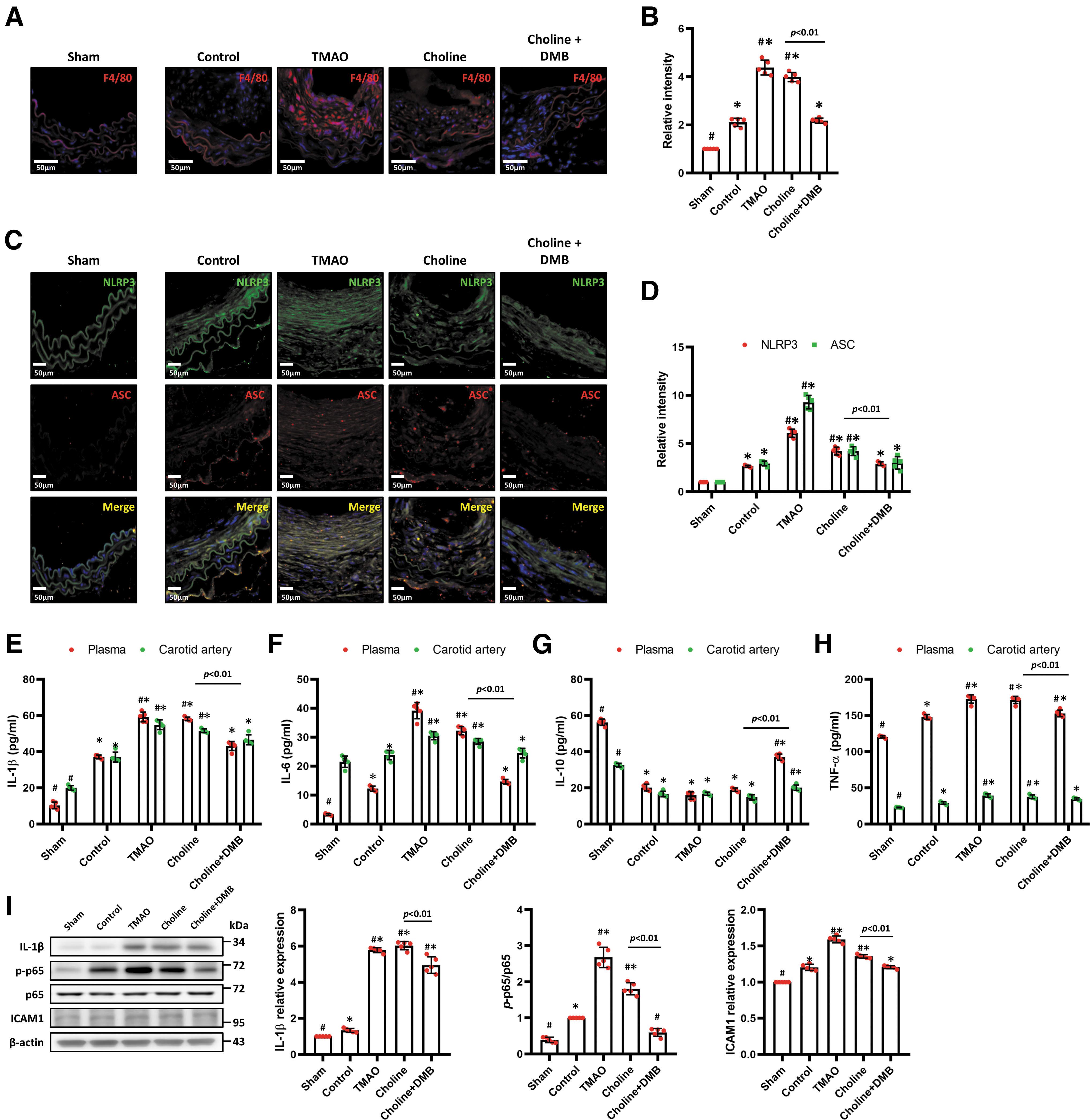
Partial left carotid artery (LCA) ligation upregulated the expression of the proinflammatory cytokines IL-1β, IL-6, and TNF-α and downregulated the expression of the anti-inflammatory cytokine IL-10 in plasma and carotid arteries (Fig. 2E–H). The high-TMAO and high-choline diets further upregulated proinflammatory cytokine (IL-1β, IL-6, and TNF-α) secretion, whereas treatment with DMB reversed these effects. Moreover, the protein expression of IL-1β and ICAM-1 and the levels of p-NF-κB p65/NF-κB p65 in the LCA were significantly increased in the high-TMAO and high-choline groups compared with the control group, and DMB attenuated these changes (Fig. 2I).
Effects of TMAO on mitochondrial function, ER stress, and ROS production after carotid artery ligation
Mitochondrial dysfunction leads to apoptosis and ROS production. The Bcl-2 family is the most notable for promoting mitochondrial stabilization. Immunofluorescence staining and Western blotting revealed upregulated Bax expression in arterial tissues in the high-TMAO and high-choline groups; in contrast, inhibition of Bax upregulation was observed in the DMB group (Fig. 3A, C). In addition, Bcl-2 expression was decreased in the high-TMAO and high-choline groups, but DMB treatment reversed this effect (Fig. 3C).

The ER stress is essential for promoting neointimal hyperplasia and vascular remodeling (Ishimura et al, 2014; Noda et al, 2015). Accumulation of unfolded or misfolded proteins in the ER causes ER stress followed by increased ROS (Ochoa et al, 2018). Indeed, treatment with TMAO may promote ER stress (Chen et al, 2019b; Mohammadi et al, 2018). Therefore, we assessed whether increased TMAO levels promoted ER stress after carotid artery ligation. Immunofluorescence staining showed upregulated expression of PERK and IRE-1α in arterial tissues in the high-TMAO and high-choline groups, whereas inhibition of PERK and IRE-1α upregulation was observed in the DMB group (Fig. 3B).
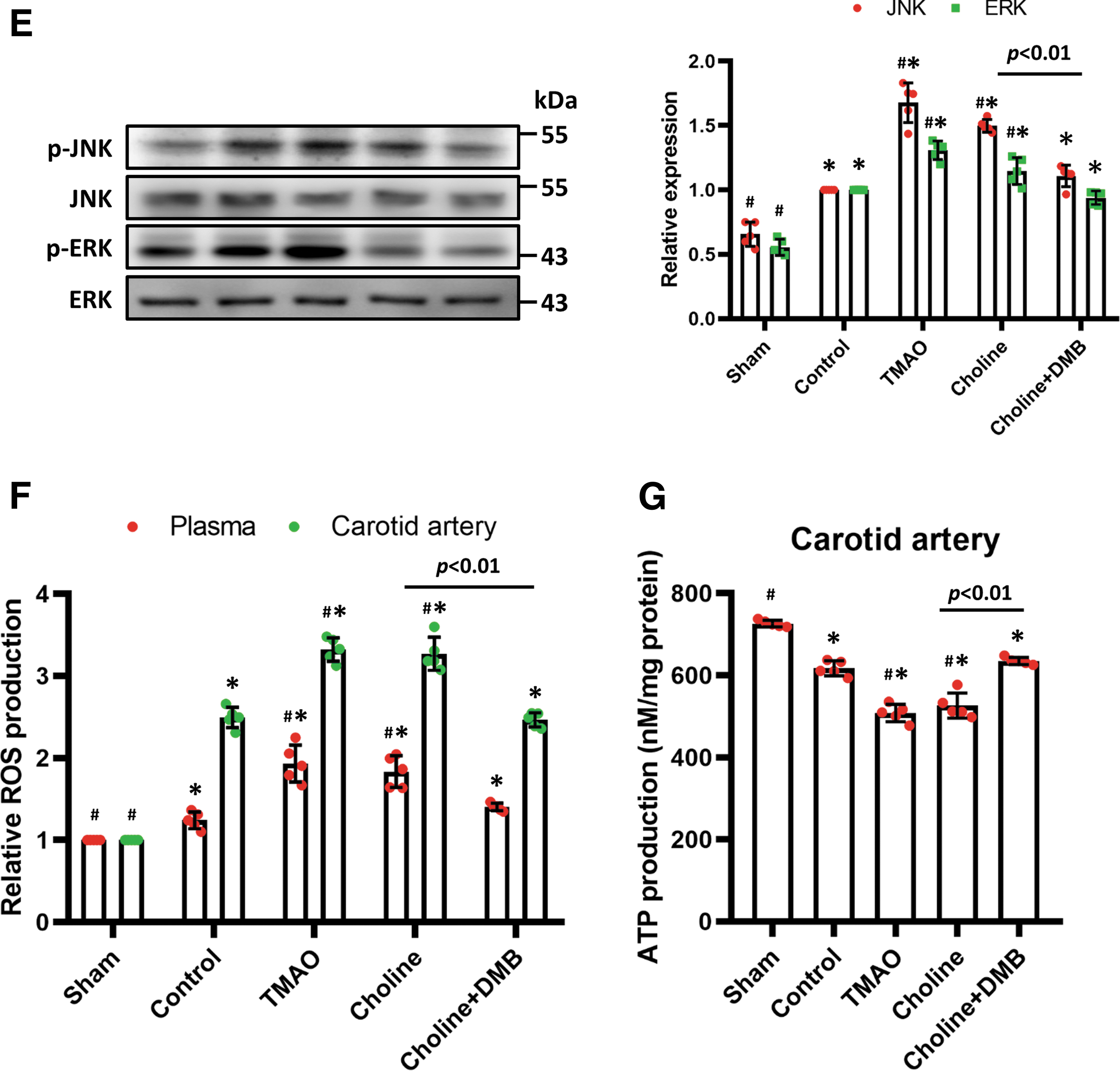
Western blotting revealed that the phospho-PERK/PERK and phospho-IRE-1α/IRE-1α ratios were higher in the high-choline and high-TMAO groups, but DMB administration reduced the expression of phospho-PERK and phospho-IRE-1α (Fig. 3D) and the expression of downstream markers, such as BiP, p-eIF2α, TRAF2, and ASK1 (Supplementary Fig. S1A). Activation of ER stress induces inflammation via mitogen-activated protein kinase (MAPK) pathways (Darling and Cook, 2014). ERK and JNK phosphorylation was significantly upregulated in the high-TMAO and high-choline groups, and DMB administration lessened this effect (Fig. 3E).
As shown in Figure 3F, partial carotid artery ligation significantly augmented ROS levels in plasma and the LCA, and the administration of high-TMAO and high-choline diets further upregulated ROS levels. In addition, treatment with DMB attenuated high-choline diet-induced ROS generation. Mitochondrial dysfunction also impairs adenosine triphosphate (ATP) production. Administration of TMAO significantly decreased ATP production; however, this damage was attenuated in response to DMB treatment (Fig. 3G).
TMAO increases activation of the NLRP3 inflammasome in vitro
To investigate whether TMAO activates the NLRP3 inflammasome in HASMCs, we assessed the expression of NLRP3 and caspase 1 in vitro. As shown in Figure 4A, TMAO increased the expression of NLRP3, ASC, and caspase 1 in a dose-dependent manner. The TMAO also activated the expression of IL-1β and ICAM-1 in HASMCs in a dose-dependent manner (Fig. 4B). To determine whether TMAO regulates the levels of phosphorylated NF-κB p65, HASMCs were treated with different doses of TMAO for 30 min.
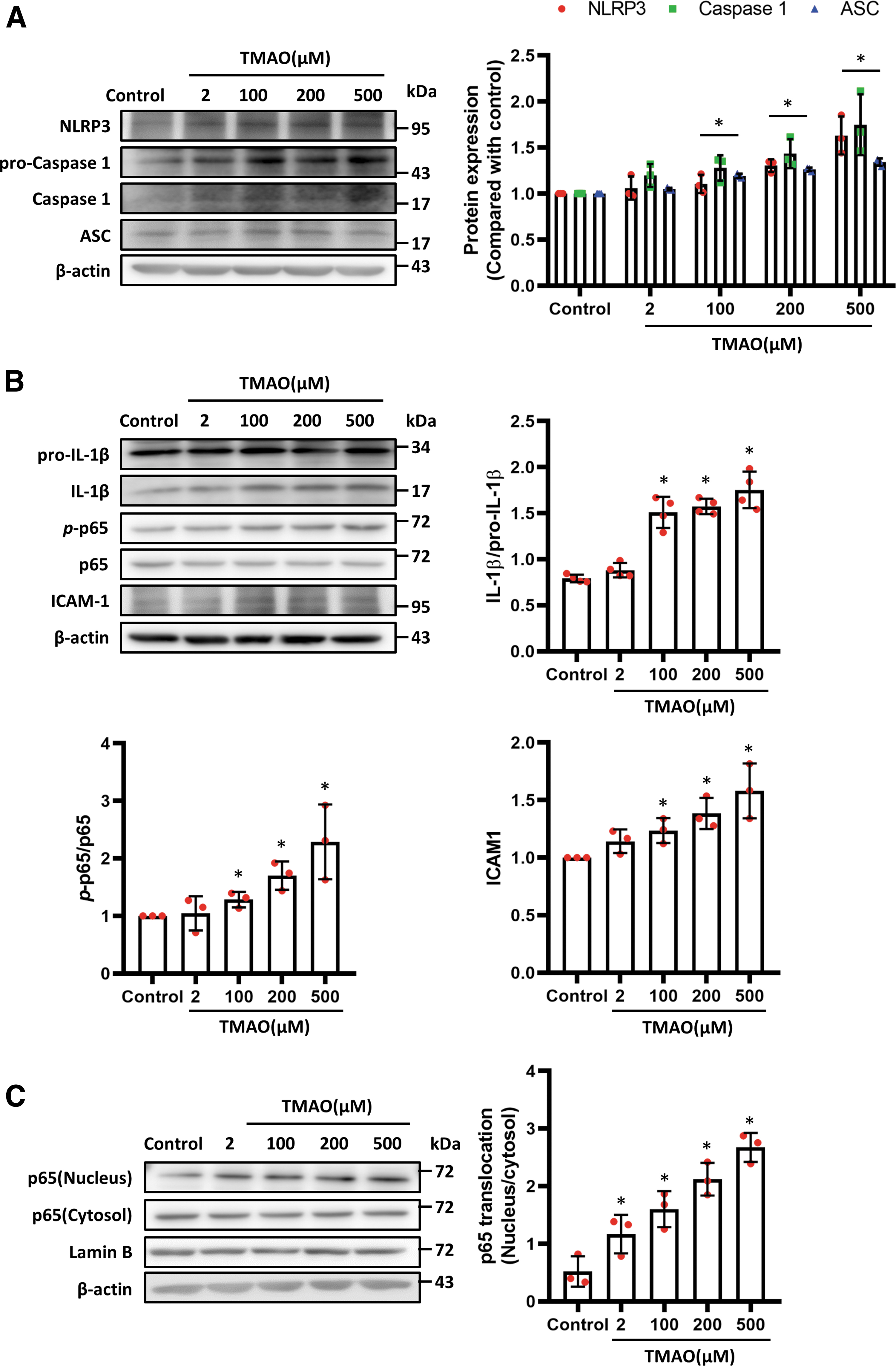
The results revealed that TMAO activated the phosphorylation of NF-κB p65 (Fig. 4B). Next, we tested the dynamics of NF-κB p65 subcellular localization after TMAO administration. Two hours after TMAO administration, NF-κB p65 was translocated from the cytosol to the nucleus (Fig. 4C; Supplementary Fig. S1E). These data indicate that high TMAO concentrations promote the phosphorylation and cytosolic-nuclear translocation of NF-κB p65.
TMAO attenuates mitochondrial function and increases ER stress in vitro
The TMAO administration increased the ratio of mitochondrial depolarization and reduced oxygen consumption in a dose-dependent manner (Fig. 5A, B) and decreased ATP generation (Fig. 5C). Moreover, TMAO induced elevated proapoptotic protein and Bax levels and downregulated Bcl-2α in HAMSCs (Fig. 5D).
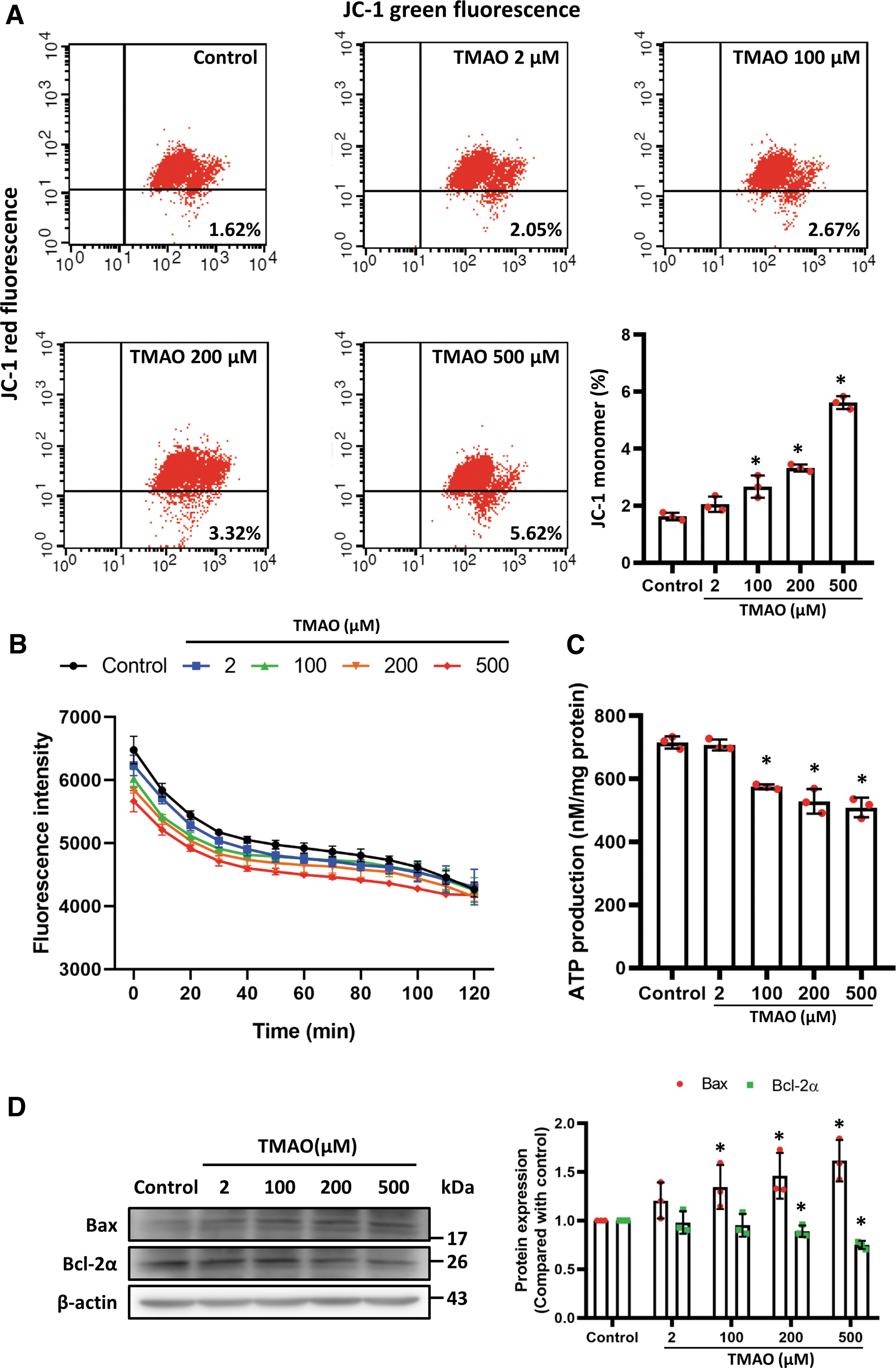
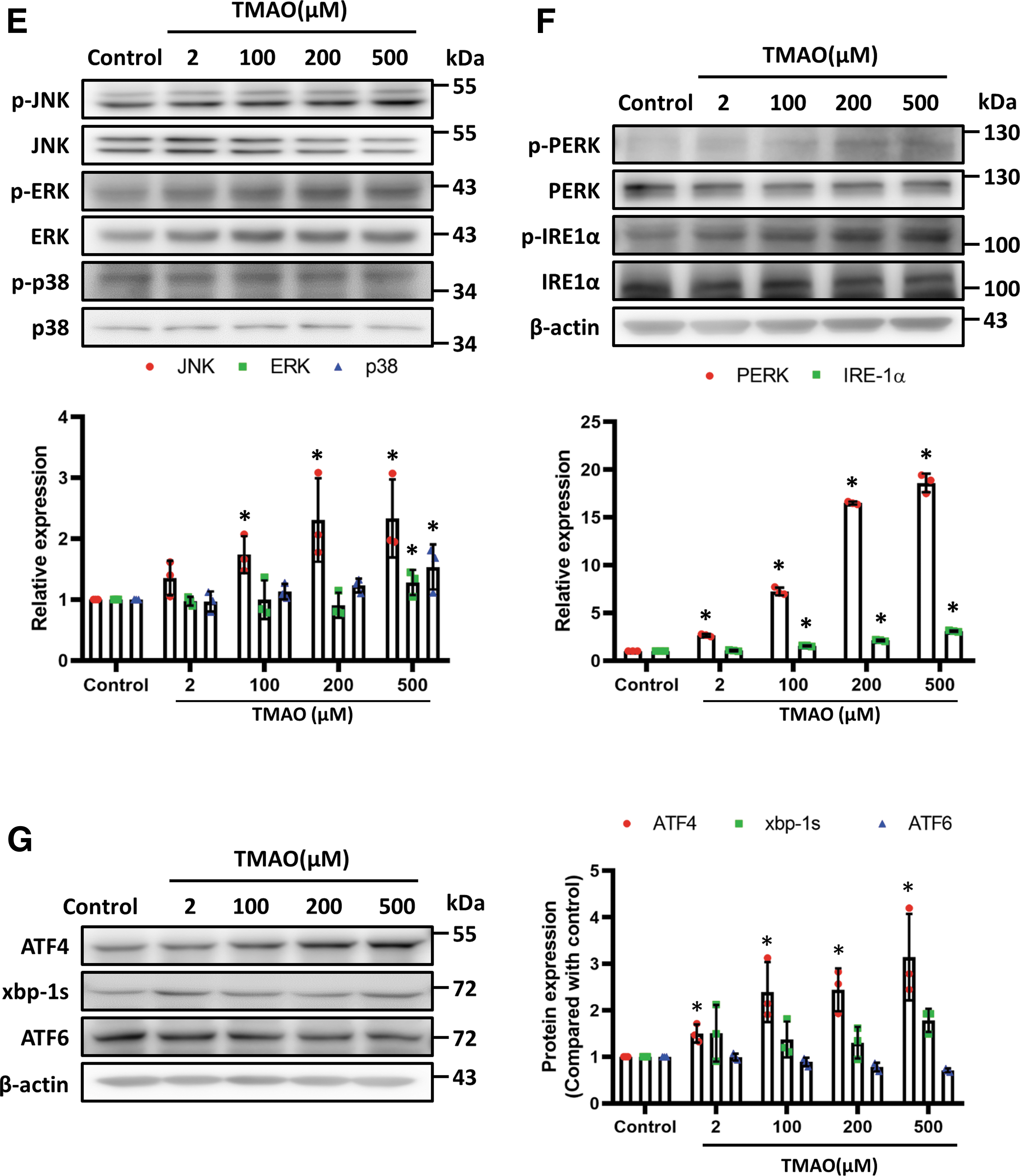
The ER stress contributes to mitochondrial dysfunction and inflammation. To determine whether TMAO affects ER stress and to elucidate the related signaling pathways, we examined the phosphorylation of JNK, ERK1/2, and p38 induced by different concentrations of TMAO in HASMCs. The TMAO significantly increased the phosphorylation of JNK and the ratios of phospho-PERK/PERK and phospho-IRE-1α/IRE-1α (Fig. 5E, F) and affected mediators downstream of ER stress, such as phospho-eIF2α, TRAF2, and ASK1. Of note, TMAO did not alter the expression levels of BiP (Supplementary Fig. S1B).
To clarify which ER stress signal TMAO participates in, we further examined factors downstream of ER stress. Interestingly, TMAO significantly increased the expression of activating transcription factor 4 (ATF4) (Fig. 5G), whereas there were no significant differences in the expression of xbp-1s or ATF6.
Moreover, the administration of TUDCA, an ER stress inhibitor, prevented TMAO-induced Bax activation and upregulated Bcl-2α in HASMCs (Fig. 6A). The TMAO induced phosphorylation of the ER stress-related proteins PERK and IRE-1α and the downstream proteins JNK, ERK, and p38. Importantly, TUDCA attenuated TMAO-induced activation of JNK and ERK (Fig. 6B). Moreover, TUDCA markedly attenuated the TMAO-induced ER stress response by suppressing PERK and IRE-1α phosphorylation in HASMCs (Fig. 6C).

Treatment with TUDCA attenuated TMAO-stimulated NLRP3/Caspase 1 activation and the subsequent release of mature IL-1β (Fig. 6D) and reduced ROS levels in HASMCs (Fig. 6E). We further assessed the effects of ER stress inhibition in TMAO-treated mice. The inhibition of ER stress by TUDCA significantly attenuated TMAO-enhanced neointimal formation (Fig. 6F) and macrophage infiltration (Supplementary Fig. S1C).
Discussion
The primary findings of this study can be summarized as follows. Mice fed high-TMAO and high-choline diets exhibited significantly enhanced neointimal hyperplasia and advanced plaques in response to carotid artery ligation. The DMB treatment attenuated this disturbed flow-induced atherosclerosis by inhibiting TMAO. Moreover, high-TMAO and high-choline diets promoted arterial elastin fragmentation, increased tissue macrophage infiltration and inflammatory cytokine secretion, and enhanced activation of NF-κB and the NLRP3 inflammasome in response to carotid ligation. Inhibition of TMAO levels by DMB treatment substantially reduced elastin fragmentation, inhibited inflammasome activation, attenuated mitochondrial dysfunction and ER stress, and reduced ROS levels (Fig. 6F).
These findings highlight the harmful effects of TMAO elevation in disturbed flow-induced neointimal hyperplasia and provide a rationale for targeting TMAO in the treatment of atherosclerosis. One of the risk factors for atherosclerosis is alterations in the composition and organization of the arterial extracellular matrix (ECM) (Lansing et al, 1950). Several proteins, such as elastic fibers, collagen, proteoglycans, hyaluronate, and glycoproteins, compose the ECM in the arterial wall (Halper and Kjaer, 2014). Atherosclerosis initially injures the arterial wall, followed by impairment of the elastic network.
The elastic structure becomes fragmented and disorganized, and collagen accumulates (Kuzan et al, 2021). Elastic fibers are composed of elastin and several microfibers and are the major components of the internal and external elastic lamina that separate the media from the intima and the adventitia (Thomson et al, 2019). There are several types of elastases, including matrix metalloproteinases, serine proteases, and cysteine proteases, which cleave insoluble and soluble elastin.
The concept that inflammation is a central part of the pathogenesis of atherosclerosis has attracted considerable attention. Emerging evidence indicates an essential role of TMAO in atherosclerosis and that it is an independent cardiovascular risk factor (Koeth et al, 2013; Wang et al, 2015). Some reports (Kurilshikov et al, 2019; Yang et al, 2019) suggest that TMAO elevation increases the expression of inflammatory cytokines and chemokines, promoting activation of the NF-κB pathway. Macrophages residing in body tissues trigger immune activation in response to injurious stimuli (Dos Anjos Cassado, 2017).
Of interest, our partial carotid ligation mouse model provides clear evidence that high-choline and high-TMAO diets substantially enhance tissue macrophage infiltration and activate inflammasomes, which may partly explain the adverse effects of high-TMAO conditions on neointimal hyperplasia and the subsequent negative vascular remodeling. These adverse effects are likely the result of marked enhancement of macrophage infiltration and vascular inflammation.
In addition, to investigate whether cellular infiltration is dependent on TMAO-induced ER stress, we demonstrated that an ER stress inhibitor reduced TMAO-promoted neointimal hyperplasia and F4/80 macrophage infiltration (Supplementary Fig. S1C). We used male mice to avoid sex differences in the current animal study. In previous studies, the expression levels of FMO in male mice differed from those in female mice (Veeravalli et al, 2018). Hasegawa et al (2020) showed that different fecal and cecal gut microbiota compositions between the sexes resulted in differential production of TMA and TMAO.
Moreover, female mice respond differently to intimal and medial thickness in a carotid artery ligation model (Patten, 2007). Through in vitro studies, we further demonstrated increased JNK phosphorylation, ICAM-1, NF-кB activation, and ROS elevation in TMAO-treated vascular SMCs, indicating that high TMAO concentrations activate inflammation in SMCs.
Mitochondrial function affects the proliferation and migration of vascular SMCs in atherosclerosis and neointimal formation (Ross and Glomset, 1973). Multiple pathways regulate the electron transport chain (ETC) in mitochondria (Grumbach and Nguyen, 2019). Mitochondria undergo oxidative phosphorylation (OXPHOS) to generate adenosine triphosphate (ATP) by consuming oxygen and sugars. Electrons are transferred from complexes I to IV through the electron transport chain. Simultaneously, positively charged protons are pumped from the mitochondrial matrix into the intermembrane space, creating the mitochondrial membrane potential.
Leakage of electrons at complexes I and III results in the reduction of oxygen to form superoxide during OXPHOS (Madamanchi and Runge, 2007). Several direct and indirect signaling pathways regulate ROS production in mitochondria, such as Ca2+ influx, energy demand, cellular redox status, hypoxia, ER stress, inflammation, immune responses, autophagy, and mitochondrial biogenesis (Bolisetty and Jaimes, 2013). Abnormal OXPHOS and ROS accumulation contribute to atherosclerosis, neointimal formation, and other vascular disorders (Peng et al, 2019).
The expression, localization, and binding of anti- and proapoptotic Bcl-2 family proteins are vital for the regulation of intrinsic apoptosis. We also demonstrated that enhanced TMAO levels result in the elevation of proapoptotic proteins and Bax levels and downregulation of Bcl-2α in HAMSCs, suggesting that elevated TMAO levels cause leakage of mitochondrial proteins into the cytosol, leading to caspase activation and enhancing cell death.
Regulation of the MAPK signaling pathway plays a critical role in neointimal formation and participates in cell proliferation, apoptosis, and inflammation. The inhibition of MAPK activation reduces neointimal formation via cell cycle arrest (Gennaro et al, 2004). Many factors, such as oxidative stress and cytokines, induce apoptosis and activation of the MAPK signaling pathway during the early stage of neointimal formation (Gomez et al, 2015). In line with previous findings, the activation of ERK under high-TMAO conditions upregulates vascular SMC inflammation (Lim and Park, 2014) and contributes to atherosclerosis.
The ERK activation is also essential for mitochondrial dysfunction, leading to increased oxidative stress and NF-κB activation (Huang et al, 2018a). Similarly, we found that mitochondrial dysfunction might be at least partially responsible for TMAO-induced vascular inflammation. Mitochondria absorb Ca2+ released from the ER in ER-stressed cells, causing opening of the permeability transition pore and the release of cytochrome c from the mitochondrial matrix.
The loss of cytochrome c inhibits complex III of the electron transport chain and enhances ROS production (Brand, 2010). Elevated TMAO concentrations increased the phosphorylation of JNK and upregulated the ratios of phospho-PERK/PERK and phospho-IRE-1α/IRE-1α, the major proteins involved in unfolded protein response (UPR) signaling in the cytosol and other organelles. These results provide crucial evidence that high TMAO levels enhance ER stress, leading to SMC apoptosis and inflammation in advanced lesions.
The TMAO elevation has been shown to cause vascular injury and promote endothelial cell inflammation by triggering NLRP3 inflammasome formation and activation (Boini et al, 2017; Ma et al, 2017) (Supplementary Fig. S1F). The TMAO activates the NLRP3 inflammasome (Boini et al, 2017) and NF-κB inflammation (Seldin et al, 2016); increases the levels of inflammatory cytokines and surface markers, including IL-1β, IL-6, TNF-α, ICAM-1, and VCAM-1 (Seldin et al, 2016); increases ROS production (Sun et al, 2016); and decreases NO production (Chou et al, 2019). Treatment with TMAO impairs the functions of endothelial cells, including tube formation, migration, and permeability (Boini et al, 2017; Chou et al, 2019; Seldin et al, 2016).
In previous studies, TMAO induced the dissociation of thioredoxin-interactive protein from thioredoxin (Sun et al, 2016) through inhibition of the NAD-dependent protein deacetylase sirtuin-3 (SIRT3)-superoxide dismutase 2 (SOD2)-mitochondrial ROS signaling pathway (Chen et al, 2017), resulting in activation of the NLRP3 inflammasome. Our study results confirm these findings and further suggest that TMAO promotes the NLRP3 inflammasome and activates ASC and caspase 1 in a dose-dependent manner in HASMCs.
Moreover, TMAO inhibition significantly downregulated NLRP3 inflammasome expression and prevented neointimal hyperplasia. These findings indicate that TMAO is a novel therapeutic target for atherosclerosis, particularly in patients with elevated TMAO concentrations, those who have undergone PCI, and those with arterial lesions with disturbed flow.
Recently, the evaluation of ER status has become an important pathophysiological paradigm in research on chronic metabolic disease. The ER stress may lead to cellular dysfunction and death, primarily through apoptosis. A recent report demonstrated that activation of the UPR, including the phosphorylation of PERK and IRE-1, the translocation of activating transcription factor 6, and accompanying inflammation, is associated with neointimal formation in arteries after wire-induced injury (Noda et al, 2015).
In addition, extracellular heat shock proteins (HSPs) activate immunoinflammatory reactions via cell surface receptors in response to atherosclerosis, whereas HSP and GRP94 regulate the degradation of misfolded proteins in the ER. Increased GRP94 is known to induce ER stress and UPR activation. High levels of TMAO increased the expression of GRP94 and HSP70 in the J774.A1 macrophage cell line (Mohammadi et al, 2018). Therefore, regulation of HSP or inhibition of ER stress may reduce the impact of neointimal formation (Ishimura et al, 2014; Kim et al, 2011). We demonstrated that a reduction in ER stress significantly decreased TMAO-enhanced neointimal formation.
In analyses of gene expression and gene set enrichment in rat hepatocytes, Chen et al. (2019b) showed that TMAO interacts with and activates PERK in the setting of metabolic dysfunction. Our data are consistent with those of a previous study in which the downstream PERK branch, ATF4, was increased after TMAO treatment. In our study, we showed that high-TMAO conditions further promoted ER stress and increased ROS generation after carotid artery ligation and that these adverse effects were ameliorated by TMAO suppression using DMB.
Previous studies have shown that the induction of XBP1 mRNA is initiated by ATF6 (Sharma et al, 2020; Yoshida et al, 2001). In this study, we found that TMAO increased the expression of p-PERK and p-IRE-1α without changing the level of XBP1s. Interestingly, treatment with TMAO did not increase the level of ATF6 (Fig. 5G). Chen et al. demonstrated that TMAO promotes metabolic dysfunction by binding to and activating PERK. The TMAO was found to activate the PERK promoter but not the IRE-1 or ATF6 promotor (Chen et al, 2019b).
Therefore, we believe that TMAO initially induces PERK/ATF4 signaling and that ATF6 and XBP1s might be subsequently activated in response to ROS or mitochondrial dysfunction (Delmotte and Sieck, 2019). We found that high TMAO concentrations not only irreversibly increased ER stress but also attenuated the expression of antioxidant proteins.
There was no significant difference in protein expression in response to choline treatment in HASMCs (Supplementary Fig. S1D). The inhibition of FMO3 might be a more effective approach for studying TMA metabolism. FMO3 knockdown has some therapeutic effects, including decreasing TMAO levels, suppressing FoxO1 via SREBP-2, improving glucose tolerance, and preventing hypercholesterolemia and atherosclerosis, in LIRKO mice (Shih et al, 2015). However, the knockdown of FMO3 affects cholesterol homeostasis and regulates LXR activity to impact hepatic inflammation and ER stress (Warrier et al, 2015).
The mechanisms of response to neointimal formation in murine models, such as that used in this study, may not necessarily reflect or predict those observed in humans.
This study has some limitations. Our data showed that TMAO regulates inflammation via the ER stress signaling pathway in SMCs. Of note, evaluation of a cell culture in the static state may not reflect the in vivo conditions in mouse studies. The flow-mediated effects on TMAO-induced vascular inflammation should be clarified in further studies. Previous studies have revealed an association between TMAO levels and incident risk for thrombotic events, showing a large dynamic range (Zhu et al, 2016). However, the physiological effects of TMAO in endothelial and smooth muscle cells occur at TMAO concentrations of ∼100–200 μM (Seldin et al, 2016). Our previous study demonstrated that inflammatory cytokine levels were also significantly increased at TMAO concentrations of ∼100–200 μM in endothelial progenitor cells (Chou et al, 2019).
In this study, the physiological concentration of TMAO in mice was ∼100–200 μM. Our study further showed that ER stress and inflammation were significantly upregulated in response to 100–200 μM TMAO treatment in smooth muscle cells, and these levels were much higher in humans. The SMCs treated with a low dose of TMAO displayed no significant changes in NLRP3 activation, p65 phosphorylation, mitochondrial stability, or ER stress (Supplementary Fig. S2A–D).
However, the TMAO concentrations required for in vitro and in vivo studies might not be useful for directly determining the concentration required for activity in the clinic. The direct demonstration that higher TMAO levels in patients can cause vascular disease should be further investigated (Randrianarisoa et al, 2016; Wang et al, 2011).
Conclusion
Our findings partly explain the mechanisms of TMAO-induced vascular inflammation and disturbed flow-induced atherosclerosis. Multiple mechanisms underlie the atherosclerotic changes during neointimal hyperplasia in mice fed high-choline and high-TMAO diets, including increased inflammasome activation, increased ER stress, and the induction of ROS production and mitochondrial dysfunction. The protective effects of TMAO inhibition using DMB suggest that DMB could be part of a nonlethal and applicable strategy for TMAO inhibition.
However, this study has some limitations. Choline and TMAO were supplemented in a regular diet, and DMB was supplemented in drinking water. There were no significant differences in the average uptake or weight changes among the groups. The DMB has an intrinsically foul odor, resulting in reduced water intake. Another experiment should be performed to ensure that the observed effect was not due to reduced water intake throughout the study.
The effects of DMB treatment at earlier time points and initial treatment after injury might provide additional evidence. Other more potent and specific inhibitors have recently been described (Roberts et al, 2018) but were not tested in the current study. The key findings of the present study should be confirmed in further studies to determine their applicability in humans.
Materials and Methods
Animals
Six-week-old male WT mice with a C57BL/6 genetic background were used in this study. The animals were kept in microisolator cages on a 12-h day/night cycle. Mice were fed with Laboratory Rodent Diet 5010 as a chow diet. For the designed diet, the chow diet was mixed with compounds (TMAO or choline) weekly. Mixed diets were air dried and sterilized by UV light for 30 min. Two weeks before surgery, the mice were separated randomly into four groups (n = 10, each group) and fed: (1) regular chow (control group), (2) chow supplemented with 1.0% TMAO (high-TMAO group; cat. no. 317594; Sigma-Aldrich, St. Louis, MO), (3) chow supplemented with 1.2% choline (high-choline group; cat. no. 26978; Sigma-Aldrich), and (4) the high-choline diet with 1.0% DMB in drinking water (DMB group; cat. no. 183105; Sigma-Aldrich). The mice were fed these diets for 6 weeks, until sacrifice.
To establish the high-TMAO murine model, 10 mice/group were used to assess the measurement of TMAO levels. To reduce the number of the animals used in the experiment, five mice/group were used to assess tissue sections and another five mice/group were used to assess the in vivo experiments.
To investigate whether the inhibition of ER stress reduced TMAO-promoted neointimal formation, mice were fed a high-TMAO diet and given TUDCA (100 mg/kg/day) via an intraperitoneal injection (TUDCA group; cat. no. T779310; Sigma-Aldrich).
Ethics
All experimental procedures and protocols involving animals were conducted in accordance with the institutional guidelines for animal care of National Yang Ming Chiao Tung University (Taipei, Taiwan) and the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health (8th edition, 2011). All methods in this study were reported in accordance with ARRIVE guidelines.
Partial carotid artery ligation model
To establish an in vivo model of disturbed flow-induced vascular remodeling (Chen et al, 2020), we partially ligated the LCAs of all mice. Carotid artery ligation was performed at week 2 from the start of treatment. Each mouse was anesthetized by an intraperitoneal injection of 250 mg/kg avertin. Anesthetization was considered to be adequate when the mouse made no attempt to withdraw the limb on the application of pressure.
The LCA was exposed through a small midline incision in the neck. Three branches of the LCA (the external, internal, and occipital carotid arteries) were ligated with 6-0 silk sutures, and the superior thyroid artery was left open. In all groups, the right common carotid artery was left unligated as a control. At different timepoints, we isolated the ligated and contralateral unligated arteries for morphometric and Western blot analyses. The protocol is shown in Figure 1A.
Morphometric analysis
Four weeks after carotid artery ligation, the mice were euthanized with an anesthetic overdose via intraperitoneal injection followed by cardiac puncture. The left ventricle was cannulated, perfused with phosphate-buffered saline (PBS), and fixed with 4% paraformaldehyde (PFA; cat. no. 1.00496; Sigma-Aldrich). According to a previous study (Ryan et al, 2002), we fixed arterial perfusion at physiological pressure (120 mmHg in perfusion and fixation).
The left and right carotid arteries were collected and incubated in 4% PFA for 8 h. After cryopreservation in 30% sucrose/PBS at 4°C, the arteries were embedded in Tissue-Tek optimal cutting temperature compound and frozen. Cross-sections (5 μm thick) were taken 1.5 mm proximal to the bifurcation and stained with hematoxylin and eosin (H&E). Four regions (the lumen, intima, media, and total vascular area) of the H&E-stained cross sections were examined using ImageJ software (National Institutes of Health).
The areas surrounded by the luminal surface, internal elastic lamina, and external elastic lamina were calculated. The intimal area was determined by subtracting the luminal area from the area defined by the internal elastic lamina, and the medial area was calculated by subtracting the area defined by the internal elastic lamina from that defined by the external elastic lamina.
Elastic fragmentation measurement
Frozen carotid artery sections were cut and stained with Verhoeff-van Gieson (VVG) stain for visualization of elastic fibers. The sections were hydrated and stained in Verhoeff's solution for 10 min. Sections were then incubated in 2% ferric chloride for 2 min and treated with 5% sodium thiosulfate for 1 min. The sections were counterstained in Van Gieson's solution (cat. no. HT25A; Sigma-Aldrich) for 5 min and dehydrated. Fragmentation was defined as the presence of discernable breaks of continuous elastin fibers.
Images were acquired under an Olympus BX63 microscope (Olympus, Center Valley, PA), and the number of elastin fragments was counted on representative VVG images of each mouse aorta.
Measurement of plasma TMAO levels
All 10 mice were subjected to TMAO detection, and 5 mice were randomly subjected to inflammatory cytokine detection. Four weeks after partial LCA ligation, a 500-μL blood sample was obtained from each mouse for measurement of the TMAO level. Blood samples were collected into microtubes containing EDTA from the submandibular vein of each mouse. The blood samples were centrifuged, and plasma samples were collected and stored at −20°C until TMAO measurement in batched assays ∼1 week later.
For the determination of TMAO in plasma, a protein precipitation procedure was utilized for sample preparation, and the procedure is described as follows. A 200 μL aliquot of plasma and 20 μL of TMAO-d9 at a 1 mg/mL concentration as an internal standard were placed in a 1.5 mL centrifuge tube. After shaking for 1 min, 500 μL acetonitrile was added to the same centrifuge tube, which was subsequently vortexed for 1 min. After centrifugation for 10 min at 3000 rpm, the supernatant was analyzed using a liquid chromatography-tandem mass spectrometer (LC-MS/MS).
The LC-MS/MS was composed of an Agilent 1100 series HPLC system (Agilent Technologies, Palo Alto, CA) and 4000 QTrap quadrupole linear ion trap mass spectrometer (Applied Biosystems, Foster City, CA) with an electrospray ionization source. The chromatographic gradient separation was performed on a Luna 5 μm Silica, 2.0 × 150 mm, 5 μm particle size, LC column (Phenomenex, Torrance, CA) with diH2O-methanol (containing 0.1% acetic acid) as the mobile phase system. Multiple reaction monitoring was used to quantify TMAO and TMAO-d9 with the ion transition m/z 76.1 to 58.1 and 85.3 to 66.1.
Measurement of IL-1β, IL-6, IL-10, and TNF-α
Blood samples were collected into microtubes containing EDTA from the submandibular vein of each mouse. The blood samples were centrifuged at 3000 g for 15 min for plasma isolation. The levels of IL-1β (cat. no. MLB00C), IL-6 (cat. no. M6000B), IL-10 (cat. no. M1000B), and TNF-α (TNF-α; cat. no. MTA00B) were quantified using ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. The list of limit of quantification, intra- and inter-assay CV values were provided as supplementary material (Supplementary Table S1).
ROS measurement
Carotid arteries were homogenized in ice-cold Tris-HCl buffer (40 mM, pH 7.4). Tissue homogenates were incubated with reagent from a 2′,7′-dichlorofluorescin (DCF) diacetate assay kit (cat no. C6827; Life Technologies, Carlsbad, CA) for 40 min at 37°C in the dark. The HASMCs were treated with different concentrations of TMAO for 24 h. After removal of the medium, 100 μL of DCFDA was added to each well and incubated for 40 min at 37°C in the dark.
The samples were subjected to fluorospectrophotometric analysis in a Multimode microplate reader (Infinite 200 PRO; TECAN, Mannedorf, Switzerland) at an excitation wavelength of 488 nm and an emission wavelength of 520 nm. The amount of intracellular ROS was proportional to the DCF fluorescence intensity, which was recorded directly to indicate the relative amount of ROS. Relative changes in DCF fluorescence are expressed as fold increases relative to the sham group.
Cell culture
Three batches of human aortic smooth muscle cells (HASMCs) were purchased from Life Technologies (cat. no. C0075C) and ScienCell (Carlsbad, CA; cat. no. 6110). The HASMCs at the fourth through eighth passages were used. They were cultured in M231 medium supplemented with smooth muscle growth supplement in a humidified incubator (95% air and 5% CO2) at 37°C.
Measurement of mitochondrial membrane potential
The HASMCs were seeded at 2 × 105 cells/well in a six-well plate. At 80% confluence, the cells were treated with different concentrations of TMAO for 24 h. The mitochondrial membrane potential (ΔѰm) was assessed with a JC-1 assay (cat. no. 551302; BD Biosciences, San Jose, CA) according to the manufacturer's instructions. The HASMCs were stained with JC-1 for 30 min. Because of the high membrane potential in the healthy cells, JC-1 aggregated and accumulated within the mitochondrial matrix.
JC-1 formed monomers in the presence of a low membrane potential and was located in the cytosol of apoptotic cells. The amount of JC-1 retained by 10,000 cells per sample was measured at 530 nm (monomer form, green fluorescence) and 590 nm (aggregation form, red fluorescence) using a flow cytometer and CytExpert software for analysis. The change in JC-1-derived fluorescence from red to green was evaluated.
Oxygen consumption assay and ATP generation assay
The mitochondrial oxygen consumption rate was measured using an oxygen consumption rate assay kit (cat. no. 600800; Cayman chemical) following the manufacturer's instructions. The HASMCs were plated on a 96-well Viewplate® plate at 5000 cells per well and tested in medium 231. Then, TMAO was injected to achieve the indicated final concentration. Total protein was isolated from each well and quantified for normalization after the assay.
The ATP was measured in mice and HASMCs using an ATPlite luminescence ATP detection assay system (cat. no. 6016943; PerkinElmer). The ATP concentrations were calculated using a standard curve to estimate the levels of ATP.
Western blotting
Cells were lysed in buffer (62.5 mM Tris-HCl, 2% sodium dodecyl sulfate [SDS], 10% glycerol, 0.5 mM phenylmethylsulfonyl fluoride, and 2 μg/mL aprotinin, pepstatin and leupeptin), and the protein lysates were subjected to SDS polyacrylamide gel electrophoresis followed by electroblotting onto polyvinylidene difluoride membranes.
The membranes were probed with monoclonal antibodies (at 1:1000 unless noted otherwise) against IL-1β (cat. no. ab216995; Abcam, Burlingame, CA), extracellular signal-regulated kinase (ERK; cat. no. ab184699; Abcam), phospho-ERK (cat. no. ab201015; Abcam), c-Jun N-terminal kinase (JNK; cat. no. 9252; Cell Signaling Technology, Danvers, MA), phospho-JNK (cat. no. 9251; Cell Signaling Technology), p38 (cat. no. 9212; Cell Signaling Technology), phospho-p38 (cat. no. 9211; Cell Signaling Technology;), NF-κB p65 (cat. no. 8242; Cell Signaling Technology), phospho-NF-κB p65 (cat. no. 3033; Cell Signaling Technology), ICAM-1 (cat. no. sc-8439; Santa Cruz Biotechnology, Santa Cruz, CA), NACHT, LRR and PYD domains-containing protein 3 (NLRP3; cat. no. ab4207; Abcam), caspase 1 (cat. no. GTX101322; GeneTex, Hsinchu City, Taiwan), apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC; cat. no. ab155970; Abcam), Bax (cat. no. ab182733; Abcam), B cell lymphoma (Bcl)-2α (cat. no. ab182858; Abcam), protein kinase R-like ER kinase (PERK; cat. no. GTX66661; GeneTex), phospho-PERK (cat. no. GTX00673; GeneTex), inositol-requiring transmembrane kinase/endoribonuclease (IRE)-1α (cat. no. GTX30005; GeneTex), phospho-IRE-1α (cat. no. GTX132808; GeneTex), xbp-1s (cat. no. 12782; Cell Signaling Technology), ATF4 (cat. no. GTX101943; GeneTex), ATF6 (cat. no. GTX104820; GeneTex), and β-actin (1:5000; cat. no. ab8224; Abcam). For ER stress, the membranes were probed with antibodies against protein kinase R-like ER kinase (PERK; cat. no. GTX66661; GeneTex), phospho-PERK (cat. no. GTX00673; GeneTex), inositol-requiring transmembrane kinase/endoribonuclease (IRE)-1α (cat. no. GTX30005; GeneTex), phospho-IRE-1α (cat. no. GTX132808; GeneTex), xbp-1s (cat. no. 12782; Cell Signaling Technology), ATF4 (cat. no. GTX101943; GeneTex), ATF6 (cat. no. GTX104820; GeneTex).
Protein expression was visualized using chemiluminescence detection reagents (Merck Millipore, Billerica, MA) and a luminescence imaging system (Amersham Imager 680; GE Healthcare, Menlo Park, CA). The results were analyzed using ImageJ software. The full-length western blot images were provided as supplementary material (Supplementary Figure S3–S13).
Immunofluorescence
The immunofluorescence of carotid artery sections was analyzed after antigen retrieval in sodium citrate buffer (10 mM sodium citrate, pH 6) for 20 min at 95°C. Sections were blocked with bovine serum albumin for 10 min; then, they were incubated with anti-F4/80 (1:50; cat. no. 70076; Abcam), anti-NLRP3 (1:50; cat. no. ab4207; Abcam), anti-ASC (1:50; cat. no. ab155970; Abcam), anti-Bax (1:50; cat. no. ab182733; Abcam), anti-PERK (1:100; cat. no. GTX66661; GeneTex), anti-IRE-1α (1:100; cat. no. GTX30005; GeneTex), and anti-α smooth muscle actin (SMA, 1:100; cat. no. ab7817; Abcam) antibodies overnight at 4°C.
Then, the sections were incubated with goat anti-rabbit Alexa Fluor 488 and goat anti-mouse Alexa Fluor 594 secondary antibodies for 1 h at room temperature, and they were counterstained with Hoechst 33342 (1 μg/mL; cat. no. B2261; Sigma-Aldrich). Images were acquired using an Olympus BX63 microscope.
Statistical analysis
All experiments were performed independently at least three times, and data were tested for normality using a Shapiro–Wilk test. All continuous variables are presented as the mean ± standard deviation. The F test for equal variance was performed before the differences among groups were analyzed. Comparisons between two groups were analyzed using Student's t test. For multiple groups, the data were analyzed using one-way ANOVA.
For post hoc analysis, Scheffe's multiple-comparison post hoc test was applied to correct for multiple comparisons, and a Fisher Least Significant Difference test was used for planned comparisons. Target protein expression measured by immunoblotting was determined via densitometry and is expressed relative to an internal control or as phosphorylated protein relative to total protein expression. Statistical significance was defined as a p value <0.01. Analyses were performed using a statistical software package (SPSS version 14.0 for Windows; SPSS, Inc., Chicago, IL).
Footnotes
Authors' Contributions
C.Y.: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing—original draft, and Writing—review and editing. H.B.: Conceptualization, Data curation, Resources, and Validation. S.C.: Formal Analysis, Methodology, and Validation. S.H.: Methodology, Resources. R.H.: Resources, Validation. Y.W.: Validation. Y.L.: Validation. C.S.: Validation. P.H.: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, and Writing—review and editing. J.W.: Writing—review and editing, Supervision. S.J.: Writing—review and editing, Supervision.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported, in part, by research grants from the Ministry of Science and Technology of Taiwan (MOST 104-2314-B-075-047), the Novel Bioengineering and Technological Approaches to Solve Two Major Health Problems in Taiwan sponsored by the Taiwan Ministry of Science and Technology Academic Excellence Program (MOST 108-2633-B-009-001), the Ministry of Health and Welfare (MOHW 106-TDU-B-211-113001), and Taipei Veterans General Hospital (V105C-0207, V106C-045). These funding agencies had no influence on the study design, data collection or analysis, decision to publish, or preparation of the article. The funding institutions took no part in the study design, data collection or analysis, publication intent, or article preparation.
Supplementary Material
Supplementary Table S1
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Figure S11
Supplementary Figure S12
Supplementary Figure S13
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.