
Abstract
Significance:
Shear stress and metabolic disorder are the two sides of the same atherosclerotic coin. Atherosclerotic lesions are prone to develop at branches and curvatures of arteries, which are exposed to oscillatory and low shear stress exerted by blood flow. Meanwhile, metabolic disorders are pivotal contributors to the formation and advancement of atherosclerotic plaques.
Recent Advances:
Accumulated evidence has provided insight into the impact and mechanisms of biomechanical forces and metabolic disorder on atherogenesis, in association with mechanotransduction, epigenetic regulation, and so on. Moreover, recent studies have shed light on the cross talk between the two drivers of atherosclerosis.
Critical Issues:
There are extensive cross talk and interactions between shear stress and metabolic disorder during the pathogenesis of atherosclerosis. The communications may amplify the proatherogenic effects through increasing oxidative stress and inflammation. Nonetheless, the precise mechanisms underlying such interactions remain to be fully elucidated as the cross talk network is considerably complex.
Future Directions:
A better understanding of the cross talk network may confer benefits for a more comprehensive clinical management of atherosclerosis. Critical mediators of the cross talk may serve as promising therapeutic targets for atherosclerotic vascular diseases, as they can inhibit effects from both sides of the plaque. Hence, further in-depth investigations with advanced omics approaches are required to develop novel and effective therapeutic strategies against atherosclerosis. Antioxid. Redox Signal. 37, 820–841.
Introduction
Atherosclerosis is a chronic arterial disease characterized by abnormal deposition of fatty and fibrous matter in the wall of curved and bifurcated arteries that are constantly exposed to low shear stress or disturbed blood flow (107). Atherosclerosis is considered primarily to be a metabolic disease involving disorders of lipid, glucose, and amino acid metabolism. Anatomic and biomechanical evidence suggests that hemodynamics (blood flow pattern) also plays an important role in the development of atherosclerotic plaques (14, 16). In this article, we review the impact of biomechanical forces and metabolic dysregulation on atherogenesis and explore the underlying connection between the two pathogenic factors. It is emphasized that metabolic disorder and disturbed blood flow are two interconnected drivers in the atherogenic process, acting synergistically to the initiation and evolution of atherosclerotic plaques.
Shear Stress and Atherosclerosis
The vascular system is a tree-like structure in which blood vessels vary in diameter and morphology. Consequently, the friction forces generated by blood flow vary in both direction and velocity (14, 16). Shear stress is the tangential force derived from friction of the blood flow on the inner layer of the blood vessels (16, 28). The magnitude of shear stress in humans ranges from 10 to 70 dyn/cm2 in arteries (24). Atherosclerotic plaques are usually distributed at vascular curvatures and bifurcations where the flow pattern is disturbed and shear stress is low and oscillatory. In contrast to the atheroprone disturbed flow (DF), laminar flow (LF) in the straight part of an artery exerts atheroprotective effects on blood vessels (24). Numerous studies have shown that the initiation, progression, and rupture of atherosclerosis plaques are regulated by biomechanical forces (22, 88, 152) (Fig. 1). Arterial segments exposed to DF and low shear stress attain more plaque burden and undergo excessive expansive remodeling. The plaque and arterial remodeling will further disturb the blood flow and lower the shear stress (13, 88). The vicious cycle between DF and arterial structure will deteriorate the vulnerability of plaques with thin cap fibroatheroma and increase the risk of plaque rupture (88).
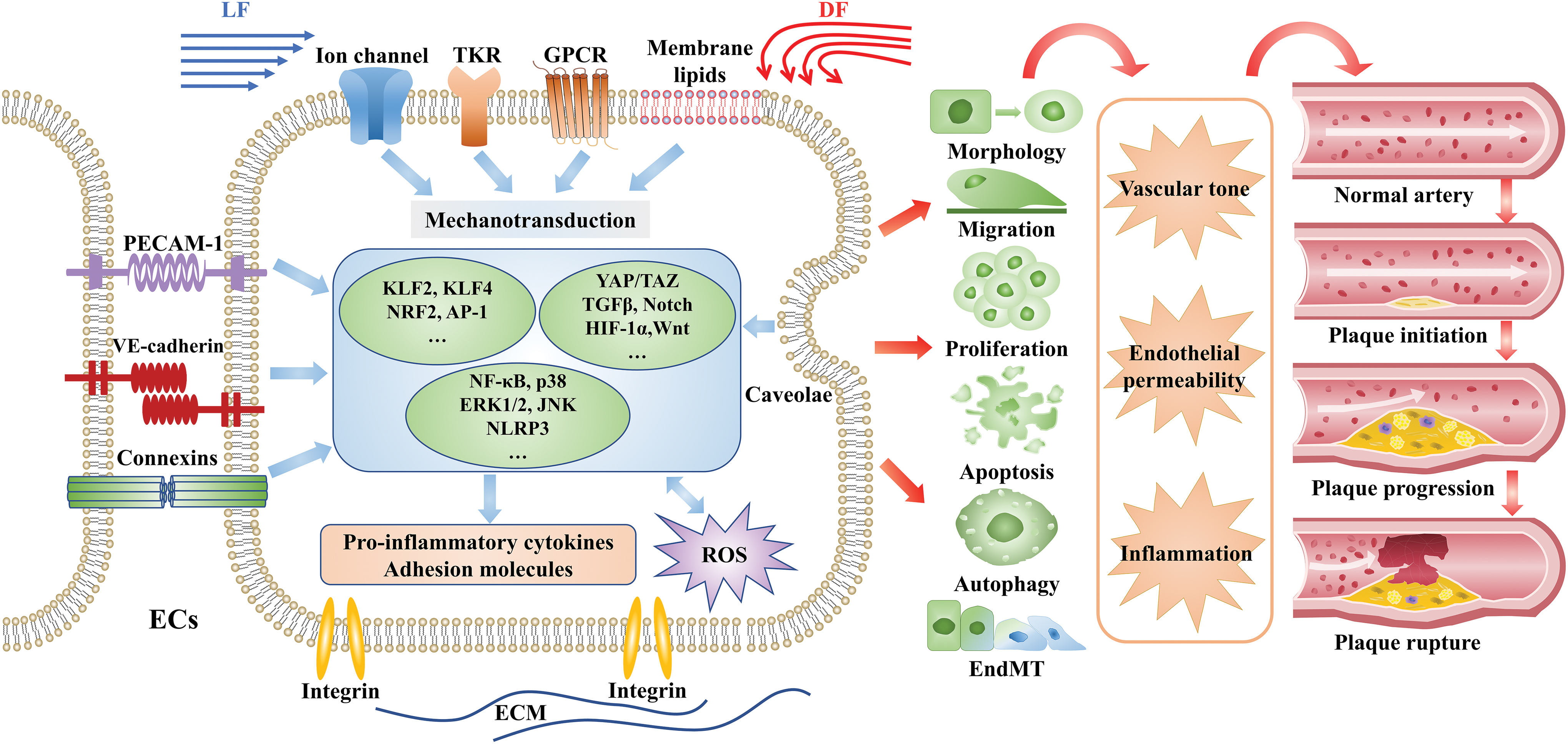
Mechanotransduction of shear stress
Hemodynamic forces are detected by a variety of mechanosensitive structures or sensors on the surface of endothelial cells (ECs), and are converted into biochemical signals. Thereby shear stress activates various downstream signaling pathways and eventually regulates multiple physiological processes in ECs.
Mechanosensors
Many surface components have been reported to function as mechanosensors, including basement surface structures (integrins); lateral surface sensors (junctional proteins, adhesion molecules, platelet EC adhesion molecule-1); and luminal sensors (tyrosine kinase receptors, caveolae, ion channels, G protein-coupled receptors (GPCRs), bilayer membrane lipids, the glycocalyx, and primary cilia) (6, 16, 41). These mechanosensors consist of a complex sensing system (Fig. 1). Some of them have been shown to be involved in the initiation and progression of atherosclerosis (52, 73). Previous study shows that LF activates integrin and enhances interaction between integrin and Gα13, resulting in inhibition of RhoA and yes-associated protein (YAP), and consequently reduces plaque formation (175). A newly identified mechanosensor, Piezo ion channel, is extensively studied in the mechanotransduction of shear stress (6). Interestingly, LF and DF have the opposite effect on Piezo. LF boosts Piezol-mediated endothelial nitric oxide synthase (eNOS) expression, which is abrogated by endothelial-specific Piezol knockout (1, 33). On the contrary, Piezol plays a role in DF-induced nuclear factor kappa-B (NF-κB) activation (176). Furthermore, activation of Piezo1 may promote shear stress-induced Ca2+ entry in megakaryocytes, thereby contributing to thrombus formation (70).
Signaling pathways
The mechanical stimulation triggers a spectrum of signaling pathways, including the mitogen-activated protein kinase (MAPK) pathway, phosphoinositide 3-kinases (PI3K)/protein kinase B (AKT) pathway, NF-κB signaling pathway, and redox signaling pathway. They subsequently regulate downstream functional target genes, such as oxidant and antioxidant genes, proatherogenic and prothrombotic genes, and inflammatory mediators to modulate EC function and phenotype (endothelial morphology, proliferation, apoptosis, migration, and secretion) (13, 24, 60).
In recent years, multiple developmental pathways associated with artery development and vascular disorders are reported to play roles in mechanotransduction linking shear stress to atherogenesis. The recently identified signaling pathways include the BMP/TGFβ pathway, Notch pathway, Hippo–YAP/transcriptional coactivator with PDZ-binding motif (TAZ) pathway, Wnt pathway, as well as other pathways mediated by hypoxia-inducible factor-1α (HIF-1α), twist-related protein 1, and homeobox (162). Low and oscillatory shear stress triggers activation of these signaling pathways and aggravates inflammation and vascular permeability, which are the hallmarks of atherogenesis. For example, DF promotes the expression of flow-sensitive BMP4 and activates the downstream targets, SMAD1 and SMAD5 (207). Likewise, Wnt signaling is reported to be activated by DF and induces EC migration and inflammatory response (162).
Shear stress and EC morphology, proliferation, migration, and apoptosis
Flow-derived mechanical forces regulate various phenotypic changes of ECs, including morphology, proliferation, migration, and apoptosis. Regarding the morphology of ECs, a multitude of studies have revealed that ECs exposed to LF and high shear stress are closely aligned along the direction of blood flow, and actin stress fibers are arranged in parallel at the center of the cells; but when ECs are exposed to DF, they convert to a round shape, and actin stress fibers are disorderly orientated (24). Compared with ECs that remain stable under LF, the perturbed ECs in the oscillatory shear-stress region have a more rapid turnover rate, and greater active DNA synthesis and proliferation (23). Cell proliferation plays a vital role in endothelial damage repair and early plaque formation. In response to LF, the antiproliferative MAPK pathway and the proliferative AKT pathway are balanced with each other, while DF breaks this balance by stimulating AKT and the mammalian target of rapamycin (mTOR) pathway, thereby promoting EC proliferation (58, 207). Biomechanical stress can regulate the migration of ECs via various mechanisms, such as alteration of cell polarity and cytoskeleton structure, formation of lamellar lipoprotein, contraction of stress fiber, and interaction between ECs and extracellular matrix (ECM). LF is shown to enhance EC migration, thereby promoting wound healing (139). In response to proinflammatory factors such as tumor necrosis factor-α (TNF-α) or oxidized low-density lipoprotein (ox-LDL), ECs under atheroprone DF trigger cell apoptosis through mechanisms involving activation of p53, protein kinase C (PKC), and c-Jun N-terminal kinase (JNK) signaling pathways (17, 64). Enhancement of EC apoptosis precipitates endothelial dysfunction, increases endothelium permeability, and thus contributes to plaque erosion and rupture.
Shear stress and endothelial/mesenchymal transition
In recent years, the interrelationship between endothelial/mesenchymal transition (EndMT) and atherosclerosis has attracted extensive attention. As an early feature of neointimal hyperplasia and atherosclerotic plaque formation, EndMT is precisely regulated by shear stresses (129). EndMT is a cellular process characterized by decreased expression of endothelial markers and increased expression of interstitial markers, presenting destruction of intercellular connections and loss of cell polarity. Moonen et al. uncovered the simultaneous expression of endothelial and interstitial markers in atherosclerotic plaques in mice, pigs, and humans, suggesting that EndMT is ubiquitous in the process of plaque formation. DF can promote atherosclerosis through extracellular signal-regulated kinases (ERK) 5-dependent EndMT both in vitro and in vivo (129). Vanchin et al. revealed that EndMT plays a critical role in early plaque formation and intimal hyperplasia via the transforming growth factor-beta miR-374b-MAPK7 axis (167). Lai et al. proved that pulsatile blood flow and disturbed blood flow have opposite effects in regulating EndMT-related genes. Reactive oxygen species (ROS) promote EndMT caused by oscillatory shear stress, while AMP-activated protein kinase (AMPK) and sirtuin-1 inhibit EndMT (89). In addition, shear stress also regulates EndMT through DNA methylation of related gene promoters (89). Taken together, targeting EndMT and shear stress might bring novel strategies for the treatment of atherosclerosis.
Shear stress and EC autophagy
Autophagy maintains intracellular homeostasis in response to a range of stressors through removal of dysfunctional components. Previous studies indicate that autophagy is involved in shear stress-induced cellular responses and atherogenesis. Vion et al. uncovered that autophagy functions as an atheroprotective mechanism by sustaining the orderly alignment of ECs under physiological mechanical stress (170). Kheloufi et al. further revealed that high shear force boosts autophagy in ECs, and endothelial autophagy inhibits EC apoptosis, aging, and inflammation, collectively suppressing the formation of atherosclerotic plaques (81). The latest study demonstrates that LF induces YAP nuclear export mediated by SIRT1-dependent deacetylation and degradation through autophagy, and subsequently inhibits atherogenesis (193).
Shear stress and endothelial permeability
A large amount of evidence unveils that DF increases endothelial permeability and accelerates atherosclerotic plaque formation. Junctional complexes, including connexin and vascular endothelial cadherins (VE-cadherins), play a critical role in endothelium permeability (84). It was reported that at rat aorta bifurcations, the distribution of connexin at the periphery of ECs was discontinuous; the expression of VE-cadherin was significantly upregulated in the narrow downstream of DF. Consistently, discontinuous distribution of connexin was also observed in the atherosclerotic lesions of humans and rabbits (24). In vitro experiments also confirmed that when ECs were exposed to oscillatory flow, VE-cadherin showed an intermittent distribution (126). In addition, ECs affected by oscillatory and low shear stress alter from spindle to round, and these morphology changes also increase endothelial permeability in the atherosclerotic arterial area (24). In the arterial segments exposed to DF, the velocity of blood flow is slowed down, whereas the permeability of vascular endothelium is increased, which causes the accumulation of macromolecular substances (such as lipids) and infiltration of inflammatory cells and ultimately promotes plaque formation and progression.
Shear stress and inflammation
Atherosclerosis is a chronic inflammatory disease while unfavorable hemodynamic forces such as DF are essential for the inflammatory response in the vascular endothelium. Both NF-κB and MAPK pathways play extremely important roles in mechanical force-induced inflammation in ECs (27). For example, unidirectional LF can act on the promoter of Kruppel-like factor 2 (KLF2) through the ERK5-myocyte enhancer factor 2 (MEF2) pathway, thereby increasing KLF2 expression in ECs. KLF2 is a negative regulatory molecule upstream of the NF-κB pathway, which supports that unidirectional LF can inhibit NF-κB activation. We have recently demonstrated that KLF2 is required for unidirectional LF-induced upregulation of transcription factor EB (TFEB), and TFEB is an anti-inflammatory effector downstream of LF-KLF2 signaling cascade in ECs (161). High shear force can also upregulate the expression of nuclear factor erythroid 2-related factor 2 (NRF2), dephosphorylate p38 and JNK, and exert anti-inflammatory effects (88). On the contrary, when ECs are subjected to low or oscillatory shear stress, the NF-κB pathway or the MAPK pathway is activated to increase the expression of proinflammatory molecules (e.g., TNF-α and interleukin [IL]-1), chemical chemokines (e.g., monocyte chemoattractant protein-1 [MCP-1]), and adhesion molecules (e.g., P-selectin, intercellular adhesion molecule-1 [ICAM-1], vascular cell adhesion protein-1 [VCAM-1], and E-selectin) (16). These adhesion molecules and chemokines promote adhesion and migration of inflammatory cells toward vascular endothelium, which is a critical step for the initiation of atherosclerosis (106). We have recently demonstrated that a significant role of toll-like receptor 4 (TLR4) in DF-induced endothelial inflammation and fibronectin containing the extra domain A may act as the endogenous TLR4 activator, thus adding another novel mechanism of vascular inflammation under DF condition in relation to atherogenesis (143).
Shear stress and ROS
ROS play a pivotal role during the initiation and advancement of atherosclerotic plaques (49). Distinct blood flow patterns exert different effects on redox homeostasis of ECs. LF transiently induces ROS production, and subsequently promotes the expression of antioxidant enzymes, neutralizing ROS and forming an antioxidant environment (132). On the contrary, DF promotes ROS generation steadily through inducing the expression of oxidases such as NADPH oxidase (NOX) and xanthine oxidase, while it also inhibits the ROS clearance via downregulation of superoxide dismutase and glutathione (16). Under DF, excessive ROS promote the oxidative modification of low-density lipoprotein (LDL) to form ox-LDL; meanwhile, the atheroprotective nitric oxide (NO) is continuously diminished due to impaired eNOS activity and formation of peroxynitrite anion, eventually promoting the formation of atherosclerotic plaques (69).
Shear stress and epigenetic regulation
In recent years, growing evidence suggests that epigenetic signatures, including DNA methylation, histone modification, and noncoding RNA regulation, are involved in the control of vascular function and formation of atherosclerotic plaques (61, 92, 203).
Shear stress affects the cellular translocation and expression of class I histone deacetylases (HDACs) (including HDAC-1, HDAC-2, and HDAC-3) (93, 195 –197) and class II HDACs (including HDAC-4, HDAC-5, and HDAC-7) (93, 177, 178) (Table 1). These HDACs can modulate antioxidant and anti-inflammatory pathways via deacetylation of specific transcription factors (e.g., NRF2, MEF2/KLF2). In addition, oscillatory shear stress facilitates the forming of HDAC-1/-2/-3 and HDAC-3/-5/-7 complex in ECs and thereby promotes local oxidative stress and inflammatory responses (92). Under oscillatory shear stress, class I HDACs can promote the proliferation of ECs by regulating p21 and cyclin A. By contrast, under a steady LF, class II HDACs are shuttled from the nucleus to the cytoplasm. Decreased levels of nuclear HDACs inhibit EC proliferation, oxidative stress, and vascular inflammation, thereby bringing antiatherosclerotic benefits (93, 196).
Epigenetic Regulation of Endothelial Cell Functions Under Shear Stress
DNMT, DNA methyltransferase; EC, endothelial cell; HDAC, histone deacetylase; NO, nitric oxide.
Recent studies have shown that abnormal expression of DNA methyltransferase (DNMT) in ECs can affect DNA methylation levels and cause vascular endothelial dysfunction (Table 1) (36, 63, 92). Under different flow patterns, DNMT1 and DNMT3a regulate inflammatory responses through various mechanisms. Oscillatory shear stress upregulates the expression of DNMT1 in ECs, promotes the inflammatory response, and subsequently compromises endothelial function. Besides, DF-induced DNMT1 can upregulate cyclin A to increase EC proliferation (35, 202). DNMT3a is another identified mechanosensitive methyltransferase involved in atherogenesis. DF upregulates DNMT3a, which promotes DNA methylation of the promoter of KLF-4, and thereby regulates the expression of downstream genes, such as MCP-1 and NOS3, and ultimately contributes to the formation of atherosclerotic plaque (76).
Noncoding RNAs participate in regulating EC function under the stimulation of shear stress (Table 1). MicroRNAs, including miR-10a, miR-34a, miR-21, miR-92a, miR-98, miR-146a, miR-451, miR-708, and miR-712, are associated with shear stress-regulated EC inflammation, oxidation, and NO production. For example, the flow-sensitive miR-10a is reported to inhibit the NF-κB pathway and downregulate proinflammatory gene expression in the endothelium (40). Another set of microRNAs related to mechanical regulation of EC proliferation include miR-101, miR-23b, miR-19a, miR-126-5p, and miR-155 (19, 40, 92, 120, 142, 156). Likewise, a variety of long-noncoding RNAs are mechanosensitive and involved in atherogenesis; they include STEEL, LASSIE, SENCR, and MANTIS (78, 97, 119). For instance, LF induces the expression of MANTIS via KLF2 and Kruppel-like factor 4 (KLF4), and MANTIS in turn inhibits ICAM-1-induced monocyte adhesion, suggesting its antiatherosclerotic activity (96). A most recent study shows that laminar shear stress protects vascular endothelial tight junctions and barrier function through maintaining the expression of long-noncoding RNA MALAT1 in ECs (188).
Metabolic Disorders and Atherosclerosis
Lipid metabolism and atherosclerosis
Low-density lipoprotein
Numerous clinical studies verify a causal link between LDL and atherosclerosis (11). Familial hypercholesterolemia, an autosomal dominant inherited disorder on account of genetic mutations (e.g., mutations of LDLR gene), is associated with cumulative LDL cholesterol (LDL-C) burden and increased risk of developing premature atherosclerotic cardiovascular disease (ASCVD) (11). Large randomized-controlled clinical trials of LDL-lowering drugs such as statins suggest a reduction of risks of cardiovascular disease (CVD) events, providing compelling evidence for linking LDL-C to atherosclerosis (43). Based on the critical role of LDL-C in the pathogenesis of atherosclerosis, clinical guidelines recommended the LDL-C level for risk assessment, classification of CVD risks, and the primary target of lipid-lowering treatment (11, 107).
LDL oxidation is one of the important hallmarks of the initiation of atherosclerotic lesions. Ox-LDL induces inflammation in ECs, enhances the release of chemokines, and increases the expression of adhesion molecules (62, 107). These molecules promote monocytes infiltrating into the subendothelial space (179). The invaded monocytes convert into macrophages. With scavenger receptors, macrophages can recognize and uptake ox-LDL, forming foam cells to initiate formation of the atherosclerotic lesion (fatty streak) (110). Undoubtedly, ox-LDL has a variety of regulatory effects on macrophages (Table 2), such as polarizing macrophages into an M1-like or M2-like phenotype (158), inducing macrophage apoptosis (57), and impeding the migration of macrophages from plaque lesions (144). Moreover, ox-LDL triggers the inflammatory response of macrophages and induces macrophages to secrete cytokines, for example, MCP-1, IL-1, IL-8, and TNF-a (164), thereby activating dendritic cells, T cells, and smooth muscle cells, and jointly promoting plaque formation (83, 112, 118).
Different Functions of Oxidized Low-Density Lipoprotein, High-Density Lipoprotein, Triglyceride in Atherosclerosis
HDL, high-density lipoprotein; LDL, low-density lipoprotein; oxLDL, oxidized low-density lipoprotein; RCT, reverse cholesterol transport; VSMC, vascular smooth muscle cell.
Triglyceride
Randomized prospective trials provide strong evidence that triglyceride is another risk factor for ASCVD after adjusting for confounding factors, LDL-C, total cholesterol, and high-density lipoprotein cholesterol (HDL-C) (138). Moreover, interventions targeting to lower the triglyceride level can reduce CVD risks, thus establishing the necessity of assessing triglyceride levels in patients at risk (123). In recent years, omega-3 polyunsaturated fatty acids have been shown to decrease plasma triglyceride levels and the risk of CVD in several clinical studies (160). It is also recommended that for patients with hypertriglyceridemia and other ASCVD risk factors, clinicians should consider strengthening statin therapy or starting the use of omega-3 fish oil to prevent pancreatitis (55).
As the major component of very low-density lipoprotein (VLDL) and chylomicrons, triglyceride promotes the cholesterol deposition in the atherosclerotic lesion (135). Monoglycerides and free fatty acids, two products derived from the hydrolysis of triglyceride-rich particles, can induce cytotoxic and inflammatory responses and even cell death (111, 135). In addition, patients with hypertriglyceridemia have elevated plasma levels of inflammatory molecules (e.g., MCP-1, VCAM-1, and TNF-α) and thrombogenic factors (Table 2) (50, 134). Therefore, high triglyceride levels have been suggested to be associated with ASCVD by raising vascular inflammation and thrombosis (135).
High-density lipoprotein
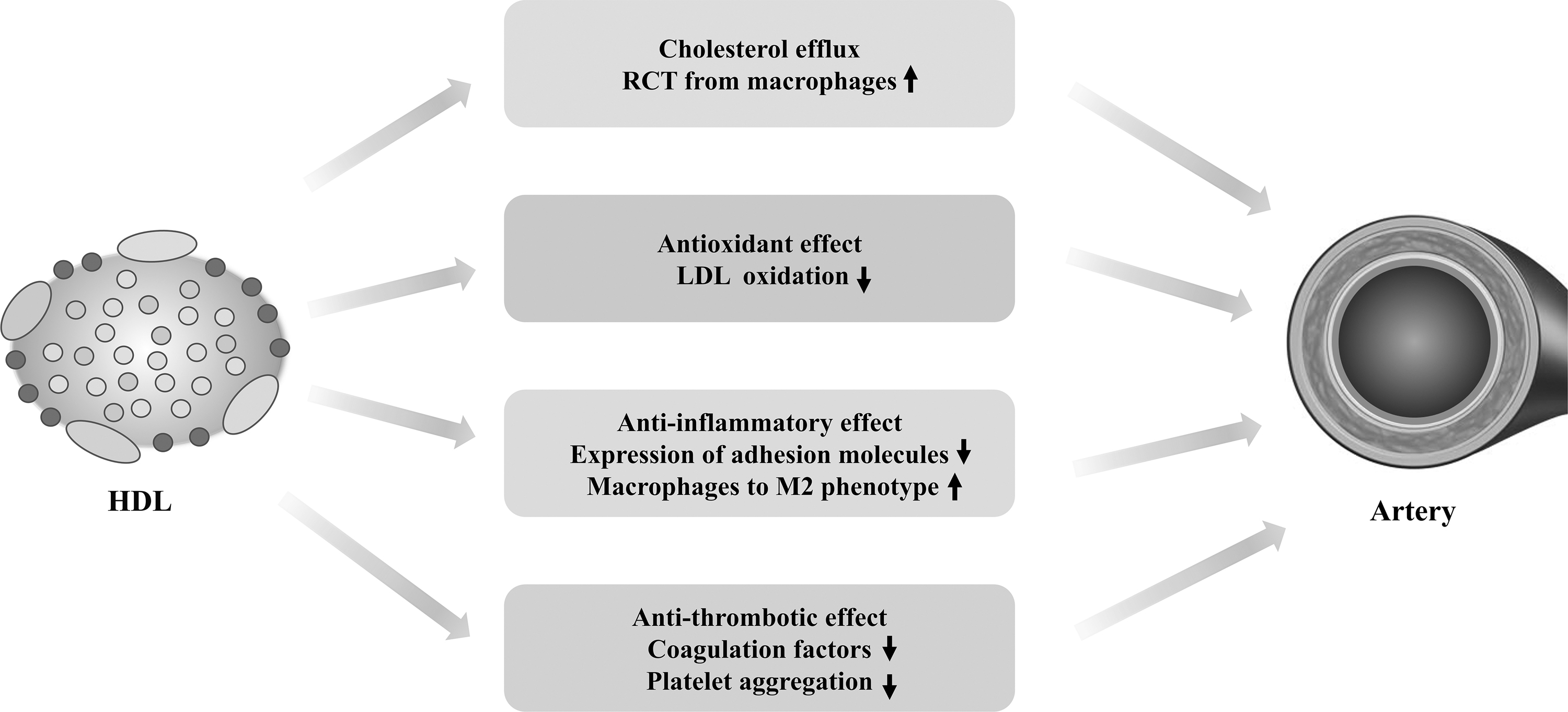
Epidemiological studies indicate an inverse correlation of HDL-C levels with the risk of ASCVD. Nevertheless, the genetic evidence from a genome-wide association study shows that high HDL-C levels do not have protective roles against CVD (171). Furthermore, clinical studies with high-density lipoprotein (HDL)-raising drugs failed to lower the risks of ASCVD (136). It is worth noting that the HDL-C level is not equivalent to the abundance of HDL particles, cholesterol efflux capacity, and HDL subtype distribution, all of which are shown to be better biomarkers than HDL-C levels in assessing CVD risks (136). Apolipoprotein A-I (apoA-I) is the main protein component of HDL, which possesses several biological functions. Mature HDL and lipid-poor apoA-I prevent the forming of foam cells by transporting cholesterol from macrophages (107).
Conventionally, the primary atheroprotective effect of HDL arises from its ability to mediate reverse cholesterol transport (RCT) from macrophages to the liver (107, 136). Besides RCT, HDL is reported to mediate antioxidant, anti-inflammatory, and antithrombotic effects (Fig. 2) (59). In terms of its antioxidant property, HDL reduces lipid hydroperoxides through apoA-I and protects LDL from oxidation by antioxidant enzymes such as Lp-PLA2 or LCAT (5). The anti-inflammatory function of HDL is much more complicated. HDL reduces the recruitment of monocytes by repressing EC activation (166). In addition, HDL can also promote the transformation from M1 proinflammatory macrophages to M2 anti-inflammatory macrophages (111, 163).
Glucose metabolism and atherosclerosis
It is well established that diabetes is one of the major independent risk factors for CVD. A meta-analysis of 102 prospective studies suggests that patients with diabetes had twice the risk of developing coronary heart disease than nondiabetics and that the risk increased as blood glucose levels rose (153). A national registry study of type 1 diabetes in Sweden found that patients who developed type 1 diabetes by the age of 10 carried a 30-fold increased risk of CVD (145). Another registered study revealed that the average cardiovascular mortality of diabetic patients was higher, and subgroup analysis suggested that age and blood glucose control levels were important predictors for all-cause mortality (154).
Large quantities of studies provide compelling evidence that glucose control is beneficial for the reduction of CVD risks in patients with diabetes. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study (DCCT/EDIC) (type 1 diabetes), the Veterans Affairs Diabetes Trial (VADT) (type 2 diabetes), and the UK Prospective Diabetes Study (UKPDS) showed that glucose-lowering intervention reduced the incidence of CVD events during the long-term follow-up (65, 130). In recent years, a number of randomized-controlled trials on sodium/glucose cotransporter-2 (SGLT2) inhibitors or glucagon-like peptide-1 (GLP-1) analogues for diabetic patients at high CVD risk have demonstrated promising results. Three randomized-controlled trials, CANVAS, EMPA-REG OUTCOME, and DECLARE-TIMI 58, revealed that SGLT2 inhibitors could reduce the risk of cardiovascular mortality by 17%–38% (131, 183, 209). Studies of SUSTAIN-6, PIONEER 6, and LEADER suggested that GLP-1 analogues reduced the incidence of major adverse events (nonfatal myocardial infarction, death of CVD, and nonfatal stroke) by 12% (68, 121, 122). Given the significant cardiovascular benefits, SGLT2 inhibitors and GLP-1 analogues are recommended for patients with type 2 diabetes and CVD or at high/very high CVD risk (26).
Uncontrolled hyperglycemia is the most important feature of diabetes, and it is the major pathological condition related to cardiovascular events. Advanced glycosylation end products (AGEs) are produced by nonenzymatic reactions between reducing sugars and lipids, proteins, or nucleic acids in the body. Previous studies confirm that persistent hyperglycemia significantly increases the production of AGEs; the latter known to participate in the initiation and development of atherosclerosis (80) (Fig. 3). AGEs accelerate the expression of VCAM-1 and ICAM-1, and facilitate monocyte migration to the subendothelial layer and the conversion into macrophages. AGE-modified LDL increases macrophage-foam cell transformation (186). AGEs can also reduce the expression of ABCA1 and ABCG1 in monocytes, thereby inhibiting RCT. In addition, AGEs promote vasoconstriction via upregulating endothelin-1, impede vasodilation by reducing NO production, and trigger modification of ECM, thereby accelerating the development of atherosclerosis (189).
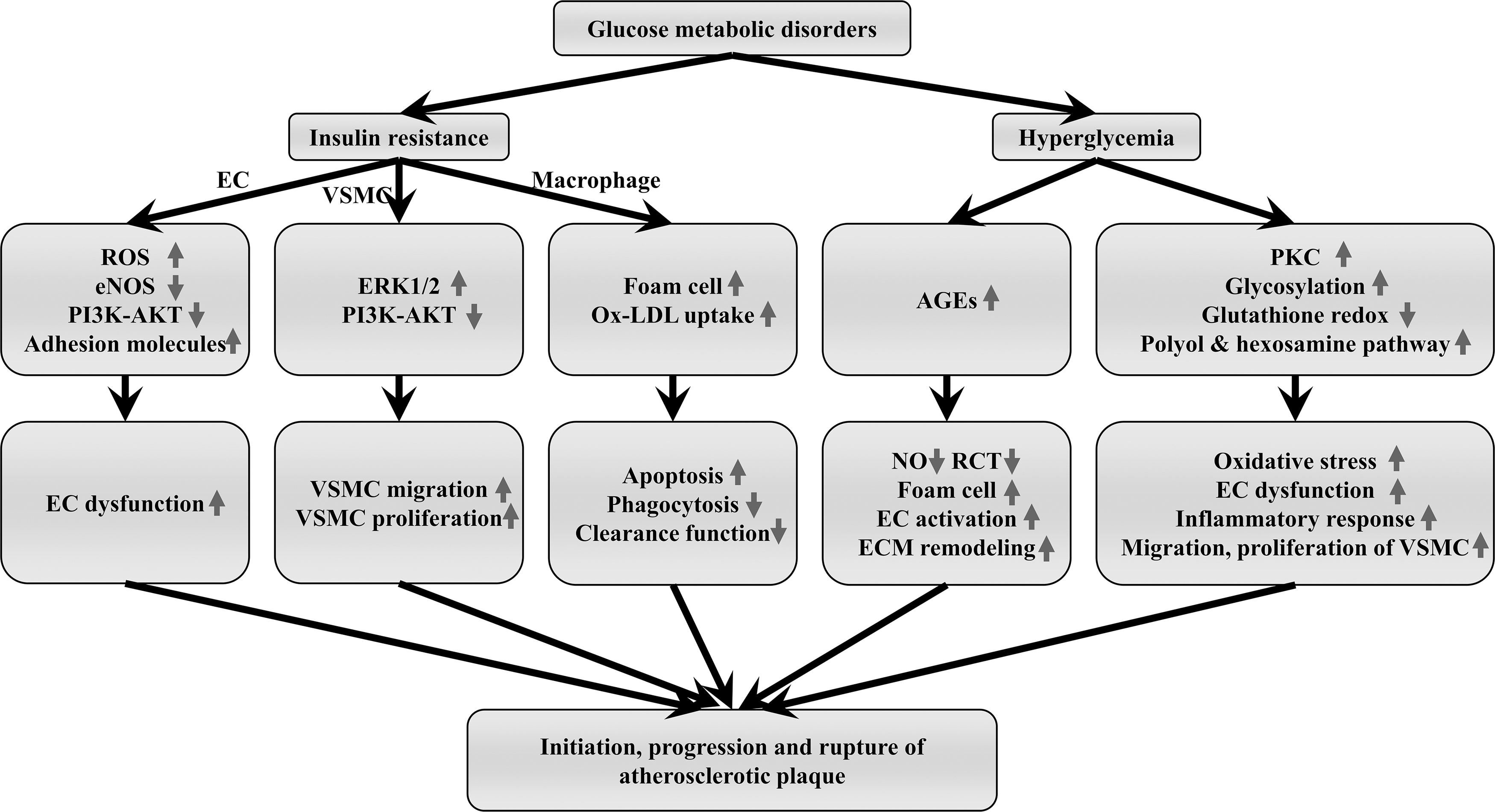
Hyperglycemia also promotes the progression of atherosclerotic plaques by overproduction of ROS from multiple metabolic pathways and processes, such as glucose auto-oxidation, enhanced glycosylation, AGE-RAGE axis activation, enhanced polyol and hexosamine pathways, impaired glutathione redox cycle, and PKC activation. (80). Excessive ROS participate in atherogenesis through the incorrect expression of redox-regulated genes, induction of inflammatory responses, aggravated endothelial dysfunction, migration, proliferation, and transformation of vascular smooth muscle cell (VSMC), and increased thrombotic tendency and plaque instability (46).
Insulin resistance refers to the phenomenon that insulin failed to function normally in insulin-sensitive target organs or tissues (e.g., adipose tissue, liver, skeletal muscle), resulting in a decrease in target tissue glucose uptake and utilization. Insulin resistance is attributed to defective insulin receptor (IR) signaling and it plays a critical role in the formation of atherosclerosis plaque and progression of advanced plaque by involving ECs, VSMCs, and macrophages (Fig. 3) (10, 12). Endothelial dysfunction is an initial driving factor for plaque formation. Endothelium-specific disruption of the insulin signaling through EC-specific knockout of IR or EC-specific overexpression of the mutant form of IR suppressed eNOS phosphorylation by PI3K-AKT, reduced NO production, and subsequently led to increased ROS production, inflammation, and eventually endothelial dysfunction and atherogenesis (34).
VSMCs are also involved in the disruption of insulin signaling and atherogenesis. VSMCs express both IR and insulin-like growth factor 1 receptor (IGF1R). IR deficiency reduced insulin activation and promoted VSMC proliferation and migration, which might be mediated through the IGF1R-ERK-1/2 signaling pathway. In the process of atherogenesis, enhancement of VSMC proliferation and migration may convert early plaques into irreversible plaques that contain more VSMCs (12, 44, 108).
Insulin resistance also contributes to the macrophage-related atherogenic process. Reardon et al. identified that “macrophage-sterol response network” disorders were induced by insulin resistance through a high-throughput proteomic study (146). Liang et al. further revealed that insulin-deficient macrophages increased uptake of ox-LDL and cholesterol esters, which deteriorated the formation of foam cells. On the contrary, insulin-resistant macrophages are prone to apoptosis when exposed to nutrition deprivation, free cholesterol load, and ox-LDL (104). Impairment of phagocytosis and clearance functions makes the lipid-rich core larger and enhances inflammation, eventually leading to plaque fragility and even rupture (105).
Amino acid metabolism and atherosclerosis
Branched-chain amino acids
Branched-chain amino acids (BCAAs) refer to three amino acids, leucine, isoleucine, and valine, with a branched structure in the side chain. In observational studies, patients with diabetes and CVD are associated with increased plasma levels of BCAAs, but this causal relationship remains undetermined (48, 191). Shah's team performed a mass spectrometry analysis of 63 metabolites in patients' plasma and found that an increase of BCAAs was a risk factor for coronary heart disease (9). More importantly, a cross-sectional study showed that leucine supplement was beneficial to lower vascular stiffness and CVD (74). In line with this, Grajeda-Iglesias and colleagues reported that leucine reduced VLDL uptake by macrophages and inhibited formation of foam cells (150). On the contrary, Zhenyukh et al. revealed that BCAAs increased the expression of E-selectin and ICAM-1 by increasing NF-κB activity, thereby promoting the recruitment of inflammatory cells; meanwhile, BCAAs could enhance local oxidative stress, causing endothelial dysfunction and promoting atherogenesis (205).
Arginine and homoarginine
Arginine is oxidized to produce NO by NOS. Clinical trials showed that exogenous arginine supplementation could benefit patients with NO deficiency and endothelial dysfunction in the short term (159), but this does not affect long-term vascular function and clinical cardiovascular outcomes (182). A registry study on acute coronary syndromes unveiled that oral arginine administration actually increased mortality in patients with myocardial infarction (157). The explanation for this phenomenon is that mere supplementation with arginine may deprive the eNOS cofactor of tetrahydrobiopterin, prompting uncoupling of eNOS, which intensifies the progression of the disease course.
Homoarginine is an amino acid similar to arginine in chemical structure, which can also serve as a substrate for NOS to generate NO. Endogenous homoarginine is synthesized from arginine and lysine with the action of L-arginine: glycine amidinotransferase. Existing evidence suggests that a lower homoarginine level is associated with poor prognosis of CVD (4). Administration of homoarginine to ApoE-knockout mice reduced the area of atherosclerotic plaques (133), and oral homoarginine treatment in patients dramatically increased the plasma level of homoarginine with good safety and tolerance (3). Exogenous supplementation of homoarginine may be a promising strategy for improving the prognosis of patients with CVD, although the in-depth mechanisms between homoarginine and CVD need to be further investigated.
Glutamine and glutamic acid
Glutamine and its intermediate metabolite glutamic acid are the most abundant amino acids in the human body. Previous studies identified a negative correlation between glutamine levels and plaque progression (18). Furthermore, supplement with glutamine has cardioprotective effects in patients with CVD and diabetes and can reduce CVD mortality (116). Qi et al. found that mutations of genes involved in glutamine metabolism were correlated with a growing incidence of coronary heart disease in diabetic patients (141). Metabolomic analysis confirmed that glutamic acid and glutamine are closely related to hyperlipidemia, diabetes, hypertension, obesity, and coronary heart disease, and they can serve as a biomarker for subclinical atherosclerotic plaques (56). Moreover, glutamine plays an antioxidant and anti-inflammatory role by upregulating glutathione, heme oxygenase-1, and heat shock protein (140). In addition, glutamine promotes M2 macrophage polarization, thereby ameliorating the progression of atherosclerotic plaques (75).
Aromatic amino acids
Aromatic amino acids consist of tryptophan, phenylalanine, and tyrosine. As an essential aromatic amino acid, tryptophan's metabolites play crucial roles in the development of atherosclerotic plaques. 3-hydroxyanthranilic acid (3-HAA), a metabolite derived from tryptophan, can mitigate plaques by modulating lipid metabolism and suppressing vascular inflammation in LDLR−/− mice (200). 3-HAA analog tranilast suppressed proinflammatory cytokines and retarded plaque progression in ApoE-deficient mice (25). In a large-scale randomized-controlled study, tranilast treatment decreased the recurrence of myocardial infarction in patients receiving PCI surgery, but it failed to reduce the in-stent restenosis, suggesting that tranilast works mainly by suppressing inflammation rather than inhibiting VSMC proliferation (66).
With regard to the effects of phenylalanine and tyrosine, a cohort study of 3587 patients with diabetes unveiled the link between phenylalanine, tyrosine, and vascular diseases (180). Phenylalanine content was positively correlated with the risk of macrovascular disease, while higher tyrosine levels were correlated with lower risks of microvascular disease (180). Interestingly, a recent study revealed that phenylacetylglutamine, a gut microbial metabolite of phenylalanine, could promote platelet function and thrombotic risk, thereby contributing to exacerbation of CVD. Notably, the gut microbial regulation of amino acids has aroused significant interest in connection to CVD. Other gut microbial metabolites from amino acids, such as trimethylamine N-oxide, are also involved in the pathogenic mechanism of CVD.
Homocysteine
Homocysteine refers to a sulfur-containing amino acid generated in the course of methionine and cysteine metabolism. Hyperhomocysteinemia can be induced by genetic disorders (C677T mutation of methylenetetrahydrofolate reductase, cystathionine β-synthase deficiencies), vitamin insufficiency (vitamin B12, vitamin B6, folate), drugs (nicotinic acid, metformin), and smoking (151). Clinical studies showed that moderately elevated homocysteine levels increased the risk of CVD (67). Other studies suggested that homocysteine mediates the inflammatory response of VSMCs through MAPK and NF-κB signaling pathways, exacerbating atherosclerosis (137). Moreover, homocysteine impairs endothelial function and subsequently promotes atherosclerotic plaque formation through oxidative stress, upregulation of NF-κB, and downregulation of eNOS (38). For patients with hyperhomocysteinemia, whether exogenous supplementation of folic acid and vitamins B6 and B12 can delay the progress of atherosclerotic plaques and reduce cardiovascular events is inconclusive, and further investigation is needed (124).
Cross Talk Between Shear Stress and Metabolic Drivers in Atherosclerosis
Cross talk between shear stress and lipid metabolism
There is a link between shear stress and lipid metabolism. In a pig model of atherosclerosis, hypercholesterolemia and endothelial shear stress are reported to be synergistic on rapid plaque progression (86). On the one hand, lipids can modulate the impact of shear stress. For example, cholesterol loading repressed the shear stress-induced expression of heme oxygenase 1 and NOS2, prostaglandin E2, and cyclooxygenase-2 in both in vivo and in vitro studies (114, 147). Hypercholesterolemia reduced NO production and vasodilatation in response to shear stress, thus promoting plaque formation in ApoE knockout mice (39, 91).On the other hand, shear stress impacts lipid metabolism. For instance, low shear stress disrupted cholesterol metabolism in ECs and upregulated the expression of LDL receptor (168). In ECs, DF and low shear stress activated sterol regulatory element-binding protein (SREBP); the latter is required to increase LDL uptake and fatty acid or cholesterol synthesis (16, 113). Lin et al. showed that the Rho-cofilin pathway mediated the DF-induced SREBP activation (109). The extended studies further revealed the role of SREBP in DF-induced inflammation. DF activated the NOD-like receptor family and pyrin domain containing 3 (NLRP3) inflammasome mediated by SREBP2, and promoted IL-1β production, thereby contributing to inflammation and atherogenesis (148, 185). In addition, miR-92a was involved in the SREBP2-inflammasome signaling pathway to exacerbate endothelial dysfunction and atherogenesis (20). Likewise, shear stress-activated SREBPs increased the expression of IL-8 that also involves the inflammatory process of atherosclerosis (192).
Lectin-like ox-LDL receptor-1 (LOX-1) has been widely described to promote atherogenesis. Shear stress regulates the LOX-1 expression through mediation of KLF2 and activator protein-1 (AP-1) (95). Liver X receptors (LXRs), the important regulators of lipid metabolism, were shown to be regulated by shear force. Laminar shear stress can activate LXRs in a PPAR-dependent manner, resulting in augmented cholesterol efflux and protected endothelial function (208). Under mechanical stimuli, anti-inflammatory transcription factor KLF4 activates LXRs and subsequently promotes cholesterol efflux and M1 to M2 transition in macrophages, and mitigates plague susceptibility (103). Proprotein convertase subtilisin/kexin type 9 (PCSK9) participates in both shear stress-induced mechanotransduction and lipid metabolism. Elevated PCSK9 expression and ROS overproduction occurred concomitantly in vascular regions that experience low shear stress (30).
In addition, the cross talk of shear stress and lipid metabolism also manifests in the barrier function of endothelium (16). DF increases endothelial permeability to facilitate uptake of LDL and migration of inflammatory monocytes into the subendothelial layer. Subsequently, the ox-LDL initiates the onset of atherosclerotic plaque formation. DF and low shear stress increase mitosis and apoptosis of ECs and impair the endothelial structural integrity (widening gaps and increased leaky spots) as observed in atheroprone regions (16, 58). DF prolongs the contact between circulating blood and ECs, and plaques with matrix remodeling also enhance the retention of LDL, both of which exacerbate the transportation of LDL and infiltration of monocytes to the vascular wall (60).
Cross talk between shear stress and glucose metabolism
Hyperglycemia modulates the effect of shear stress on vascular biology. Gresele et al. revealed that hyperglycemia promoted high shear stress-induced platelet activation and raised the risk of thrombosis in arteries with atherosclerotic stenosis (54). This result also indicated that glycemic control may be of significance for vascular stenotic lesions, particularly in-stent restenosis. Clinical studies showed that the antidiabetic drug, SGLT2 inhibitor, increased blood viscosity and wall shear stress, while reducing intima-media thickness in diabetic patients (71). Notably, KLF2 is a pivotal mechanosensitive regulator of shear stress and mediates the flow-dependent atheroprotective effect against endothelial dysfunction. High glucose exposure decreases the KLF2 expression in ECs, and an in vivo study further revealed that high glucose downregulates the KLF2 expression, thereby exacerbating endothelial injury in diabetic vascular complications (206). Besides, endothelial glycolysis is also involved in mechanical stress-induced production of NO via regulation of purinergic signaling and PKCδ (8).
Shear stress also regulates glucose metabolism. Recent studies highlight the critical bridge role of KLF2 in shear stress-induced impact on EC glucose metabolism. Uniform flow suppresses endothelial glycolysis and mitochondrial respiration via downregulation of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) in a KLF2-dependent manner to maintain vascular homeostasis and endothelial cellular metabolism (32). DF can reduce the KLF2 expression, thereby increasing glycolysis and angiogenesis (94). Consistently, ECs exposed to oscillatory shear stress exhibited elevated glycolysis, accompanied by increased inflammation (42, 184). Further evidence shows that shear stress modulates the biosynthesis of hyaluronan (the main structural component of glycocalyx in the endothelium) through KLF2-mediated endothelial glycolysis, while glycocalyx confers an antiatherosclerotic property by maintaining normal vascular permeability (173).
In vivo and in vitro studies demonstrated that higher shear stress on endothelium promoted insulin-induced vasodilatation in skeletal muscle (172). DF is associated with increased expression of RAGE. More significantly, oscillatory shear stress aggravates the inflammatory responses of ECs in an AGE-dependent manner (29). Notably, hyperglycemia increases subendothelial ECM remodeling at the early stage of plaque formation in diabetic ApoE-deficient mice (53). At this stage, excessive accumulation of matrix proteins such as fibronectin can induce the expression of inflammatory genes and lead to endothelial dysfunction. Arterial regions exposed to DF are prone to the buildup of matrix proteins under hyperglycemic condition. By contrast, unidirectional shear stress alleviates the hyperglycemia-induced deposition of fibronectin and fibrillogenesis in ECs (53). Hyperglycemia is shown to induce activation of NF-κB; the latter is further activated by low shear stress through suppression of eNOS expression and activity. On the contrary, high shear stress blunts the proatherogenic effect of hyperglycemia by suppression of NF-κB activity and ameliorating the development of atherosclerotic lesions (128). Taken together, flow patterns regulate the impact of hyperglycemia on atherogenesis.
Signaling Pathways in the Cross Talk Between Shear Stress and Metabolic Disorder
Inflammatory pathways
Biomechanical stress and metabolic disorder modulate inflammatory responses by regulation of several inflammatory pathways, such as the NF-κB, MAPK, and NLRP3/IL-1β pathways (Table 3).
Signaling Pathways in the Cross Talk Between Shear Stress and Metabolic Disorders
AKT, protein kinase B; AMPK, AMP-activated protein kinase; eNOS, endothelial nitric oxide synthase; FoxO1, forkhead box protein O1; HIF-1α, hypoxia-inducible factor-1α; IL, interleukin; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor -κB; NLRP3, NOD-like receptor family, pyrin domain containing 3; PI3K, phosphoinositide 3-kinases; TAZ, transcriptional coactivator with PDZ-binding motif; YAP, yes-associated protein.
The role of NF-κB signaling in inflammatory activation in response to low and oscillatory shear stress was elucidated before (13). NF-κB activation produces proinflammatory effects in ECs, macrophages, and VSMCs exposed to high glucose, ox-LDL, or free fatty acid (7, 179). In addition, a link is observed between NF-κB inflammatory pathway and enhanced glycolysis in ECs. As discussed above, ECs exposed to oscillatory shear stress exhibit increasing glycolysis and proinflammatory responses. Low shear stress drives endothelial inflammation through induction of glycolysis in an NF-κB/HIF-1α-dependent mechanism in the absence of hypoxia (42). Furthermore, proinflammatory cytokines enhance endothelial glucose metabolism, particularly glycolysis, which in turn aggravates cytokine-induced activation of NF-κB signaling (165). Overall, the interaction between NF-κB inflammatory pathway and glycolysis may contribute to maintaining the DF-induced proinflammatory process (165).
Similar to NF-κB, the MAPK (ERK1/2, p38, JNK) signaling pathway and the downstream AP-1 also play important roles in mediating the inflammatory programs in response to shear stress. Several mechanosensitive regulators, such as KLF2 and NRF2, involve extensive links between shear stress and MAPK pathway (13). On the contrary, ox-LDL, triglyceride, and high glucose also trigger the activation of MAPKs and increase the expression of inflammatory genes in ECs (179, 194).
NLRP3 inflammasome can be activated by cholesterol crystals and ox-LDL to increase the expression of IL-1β and exacerbate atherogenesis (77). Oscillatory shear stress represents an initial trigger for the activation of NLRP3 during atherogenesis (185). As discussed in the previous sections, SREBP2 exacerbates the initiation and progression of atherosclerosis. Intriguingly, DF-activated SREBP2 can induce the NLRP3 inflammasome via transactivation of NLRP3 in ECs (185).
Metabolic signaling pathways
PI3K-AKT pathway
Unidirectional blood flow stimulates PI3K-AKT pathway, inhibits apoptosis, and maintains the survival of ECs. Flow-induced activation of PI3K-AKT axis impacts the activity of eNOS. Mechanosensitive AKT can increase the phosphorylation of eNOS at Ser1177, giving rise to an increased formation of NO that confers an antiatherogenic effect (58). However, cessation of mechanical stress was reported to trigger PI3K/AKT activation and lead to NOX assembly and ROS generation, which might bring about harmful effects (15). On the contrary, PI3K-AKT pathway acts as a vital mediator of metabolic homeostasis, especially insulin-mediated glucose metabolism. In diabetes and obesity, insulin resistance results in downregulation of PI3K-AKT and suppression of eNOS phosphorylation, leading to EC dysfunction (198). Not only hyperglycemia and insulin resistance, but other metabolic factors can also regulate the activity of PI3K-AKT during the course of atherosclerosis. Free fatty acids are known to blunt insulin-induced NO production via inhibiting PI3K-AKT pathway (51).
HIF-1α pathway
HIF-1α is reported to be involved in intraplaque neovascularization and hemorrhage, one of the main features of rupture-prone atherosclerotic lesions. Intraplaque hypoxia-induced activation of HIF-1α signaling pathway gives rise to an increased secretion of VEGF that in turn increases intraplaque angiogenesis and endothelial permeability (165). Hypoxia is the established major stimulus for HIF-1α activity. Interestingly, accumulating evidence indicates that shear stress is also an important stimulator of HIF-1α. At atheroprone sites along the arterial tree, oscillatory and low shear stress elevates HIF-1α activity in the presence of sufficient oxygen supply. During this process, NF-κB contributes to an increase of HIF-1α mRNA, while Cezanne, a ubiquitin-editing enzyme, acts to stabilize HIF-1α protein. Accumulation and activation of excessive HIF-1α increase endothelial glycolysis and subsequently trigger EC proliferation and focal inflammation (42). Disturbed shear stress stabilizes HIF-1α by stimulating NOX4-derived ROS. HIF-1α also increases the expression of pyruvate dehydrogenase kinase-1 and glycolytic enzymes, and thereby promotes glycolysis and impairs mitochondrial respiratory capacity. This metabolic reprogram increases the expression of proinflammatory genes, leading to endothelial activation (184).
Forkhead box protein O1 pathway
Forkhead box protein O1 (FoxO1) is one of the predominant isoforms of FoxO family in vascular tissues, and it can act as a mediator of metabolic diseases (e.g., diabetes, CVD) by regulating cell cycle, proliferation, apoptosis, metabolism, oxidative stress resistance, and so on (98). FoxO1 functions as a nutrient sensor and participates in the modulation of energy homeostasis. Growth signals such as insulin and insulin-like growth factors inactivate FoxO1 via activation of PI3K-AKT and MAPK pathway (98). In ECs, FoxO1 can regulate the level of HMG-CoA reductase which serves as a crucial enzyme in the biosynthesis of cholesterol. FoxO1 also enhances vascular remodeling and leakage via upregulation of angiopoietin-2 (82). Biomechanical shear stress is reported to regulate phosphorylation and degradation of FoxO1 in ECs via an AMPK-dependent manner (31, 98). Decreased and inactivated FoxO1 can lead to downregulation of HMG-CoA and angiopoietin-2, thus protecting endothelial function (31, 47). Furthermore, recent evidence shows that FoxO1 serves as a link between expansion of blood vessels and EC metabolism, and maintains a quiescent phenotype of ECs by lowering glycolysis and mitochondrial respiration (101, 181).
AMPK pathway
Notably, AMPK is an essential metabolic regulator, and AMPK activation impacts lipid and glucose metabolism as well as protein synthesis in various organs. AMPK activation exerts atheroprotective effects via modulating lipid (mainly cholesterol) homeostasis, increasing NO production and decreasing vascular inflammation (Table 3) (194). Under hyperglycemia, AMPK phosphorylates SREBP-1c and inhibits cleavage and translocation of SREBP-1c, thereby suppressing lipogenesis and lipid accumulation in hepatocytes (102, 194). In vivo studies also demonstrate that activation of AMPK strengthens the antiatherosclerotic effect of HDL. In ApoE–/– mice, AMPK protects macrophages against cholesterol accumulation by increasing cholesterol efflux, which remarkably mitigates the progression of atherosclerosis (100, 115). Moreover, AMPK is a vital modulator of inflammatory activation in atherosclerosis. AMPK promotes the expression of anti-inflammatory cytokines in macrophages. The mechanism involves the inhibition of NF-κB mediated by sirtuin 1 (100). Another study suggested that monounsaturated fatty acid conferred anti-inflammatory effects through suppression of AMPK-NLRP3 axis in macrophages (45). As discussed in the section of FoxO1 pathway, shear stress-activated AMPK can regulate FoxO1 to exert an anti-inflammatory and antiatherosclerotic effect (47). As another response to shear stress, AMPK plays a crucial role in the regulation of vascular tone by elevating eNOS activity and the subsequent NO production (201). DF upregulates PRAK/AMPKs in ECs in vivo, which increases glycolysis and proliferation of ECs, and hence maintains the integrity of endothelial barrier and protects hyperlipidemic mice against progression of atherosclerotic lesions (190).
mTOR pathway
mTORC1 is involved in cell growth, synthesis of protein, lipid, and nucleotide, and autophagy, while mTORC2 is engaged in cell proliferation and survival (155). Notably, the mTOR pathway plays a role in atherogenesis. mTOR inhibitors such as rapamycin inhibit VSMC proliferation and rapamycin-eluting stents have been used for prevention of in-stent restenosis (87). mTORC1 increases the expression of HIF-1α that can accelerate intraplaque neovascularization and destabilizes plaques (125). In contrast to MAPK, mTOR signaling promotes lipid accumulation by inducing SREBP-dependent lipid synthesis (90). Moreover, mTOR activation exerts proinflammatory effects, boosts formation of foam cells, and blunts foam cell egression through NF-κB pathway and SIRT1-liver X receptor-CCR7 axis, exacerbating plaque formation (204). By contrast, inhibition of mTOR suppresses MCP-1-induced recruitment and migration of monocytes (125).
Similar to AMPK, mTOR is also regulated by shear stress. Cheng et al. showed that the mTOR inhibitor rapamycin protects ECs subjected to low shear stress by increasing eNOS expression (21). Li et al. further explained that within a short period, low shear stress activated mTORC1/eNOS, whereas it inhibited mTORC2/AKT, which triggered endothelial inflammation and increased ROS production (99). Inhibition of mTOR signaling and downstream sestrin1 mitigated oxidative stress (production of ROS and reactive nitrogen species) and low shear stress-induced EC apoptosis (199). By contrast, mTOR activation by low shear stress also impairs endothelial autophagy and EC alignment in response to shear stress, and results in endothelial inflammation and apoptosis (170). Besides, mTOR-S6K signaling pathway mediates the distinct effects of different flow patterns on cell cycle. DF exerts its proliferative effects via activation of mTOR-S6K signaling (58). In vitro and in intro evidence shows that mTOR mediates the proatherogenic effect of DF by increasing the level of DNMTs, which act on the promoter methylation of connective tissue growth factor and cyclin A (202).
Developmental signaling pathways
Notch pathway
Notch pathway impacts cell proliferation, differentiation, and apoptosis (169). Substantial evidence shows that the Notch pathway is a critical player in the pathophysiology of atherosclerosis depending on the type(s) of vascular cells affected. For example, Notch activation protects endothelial function against inflammation and sustains the contractile state of VSMCs. By contrast, Notch activation in macrophages augments inflammation by favoring the conversion to proinflammatory M1 phenotype (149, 169). Importantly, Notch signaling is required for the communication between shear stress and metabolic disorders. Notch signaling is an established protector of endothelial function against atherogenic stressors such as dyslipidemia and DF. In ECs, high shear stress-activated Notch1 stabilizes cellular junctions and maintains junctional integrity (101, 117). Notch signaling increases fatty acid oxidation in ECs, while increased fatty acid oxidation promotes regeneration of NADPH, which is consumed by vasoprotective enzymes for maintaining redox homeostasis and thus preventing endothelial dysfunction (79). Notch pathway upregulates the expression of CD36, endothelial lipase, and FABP4, which are essential for transportation of fatty acid across the endothelium. Inhibition of this signaling in ECs impairs fatty acid transport accompanied by disorders of both lipid and glucose metabolism, subsequently leading to angiogenesis and vascular remodeling (72). Notch1 activation enhances both oxidative phosphorylation and glycolysis by increasing mitochondrial mass and inducing PFKFB3 to maintain proliferative capacity. PFKFB3 increases generation of acetyl-CoA to mediate histone acetylation at enhancer sites of proliferative genes. This mechanism enables EC regeneration and helps maintain monolayer integrity and vascular homeostasis in response to injury (127).
Hippo pathway
Hippo pathway is another novel mediator between shear stress and metabolism in atherosclerosis. Hippo pathway is engaged in suppression of cell proliferation and promotion of apoptosis. This pathway has been recently demonstrated to play an essential role in atherogenesis, angiogenesis, and vascular remodeling. YAP and TAZ, the effectors of Hippo pathway, are regulated by shear stress in ECs (174, 187). A previous study unveiled that LF activated integrin and enhanced interaction between integrin and Gα13, resulting in suppression of RhoA. As a result, Yap was phosphorylated and retained in the cytosol for degradation; inhibition of YAP nuclear translocation suppressed the expression of inflammatory genes and reduced plaque formation in ApoE−/− mice. On the contrary, DF activated YAP/TAZ, augmented inflammation, and atherogenesis mediated by JNK activation (175). On the contrary, Hippo pathway is reported to attenuate insulin resistance and glucose intolerance via downregulation of peroxisome proliferator-activated receptor γ activity (37). Increasing evidence also indicates that YAP and TAZ are involved in cellular metabolism, such as glycolysis, amino acid metabolism, lipogenesis, and lipid oxidation, suggesting that YAP and TAZ act as a key mediator of vascular homeostasis via coordinating nutrients and energy (2). In response to energy stress, AMPK activation inhibits YAP/TAZ by direct phosphorylation. Unsaturated fatty acids have been proposed to inactive β-catenin and protect YAP and TAZ from degradation (85).
Conclusion and Clinical Implications
A thorough understating of how biomechanical forces and metabolic dysregulation impact atherosclerosis will promote the development of clinical prevention, early diagnosis, and therapeutic interventions (surgeries and drugs) on atherosclerosis.
As discussed in previous sections, dyslipidemia and hyperglycemia have been identified as risk factors of atherosclerosis and clinical guidelines recommend therapeutic interventions on these risk factors. As for biomechanical forces, current clinical examination techniques especially state-of-the-art imaging techniques (e.g., ultrasound, computed tomography, magnetic resonance imaging, intravascular ultrasound, computational modeling) can assay or calculate shear stress in the arteries of patients. Prospective clinical studies are needed to evaluate the accuracy and specificity of magnitudes of shear stress and flow patterns in detecting potential plaque initiation, progression, and rupture. Moreover, future clinical investigations are also required to ascertain whether integrating metabolic disorder with mechanical parameters can improve the precise predictive capacity of clinical examination techniques to identify patients at high risk of atherosclerosis or acute cardiovascular events.
The mechanisms of mechanical forces on vascular function and atherogenesis have also some implications on surgical interventions. Some strategies are mainly adopted to maintain or modify mechanical forces to prevent endothelial dysfunction and neointimal hyperplasia. For example, distal arteriovenous fistulas and flow dividers have been applied in surgeries (bypass graft, stenting, etc.) to increase shear stress and vascular patency.
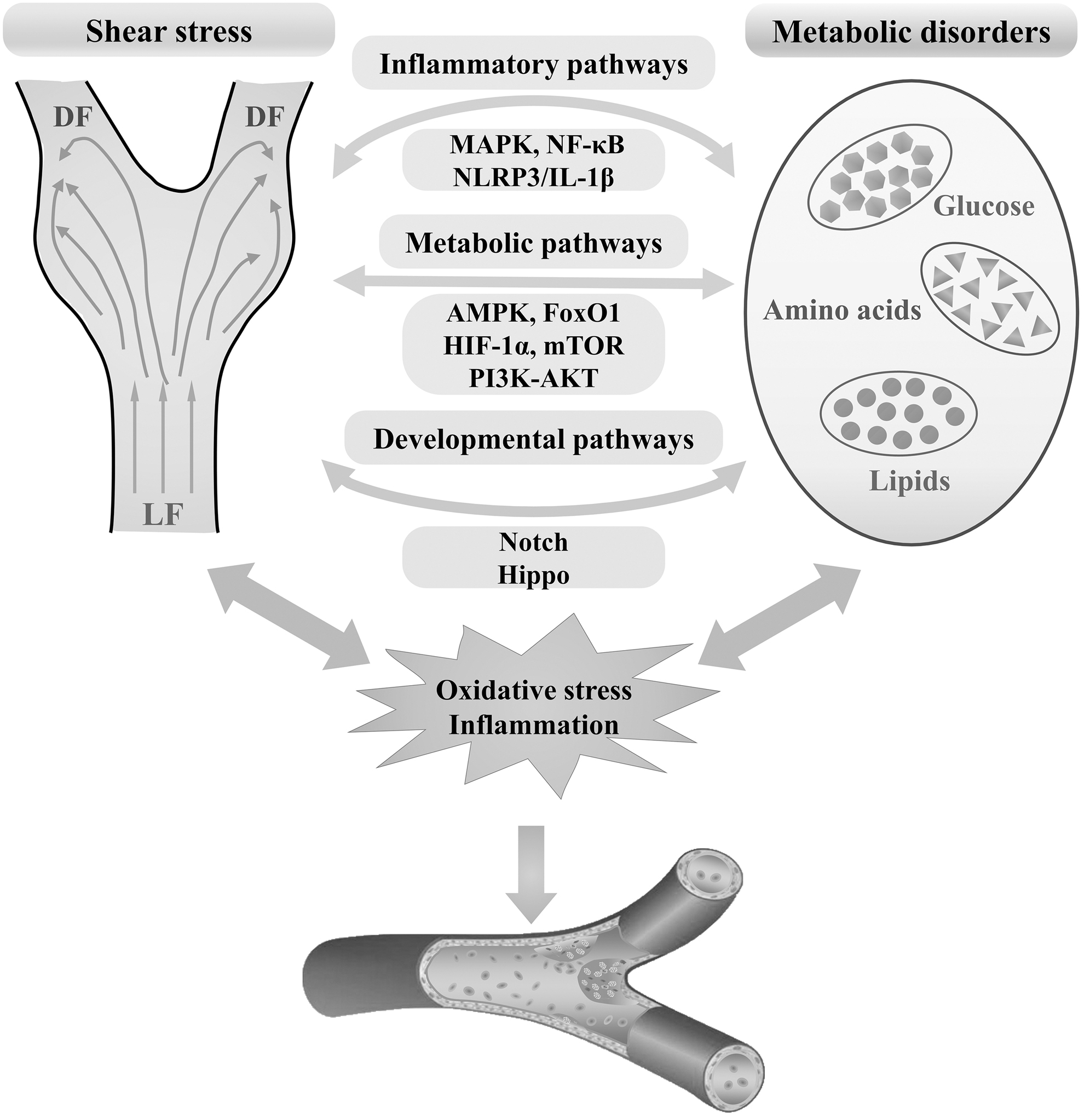
Shear stress (DF) and metabolic disorders are established drivers of atherosclerosis, and emerging evidence unveils extensive cross talk between these “culprits” during the pathophysiological process of this disease (Fig. 4). The communication may amplify the proatherogenic effect for each other. Identification of critical mediators of the cross talk may serve as promising therapeutic targets for the treatment of atherosclerotic vascular diseases by controlling both sides of the plaque. Undoubtedly, elucidation of the complicated interactions and cross talk requiring further in-depth investigation with advanced multiple omics approaches shall provide new insights into developing more effective therapeutic strategies against atherosclerosis.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported by Research Grants Council Collaborative Research Fund (C4024-16W), Research Grants Council General Research Fund (14112919), Health and Medical Research Fund (05161746), National Natural Science Foundation of China (91939302, 81561128017), Research Grants Council Senior Research Fellow Scheme (2021-4S04) and Hong Kong PhD Fellowship.